Label: READYSHARP ANESTHETICS PLUS KETOROLAC- lidocaine hydrochloride, bupivacaine hydrochloride, and ketorolac tromethamine kit



-
Contains inactivated NDC Code(s)
NDC Code(s): 0409-1560-10, 0409-3793-19, 0409-4713-42, 53225-4000-1 - Packager: Terrain Pharmaceuticals
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 19, 2017
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Lidocaine Hydrochloride Injection, USP is a sterile, nonpyrogenic solution of lidocaine hydrochloride in water for injection for parenteral administration in various concentrations with characteristics as follows:
Concentration
0.5%
1%
1.5%
2%
mg/mL lidocaine HCl (anhyd.)
5
10
15
20
mg/mL sodium chloride
8
7
6.5
6
Multiple-dose vials contain 0.1% of methylparaben added as preservative. May contain sodium hydroxide and/or hydrochloric acid for pH adjustment. The pH is 6.5 (5.0 to 7.0). See HOW SUPPLIED section for various sizes and strengths.
Lidocaine is a local anesthetic of the amide type.
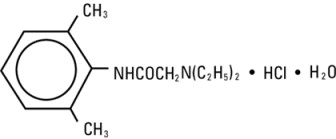
Lidocaine Hydrochloride, USP is chemically designated 2-(diethylamino)-N-(2,6-dimethylphenyl)-acetamide monohydrochloride monohydrate, a white powder freely soluble in water. The molecular weight is 288.82. It has the following structural formula:
The semi-rigid vial used for the plastic vials is fabricated from a specially formulated polyolefin. It is a copolymer of ethylene and propylene. The safety of the plastic has been confirmed by tests in animals according to USP biological standards for plastic containers. The container requires no vapor barrier to maintain the proper drug concentration.
-
CLINICAL PHARMACOLOGY
Mechanism of action: Lidocaine stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses, thereby effecting local anesthetic action.
Hemodynamics: Excessive blood levels may cause changes in cardiac output, total peripheral resistance, and mean arterial pressure. With central neural blockade these changes may be attributable to block of autonomic fibers, a direct depressant effect of the local anesthetic agent on various components of the cardiovascular system and/or the beta-adrenergic receptor stimulating action of epinephrine when present. The net effect is normally a modest hypotension when the recommended dosages are not exceeded.
Pharmacokinetics and metabolism: Information derived from diverse formulations, concentrations and usages reveals that lidocaine is completely absorbed following parenteral administration, its rate of absorption depending, for example, upon various factors such as the site of administration and the presence or absence of a vasoconstrictor agent. Except for intravascular administration, the highest blood levels are obtained following intercostal nerve block and the lowest after subcutaneous administration.
The plasma binding of lidocaine is dependent on drug concentration, and the fraction bound decreases with increasing concentration. At concentrations of 1 to 4 mcg of free base per mL, 60 to 80 percent of lidocaine is protein bound. Binding is also dependent on the plasma concentration of the alpha-1-acid glycoprotein.
Lidocaine crosses the blood-brain and placental barriers, presumably by passive diffusion.
Lidocaine is metabolized rapidly by the liver, and metabolites and unchanged drug are excreted by the kidneys. Biotransformation includes oxidative N-dealkylation, ring hydroxylation, cleavage of the amide linkage, and conjugation. N-dealkylation, a major pathway of biotransformation, yields the metabolites monoethylglycinexylidide and glycinexylidide. The pharmacological/toxicological actions of these metabolites are similar to, but less potent than, those of lidocaine. Approximately 90% of lidocaine administered is excreted in the form of various metabolites, and less than 10% is excreted unchanged. The primary metabolite in urine is a conjugate of 4-hydroxy-2, 6-dimethylaniline.
The elimination half-life of lidocaine following an intravenous bolus injection is typically 1.5 to 2.0 hours. Because of the rapid rate at which lidocaine is metabolized, any condition that affects liver function may alter lidocaine kinetics. The half-life may be prolonged two-fold or more in patients with liver dysfunction. Renal dysfunction does not affect lidocaine kinetics but may increase the accumulation of metabolites.
Factors such as acidosis and the use of CNS stimulants and depressants affect the CNS levels of lidocaine required to produce overt systemic effects. Objective adverse manifestations become increasingly apparent with increasing venous plasma levels above 6.0 mcg free base per mL. In the rhesus monkey arterial blood levels of 18-21 mcg/mL have been shown to be threshold for convulsive activity.
-
INDICATIONS AND USAGE
Lidocaine Hydrochloride Injection, USP is indicated for production of local or regional anesthesia by infiltration techniques such as percutaneous injection and intravenous regional anesthesia by peripheral nerve block techniques such as brachial plexus and intercostal and by central neural techniques such as lumbar and caudal epidural blocks, when the accepted procedures for these techniques as described in standard textbooks are observed.
- CONTRAINDICATIONS
-
WARNINGS
LIDOCAINE HYDROCHLORIDE INJECTION, FOR INFILTRATION AND NERVE BLOCK, SHOULD BE EMPLOYED ONLY BY CLINICIANS WHO ARE WELL VERSED IN DIAGNOSIS AND MANAGEMENT OF DOSE-RELATED TOXICITY AND OTHER ACUTE EMERGENCIES THAT MIGHT ARISE FROM THE BLOCK TO BE EMPLOYED AND THEN ONLY AFTER ENSURING THE IMMEDIATE AVAILABILITY OF OXYGEN, OTHER RESUSCITATIVE DRUGS, CARDIOPULMONARY EQUIPMENT, AND THE PERSONNEL NEEDED FOR PROPER MANAGEMENT OF TOXIC REACTIONS AND RELATED EMERGENCIES (See also ADVERSE REACTIONS and PRECAUTIONS). DELAY IN PROPER MANAGEMENT OF DOSE-RELATED TOXICITY, UNDERVENTILATION FROM ANY CAUSE AND/OR ALTERED SENSITIVITY MAY LEAD TO THE DEVELOPMENT OF ACIDOSIS, CARDIAC ARREST AND, POSSIBLY, DEATH.
Intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures is an unapproved use, and there have been post-marketing reports of chondrolysis in patients receiving such infusions. The majority of reported cases of chondrolysis have involved the shoulder joint; cases of gleno-humeral chondrolysis have been described in pediatric and adult patients following intra-articular infusions of local anesthetics with and without epinephrine for periods of 48 to 72 hours. There is insufficient information to determine whether shorter infusion periods are not associated with these findings. The time of onset of symptoms, such as joint pain, stiffness and loss of motion can be variable, but may begin as early as the 2nd month after surgery. Currently, there is no effective treatment for chondrolysis; patients who experienced chondrolysis have required additional diagnostic and therapeutic procedures and some required arthroplasty or shoulder replacement.
To avoid intravascular injection, aspiration should be performed before the local anesthetic solution is injected. The needle must be repositioned until no return of blood can be elicited by aspiration. Note, however, that the absence of blood in the syringe does not guarantee that intravascular injection has been avoided.
Local anesthetic solutions containing antimicrobial preservatives (e.g., methylparaben) should not be used for epidural or spinal anesthesia because the safety of these agents has not been established with regard to intrathecal injection, either intentional or accidental.
-
PRECAUTIONS
General:
The safety and effectiveness of lidocaine depend on proper dosage, correct technique, adequate precautions, and readiness for emergencies. Standard textbooks should be consulted for specific techniques and precautions for various regional anesthetic procedures.
Resuscitative equipment, oxygen, and other resuscitative drugs should be available for immediate use. (See WARNINGS and ADVERSE REACTIONS). The lowest dosage that results in effective anesthesia should be used to avoid high plasma levels and serious adverse effects. Syringe aspirations should also be performed before and during each supplemental injection when using indwelling catheter techniques. During the administration of epidural anesthesia, it is recommended that a test dose be administered initially and that the patient be monitored for central nervous system toxicity and cardiovascular toxicity, as well as for signs of unintended intrathecal administration before proceeding. When clinical conditions permit, consideration should be given to employing local anesthetic solutions that contain epinephrine for the test dose because circulatory changes compatible with epinephrine may also serve as a warning sign of unintended intravascular injection. An intravascular injection is still possible even if aspirations for blood are negative. Repeated doses of lidocaine may cause significant increases in blood levels with each repeated dose because of slow accumulation of the drug or its metabolites. Tolerance to elevated blood levels varies with the status of the patient. Debilitated, elderly patients, acutely ill patients and children should be given reduced doses commensurate with their age and physical condition. Lidocaine should also be used with caution in patients with severe shock or heart block. Lumbar and caudal epidural anesthesia should be used with extreme caution in persons with the following conditions: existing neurological disease, spinal deformities, septicemia and severe hypertension.
Local anesthetic solutions containing a vasoconstrictor should be used cautiously and in carefully circumscribed quantities in areas of the body supplied by end arteries or having otherwise compromised blood supply. Patients with peripheral vascular disease and those with hypertensive vascular disease may exhibit exaggerated vasoconstrictor response. Ischemic injury or necrosis may result. Preparations containing a vasoconstrictor should be used with caution in patients during or following the administration of potent general anesthetic agents, since cardiac arrhythmias may occur under such conditions.
Careful and constant monitoring of cardiovascular and respiratory (adequacy of ventilation) vital signs and the patient’s state of consciousness should be accomplished after each local anesthetic injection. It should be kept in mind at such times that restlessness, anxiety, tinnitus, dizziness, blurred vision, tremors, depression or drowsiness may be early warning signs of central nervous system toxicity.
Since amide-type local anesthetics are metabolized by the liver, lidocaine should be used with caution in patients with hepatic disease. Patients with severe hepatic disease, because of their inability to metabolize local anesthetics normally, are at greater risk of developing toxic plasma concentrations. Lidocaine should also be used with caution in patients with impaired cardiovascular function since they may be less able to compensate for functional changes associated with the prolongation of A-V conduction produced by these drugs. Many drugs used during the conduct of anesthesia are considered potential triggering agents for familial malignant hyperthermia. Since it is not known whether amide-type local anesthetics may trigger this reaction and since the need for supplemental general anesthesia cannot be predicted in advance, it is suggested that a standard protocol for the management of malignant hyperthermia should be available. Early unexplained signs of tachycardia, tachypnea, labile blood pressure and metabolic acidosis may precede temperature elevation. Successful outcome is dependent on early diagnosis, prompt discontinuance of the suspect triggering agent(s) and institution of treatment, including oxygen therapy, indicated supportive measures and dantrolene (consult dantrolene sodium intravenous package insert before using).
Proper tourniquet technique, as described in publications and standard textbooks, is essential in the performance of intravenous regional anesthesia. Solutions containing epinephrine or other vasoconstrictors should not be used for this technique.
Lidocaine should be used with caution in persons with known drug sensitivities. Patients allergic to para-aminobenzoic acid derivatives (procaine, tetracaine, benzocaine, etc.) have not shown cross sensitivity to lidocaine.
Use in the Head and Neck Area: Small doses of local anesthetics injected into the head and neck area, including retrobulbar, dental and stellate ganglion blocks, may produce adverse reactions similar to systemic toxicity seen with unintentional intravascular injections of larger doses. Confusion, convulsions, respiratory depression and/or respiratory arrest and cardiovascular stimulation or depression have been reported. These reactions may be due to intra-arterial injections of the local anesthetic with retrograde flow to the cerebral circulation. Patients receiving these blocks should have their circulation and respiration monitored and be constantly observed. Resuscitative equipment and personnel for treating adverse reactions should be immediately available. Dosage recommendations should not be exceeded. (See DOSAGE AND ADMINISTRATION).
Information for Patients:
When appropriate, patients should be informed in advance that they may experience temporary loss of sensation and motor activity, usually in the lower half of the body following proper administration of epidural anesthesia.
Clinically Significant Drug Interactions:
The administration of local anesthetic solutions containing epinephrine or norepinephrine to patients receiving monoamine oxidase inhibitors or tricyclic antidepressants may produce severe prolonged hypertension.
Phenothiazines and butyrophenones may reduce or reverse the pressor effect of epinephrine.
Concurrent use of these agents should generally be avoided. In situations when concurrent therapy is necessary, careful patient monitoring is essential.
Concurrent administration of vasopressor drugs (for the treatment of hypotension related to obstetric blocks) and ergot-type oxytoxic drugs may cause severe persistent hypertension or cerebrovascular accidents.
Drug Laboratory Test Interactions:
The intramuscular injection of lidocaine may result in an increase in creatine phosphokinase levels. Thus, the use of this enzyme determination without isoenzyme separation as a diagnostic test for the presence of acute myocardial infarction may be compromised by the intramuscular injection of lidocaine.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Studies of lidocaine in animals to evaluate the carcinogenic and mutagenic potential or the effect on fertility have not been conducted.
Pregnancy:
Teratogenic Effects. Pregnancy Category B. Reproduction studies have been performed in rats at doses up to 6.6 times the human dose and have revealed no evidence of harm to the fetus caused by lidocaine. There are, however, no adequate and well-controlled studies in pregnant women. Animal reproduction studies are not always predictive of human response. General consideration should be given to this fact before administering lidocaine to women of childbearing potential, especially during early pregnancy when maximum organogenesis takes place.
Labor and Delivery:
Local anesthetics rapidly cross the placenta and when used for epidural, paracervical, pudendal or caudal block anesthesia, can cause varying degrees of maternal, fetal and neonatal toxicity (See CLINICAL PHARMACOLOGY— Pharmacokinetics). The potential for toxicity depends upon the procedure performed, the type and amount of drug used, and the technique of drug administration. Adverse reactions in the parturient, fetus and neonate involve alterations of the central nervous system peripheral vascular tone and cardiac function.
Maternal hypotension has resulted from regional anesthesia. Local anesthetics produce vasodilation by blocking sympathetic nerves. Elevating the patient’s legs and positioning her on her left side will help prevent decreases in blood pressure. The fetal heart rate also should be monitored continuously, and electronic fetal monitoring is highly advisable.
Epidural, spinal, paracervical, or pudendal anesthesia may alter the forces of parturition through changes in uterine contractility or maternal expulsive efforts. In one study, paracervical block anesthesia was associated with a decrease in the mean duration of first stage labor and facilitation of cervical dilation. However, spinal and epidural anesthesia have also been reported to prolong the second stage of labor by removing the parturient’s reflex urge to bear down or by interfering with motor function. The use of obstetrical anesthesia may increase the need for forceps assistance.
The use of some local anesthetic drug products during labor and delivery may be followed by diminished muscle strength and tone for the first day or two of life. The long-term significance of these observations is unknown. Fetal bradycardia may occur in 20 to 30 percent of patients receiving paracervical nerve block anesthesia with the amide-type local anesthetics and may be associated with fetal acidosis. Fetal heart rate should always be monitored during paracervical anesthesia. The physician should weigh the possible advantages against risks when considering paracervical block in prematurity, toxemia of pregnancy and fetal distress. Careful adherence to recommended dosage is of the utmost importance in obstetrical paracervical block. Failure to achieve adequate analgesia with recommended doses should arouse suspicion of intravascular or fetal intracranial injection. Cases compatible with unintended fetal intracranial injection of local anesthetic solution have been reported following intended paracervical or pudendal block or both. Babies so affected present with unexplained neonatal depression at birth, which correlates with high local anesthetic serum levels, and often manifest seizures within six hours. Prompt use of supportive measures combined with forced urinary excretion of the local anesthetic has been used successfully to manage this complication.
Case reports of maternal convulsions and cardiovascular collapse following use of some local anesthetics for paracervical block in early pregnancy (as anesthesia for elective abortion) suggest that systemic absorption under these circumstances may be rapid. The recommended maximum dose of each drug should not be exceeded. Injection should be made slowly and with frequent aspiration. Allow a 5-minute interval between sides.
-
ADVERSE REACTIONS
Systemic: Adverse experiences following the administration of lidocaine are similar in nature to those observed with other amide local anesthetic agents. These adverse experiences are, in general, dose-related and may result from high plasma levels caused by excessive dosage, rapid absorption or inadvertent intravascular injection, or may result from a hypersensitivity, idiosyncrasy or diminished tolerance on the part of the patient. Serious adverse experiences are generally systemic in nature. The following types are those most commonly reported:
Central Nervous System: CNS manifestations are excitatory and/or depressant and may be characterized by lightheadedness, nervousness, apprehension, euphoria, confusion, dizziness, drowsiness, tinnitus, blurred or double vision, vomiting, sensations of heat, cold or numbness, twitching, tremors, convulsions, unconsciousness, respiratory depression and arrest. The excitatory manifestations may be very brief or may not occur at all, in which case the first manifestation of toxicity may be drowsiness merging into unconsciousness and respiratory arrest.
Drowsiness following the administration of lidocaine is usually an early sign of a high blood level of the drug and may occur as a consequence of rapid absorption.
Cardiovascular System: Cardiovascular manifestations are usually depressant and are characterized by bradycardia, hypotension, and cardiovascular collapse, which may lead to cardiac arrest.
Allergic: Allergic reactions are characterized by cutaneous lesions, urticaria, edema or anaphylactoid reactions. Allergic reactions may occur as a result of sensitivity either to local anesthetic agents or to the methylparaben used as a preservative in multiple dose vials. Allergic reactions as a result of sensitivity to lidocaine are extremely rare and, if they occur, should be managed by conventional means. The detection of sensitivity by skin testing is of doubtful value.
Neurologic: The incidences of adverse reactions associated with the use of local anesthetics may be related to the total dose of local anesthetic administered and are also dependent upon the particular drug used, the route of administration and the physical status of the patient. In a prospective review of 10,440 patients who received lidocaine for spinal anesthesia, the incidences of adverse reactions were reported to be about 3 percent each for positional headaches, hypotension and backache; 2 percent for shivering; and less than 1 percent each for peripheral nerve symptoms, nausea, respiratory inadequacy and double vision. Many of these observations may be related to local anesthetic techniques, with or without a contribution from the local anesthetic.
In the practice of caudal or lumbar epidural block, occasional unintentional penetration of the subarachnoid space by the catheter may occur. Subsequent adverse effects may depend partially on the amount of drug administered subdurally.
These may include spinal block of varying magnitude (including total spinal block), hypotension secondary to spinal block, loss of bladder and bowel control, and loss of perineal sensation and sexual function. Persistent motor, sensory and/or autonomic (sphincter control) deficit of some lower spinal segments with slow recovery (several months) or incomplete recovery have been reported in rare instances when caudal or lumbar epidural block has been attempted. Backache and headache have also been noted following use of these anesthetic procedures.
There have been reported cases of permanent injury to extraocular muscles requiring surgical repair following retrobulbar administration.
-
OVERDOSAGE
Acute emergencies from local anesthetics are generally related to high plasma levels encountered during therapeutic use of local anesthetics or to unintended subarachnoid injection of local anesthetic solution (see ADVERSE REACTIONS, WARNINGS and PRECAUTIONS).
Management of Local Anesthetic Emergencies: The first consideration is prevention, best accomplished by careful monitoring of cardiovascular and respiratory vital signs and the patient’s state of consciousness after each local anesthetic injection. At the first sign of change, oxygen should be administered.
The first step in the management of convulsions, as well as underventilation or apnea due to unintended subarachnoid injection of drug solution, consists of immediate attention to the maintenance of a patent airway and assisted or controlled ventilation with oxygen and a delivery system capable of permitting immediate positive airway pressure by mask. Immediately after the institution of these ventilatory measures, the adequacy of the circulation should be evaluated, keeping in mind that drugs used to treat convulsions sometimes depress the circulation when administered intravenously. Should convulsions persist despite adequate respiratory support, and if the status of the circulation permits, small increments of an ultra-short acting barbiturate (such as thiopental or thiamylal) or a benzodiazepine (such as diazepam) may be administered intravenously. The clinician should be familiar, prior to use of local anesthetics, with these anticonvulsant drugs. Supportive treatment of circulatory depression may require administration of intravenous fluids and, when appropriate, a vasopressor as directed by the clinical situation (e.g., ephedrine).
If not treated immediately, both convulsions and cardiovascular depression can result in hypoxia, acidosis, bradycardia, arrhythmias and cardiac arrest. Underventilation or apnea due to unintentional subarachnoid injection of local anesthetic solution may produce these same signs and also lead to cardiac arrest if ventilatory support is not instituted. If cardiac arrest should occur standard cardiopulmonary resuscitative measures should be instituted.
Endotracheal intubation, employing drugs and techniques familiar to the clinician, may be indicated, after initial administration of oxygen by mask, if difficulty is encountered in the maintenance of a patent airway or if prolonged ventilatory support (assisted or controlled) is indicated.
Dialysis is of negligible value in the treatment of acute overdosage with lidocaine.
The oral LD 50 of lidocaine HCl in non-fasted female rats is 459 (346−773) mg/kg (as the salt) and 214 (159−324) mg/kg (as the salt) in fasted female rats.
-
DOSAGE AND ADMINISTRATION
Table 1 (Recommended Dosages) summarizes the recommended volumes and concentrations of Lidocaine Hydrochloride Injection, USP for various types of anesthetic procedures. The dosages suggested in this table are for normal healthy adults and refer to the use of epinephrine-free solutions. When larger volumes are required only solutions containing epinephrine should be used, except in those cases where vasopressor drugs may be contraindicated.
There have been adverse event reports of chondrolysis in patients receiving intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures. Lidocaine is not approved for this use (see WARNINGS and DOSAGE AND ADMINISTRATION).
These recommended doses serve only as a guide to the amount of anesthetic required for most routine procedures. The actual volumes and concentrations to be used depend on a number of factors such as type and extent of surgical procedure, depth of anesthesia and degree of muscular relaxation required, duration of anesthesia required, and the physical condition of the patient. In all cases the lowest concentration and smallest dose that will produce the desired result should be given. Dosages should be reduced for children and for elderly and debilitated patients and patients with cardiac and/or liver disease.
The onset of anesthesia, the duration of anesthesia and the degree of muscular relaxation are proportional to the volume and concentration (i.e., total dose) of local anesthetic used. Thus, an increase in volume and concentration of Lidocaine Hydrochloride Injection will decrease the onset of anesthesia, prolong the duration of anesthesia, provide a greater degree of muscular relaxation and increase the segmental spread of anesthesia. However, increasing the volume and concentration of Lidocaine Hydrochloride Injection may result in a more profound fall in blood pressure when used in epidural anesthesia. Although the incidence of side effects with lidocaine is quite low, caution should be exercised when employing large volumes and concentrations, since the incidence of side effects is directly proportional to the total dose of local anesthetic agent injected.
For intravenous regional anesthesia, only the 50 mL single-dose vial containing 0.5% Lidocaine Hydrochloride Injection, USP should be used.
Epidural Anesthesia
For epidural anesthesia, only the following available specific products of Lidocaine Hydrochloride Injection by Hospira are recommended:
1%. . . . . . . . . . . . . . . . . . . . 30 mL single-dose teartop vials
1.5%. . . . . . . . . . . . . . . . . . . . . . . 20 mL single-dose ampuls
2%. . . . . . . . . . . . . . . . . . . . . . . . . 10 mL single-dose ampuls
Although these solutions are intended specifically for epidural anesthesia, they may also be used for infiltration and peripheral nerve block provided they are employed as single dose units. These solutions contain no bacteriostatic agent. In epidural anesthesia, the dosage varies with the number of dermatomes to be anesthetized (generally 2−3 mL of the indicated concentration per dermatome).
Caudal and Lumbar Epidural Block: As a precaution against the adverse experiences sometimes observed following unintentional penetration of the subarachnoid space, a test dose such as 2−3 mL of 1.5% lidocaine hydrochloride should be administered at least 5 minutes prior to injecting the total volume required for a lumbar or caudal epidural block. The test dose should be repeated if the patient is moved in a manner that may have displaced the catheter. Epinephrine, if contained in the test dose (10−15 mcg have been suggested), may serve as a warning of unintentional intravascular injection. If injected into a blood vessel, this amount of epinephrine is likely to produce a transient "epinephrine response" within 45 seconds, consisting of an increase in heart rate and systolic blood pressure, circumoral pallor, palpitations and nervousness in the unsedated patient. The sedated patient may exhibit only a pulse rate increase of 20 or more beats per minute for 15 or more seconds. Patients on beta-blockers may not manifest changes in heart rate, but blood pressure monitoring can detect an evanescent rise in systolic blood pressure. Adequate time should be allowed for onset of anesthesia after administration of each test dose. The rapid injection of a large volume of Lidocaine Hydrochloride Injection through the catheter should be avoided, and, when feasible, fractional doses should be administered.
In the event of the known injection of a large volume of local anesthetic solutions into the subarachnoid space, after suitable resuscitation and if the catheter is in place, consider attempting the recovery of drug by draining a moderate amount of cerebrospinal fluid (such as 10 mL) through the epidural catheter.
Maximum Recommended Dosages
NOTE: The products accompanying this insert do not contain epinephrine.
Adults: For normal healthy adults, the individual maximum recommended dose of lidocaine HCl with epinephrine should not exceed 7 mg/kg (3.5 mg/lb) of body weight and in general it is recommended that the maximum total dose not exceed 500 mg. When used without epinephrine, the maximum individual dose should not exceed 4.5 mg/kg (2 mg/lb) of body weight and in general it is recommended that the maximum total dose does not exceed 300 mg. For continuous epidural or caudal anesthesia, the maximum recommended dosage should not be administered at intervals of less than 90 minutes. When continuous lumbar or caudal epidural anesthesia is used for non-obstetrical procedures, more drug may be administered if required to produce adequate anesthesia.
The maximum recommended dose per 90 minute period of lidocaine hydrochloride for paracervical block in obstetrical patients and non-obstetrical patients is 200 mg total. One-half of the total dose is usually administered to each side. Inject slowly five minutes between sides. (See also discussion of paracervical block in PRECAUTIONS).
For intravenous regional anesthesia, the dose administered should not exceed 4 mg/kg in adults.
Children: It is difficult to recommend a maximum dose of any drug for children, since this varies as a function of age and weight. For children over 3 years of age who have a normal lean body mass and normal body development, the maximum dose is determined by the child’s age and weight. For example, in a child of 5 years weighing 50 lbs., the dose of lidocaine HCl should not exceed 75 — 100 mg (1.5 — 2 mg/lb). The use of even more dilute solutions (i.e., 0.25 — 0.5%) and total dosages not to exceed 3 mg/kg (1.4 mg/lb) are recommended for induction of intravenous regional anesthesia in children.
In order to guard against systemic toxicity, the lowest effective concentration and lowest effective dose should be used at all times. In some cases it will be necessary to dilute available concentrations with 0.9% sodium chloride injection in order to obtain the required final concentration.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. Solutions that are discolored and/or contain particulate matter should not be used.
Table 1
Recommended Dosages of Lidocaine Hydrochloride Injection, USP for Various Anesthetic
Procedures in Normal Healthy Adults
Lidocaine Hydrochloride Injection, USP (without Epinephrine)
Procedure
Conc. (%)
Vol. (mL)
Total Dose (mg)
Infiltration
Percutaneous
0.5 or 1.0
1−60
5−300
Intravenous Regional
0.5
10−60
50−300
Peripheral Nerve Blocks, e.g.
Brachial
1.5
15−20
225−300
Dental
2.0
1−5
20−100
Intercostal
1.0
3
30
Paravertebral
1.0
3−5
30−50
Pudendal (each side)
1.0
10
100
Paracervical
Obstetrical Analgesia
(each side)
1.0
10
100
Sympathetic Nerve Blocks, e.g.
Cervical (stellate ganglion)
1.0
5
50
Lumbar
1.0
5−10
50−100
Central Neural Blocks
Epidural*
Thoracic
1.0
20−30
200−300
Lumbar
Analgesia
1.0
25−30
250−300
Anesthesia
1.5
15−20
225−300
2.0
10−15
200−300
Caudal
Obstetrical Analgesia
1.0
20−30
200−300
Surgical Anesthesia
1.5
15−20
225−300
*Dose determined by number of dermatomes to be anesthetized (2 to 3 mL/dermatome).
THE ABOVE SUGGESTED CONCENTRATIONS AND VOLUMES SERVE ONLY AS A GUIDE. OTHER VOLUMES AND CONCENTRATIONS MAY BE USED PROVIDED THE TOTAL MAXIMUM RECOMMENDED DOSE IS NOT EXCEEDED.
Sterilization, Storage and Technical Procedures: Disinfecting agents containing heavy metals, which cause release of respective ions (mercury, zinc, copper, etc.) should not be used for skin or mucous membrane disinfection as they have been related to incidence of swelling and edema. When chemical disinfection of multi-dose vials is desired, either isopropyl alcohol (91%) or 70% ethyl alcohol is recommended. Many commercially available brands of rubbing alcohol, as well as solutions of ethyl alcohol not of USP grade, contain denaturants which are injurious to rubber and, therefore, are not to be used. It is recommended that chemical disinfection be accomplished by wiping the vial stopper thoroughly with cotton or gauze that has been moistened with the recommended alcohol just prior to use.
-
HOW SUPPLIED
Lidocaine Hydrochloride Injection, USP is supplied as follows:
NDC
Container
Concentration
Size
Total (mg)
Single-dose:
0409-4278-01
Glass Teartop Vial
0.5% (5 mg/mL)
50 mL
250
0409-4713-01
Glass Ampul
1% (10 mg/mL)
2 mL (bulk – 400 units)
20
0409-4713-02
Glass Ampul
1% (10 mg/mL)
5 mL
50
0409-4713-05
Glass Ampul
1% (10 mg/mL)
5 mL (bulk – 400 units)
50
0409-4713-20
Glass Ampul
1% (10 mg/mL)
20 mL
200
0409-4713-32
Glass Ampul
1% (10 mg/mL)
2 mL
20
0409-4713-62
Glass Ampul
1% (10 mg/mL)
2 mL (bulk – 800 units)
20
0409-4713-65
Glass Ampul
1% (10 mg/mL)
5 mL (bulk – 800 units)
50
0409-4279-02
Glass Teartop Vial
1% (10 mg/mL)
30 mL
300
0409-4270-01
Sterile Glass Teartop Vial
1% (10 mg/mL)
30 mL
300
0409-4776-01
Glass Ampul
1.5% (15 mg/mL)
20 mL
300
0409-4056-01
Sterile Glass Ampul
1.5% (15 mg/mL)
20 mL
300
0409-4282-01
Glass Ampul
2% (20 mg/mL)
2 mL
40
0409-4282-02
Glass Ampul
2% (20 mg/mL)
10 mL
200
Multiple-dose:
0409-4275-01
Plastic Fliptop Vial
0.5% (5 mg/mL)
50 mL
250
0409-4276-01
Plastic Fliptop Vial
1% (10 mg/mL)
20 mL
200
0409-4276-02
Plastic Fliptop Vial
1% (10 mg/mL)
50 mL
500
0409-4277-01
Plastic Fliptop Vial
2% (20 mg/mL)
20 mL
400
0409-4277-02
Plastic Fliptop Vial
2% (20 mg/mL)
50 mL
1000
Single-dose products are preservative-free.
Store at 20 to 25°C (68 to 77°F). [See USP Controlled Room Temperature.]
Lidocaine Hydrochloride Injection, USP solutions packaged in ampuls and glass teartop vials may be autoclaved one time only. Autoclave at 15 pounds pressure, 121°C (250°F) for 15 minutes. DO NOT AUTOCLAVE PRODUCT IN PLASTIC VIALS.
Revised: February, 2010
Printed in USA EN-2421
Hospira, Inc., Lake Forest, IL 60045 USA
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
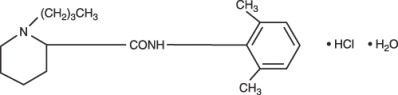
Bupivacaine hydrochloride is 2-Piperidinecarboxamide, 1-butyl- N-(2,6-dimethylphenyl)-, monohydrochloride, monohydrate, a white crystalline powder that is freely soluble in 95 percent ethanol, soluble in water, and slightly soluble in chloroform or acetone. It has the following structural formula:
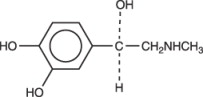
Epinephrine is (-)-3,4-Dihydroxy-α-[(methylamino)methyl] benzyl alcohol. It has the following structural formula:
MARCAINE is available in sterile isotonic solutions with and without epinephrine (as bitartrate) 1:200,000 for injection via local infiltration, peripheral nerve block, and caudal and lumbar epidural blocks. Solutions of MARCAINE may be autoclaved if they do not contain epinephrine. Solutions are clear and colorless.
Bupivacaine is related chemically and pharmacologically to the aminoacyl local anesthetics. It is a homologue of mepivacaine and is chemically related to lidocaine. All three of these anesthetics contain an amide linkage between the aromatic nucleus and the amino, or piperidine group. They differ in this respect from the procaine-type local anesthetics, which have an ester linkage.
MARCAINE — Sterile isotonic solutions containing sodium chloride. In multiple-dose vials, each mL also contains 1 mg methylparaben as antiseptic preservative. The pH of these solutions is adjusted to between 4 and 6.5 with sodium hydroxide or hydrochloric acid.
MARCAINE with epinephrine 1:200,000 (as bitartrate)—Sterile isotonic solutions containing sodium chloride. Each mL contains bupivacaine hydrochloride and 0.0091 mg epinephrine bitartrate, with 0.5 mg sodium metabisulfite, 0.001 mL monothioglycerol, and 2 mg ascorbic acid as antioxidants, 0.0017 mL 60% sodium lactate buffer, and 0.1 mg edetate calcium disodium as stabilizer. In multiple-dose vials, each mL also contains 1 mg methylparaben as antiseptic preservative. The pH of these solutions is adjusted to between 3.4 and 4.5 with sodium hydroxide or hydrochloric acid. The specific gravity of MARCAINE 0.5% with epinephrine 1:200,000 (as bitartrate) at 25°C is 1.008 and at 37°C is 1.008.
-
CLINICAL PHARMACOLOGY
Local anesthetics block the generation and the conduction of nerve impulses, presumably by increasing the threshold for electrical excitation in the nerve, by slowing the propagation of the nerve impulse, and by reducing the rate of rise of the action potential. In general, the progression of anesthesia is related to the diameter, myelination, and conduction velocity of affected nerve fibers. Clinically, the order of loss of nerve function is as follows: (1) pain, (2) temperature, (3) touch, (4) proprioception, and (5) skeletal muscle tone.
Systemic absorption of local anesthetics produces effects on the cardiovascular and central nervous systems (CNS). At blood concentrations achieved with normal therapeutic doses, changes in cardiac conduction, excitability, refractoriness, contractility, and peripheral vascular resistance are minimal. However, toxic blood concentrations depress cardiac conduction and excitability, which may lead to atrioventricular block, ventricular arrhythmias, and cardiac arrest, sometimes resulting in fatalities. In addition, myocardial contractility is depressed and peripheral vasodilation occurs, leading to decreased cardiac output and arterial blood pressure. Recent clinical reports and animal research suggest that these cardiovascular changes are more likely to occur after unintended intravascular injection of bupivacaine. Therefore, incremental dosing is necessary.
Following systemic absorption, local anesthetics can produce central nervous system stimulation, depression, or both. Apparent central stimulation is manifested as restlessness, tremors and shivering progressing to convulsions, followed by depression and coma progressing ultimately to respiratory arrest. However, the local anesthetics have a primary depressant effect on the medulla and on higher centers. The depressed stage may occur without a prior excited state.
Pharmacokinetics: The rate of systemic absorption of local anesthetics is dependent upon the total dose and concentration of drug administered, the route of administration, the vascularity of the administration site, and the presence or absence of epinephrine in the anesthetic solution. A dilute concentration of epinephrine (1:200,000 or 5 mcg/mL) usually reduces the rate of absorption and peak plasma concentration of MARCAINE, permitting the use of moderately larger total doses and sometimes prolonging the duration of action.
The onset of action with MARCAINE is rapid and anesthesia is long lasting. The duration of anesthesia is significantly longer with MARCAINE than with any other commonly used local anesthetic. It has also been noted that there is a period of analgesia that persists after the return of sensation, during which time the need for strong analgesics is reduced.
The onset of action following dental injections is usually 2 to 10 minutes and anesthesia may last two or three times longer than lidocaine and mepivacaine for dental use, in many patients up to 7 hours. The duration of anesthetic effect is prolonged by the addition of epinephrine 1:200,000.
Local anesthetics are bound to plasma proteins in varying degrees. Generally, the lower the plasma concentration of drug the higher the percentage of drug bound to plasma proteins.
Local anesthetics appear to cross the placenta by passive diffusion. The rate and degree of diffusion is governed by (1) the degree of plasma protein binding, (2) the degree of ionization, and (3) the degree of lipid solubility. Fetal/ maternal ratios of local anesthetics appear to be inversely related to the degree of plasma protein binding, because only the free, unbound drug is available for placental transfer. MARCAINE with a high protein binding capacity (95%) has a low fetal/maternal ratio (0.2 to 0.4). The extent of placental transfer is also determined by the degree of ionization and lipid solubility of the drug. Lipid soluble, nonionized drugs readily enter the fetal blood from the maternal circulation.
Depending upon the route of administration, local anesthetics are distributed to some extent to all body tissues, with high concentrations found in highly perfused organs such as the liver, lungs, heart, and brain.
Pharmacokinetic studies on the plasma profile of MARCAINE after direct intravenous injection suggest a three-compartment open model. The first compartment is represented by the rapid intravascular distribution of the drug. The second compartment represents the equilibration of the drug throughout the highly perfused organs such as the brain, myocardium, lungs, kidneys, and liver. The third compartment represents an equilibration of the drug with poorly perfused tissues, such as muscle and fat. The elimination of drug from tissue distribution depends largely upon the ability of binding sites in the circulation to carry it to the liver where it is metabolized.
After injection of MARCAINE for caudal, epidural, or peripheral nerve block in man, peak levels of bupivacaine in the blood are reached in 30 to 45 minutes, followed by a decline to insignificant levels during the next three to six hours.
Various pharmacokinetic parameters of the local anesthetics can be significantly altered by the presence of hepatic or renal disease, addition of epinephrine, factors affecting urinary pH, renal blood flow, the route of drug administration, and the age of the patient. The half-life of MARCAINE in adults is 2.7 hours and in neonates 8.1 hours.
In clinical studies, elderly patients reached the maximal spread of analgesia and maximal motor blockade more rapidly than younger patients. Elderly patients also exhibited higher peak plasma concentrations following administration of this product. The total plasma clearance was decreased in these patients.
Amide-type local anesthetics such as MARCAINE are metabolized primarily in the liver via conjugation with glucuronic acid. Patients with hepatic disease, especially those with severe hepatic disease, may be more susceptible to the potential toxicities of the amide-type local anesthetics. Pipecoloxylidine is the major metabolite of MARCAINE.
The kidney is the main excretory organ for most local anesthetics and their metabolites. Urinary excretion is affected by urinary perfusion and factors affecting urinary pH. Only 6% of bupivacaine is excreted unchanged in the urine.
When administered in recommended doses and concentrations, MARCAINE does not ordinarily produce irritation or tissue damage and does not cause methemoglobinemia.
-
INDICATIONS AND USAGE
MARCAINE is indicated for the production of local or regional anesthesia or analgesia for surgery, dental and oral surgery procedures, diagnostic and therapeutic procedures, and for obstetrical procedures. Only the 0.25% and 0.5% concentrations are indicated for obstetrical anesthesia. (See WARNINGS.)
Experience with nonobstetrical surgical procedures in pregnant patients is not sufficient to recommend use of 0.75% concentration of MARCAINE in these patients.
MARCAINE is not recommended for intravenous regional anesthesia (Bier Block). See WARNINGS.
The routes of administration and indicated MARCAINE concentrations are:
∙ local infiltration 0.25%
∙ peripheral nerve block 0.25% and 0.5%
∙ retrobulbar block 0.75%
∙ sympathetic block 0.25%
∙ lumbar epidural 0.25%, 0.5%, and 0.75%
(0.75% not for obstetrical anesthesia)
∙ caudal 0.25% and 0.5%
∙ epidural test dose 0.5% with epinephrine 1:200,000
∙ dental blocks 0.5% with epinephrine 1:200,000
(See DOSAGE AND ADMINISTRATION for additional information.)
Standard textbooks should be consulted to determine the accepted procedures and techniques for the administration of MARCAINE.
-
CONTRAINDICATIONS
MARCAINE is contraindicated in obstetrical paracervical block anesthesia. Its use in this technique has resulted in fetal bradycardia and death.
MARCAINE is contraindicated in patients with a known hypersensitivity to it or to any local anesthetic agent of the amide-type or to other components of MARCAINE solutions.
-
WARNINGS
THE 0.75% CONCENTRATION OF MARCAINE IS NOT RECOMMENDED FOR OBSTETRICAL ANESTHESIA. THERE HAVE BEEN REPORTS OF CARDIAC ARREST WITH DIFFICULT RESUSCITATION OR DEATH DURING USE OF MARCAINE FOR EPIDURAL ANESTHESIA IN OBSTETRICAL PATIENTS. IN MOST CASES, THIS HAS FOLLOWED USE OF THE 0.75% CONCENTRATION. RESUSCITATION HAS BEEN DIFFICULT OR IMPOSSIBLE DESPITE APPARENTLY ADEQUATE PREPARATION AND APPROPRIATE MANAGEMENT. CARDIAC ARREST HAS OCCURRED AFTER CONVULSIONS RESULTING FROM SYSTEMIC TOXICITY, PRESUMABLY FOLLOWING UNINTENTIONAL INTRAVASCULAR INJECTION. THE 0.75% CONCENTRATION SHOULD BE RESERVED FOR SURGICAL PROCEDURES WHERE A HIGH DEGREE OF MUSCLE RELAXATION AND PROLONGED EFFECT ARE NECESSARY.
LOCAL ANESTHETICS SHOULD ONLY BE EMPLOYED BY CLINICIANS WHO ARE WELL VERSED IN DIAGNOSIS AND MANAGEMENT OF DOSE-RELATED TOXICITY AND OTHER ACUTE EMERGENCIES WHICH MIGHT ARISE FROM THE BLOCK TO BE EMPLOYED, AND THEN ONLY AFTER INSURING THE IMMEDIATE AVAILABILITY OF OXYGEN, OTHER RESUSCITATIVE DRUGS, CARDIOPULMONARY RESUSCITATIVE EQUIPMENT, AND THE PERSONNEL RESOURCES NEEDED FOR PROPER MANAGEMENT OF TOXIC REACTIONS AND RELATED EMERGENCIES. (See also ADVERSE REACTIONS, PRECAUTIONS, and OVERDOSAGE.) DELAY IN PROPER MANAGEMENT OF DOSE-RELATED TOXICITY, UNDERVENTILATION FROM ANY CAUSE, AND/OR ALTERED SENSITIVITY MAY LEAD TO THE DEVELOPMENT OF ACIDOSIS, CARDIAC ARREST AND, POSSIBLY, DEATH.
Local anesthetic solutions containing antimicrobial preservatives, i.e., those supplied in multiple-dose vials, should not be used for epidural or caudal anesthesia because safety has not been established with regard to intrathecal injection, either intentionally or unintentionally, of such preservatives.
Intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures is an unapproved use, and there have been post-marketing reports of chondrolysis in patients receiving such infusions. The majority of reported cases of chondrolysis have involved the shoulder joint; cases of gleno-humeral chondrolysis have been described in pediatric and adult patients following intra-articular infusions of local anesthetics with and without epinephrine for periods of 48 to 72 hours. There is insufficient information to determine whether shorter infusion periods are not associated with these findings. The time of onset of symptoms, such as joint pain, stiffness and loss of motion can be variable, but may begin as early as the 2nd month after surgery. Currently, there is no effective treatment for chondrolysis; patients who experienced chondrolysis have required additional diagnostic and therapeutic procedures and some required arthroplasty or shoulder replacement.
It is essential that aspiration for blood or cerebrospinal fluid (where applicable) be done prior to injecting any local anesthetic, both the original dose and all subsequent doses, to avoid intravascular or subarachnoid injection. However, a negative aspiration does not ensure against an intravascular or subarachnoid injection.
MARCAINE with epinephrine 1:200,000 or other vasopressors should not be used concomitantly with ergot-type oxytocic drugs, because a severe persistent hypertension may occur. Likewise, solutions of MARCAINE containing a vasoconstrictor, such as epinephrine, should be used with extreme caution in patients receiving monoamineoxidase inhibitors (MAOI) or antidepressants of the triptyline or imipramine types, because severe prolonged hypertension may result.
Until further experience is gained in pediatric patients younger than 12 years, administration of MARCAINE in this age group is not recommended.
Mixing or the prior or intercurrent use of any other local anesthetic with MARCAINE cannot be recommended because of insufficient data on the clinical use of such mixtures.
There have been reports of cardiac arrest and death during the use of MARCAINE for intravenous regional anesthesia (Bier Block). Information on safe dosages and techniques of administration of MARCAINE in this procedure is lacking. Therefore, MARCAINE is not recommended for use in this technique.
MARCAINE with epinephrine 1:200,000 contains sodium metabisulfite, a sulfite that may cause allergic-type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people. Single-dose ampuls and single-dose vials of MARCAINE without epinephrine do not contain sodium metabisulfite.
-
PRECAUTIONS
General: The safety and effectiveness of local anesthetics depend on proper dosage, correct technique, adequate precautions, and readiness for emergencies. Resuscitative equipment, oxygen, and other resuscitative drugs should be available for immediate use. (See WARNINGS, ADVERSE REACTIONS, and OVERDOSAGE.) During major regional nerve blocks, the patient should have IV fluids running via an indwelling catheter to assure a functioning intravenous pathway. The lowest dosage of local anesthetic that results in effective anesthesia should be used to avoid high plasma levels and serious adverse effects. The rapid injection of a large volume of local anesthetic solution should be avoided and fractional (incremental) doses should be used when feasible.
Epidural Anesthesia: During epidural administration of MARCAINE, 0.5% and 0.75% solutions should be administered in incremental doses of 3 mL to 5 mL with sufficient time between doses to detect toxic manifestations of unintentional intravascular or intrathecal injection. Injections should be made slowly, with frequent aspirations before and during the injection to avoid intravascular injection. Syringe aspirations should also be performed before and during each supplemental injection in continuous (intermittent) catheter techniques. An intravascular injection is still possible even if aspirations for blood are negative.
During the administration of epidural anesthesia, it is recommended that a test dose be administered initially and the effects monitored before the full dose is given. When using a “continuous” catheter technique, test doses should be given prior to both the original and all reinforcing doses, because plastic tubing in the epidural space can migrate into a blood vessel or through the dura. When clinical conditions permit, the test dose should contain epinephrine (10 mcg to 15 mcg has been suggested) to serve as a warning of unintended intravascular injection. If injected into a blood vessel, this amount of epinephrine is likely to produce a transient “epinephrine response” within 45 seconds, consisting of an increase in heart rate and/or systolic blood pressure, circumoral pallor, palpitations, and nervousness in the unsedated patient. The sedated patient may exhibit only a pulse rate increase of 20 or more beats per minute for 15 or more seconds. Therefore, following the test dose, the heart rate should be monitored for a heart rate increase. Patients on beta-blockers may not manifest changes in heart rate, but blood pressure monitoring can detect a transient rise in systolic blood pressure. The test dose should also contain 10 mg to 15 mg of MARCAINE or an equivalent amount of another local anesthetic to detect an unintended intrathecal administration. This will be evidenced within a few minutes by signs of spinal block (e.g., decreased sensation of the buttocks, paresis of the legs, or, in the sedated patient, absent knee jerk). The Test Dose formulation of MARCAINE contains 15 mg of bupivacaine and 15 mcg of epinephrine in a volume of 3 mL. An intravascular or subarachnoid injection is still possible even if results of the test dose are negative. The test dose itself may produce a systemic toxic reaction, high spinal or epinephrine-induced cardiovascular effects.
Injection of repeated doses of local anesthetics may cause significant increases in plasma levels with each repeated dose due to slow accumulation of the drug or its metabolites, or to slow metabolic degradation. Tolerance to elevated blood levels varies with the status of the patient. Debilitated, elderly patients and acutely ill patients should be given reduced doses commensurate with their age and physical status. Local anesthetics should also be used with caution in patients with hypotension or heartblock.
Careful and constant monitoring of cardiovascular and respiratory (adequacy of ventilation) vital signs and the patient’s state of consciousness should be performed after each local anesthetic injection. It should be kept in mind at such times that restlessness, anxiety, incoherent speech, lightheadedness, numbness and tingling of the mouth and lips, metallic taste, tinnitus, dizziness, blurred vision, tremors, twitching, depression, or drowsiness may be early warning signs of central nervous system toxicity.
Local anesthetic solutions containing a vasoconstrictor should be used cautiously and in carefully restricted quantities in areas of the body supplied by end arteries or having otherwise compromised blood supply such as digits, nose, external ear, or penis. Patients with hypertensive vascular disease may exhibit exaggerated vasoconstrictor response. Ischemic injury or necrosis may result.
Because amide-local anesthetics such as MARCAINE are metabolized by the liver, these drugs, especially repeat doses, should be used cautiously in patients with hepatic disease. Patients with severe hepatic disease, because of their inability to metabolize local anesthetics normally, are at a greater risk of developing toxic plasma concentrations. Local anesthetics should also be used with caution in patients with impaired cardiovascular function because they may be less able to compensate for functional changes associated with the prolongation of AV conduction produced by these drugs.
Serious dose-related cardiac arrhythmias may occur if preparations containing a vasoconstrictor such as epinephrine are employed in patients during or following the administration of potent inhalation anesthetics. In deciding whether to use these products concurrently in the same patient, the combined action of both agents upon the myocardium, the concentration and volume of vasoconstrictor used, and the time since injection, when applicable, should be taken into account.
Many drugs used during the conduct of anesthesia are considered potential triggering agents for familial malignant hyperthermia. Because it is not known whether amide-type local anesthetics may trigger this reaction and because the need for supplemental general anesthesia cannot be predicted in advance, it is suggested that a standard protocol for management should be available. Early unexplained signs of tachycardia, tachypnea, labile blood pressure, and metabolic acidosis may precede temperature elevation. Successful outcome is dependent on early diagnosis, prompt discontinuance of the suspect triggering agent(s) and prompt institution of treatment, including oxygen therapy, indicated supportive measures and dantrolene. (Consult dantrolene sodium intravenous package insert before using.)
Use in Head and Neck Area: Small doses of local anesthetics injected into the head and neck area, including retrobulbar, dental, and stellate ganglion blocks, may produce adverse reactions similar to systemic toxicity seen with unintentional intravascular injections of larger doses. The injection procedures require the utmost care. Confusion, convulsions, respiratory depression, and/or respiratory arrest, and cardiovascular stimulation or depression have been reported. These reactions may be due to intra-arterial injection of the local anesthetic with retrograde flow to the cerebral circulation. They may also be due to puncture of the dural sheath of the optic nerve during retrobulbar block with diffusion of any local anesthetic along the subdural space to the midbrain. Patients receiving these blocks should have their circulation and respiration monitored and be constantly observed. Resuscitative equipment and personnel for treating adverse reactions should be immediately available. Dosage recommendations should not be exceeded. (See DOSAGE AND ADMINISTRATION.)
Use in Ophthalmic Surgery: Clinicians who perform retrobulbar blocks should be aware that there have been reports of respiratory arrest following local anesthetic injection. Prior to retrobulbar block, as with all other regional procedures, the immediate availability of equipment, drugs, and personnel to manage respiratory arrest or depression, convulsions, and cardiac stimulation or depression should be assured (see also WARNINGS and Use In Head and Neck Area, above). As with other anesthetic procedures, patients should be constantly monitored following ophthalmic blocks for signs of these adverse reactions, which may occur following relatively low total doses.
A concentration of 0.75% bupivacaine is indicated for retrobulbar block; however, this concentration is not indicated for any other peripheral nerve block, including the facial nerve, and not indicated for local infiltration, including the conjunctiva (see INDICATIONS AND USAGE and PRECAUTIONS, General). Mixing MARCAINE with other local anesthetics is not recommended because of insufficient data on the clinical use of such mixtures.
When MARCAINE 0.75% is used for retrobulbar block, complete corneal anesthesia usually precedes onset of clinically acceptable external ocular muscle akinesia. Therefore, presence of akinesia rather than anesthesia alone should determine readiness of the patient for surgery.
Use in Dentistry: Because of the long duration of anesthesia, when MARCAINE 0.5% with epinephrine is used for dental injections, patients should be cautioned about the possibility of inadvertent trauma to tongue, lips, and buccal mucosa and advised not to chew solid foods or test the anesthetized area by biting or probing.
Information for Patients: When appropriate, patients should be informed in advance that they may experience temporary loss of sensation and motor activity, usually in the lower half of the body, following proper administration of caudal or epidural anesthesia. Also, when appropriate, the physician should discuss other information including adverse reactions in the package insert of MARCAINE.
Patients receiving dental injections of MARCAINE should be cautioned not to chew solid foods or test the anesthetized area by biting or probing until anesthesia has worn off (up to 7 hours).
Clinically Significant Drug Interactions: The administration of local anesthetic solutions containing epinephrine or norepinephrine to patients receiving monoamine oxidase inhibitors or tricyclic antidepressants may produce severe, prolonged hypertension. Concurrent use of these agents should generally be avoided. In situations when concurrent therapy is necessary, careful patient monitoring is essential.
Concurrent administration of vasopressor drugs and of ergot-type oxytocic drugs may cause severe, persistent hypertension or cerebrovascular accidents.
Phenothiazines and butyrophenones may reduce or reverse the pressor effect of epinephrine.
Carcinogenesis, Mutagenesis, Impairment of Fertility: Long-term studies in animals to evaluate the carcinogenic potential of bupivacaine hydrochloride have not been conducted. The mutagenic potential and the effect on fertility of bupivacaine hydrochloride have not been determined.
Pregnancy Category C: There are no adequate and well-controlled studies in pregnant women. MARCAINE should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Bupivacaine hydrochloride produced developmental toxicity when administered subcutaneously to pregnant rats and rabbits at clinically relevant doses. This does not exclude the use of MARCAINE at term for obstetrical anesthesia or analgesia. (See Labor and Delivery)
Bupivacaine hydrochloride was administered subcutaneously to rats at doses of 4.4, 13.3, & 40 mg/kg and to rabbits at doses of 1.3, 5.8, & 22.2 mg/kg during the period of organogenesis (implantation to closure of the hard palate). The high doses are comparable to the daily maximum recommended human dose (MRHD) of 400 mg/day on a mg/m 2 body surface area (BSA) basis. No embryo-fetal effects were observed in rats at the high dose which caused increased maternal lethality. An increase in embryo-fetal deaths was observed in rabbits at the high dose in the absence of maternal toxicity with the fetal No Observed Adverse Effect Level representing approximately 1/5th the MRHD on a BSA basis.
In a rat pre- and post-natal development study (dosing from implantation through weaning) conducted at subcutaneous doses of 4.4, 13.3, & 40 mg/kg mg/kg/day, decreased pup survival was observed at the high dose. The high dose is comparable to the daily MRHD of 400 mg/day on a BSA basis.
Labor and Delivery: SEE BOXED WARNING REGARDING OBSTETRlCAL USE OF 0.75% MARCAINE.
MARCAINE is contraindicated for obstetrical paracervical block anesthesia.
Local anesthetics rapidly cross the placenta, and when used for epidural, caudal, or pudendal block anesthesia, can cause varying degrees of maternal, fetal, and neonatal toxicity. (See CLINICAL PHARMACOLOGY, Pharmacokinetics.) The incidence and degree of toxicity depend upon the procedure performed, the type, and amount of drug used, and the technique of drug administration. Adverse reactions in the parturient, fetus, and neonate involve alterations of the central nervous system, peripheral vascular tone, and cardiac function.
Maternal hypotension has resulted from regional anesthesia. Local anesthetics produce vasodilation by blocking sympathetic nerves. Elevating the patient’s legs and positioning her on her left side will help prevent decreases in blood pressure. The fetal heart rate also should be monitored continuously and electronic fetal monitoring is highly advisable.
Epidural, caudal, or pudendal anesthesia may alter the forces of parturition through changes in uterine contractility or maternal expulsive efforts. Epidural anesthesia has been reported to prolong the second stage of labor by removing the parturient’s reflex urge to bear down or by interfering with motor function. The use of obstetrical anesthesia may increase the need for forceps assistance.
The use of some local anesthetic drug products during labor and delivery may be followed by diminished muscle strength and tone for the first day or two of life. This has not been reported with bupivacaine.
It is extremely important to avoid aortocaval compression by the gravid uterus during administration of regional block to parturients. To do this, the patient must be maintained in the left lateral decubitus position or a blanket roll or sandbag may be placed beneath the right hip and gravid uterus displaced to the left.
Nursing Mothers: Bupivacaine has been reported to be excreted in human milk suggesting that the nursing infant could be theoretically exposed to a dose of the drug. Because of the potential for serious adverse reactions in nursing infants from bupivacaine, a decision should be made whether to discontinue nursing or not administer bupivacaine, taking into account the importance of the drug to the mother.
Pediatric Use: Until further experience is gained in pediatric patients younger than 12 years, administration of MARCAINE in this age group is not recommended. Continuous infusions of bupivacaine in children have been reported to result in high systemic levels of bupivacaine and seizures; high plasma levels may also be associated with cardiovascular abnormalities. (See WARNINGS, PRECAUTIONS, and OVERDOSAGE.)
Geriatric Use: Patients over 65 years, particularly those with hypertension, may be at increased risk for developing hypotension while undergoing anesthesia with MARCAINE. (See ADVERSE REACTIONS.)
Elderly patients may require lower doses of MARCAINE. (See PRECAUTIONS, Epidural Anesthesia and DOSAGE AND ADMINISTRATION.)
In clinical studies, differences in various pharmacokinetic parameters have been observed between elderly and younger patients. (See CLINICAL PHARMACOLOGY.)
This product is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. (See CLINICAL PHARMACOLOGY.)
-
ADVERSE REACTIONS
Reactions to MARCAINE are characteristic of those associated with other amide-type local anesthetics. A major cause of adverse reactions to this group of drugs is excessive plasma levels, which may be due to overdosage, unintentional intravascular injection, or slow metabolic degradation.
The most commonly encountered acute adverse experiences which demand immediate counter-measures are related to the central nervous system and the cardiovascular system. These adverse experiences are generally dose related and due to high plasma levels which may result from overdosage, rapid absorption from the injection site, diminished tolerance, or from unintentional intravascular injection of the local anesthetic solution. In addition to systemic dose-related toxicity, unintentional subarachnoid injection of drug during the intended performance of caudal or lumbar epidural block or nerve blocks near the vertebral column (especially in the head and neck region) may result in underventilation or apnea (“Total or High Spinal”). Also, hypotension due to loss of sympathetic tone and respiratory paralysis or underventilation due to cephalad extension of the motor level of anesthesia may occur. This may lead to secondary cardiac arrest if untreated. Patients over 65 years, particularly those with hypertension, may be at increased risk for experiencing the hypotensive effects of MARCAINE. Factors influencing plasma protein binding, such as acidosis, systemic diseases which alter protein production, or competition of other drugs for protein binding sites, may diminish individual tolerance.
Central Nervous System Reactions: These are characterized by excitation and/or depression. Restlessness, anxiety, dizziness, tinnitus, blurred vision, or tremors may occur, possibly proceeding to convulsions. However, excitement may be transient or absent, with depression being the first manifestation of an adverse reaction. This may quickly be followed by drowsiness merging into unconsciousness and respiratory arrest. Other central nervous system effects may be nausea, vomiting, chills, and constriction of the pupils.
The incidence of convulsions associated with the use of local anesthetics varies with the procedure used and the total dose administered. In a survey of studies of epidural anesthesia, overt toxicity progressing to convulsions occurred in approximately 0.1% of local anesthetic administrations.
Cardiovascular System Reactions: High doses or unintentional intravascular injection may lead to high plasma levels and related depression of the myocardium, decreased cardiac output, heartblock, hypotension, bradycardia, ventricular arrhythmias, including ventricular tachycardia and ventricular fibrillation, and cardiac arrest. (See WARNINGS, PRECAUTIONS, and OVERDOSAGE.)
Allergic: Allergic-type reactions are rare and may occur as a result of sensitivity to the local anesthetic or to other formulation ingredients, such as the antimicrobial preservative methylparaben contained in multiple-dose vials or sulfites in epinephrine-containing solutions. These reactions are characterized by signs such as urticaria, pruritus, erythema, angioneurotic edema (including laryngeal edema), tachycardia, sneezing, nausea, vomiting, dizziness, syncope, excessive sweating, elevated temperature, and possibly, anaphylactoid-like symptomatology (including severe hypotension). Cross sensitivity among members of the amide-type local anesthetic group has been reported. The usefulness of screening for sensitivity has not been definitely established.
Neurologic: The incidences of adverse neurologic reactions associated with the use of local anesthetics may be related to the total dose of local anesthetic administered and are also dependent upon the particular drug used, the route of administration, and the physical status of the patient. Many of these effects may be related to local anesthetic techniques, with or without a contribution from the drug.
In the practice of caudal or lumbar epidural block, occasional unintentional penetration of the subarachnoid space by the catheter or needle may occur. Subsequent adverse effects may depend partially on the amount of drug administered intrathecally and the physiological and physical effects of a dural puncture. A high spinal is characterized by paralysis of the legs, loss of consciousness, respiratory paralysis, and bradycardia.
Neurologic effects following epidural or caudal anesthesia may include spinal block of varying magnitude (including high or total spinal block); hypotension secondary to spinal block; urinary retention; fecal and urinary incontinence; loss of perineal sensation and sexual function; persistent anesthesia, paresthesia, weakness, paralysis of the lower extremities and loss of sphincter control all of which may have slow, incomplete, or no recovery; headache; backache; septic meningitis; meningismus; slowing of labor; increased incidence of forceps delivery; and cranial nerve palsies due to traction on nerves from loss of cerebrospinal fluid.
Neurologic effects following other procedures or routes of administration may include persistent anesthesia, paresthesia, weakness, paralysis, all of which may have slow, incomplete, or no recovery.
-
OVERDOSAGE
Acute emergencies from local anesthetics are generally related to high plasma levels encountered during therapeutic use of local anesthetics or to unintended subarachnoid injection of local anesthetic solution. (See ADVERSE REACTIONS, WARNINGS, and PRECAUTIONS.)
Management of Local Anesthetic Emergencies: The first consideration is prevention, best accomplished by careful and constant monitoring of cardiovascular and respiratory vital signs and the patient’s state of consciousness after each local anesthetic injection. At the first sign of change, oxygen should be administered.
The first step in the management of systemic toxic reactions, as well as underventilation or apnea due to unintentional subarachnoid injection of drug solution, consists of immediate attention to the establishment and maintenance of a patent airway and effective assisted or controlled ventilation with 100% oxygen with a delivery system capable of permitting immediate positive airway pressure by mask. This may prevent convulsions if they have not already occurred.
If necessary, use drugs to control the convulsions. A 50 mg to 100 mg bolus IV injection of succinylcholine will paralyze the patient without depressing the central nervous or cardiovascular systems and facilitate ventilation. A bolus IV dose of 5 mg to 10 mg of diazepam or 50 mg to 100 mg of thiopental will permit ventilation and counteract central nervous system stimulation, but these drugs also depress central nervous system, respiratory, and cardiac function, add to postictal depression and may result in apnea. Intravenous barbiturates, anticonvulsant agents, or muscle relaxants should only be administered by those familiar with their use. Immediately after the institution of these ventilatory measures, the adequacy of the circulation should be evaluated. Supportive treatment of circulatory depression may require administration of intravenous fluids, and when appropriate, a vasopressor dictated by the clinical situation (such as ephedrine or epinephrine to enhance myocardial contractile force).
Endotracheal intubation, employing drugs and techniques familiar to the clinician, may be indicated after initial administration of oxygen by mask if difficulty is encountered in the maintenance of a patent airway, or if prolonged ventilatory support (assisted or controlled) is indicated.
Recent clinical data from patients experiencing local anesthetic-induced convulsions demonstrated rapid development of hypoxia, hypercarbia, and acidosis with bupivacaine within a minute of the onset of convulsions. These observations suggest that oxygen consumption and carbon dioxide production are greatly increased during local anesthetic convulsions and emphasize the importance of immediate and effective ventilation with oxygen which may avoid cardiac arrest.
If not treated immediately, convulsions with simultaneous hypoxia, hypercarbia, and acidosis plus myocardial depression from the direct effects of the local anesthetic may result in cardiac arrhythmias, bradycardia, asystole, ventricular fibrillation, or cardiac arrest. Respiratory abnormalities, including apnea, may occur. Underventilation or apnea due to unintentional subarachnoid injection of local anesthetic solution may produce these same signs and also lead to cardiac arrest if ventilatory support is not instituted. If cardiac arrest should occur, successful outcome may require prolonged resuscitative efforts.
The supine position is dangerous in pregnant women at term because of aortocaval compression by the gravid uterus. Therefore during treatment of systemic toxicity, maternal hypotension or fetal bradycardia following regional block, the parturient should be maintained in the left lateral decubitus position if possible, or manual displacement of the uterus off the great vessels be accomplished.
The mean seizure dosage of bupivacaine in rhesus monkeys was found to be 4.4 mg/kg with mean arterial plasma concentration of 4.5 mcg/mL. The intravenous and subcutaneous LD 50 in mice is 6 mg/kg to 8 mg/kg and 38 mg/kg to 54 mg/kg respectively.
-
DOSAGE AND ADMINISTRATION
The dose of any local anesthetic administered varies with the anesthetic procedure, the area to be anesthetized, the vascularity of the tissues, the number of neuronal segments to be blocked, the depth of anesthesia and degree of muscle relaxation required, the duration of anesthesia desired, individual tolerance, and the physical condition of the patient. The smallest dose and concentration required to produce the desired result should be administered. Dosages of MARCAINE should be reduced for elderly and/or debilitated patients and patients with cardiac and/or liver disease. The rapid injection of a large volume of local anesthetic solution should be avoided and fractional (incremental) doses should be used when feasible.
For specific techniques and procedures, refer to standard textbooks.
There have been adverse event reports of chondrolysis in patients receiving intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures. MARCAINE is not approved for this use (see WARNINGS and DOSAGE AND ADMINISTRATION).
In recommended doses, MARCAINE produces complete sensory block, but the effect on motor function differs among the three concentrations.
0.25%—when used for caudal, epidural, or peripheral nerve block, produces incomplete motor block. Should be used for operations in which muscle relaxation is not important, or when another means of providing muscle relaxation is used concurrently. Onset of action may be slower than with the 0.5% or 0.75% solutions.
0.5%— provides motor blockade for caudal, epidural, or nerve block, but muscle relaxation may be inadequate for operations in which complete muscle relaxation is essential.
0.75%—produces complete motor block. Most useful for epidural block in abdominal operations requiring complete muscle relaxation, and for retrobulbar anesthesia. Not for obstetrical anesthesia.
The duration of anesthesia with MARCAINE is such that for most indications, a single dose is sufficient.
Maximum dosage limit must be individualized in each case after evaluating the size and physical status of the patient, as well as the usual rate of systemic absorption from a particular injection site. Most experience to date is with single doses of MARCAINE up to 225 mg with epinephrine 1:200,000 and 175 mg without epinephrine; more or less drug may be used depending on individualization of each case.
These doses may be repeated up to once every three hours. In clinical studies to date, total daily doses have been up to 400 mg. Until further experience is gained, this dose should not be exceeded in 24 hours. The duration of anesthetic effect may be prolonged by the addition of epinephrine.
The dosages in Table 1 have generally proved satisfactory and are recommended as a guide for use in the average adult. These dosages should be reduced for elderly or debilitated patients. Until further experience is gained, MARCAINE is not recommended for pediatric patients younger than 12 years. MARCAINE is contraindicated for obstetrical paracervical blocks, and is not recommended for intravenous regional anesthesia (Bier Block).
Use in Epidural Anesthesia: During epidural administration of MARCAINE, 0.5% and 0.75% solutions should be administered in incremental doses of 3 mL to 5 mL with sufficient time between doses to detect toxic manifestations of unintentional intravascular or intrathecal injection. In obstetrics, only the 0.5% and 0.25% concentrations should be used; incremental doses of 3 mL to 5 mL of the 0.5% solution not exceeding 50 mg to 100 mg at any dosing interval are recommended. Repeat doses should be preceded by a test dose containing epinephrine if not contraindicated. Use only the single-dose ampuls and single-dose vials for caudal or epidural anesthesia; the multiple-dose vials contain a preservative and therefore should not be used for these procedures.
Test Dose for Caudal and Lumbar Epidural Blocks: The Test Dose of MARCAINE (0.5% bupivacaine with 1:200,000 epinephrine in a 3 mL ampul) is recommended for use as a test dose when clinical conditions permit prior to caudal and lumbar epidural blocks. This may serve as a warning of unintended intravascular or subarachnoid injection. (See PRECAUTIONS.) The pulse rate and other signs should be monitored carefully immediately following each test dose administration to detect possible intravascular injection, and adequate time for onset of spinal block should be allotted to detect possible intrathecal injection. An intravascular or subarachnoid injection is still possible even if results of the test dose are negative. The test dose itself may produce a systemic toxic reaction, high spinal or cardiovascular effects from the epinephrine. (See WARNINGS and OVERDOSAGE.)
Use in Dentistry: The 0.5% concentration with epinephrine is recommended for infiltration and block injection in the maxillary and mandibular area when a longer duration of local anesthetic action is desired, such as for oral surgical procedures generally associated with significant postoperative pain. The average dose of 1.8 mL (9 mg) per injection site will usually suffice; an occasional second dose of 1.8 mL (9 mg) may be used if necessary to produce adequate anesthesia after making allowance for 2 to 10 minutes onset time. (See CLINICAL PHARMACOLOGY.) The lowest effective dose should be employed and time should be allowed between injections; it is recommended that the total dose for all injection sites, spread out over a single dental sitting, should not ordinarily exceed 90 mg for a healthy adult patient (ten 1.8 mL injections of 0.5% MARCAINE with epinephrine). Injections should be made slowly and with frequent aspirations. Until further experience is gained, MARCAINE in dentistry is not recommended for pediatric patients younger than 12 years.
Unused portions of solution not containing preservatives, i.e., those supplied in single-dose ampuls and single-dose vials, should be discarded following initial use.
This product should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Solutions which are discolored or which contain particulate matter should not be administered.
Table 1. Recommended Concentrations and Doses of MARCAINE Type of
BlockConc. Each Dose Motor
Block 1(mL) (mg) Local
infiltration
0.25% 4
up to
max.up to
max.— Epidural
0.75% 2,4 10-20 75-150 complete 0.5% 4
10-20
50-100
moderate
to complete0.25% 4
10-20
25-50
partial
to moderateCaudal
0.5% 4
15-30
75-150
moderate
to complete0.25% 4
15-30
37.5-75
moderate Peripheral
nerves
0.5% 4
5 to
max.25 to
max.moderate
to complete0.25% 4
5 to
max.12.5 to
max.moderate
to completeRetrobulbar 3 0.75% 4 2-4 15-30 complete Sympathetic 0.25% 20-50 50-125 — Dental 3 0.5%
w/epi1.8-3.6
per site9-18
per site— Epidural 3
Test Dose0.5%
w/epi2-3 10-15
(10-15 micrograms epinephrine)— 1With continuous (intermittent) techniques, repeat doses increase the degree of motor block. The first repeat dose of 0.5% may produce complete motor block. Intercostal nerve block with 0.25% may also produce complete motor block for intra-abdominal surgery.
2For single-dose use, not for intermittent epidural technique. Not for obstetrical anesthesia.
3See PRECAUTIONS.
4Solutions with or without epinephrine.
-
HOW SUPPLIED
These solutions are not for spinal anesthesia.
Store at 20 to 25°C (68 to 77°F). [See USP Controlled Room Temperature.]
MARCAINE ―Solutions of MARCAINE that do not contain epinephrine may be autoclaved. Autoclave at 15-pound pressure, 121°C (250°F) for 15 minutes.
NDC No. Container Fill Quantity 0.25%—Contains 2.5 mg bupivacaine hydrochloride per mL. 0409-1559-10 Single-dose vials 10 mL box of 10 0409-1559-30 Single-dose vials 30 mL box of 10 0409-1587-50 Multiple-dose vials 50 mL box of 1
0.5%—Contains 5 mg bupivacaine hydrochloride per mL. 0409-1560-10 Single-dose vials 10 mL box of 10 0409-1560-29 Single-dose vials 30 mL box of 10 0409-1610-50 Multiple-dose vials 50 mL box of 1
0.75%—Contains 7.5 mg bupivacaine hydrochloride per mL. 0409-1582-10 Single-dose vials 10 mL box of 10 0409-1582-29 Single-dose vials 30 mL box of 10 MARCAINE with epinephrine 1:200,000 (as bitartrate)― Solutions of MARCAINE that contain epinephrine should not be autoclaved and should be protected from light. Do not use the solution if its color is pinkish or darker than slightly yellow or if it contains a precipitate.
NDC No. Container Fill Quantity 0.25% with epinephrine 1:200,000—Contains 2.5 mg bupivacaine hydrochloride per mL. 0409-1746-10 Single-dose vials 10 mL box of 10 0409-1746-30 Single-dose vials 30 mL box of 10 0409-1752-50 Multiple-dose vials 50 mL box of 1
0.5% with epinephrine 1:200,000—Contains 5 mg bupivacaine hydrochloride per mL.0409-1749-03 Single-dose ampuls 3 mL box of 10 0409-1749-10 Single-dose vials 10 mL box of 10 0409-1749-29 Single-dose vials 30 mL box of 10 0409-1755-50 Multiple-dose vials 50 mL box of 1 Revised: 10/2011
Printed in USA EN-2916
Hospira, Inc., Lake Forest, IL 60045 USA
-
BOXED WARNING
(What is this?)
BOXED WARNING
WARNING
Ketorolac tromethamine, a nonsteroidal anti-inflammatory drug (NSAID), is indicated for the short-term (up to 5 days in adults) management of moderately severe acute pain that requires analgesia at the opioid level. Oral ketorolac tromethamine is indicated only as continuation treatment following intravenous or intramuscular dosing of ketorolac tromethamine, if necessary. The total combined duration of use of oral ketorolac tromethamine and ketorolac tromethamine injection should not exceed 5 days.
Ketorolac tromethamine is not indicated for use in pediatric patients and it is NOT indicated for minor or chronic painful conditions. Increasing the dose of ketorolac tromethamine beyond the label recommendations will not provide better efficacy but will increase the risk of developing serious adverse events.
GASTROINTESTINAL RISK
- Ketorolac tromethamine can cause peptic ulcers, gastrointestinal bleeding and/or perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Therefore, ketorolac tromethamine is CONTRAINDICATED in patients with active peptic ulcer disease, in patients with recent gastrointestinal bleeding or perforation, and in patients with a history of peptic ulcer disease or gastrointestinal bleeding. Elderly patients are at greater risk for serious gastrointestinal events (see WARNINGS).
CARDIOVASCULAR THROMBOTIC EVENTS
- Nonsteroidal anti-inflammatory drugs (NSAIDs) cause an increased risk of serious cardiovascular thrombotic events, including myocardial infarction and stroke, which can be fatal. This risk may occur early in treatment and may increase with duration of use (see WARNINGS and PRECAUTIONS).
Ketorolac tromethamine is CONTRAINDICATED in the setting of coronary artery bypass graft (CABG) surgery (see CONTRAINDICATIONS and WARNINGS).
RENAL RISK
- Ketorolac tromethamine is CONTRAINDICATED in patients with advanced renal impairment and in patients at risk for renal failure due to volume depletion (see WARNINGS).
RISK OF BLEEDING
- Ketorolac tromethamine inhibits platelet function and is, therefore, CONTRAINDICATED in patients with suspected or confirmed cerebrovascular bleeding, patients with hemorrhagic diathesis, incomplete hemostasis and those at high risk of bleeding (see WARNINGS and PRECAUTIONS).
Ketorolac tromethamine is CONTRAINDICATED as prophylactic analgesic before any major surgery.
HYPERSENSITIVITY
- Hypersensitivity reactions, ranging from bronchospasm to anaphylactic shock, have occurred and appropriate counteractive measures must be available when administering the first dose of ketorolac tromethamine injection (see CONTRAINDICATIONS and WARNINGS). Ketorolac tromethamine is CONTRAINDICATED in patients with previously demonstrated hypersensitivity to ketorolac tromethamine or allergic manifestations to aspirin or other nonsteroidal anti-inflammatory drugs (NSAIDs).
INTRATHECAL OR EPIDURAL ADMINISTRATION
- Ketorolac tromethamine is CONTRAINDICATED for intrathecal or epidural administration due to its alcohol content.
RISK DURING LABOR AND DELIVERY
- The use of ketorolac tromethamine in labor and delivery is CONTRAINDICATED because it may adversely affect fetal circulation and inhibit uterine contractions.
CONCOMITANT USE WITH NSAIDs
- Ketorolac tromethamine is CONTRAINDICATED in patients currently receiving aspirin or NSAIDs because of the cumulative risk of inducing serious NSAID-related side effects.
SPECIAL POPULATIONS
- Dosage should be adjusted for patients 65 years or older, for patients under 50 kg (110 lbs.) of body weight (see DOSAGE AND ADMINISTRATION) and for patients with moderately elevated serum creatinine (see WARNINGS). Doses of ketorolac tromethamine injection are not to exceed 60 mg (total dose per day) in these patients.
DOSAGE AND ADMINISTRATION
Ketorolac Tromethamine Tablets
- Ketorolac tromethamine tablets are indicated only as continuation therapy to ketorolac tromethamine injection, and the combined duration of use of ketorolac tromethamine injection and ketorolac tromethamine tablets is not to exceed 5 (five) days, because of the increased risk of serious adverse events.
- The recommended total daily dose of ketorolac tromethamine tablets (maximum 40 mg) is significantly lower than for ketorolac tromethamine injection (maximum 120 mg) (see DOSAGE AND ADMINISTRATION).
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Ketorolac Tromethamine Injection, USP is a member of the pyrrolo-pyrrole group of nonsteroidal anti-inflammatory drugs (NSAIDs). The chemical name for ketorolac tromethamine is (±)-5-benzoyl-2,3-dihydro-1 H-pyrrolizine-1-carboxylic acid, compound with 2-amino-2-(hydroxymethyl)-1,3-propanediol (1:1), and the structural formula is presented in Figure 1.
Ketorolac tromethamine is a racemic mixture of [-]S and [+]R ketorolac tromethamine. Ketorolac tromethamine may exist in three crystal forms. All forms are equally soluble in water. Ketorolac tromethamine has a pKa of 3.5 and an n-octanol/water partition coefficient of 0.26. The molecular weight of ketorolac tromethamine is 376.40.
Ketorolac Tromethamine Injection, USP is available for intravenous (IV) or intramuscular (IM) administration as: 15 mg in 1 mL (1.5%) and 30 mg in 1 mL (3%) in sterile solution; 60 mg in 2 mL (3%) of ketorolac tromethamine in sterile solution is available for intramuscular administration only. The solutions contain 10% (w/v) alcohol,USP, and 6.68 mg, 4.35 mg, and 8.70 mg, respectively, of sodium chloride in sterile water. The pH range is 6.9 to 7.9 and is adjusted with sodium hydroxide and/or hydrochloric acid. The sterile solutions are clear and slightly yellow in color.
-
CLINICAL PHARMACOLOGY
Pharmacodynamics
Ketorolac tromethamine is a nonsteroidal anti-inflammatory drug (NSAID) that exhibits analgesic activity in animal models. The mechanism of action of ketorolac, like that of other NSAIDs, is not completely understood but may be related to prostaglandin synthetase inhibition. The biological activity of ketorolac tromethamine is associated with the S-form.
Ketorolac tromethamine possesses no sedative or anxiolytic properties.
The peak analgesic effect of ketorolac tromethamine occurs within 2 to 3 hours and is not statistically significantly different over the recommended dosage range of ketorolac tromethamine. The greatest difference between large and small doses of ketorolac tromethamine by either route is in the duration of analgesia.
Pharmacokinetics
Ketorolac tromethamine is a racemic mixture of [-]S- and [+]R-enantiomeric forms, with the S‑form having analgesic activity.
Comparison of Intravenous, Intramuscular and Oral Pharmacokinetics
The pharmacokinetics of ketorolac tromethamine, following intravenous, intramuscular and oral doses of ketorolac tromethamine are compared in Table 1. In adults, the extent of bioavailability following administration of the ORAL and INTRAMUSCULAR forms of ketorolac tromethamine was equal to that following an intravenous bolus.
Table 1: Table of Approximate Average Pharmacokinetic Parameters (Mean±SD) Following Oral, Intramuscular and Intravenous Doses of Ketorolac Tromethamine
Oral †
Intramuscular*
Intravenous Bolus ‡
Pharmacokinetic Parameters (units)
10 mg
15 mg
30 mg
60 mg
15 mg
30 mg
Bioavailability (extent)
100%
T max1 (min)
44±34
33±21**
44±29
33±21**
1.1±0.7**
2.9±1.8
C max2 (mcg/mL) [Single-dose]
0.87±0.22
1.14±0.32**
2.42±0.68
4.55±1.27**
2.47±0.51**
4.65±0.96
C max (mcg/mL) [steady state qid]
1.05±0.26**
1.56±0.44**
3.11±0.87**
N/A ††
3.09±1.17**
6.85±2.61
C min3 (mcg/mL) [steady state qid]
0.29±0.07**
0.47±0.13**
0.93±0.26**
N/A
0.61±0.21**
1.04±0.35
C avg4 (mcg/mL) [steady state qid]
0.59±0.2**
0.94±0.29**
1.88±0.59**
N/A
1.09±0.3**
2.17±0.59
V β5 (L/kg)
———— 0.175±0.039 ————
0.210±0.044
% Dose metabolized = <50
% Dose excreted in urine = 91
% Dose excreted in feces = 6
% Plasma protein binding = 99
1 Time-to-peak plasma
concentration2 Peak plasma concentration
3 Trough plasma concentration
4 Average plasma concentration
5 Volume of distribution
† Derived from PO pharmacokinetic studies in 77 normal fasted volunteers
* Derived from intramuscular pharmacokinetic studies in 54 normal volunteers
‡ Derived from intravenous pharmacokinetic studies in 24 normal volunteers
†† Not applicable because 60 mg is only recommended as a single dose
** Mean value was simulated from observed plasma concentration data and standard deviation was simulated from percent coefficient of variation for observed C max and T max data
Linear Kinetics
In adults, following administration of single ORAL, INTRAMUSCULAR or INTRAVENOUS doses of ketorolac tromethamine in the recommended dosage ranges, the clearance of the racemate does not change. This implies that the pharmacokinetics of ketorolac tromethamine in adults, following single or multiple intramuscular, intravenous or recommended oral doses of ketorolac tromethamine, are linear. At the higher recommended doses, there is a proportional increase in the concentrations of free and bound racemate.
Distribution
The mean apparent volume (V β) of ketorolac tromethamine following complete distribution was approximately 13 liters. This parameter was determined from single-dose data. The ketorolac tromethamine racemate has been shown to be highly protein bound (99%). Nevertheless, plasma concentrations as high as 10 mcg/mL will only occupy approximately 5% of the albumin binding sites. Thus, the unbound fraction for each enantiomer will be constant over the therapeutic range. A decrease in serum albumin, however, will result in increased free drug concentrations.
Ketorolac tromethamine is excreted in human milk (see PRECAUTIONS – Nursing Mothers).
Metabolism
Ketorolac tromethamine is largely metabolized in the liver. The metabolic products are hydroxylated and conjugated forms of the parent drug. The products of metabolism, and some unchanged drug, are excreted in the urine.
Excretion
The principal route of elimination of ketorolac and its metabolites is renal. About 92% of a given dose is found in the urine, approximately 40% as metabolites and 60% as unchanged ketorolac. Approximately 6% of a dose is excreted in the feces. A single-dose study with 10 mg ketorolac tromethamine (n = 9) demonstrated that the S-enantiomer is cleared approximately two times faster than the R-enantiomer and that the clearance was independent of the route of administration. This means that the ratio of S/R plasma concentrations decreases with time after each dose. There is little or no inversion of the R- to S- form in humans. The clearance of the racemate in normal subjects, elderly individuals and in hepatically and renally impaired patients is outlined in Table 2 (see CLINICAL PHARMACOLOGY – Kinetics in Special Populations).
The half-life of the ketorolac tromethamine S-enantiomer was approximately 2.5 hours (SD ± 0.4) compared with 5 hours (SD ± 1.7) for the R-enantiomer. In other studies, the half-life for the racemate has been reported to lie within the range of 5 to 6 hours.
Accumulation
Ketorolac tromethamine administered as an intravenous bolus, every 6 hours, for 5 days, to healthy subjects (n = 13), showed no significant difference in C max on Day 1 and Day 5. Trough levels averaged 0.29 mcg/mL (SD ± 0.13) on Day 1 and 0.55 mcg/mL (SD ± 0.23) on Day 6. Steady state was approached after the fourth dose.
Accumulation of ketorolac tromethamine has not been studied in special populations (geriatric, pediatric, renal failure patients, or hepatic disease patients).
Kinetics in Special Populations
Geriatric Patients
Based on single-dose data only, the half-life of the ketorolac tromethamine racemate increased from 5 to 7 hours in the elderly (65 to 78 years) compared with young healthy volunteers (24 to 35 years) (see Table 2). There was little difference in the C max for the two groups (elderly, 2.52 mcg/mL ± 0.77; young, 2.99 mcg/mL ± 1.03) (see PRECAUTIONS – Geriatric Use).
Pediatric Patients
Limited information is available regarding the pharmacokinetics of dosing of ketorolac tromethamine in the pediatric population. Following a single intravenous bolus dose of 0.5 mg/kg in 10 children 4 to 8 years old, the half-life was 5.8 ± 1.6 hours, the average clearance was 0.042 ± 0.01 L/hr/kg, the volume of distribution during the terminal phase (V β) was 0.34 ± 0.12 L/kg and the volume of distribution at steady state (V ss) was 0.26 ± 0.08 L/kg. The volume of distribution and clearance of ketorolac in pediatric patients was higher than those observed in adult subjects (see Table 1). There are no pharmacokinetic data available for administration of ketorolac tromethamine by the intramuscular route in pediatric patients.
Renal Insufficiency
Based on single-dose data only, the mean half-life of ketorolac tromethamine in renally impaired patients is between 6 and 19 hours, and is dependent on the extent of the impairment. There is poor correlation between creatinine clearance and total ketorolac tromethamine clearance in the elderly and populations with renal impairment (r = 0.5).
In patients with renal disease, the AUC ∞ of each enantiomer increased by approximately 100% compared with healthy volunteers. The volume of distribution doubles for the S-enantiomer and increases by 1/5th for the R-enantiomer. The increase in volume of distribution of ketorolac tromethamine implies an increase in unbound fraction.
The AUC ∞-ratio of the ketorolac tromethamine enantiomers in healthy subjects and patients remained similar, indicating there was no selective excretion of either enantiomer in patients compared to healthy subjects (see WARNINGS – Renal Effects).
Hepatic Insufficiency
There was no significant difference in estimates of half-life, AUC ∞ and C max, in 7 patients with liver disease compared to healthy volunteers (see PRECAUTIONS – Hepatic Effects and Table 2).
Race
Pharmacokinetic differences due to race have not been identified.
Table 2: The Influence of Age, Liver and Kidney Function, on the Clearance and Terminal Half-life of Ketorolac Tromethamine (INTRAMUSCULAR 1 and ORAL 2) in Adult Populations
1 Estimated from 30 mg single intramuscular doses of ketorolac tromethamine
2 Estimated from 10 mg single oral doses of ketorolac tromethamine
3 Liters/hour/kilogram
Intravenous-Administration: In normal subjects (n=37), the total clearance of 30 mg intravenous-administered Ketorolac Tromethamine was 0.030 (0.017-0.051) L/h/kg. The terminal half-life was 5.6 (4.0-7.9) hours. (See Kinetics in Special Populations for use of intravenous dosing of ketorolac tromethamine in pediatric patients.)Total Clearance
[in L/h/kg] 3Terminal Half-life
[in hours]Type of Subjects
INTRAMUSCULAR
Mean (range)
ORAL
Mean (range)
INTRAMUSCULAR
Mean (range)
ORAL
Mean (range)
Normal Subjects
Intramuscular (n = 54)
mean age = 32, range = 18-60
Oral (n = 77)
mean age = 32, range = 20-600.023
(0.010-0.046)
0.025
(0.013-0.050)
5.3
(3.5-9.2)5.3
(2.4-9)Healthy Elderly Subjects
Intramuscular (n = 13), Oral (n = 12)
mean age = 72, range = 65-780.019
(0.013-0.034)
0.024
(0.018-0.034)
7
(4.7-8.6)6.1
(4.3-7.6)Patients with Hepatic Dysfunction
Intramuscular and Oral (n = 7)
mean age = 51, range = 43-640.029
(0.013-0.066)
0.033
(0.019-0.051)
5.4
(2.2-6.9)4.5
(1.6-7.6)Patients with Renal Impairment
Intramuscular (n = 25), Oral (n = 9)
serum creatinine = 1.9-5.0 mg/dL,
mean age (Intramuscular) = 54,
range = 35-71mean age (Oral) = 57,
range = 39‑700.015
(0.005-0.043)
0.016
(0.007-0.052)
10.3
(5.9-19.2)10.8
(3.4-18.9)Renal Dialysis Patients
Intramuscular and Oral (n = 9)
mean age = 40, range = 27-630.016
(0.003-0.036)
–
13.6
(8.0-39.1)–
-
CLINICAL STUDIES
Adult Patients
In a postoperative study, where all patients received morphine by a PCA device, patients treated with ketorolac tromethamine intravenous as fixed intermittent boluses (e.g., 30 mg initial dose followed by 15 mg q3h), required significantly less morphine (26%) than the placebo group. Analgesia was significantly superior, at various postdosing pain assessment times, in the patients receiving ketorolac tromethamine intravenous plus PCA morphine as compared to patients receiving PCA-administered morphine alone.
-
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of ketorolac tromethamine and other treatment options before deciding to use ketorolac. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
Acute Pain in Adult Patients
Ketorolac tromethamine is indicated for the short-term (≤5 days) management of moderately severe acute pain that requires analgesia at the opioid level, usually in a postoperative setting. Therapy should always be initiated with intravenous or intramuscular dosing of ketorolac tromethamine, and oral ketorolac tromethamine is to be used only as continuation treatment, if necessary.
The total combined duration of use of ketorolac tromethamine injection and oral ketorolac tromethamine is not to exceed 5 days of use because of the potential of increasing the frequency and severity of adverse reactions associated with the recommended doses (see WARNINGS, PRECAUTIONS, DOSAGE AND ADMINISTRATION, and ADVERSE REACTIONS). Patients should be switched to alternative analgesics as soon as possible, but ketorolac tromethamine therapy is not to exceed 5 days.
Ketorolac tromethamine injection has been used concomitantly with morphine and meperidine and has shown an opioid-sparing effect. For breakthrough pain, it is recommended to supplement the lower end of the ketorolac tromethamine injection dosage range with low doses of narcotics prn, unless otherwise contraindicated. Ketorolac tromethamine injection and narcotics should not be administered in the same syringe (see DOSAGE AND ADMINISTRATION – Pharmaceutical Information for Ketorolac Tromethamine Injection).
-
CONTRAINDICATIONS
(see also Boxed WARNING)
Ketorolac Tromethamine is contraindicated in patients with previously demonstrated hypersensitivity to ketorolac tromethamine.
Ketorolac tromethamine is contraindicated in patients with active peptic ulcer disease, in patients with recent gastrointestinal bleeding or perforation and in patients with a history of peptic ulcer disease or gastrointestinal bleeding.
Ketorolac tromethamine should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs have been reported in such patients (see WARNINGS – Anaphylactoid Reactions, and PRECAUTIONS – Pre-existing Asthma).
Ketorolac tromethamine is contraindicated as prophylactic analgesic before any major surgery.
In the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS).
Ketorolac tromethamine is contraindicated in patients with advanced renal impairment or in patients at risk for renal failure due to volume depletion (see WARNINGS for correction of volume depletion).
Ketorolac tromethamine is contraindicated in labor and delivery because, through its prostaglandin synthesis inhibitory effect, it may adversely affect fetal circulation and inhibit uterine musculature, thus increasing the risk of uterine hemorrhage.
Ketorolac tromethamine inhibits platelet function and is, therefore, contraindicated in patients with suspected or confirmed cerebrovascular bleeding, hemorrhagic diathesis, incomplete hemostasis and those at high risk of bleeding (see WARNINGS and PRECAUTIONS).
Ketorolac tromethamine is contraindicated in patients currently receiving aspirin or NSAIDs because of the cumulative risks of inducing serious NSAID-related adverse events.
The concomitant use of ketorolac tromethamine and probenecid is contraindicated.
The concomitant use of ketorolac tromethamine and pentoxifylline is contraindicated.
Ketorolac tromethamine injection is contraindicated for neuraxial (epidural or intrathecal) administration due to its alcohol content.
-
WARNINGS
(See also Boxed WARNING.)
The total combined duration of use of oral ketorolac tromethamine and intravenous or intramuscular dosing of ketorolac tromethamine is not to exceed 5 days in adults. Ketorolac tromethamine is not indicated for use in pediatric patients.
The most serious risks associated with ketorolac tromethamine are:
Gastrointestinal Effects – Risk of Ulceration, Bleeding and Perforation: Ketorolac tromethamine is contraindicated in patients with previously documented peptic ulcers and/or gastrointestinal (GI) bleeding. Ketorolac tromethamine can cause serious GI adverse events including bleeding, ulceration and perforation, of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with ketorolac tromethamine.
Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. Minor upper gastrointestinal problems, such as dyspepsia, are common and may also occur at any time during NSAID therapy.
The incidence and severity of gastrointestinal complications increases with increasing dose of, and duration of treatment with ketorolac tromethamine. Do not use ketorolac tromethamine for more than five days.
However, even short-term therapy is not without risk. In addition to past history of ulcer disease, other factors that increase the risk for GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids, or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. This should include discontinuation of ketorolac tromethamine until a serious GI adverse event is ruled out. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
NSAIDs should be given with care to patients with a history of inflammatory bowel disease (ulcerative colitis, Crohn’s disease) as their condition may be exacerbated.
Hemorrhage
Because prostaglandins play an important role in hemostasis and NSAIDs affect platelet aggregation as well, use of ketorolac tromethamine in patients who have coagulation disorders should be undertaken very cautiously, and those patients should be carefully monitored. Patients on therapeutic doses of anticoagulants (e.g., heparin or dicumarol derivatives) have an increased risk of bleeding complications if given ketorolac tromethamine concurrently; therefore, physicians should administer such concomitant therapy only extremely cautiously. The concurrent use of ketorolac tromethamine and therapy that affects hemostasis, including prophylactic low-dose heparin (2500-5000 units q12h), warfarin and dextrans have not been studied extensively, but may also be associated with an increased risk of bleeding. Until data from such studies are available, physicians should carefully weigh the benefits against the risks, and use such concomitant therapy in these patients only extremely cautiously. Patients receiving therapy that affects hemostasis should be monitored closely.
In postmarketing experience, postoperative hematomas and other signs of wound bleeding have been reported in association with the peri-operative use of intravenous or intramuscular dosing of ketorolac tromethamine. Therefore, peri-operative use of ketorolac tromethamine should be avoided and postoperative use be undertaken with caution when hemostasis is critical (see PRECAUTIONS).
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Ketorolac tromethamine and its metabolites are eliminated primarily by the kidneys, which, in patients with reduced creatinine clearance, will result in diminished clearance of the drug (see CLINICAL PHARMACOLOGY). Therefore, ketorolac tromethamine should be used with caution in patients with impaired renal function (see DOSAGE AND ADMINISTRATION) and such patients should be followed closely. With the use of ketorolac tromethamine, there have been reports of acute renal failure, interstitial nephritis and nephrotic syndrome.
Impaired Renal Function
Ketorolac tromethamine is contraindicated in patients with serum creatinine concentrations indicating advanced renal impairment (see CONTRAINDICATIONS). Ketorolac tromethamine should be used with caution in patients with impaired renal function or a history of kidney disease because it is a potent inhibitor of prostaglandin synthesis. Because patients with underlying renal insufficiency are at increased risk of developing acute renal decompensation or failure, the risks and benefits should be assessed prior to giving ketorolac tromethamine to these patients.
Anaphylactoid Reactions
As with other NSAIDs, anaphylactoid reactions may occur in patients without known prior exposure to ketorolac tromethamine. Ketorolac tromethamine should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS and PRECAUTIONS - Pre-existing Asthma). Emergency help should be sought in cases where an anaphylactoid reaction occurs.
Cardiovascular Effects
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first few weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as ketorolac tromethamine, increases the risk of serious gastrointestinal (GI) events (see WARNINGS).
Status Post Coronary Artery Bypass Graft (CABG) Surgery
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10 to 14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG surgery (see CONTRAINDICATIONS).
Post-MI Patients
Observational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years follow-up.
Avoid the use of ketorolac tromethamine in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If ketorolac tromethamine is used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
Hypertension
NSAIDs, including ketorolac tromethamine, can lead to onset of new hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including ketorolac tromethamine, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Heart Failure and Edema
The Coxib and traditional NSAID Trialists’ Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of ketorolac tromethamine may blunt the CV effects of several therapeutic agents used to treat these medical conditions (e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers (ARBs) (see DRUG INTERACTIONS).
Avoid the use of ketorolac tromethamine in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If ketorolac tromethamine is used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
Skin Reactions
NSAIDs, including ketorolac tromethamine, can cause serious skin adverse events such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
Pregnancy
In late pregnancy, as with other NSAIDs, ketorolac tromethamine should be avoided because it may cause premature closure of the ductus arteriosus.
-
PRECAUTIONS
General
Ketorolac tromethamine cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of ketorolac tromethamine in reducing inflammation may diminish the utility of this diagnostic sign in detecting complications of presumed noninfectious, painful conditions.
Hepatic Effects
Ketorolac tromethamine should be used with caution in patients with impaired hepatic function or a history of liver disease . Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs including ketorolac tromethamine. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continuing therapy. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials with NSAIDs. In addition, rare cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, liver necrosis and hepatic failure, some of them with fatal outcomes have been reported.
A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with ketorolac tromethamine. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), ketorolac tromethamine should be discontinued.
Hematologic Effects
Anemia is sometimes seen in patients receiving NSAIDs, including ketorolac tromethamine. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including ketorolac tromethamine, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia. NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible. Patients receiving ketorolac tromethamine who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored.
Pre-existing Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and other nonsteroidal anti-inflammatory drugs has been reported in such aspirin-sensitive patients, ketorolac tromethamine should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with pre-existing asthma.
Information for Patients
Ketorolac tromethamine is a potent NSAID and may cause serious side effects such as gastrointestinal bleeding or kidney failure, which may result in hospitalization and even fatal outcome.
Physicians, when prescribing ketorolac tromethamine, should inform their patients or their guardians of the potential risks of ketorolac tromethamine treatment (see Boxed WARNING, WARNINGS, PRECAUTIONS, and ADVERSE REACTIONS sections), instruct patients to seek medical advice if they develop treatment-related adverse events, and advise patients not to give oral ketorolac tromethamine to other family members and to discard any unused drug. Remember that the total combined duration of use of oral ketorolac tromethamine and intravenous or intramuscular dosing of ketorolac tromethamine is not to exceed 5 days in adults. Ketorolac tromethamine is not indicated for use in pediatric patients. Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- Advise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their healthcare provider immediately (see WARNINGS).
- Ketorolac tromethamine, like other NSAIDs, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative sign or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal Effects: Risk of Ulceration, Bleeding, and Perforation).
- Ketorolac tromethamine, like other NSAIDs, can cause serious skin side effects such as exfoliative dermatitis, SJS, and TEN, which may result in hospitalizations and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching, and should ask for medical advice when observing any indicative signs or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- Advise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur (see WARNINGS).
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and “flu-like” symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- Patients should be informed of the signs of an anaphylactoid reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS).
- In late pregnancy, as with other NSAIDs, ketorolac tromethamine should be avoided because it will cause premature closure of the ductus arteriosus.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs, should have their CBC and a chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash etc.) or if abnormal liver tests persist or worsen, ketorolac tromethamine should be discontinued.
Drug Interactions
Ketorolac is highly bound to human plasma protein (mean 99.2%). There is no evidence in animal or human studies that ketorolac tromethamine induces or inhibits hepatic enzymes capable of metabolizing itself or other drugs.
Warfarin, Digoxin, Salicylate, and Heparin
The in vitro binding of warfarin to plasma proteins is only slightly reduced by ketorolac tromethamine (99.5% control vs 99.3%) when ketorolac plasma concentrations reach 5 to 10 mcg/mL. Ketorolac does not alter digoxin protein binding. In vitro studies indicate that, at therapeutic concentrations of salicylate (300 mcg/mL), the binding of ketorolac was reduced from approximately 99.2% to 97.5%, representing a potential twofold increase in unbound ketorolac plasma levels. Therapeutic concentrations of digoxin, warfarin, ibuprofen, naproxen, piroxicam, acetaminophen, phenytoin and tolbutamide did not alter ketorolac tromethamine protein binding.
In a study involving 12 adult volunteers, oral ketorolac tromethamine was coadministered with a single dose of 25 mg warfarin, causing no significant changes in pharmacokinetics or pharmacodynamics of warfarin. In another study, ketorolac tromethamine dosed intravenous or intramuscular was given with two doses of 5000 U of heparin to 11 healthy volunteers, resulting in a mean template bleeding time of 6 minutes (3.2 to 11.4 min) compared to a mean of 6.0 minutes (3.4 to 7.5 min) for heparin alone and 5.1 minutes (3.5 to 8.5 min) for placebo. Although these results do not indicate a significant interaction between ketorolac tromethamine and warfarin or heparin, the administration of ketorolac tromethamine to patients taking anticoagulants should be done extremely cautiously and patients should be closely monitored (see WARNINGS and PRECAUTIONS – Hematologic Effects).
The effects of warfarin and NSAIDs, in general, on GI bleeding are synergistic, such that the users of both drugs together have a risk of serious GI bleeding higher than the users of either drug alone.
Aspirin
When ketorolac tromethamine is administered with aspirin, its protein binding is reduced, although the clearance of free ketorolac tromethamine is not altered. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of ketorolac tromethamine and aspirin is not generally recommended because of the potential of increased adverse effects.
Diuretics
Clinical studies, as well as postmarketing observations, have shown that ketorolac tromethamine can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS – Renal Effects), as well as to assure diuretic efficacy.
Probenecid
Concomitant administration of oral ketorolac tromethamine and probenecid resulted in decreased clearance and volume of distribution of ketorolac and significant increases in ketorolac plasma levels (total AUC increased approximately threefold from 5.4 to 17.8 mcg/h/mL) and terminal half-life increased approximately twofold from 6.6 to 15.1 hours. Therefore, concomitant use of ketorolac tromethamine and probenecid is contraindicated.
Lithium
NSAIDs have produced an elevation of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15% and the renal clearance was decreased by approximately 20%. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity.
Methotrexate
NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This may indicate that they could enhance the toxicity of methotrexate. Caution should be used when NSAIDs are administered concomitantly with methotrexate.
ACE Inhibitors/Angiotensin II Receptor Antagonists
Concomitant use of ACE inhibitors and/or angiotensin II receptor antagonists may increase the risk of renal impairment, particularly in volume-depleted patients.
Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE inhibitors and/or angiotensin II receptor antagonists. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE inhibitors and/or angiotensin II receptor antagonists.
Antiepileptic Drugs
Sporadic cases of seizures have been reported during concomitant use of ketorolac tromethamine and antiepileptic drugs (phenytoin, carbamazepine).
Psychoactive Drugs
Hallucinations have been reported when ketorolac tromethamine was used in patients taking psychoactive drugs (fluoxetine, thiothixene, alprazolam).
Pentoxifylline
When ketorolac tromethamine is administered concurrently with pentoxifylline, there is an increased tendency to bleeding.
Nondepolarizing Muscle Relaxants
In postmarketing experience there have been reports of a possible interaction between ketorolac tromethamine intravenous/intramuscular and nondepolarizing muscle relaxants that resulted in apnea. The concurrent use of ketorolac tromethamine with muscle relaxants has not been formally studied.
Selective Serotonin Reuptake Inhibitors (SSRIs)
There is an increased risk of gastrointestinal bleeding when selective serotonin reuptake inhibitors (SSRIs) are combined with NSAIDs. Caution should be used when NSAIDs are administered concomitantly with SSRIs.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
An 18-month study in mice with oral doses of ketorolac tromethamine tablets at 2 mg/kg/day (0.9 times the human systemic exposure at the recommended intramuscular or intravenous dose of 30 mg qid, based on area-under-the-plasma-concentration curve [AUC]), and a 24-month study in rats at 5 mg/kg/day (0.5 times the human AUC) showed no evidence of tumorigenicity.
Ketorolac tromethamine was not mutagenic in the Ames test, unscheduled DNA synthesis and repair, and in forward mutation assays. Ketorolac tromethamine did not cause chromosome breakage in the in vivo mouse micronucleus assay. At 1590 mcg/mL and at higher concentrations, ketorolac tromethamine increased the incidence of chromosomal aberrations in Chinese hamster ovarian cells.
Impairment of fertility did not occur in male or female rats at oral doses of 9 mg/kg (0.9 times the human AUC) and 16 mg/kg (1.6 times the human AUC) of ketorolac tromethamine, respectively.
Pregnancy
Teratogenic Effects
Pregnancy Category C
Reproduction studies have been performed during organogenesis using daily oral doses of ketorolac tromethamine tablets at 3.6 mg/kg (0.37 times the human AUC) in rabbits and at 10 mg/kg (1.0 times the human AUC) in rats. Results of these studies did not reveal evidence of teratogenicity to the fetus. However, animal reproduction studies are not always predictive of human response.
Nonteratogenic Effects
Because of the known effects of nonsteroidal anti-inflammatory drugs on the fetal cardiovascular system (closure of ductus arteriosus), use during pregnancy (particularly late pregnancy) should be avoided. Oral doses of ketorolac tromethamine tablets at 1.5 mg/kg (0.14 times the human AUC), administered after gestation day 17, caused dystocia and higher pup mortality in rats.
There are no adequate and well-controlled studies of ketorolac tromethamine in pregnant women. Ketorolac tromethamine should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Labor and Delivery
The use of ketorolac tromethamine is contraindicated in labor and delivery because, through its prostaglandin synthesis inhibitory effect, it may adversely affect fetal circulation and inhibit uterine contractions, thus increasing the risk of uterine hemorrhage (see CONTRAINDICATIONS).
Effects on Fertility
The use of ketorolac tromethamine, as with any drug known to inhibit cyclooxygenase/ prostaglandin synthesis, may impair fertility and is not recommended in women attempting to conceive. In women who have difficulty conceiving or are undergoing investigation of infertility, withdrawal of ketorolac tromethamine should be considered.
Nursing Mothers
Limited data from one published study that included 10 breastfeeding women 2-6 days postpartum showed low levels of ketorolac in breast milk and were undetectable (less than 5 ng/mL) in 4 of the patients. After a single administration of 10 mg of ketorolac tromethamin, the maximum milk concentration observed was 7.3 ng/mL, and the maximum milk-to-plasma ratio was 0.037. After 1 day of dosing (10 mg every 6 hours), the maximum milk concentration was 7.9 ng/mL, and the maximum milk-to-plasma ratio was 0.025. Assuming a daily intake of 400-1,000 mL of human milk per day and a maternal body weight of 60 kg, the calculated maximum daily infant exposure was 0.00263 mg/kg/day, which is 0.4% of the maternal weight-adjusted dose.
Exercise caution when ketorolac is administered to a nursing woman. Available information has not shown any specific adverse events in nursing infants; however, instruct patients to contact their infant’s healthcare provider if they note any adverse events.
Pediatric Use
Ketorolac tromethamine is not indicated for use in pediatric patients. The safety and effectiveness of ketorolac tromethamine in pediatric patients below the age of 17 have not been established.
Geriatric Use (≥65 Years of Age)
Because ketorolac tromethamine may be cleared more slowly by the elderly (see CLINICAL PHARMACOLOGY) who are also more sensitive to the dose-related adverse effects of NSAIDs (see WARNINGS – Gastrointestinal Effects – Risk of Ulceration, Bleeding, and Perforation), extreme caution and reduced dosages (see DOSAGE AND ADMINISTRATION) and careful clinical monitoring must be used when treating the elderly with ketorolac tromethamine.
-
ADVERSE REACTIONS
Adverse reaction rates increase with higher doses of ketorolac tromethamine. Practitioners should be alert for the severe complications of treatment with ketorolac tromethamine, such as G.I. ulceration, bleeding and perforation, postoperative bleeding, acute renal failure, anaphylactic and anaphylactoid reactions and liver failure (see Boxed WARNING, WARNINGS, PRECAUTIONS, and DOSAGE AND ADMINISTRATION). These NSAID-related complications can be serious in certain patients for whom ketorolac tromethamine is indicated, especially when the drug is used inappropriately.
In patients taking ketorolac tromethamine or other NSAIDs in clinical trials, the most frequently reported adverse experiences in approximately 1% to 10% of patients are:
Gastrointestinal (GI) experiences including:
abdominal pain
constipation/diarrhea
dyspepsia
flatulence
GI fullness
GI ulcers (gastric/duodenal)
gross bleeding/perforation
heartburn
nausea *
stomatitis
vomiting
Other experiences:
abnormal renal function
anemia
dizziness
drowsiness
edema
elevated liver enzymes
headaches *
hypertension
increased bleeding time
injection site pain
pruritus
purpura
rashes
tinnitus
sweating
* Incidence greater than 10%
Additional adverse experiences reported occasionally (<1% in patients taking ketorolac tromethamine or other NSAIDs in clinical trials) include:
Body as a Whole: fever, infections, sepsis
Cardiovascular: congestive heart failure, palpitation, pallor, tachycardia, syncope
Dermatologic: alopecia, photosensitivity, urticaria
Gastrointestinal: anorexia, dry mouth, eructation, esophagitis, excessive thirst, gastritis, glossitis, hematemesis, hepatitis, increased appetite, jaundice, melena, rectal bleeding
Hemic and Lymphatic: ecchymosis, eosinophilia, epistaxis, leukopenia, thrombocytopenia
Metabolic and Nutritional: weight change
Nervous System: abnormal dreams, abnormal thinking, anxiety, asthenia, confusion, depression, euphoria, extrapyramidal symptoms, hallucinations, hyperkinesis, inability to concentrate, insomnia, nervousness, paresthesia, somnolence, stupor, tremors, vertigo, malaise
Reproductive, female: infertility
Respiratory: asthma, cough, dyspnea, pulmonary edema, rhinitis
Special Senses: abnormal taste, abnormal vision, blurred vision, hearing loss
Urogenital: cystitis, dysuria, hematuria, increased urinary frequency, interstitial nephritis, oliguria/polyuria, proteinuria, renal failure, urinary retention
Other rarely observed reactions (reported from postmarketing experience in patients taking ketorolac tromethamine or other NSAIDs) are:
Body as a Whole: angioedema, death, hypersensitivity reactions such as anaphylaxis, anaphylactoid reaction, laryngeal edema, tongue edema (see WARNINGS), myalgia
Cardiovascular: arrhythmia, bradycardia, chest pain, flushing, hypotension, myocardial infarction, vasculitis
Dermatologic: exfoliative dermatitis, erythema multiforme, Lyell’s syndrome, bullous reactions including Stevens-Johnson syndrome and toxic epidermal necrolysis
Gastrointestinal: acute pancreatitis, liver failure, ulcerative stomatitis, exacerbation of inflammatory bowel disease (ulcerative colitis, Crohn’s disease)
Hemic and Lymphatic: agranulocytosis, aplastic anemia, hemolytic anemia, lymphadenopathy, pancytopenia, post operative wound hemorrhage (rarely requiring blood transfusion — see Boxed WARNING, WARNINGS, and PRECAUTIONS)
Metabolic and Nutritional: hyperglycemia, hyperkalemia, hyponatremia
Nervous System: aseptic meningitis, convulsions, coma, psychosis
Respiratory: bronchospasm, respiratory depression, pneumonia
Special Senses: conjunctivitis
Urogenital: flank pain with or without hematuria and/or azotemia, hemolytic uremic syndrome
Postmarketing Surveillance Study
A large postmarketing observational, nonrandomized study, involving approximately 10,000 patients receiving ketorolac tromethamine, demonstrated that the risk of clinically serious gastrointestinal (GI) bleeding was dose-dependent (see Tables 3A and 3B). This was particularly true in elderly patients who received an average daily dose greater than 60 mg/day of ketorolac tromethamine (see Table 3A).
Table 3: Incidence of Clinically Serious G.I. Bleeding as Related to Age, Total Daily Dose, and History of G.I. Perforation, Ulcer, Bleeding (PUB) after up to 5 Days of Treatment with Ketorolac Tromethamine Injection
A. Adult Patients without History of PUB
Age of Patients
Total Daily Dose of Ketorolac Tromethamine Injection
≤60 mg
>60 to 90 mg
>90 to 120 mg
>120 mg
<65 years of age
0.4%
0.4%
0.9%
4.6%
≥65 years of age
1.2%
2.8%
2.2%
7.7%
B. Adult Patients with History of PUB
Age of Patients
Total Daily Dose of Ketorolac Tromethamine Injection
≤60 mg
>60 to 90 mg
>90 to 120 mg
>120 mg
<65 years of age
2.1%
4.6%
7.8%
15.4%
≥65 years of age
4.7%
3.7%
2.8%
25.0%
-
OVERDOSAGE
Symptoms and Signs
Symptoms following acute NSAIDs overdoses are usually limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which are generally reversible with supportive care. Gastrointestinal bleeding can occur. Hypertension, acute renal failure, respiratory depression and coma may occur, but are rare. Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose.
Treatment
Patients should be managed by symptomatic and supportive care following a NSAIDs overdose. There are no specific antidotes. Emesis and/or activated charcoal (60 g to 100 g in adults, 1 g/kg to 2 g/kg in children) and/or osmotic cathartic may be indicated in patients seen within 4 hours of ingestion with symptoms or following a large oral overdose (5 to 10 times the usual dose). Forced diuresis, alkalization of urine, hemodialysis or hemoperfusion may not be useful due to high protein binding.
Single overdoses of ketorolac tromethamine have been variously associated with abdominal pain, nausea, vomiting, hyperventilation, peptic ulcers and/or erosive gastritis and renal dysfunction which have resolved after discontinuation of dosing.
-
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of ketorolac tromethamine and other treatment options before deciding to use ketorolac tromethamine. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals. In adults, the combined duration of use of intravenous or intramuscular dosing of ketorolac tromethamine and oral ketorolac tromethamine is not to exceed 5 days. In adults, the use of oral ketorolac tromethamine is only indicated as continuation therapy to intravenous or intramuscular dosing of ketorolac tromethamine. See package insert for ketorolac tromethamine tablets for transition from intravenous or intramuscular dosing of ketorolac tromethamine (single- or multiple-dose) to multiple-dose oral ketorolac tromethamine.
Note: Oral formulation should not be given as an initial dose.
Use minimum effective dose for the individual patient.
Total duration of treatment in adult patients: the combined duration of use of intravenous or intramuscular dosing of ketorolac tromethamine and oral ketorolac tromethamine is not to exceed 5 days.
KETOROLAC TROMETHAMINE INJECTION
Ketorolac tromethamine injection may be used as a single or multiple dose on a regular or “prn” schedule for the management of moderately severe, acute pain that requires analgesia at the opioid level, usually in a postoperative setting. Hypovolemia should be corrected prior to the administration of ketorolac tromethamine (see WARNINGS – Renal Effects). Patients should be switched to alternative analgesics as soon as possible, but ketorolac tromethamine therapy is not to exceed 5 days.
When administering ketorolac tromethamine injection, the intravenous bolus must be given over no less than 15 seconds. The intramuscular administration should be given slowly and deeply into the muscle. The analgesic effect begins in ~30 minutes with maximum effect in 1 to 2 hours after dosing intravenous or intramuscular. Duration of analgesic effect is usually 4 to 6 hours.
Single-Dose Treatment: The following regimen should be limited to single administration use only
Intramuscular Dosing
- Patients <65 years of age: One dose of 60 mg.
- Patients ≥65 years of age, renally impaired and/or less than 50 kg (110 lbs) of body weight: One dose of 30 mg.
Intravenous Dosing
- Patients <65 years of age: One dose of 30 mg.
- Patients ≥65 years of age, renally impaired and/or less than 50 kg (110 lbs) of body weight: One dose of 15 mg.
Multiple-Dose Treatment (Intravenous or Intramuscular)
- Patients <65 years of age: The recommended dose is 30 mg ketorolac tromethamine injection every 6 hours. The maximum daily dose for these populations should not exceed 120 mg.
- For patients ≥65 years of age, renally impaired patients (see WARNINGS), and patients less than 50 kg (110 lbs): The recommended dose is 15 mg ketorolac tromethamine injection every 6 hours. The maximum daily dose for these populations should not exceed 60 mg.
For breakthrough pain, do not increase the dose or the frequency of ketorolac tromethamine. Consideration should be given to supplementing these regimens with low doses of opioids “prn” unless otherwise contraindicated.
Pharmaceutical Information for Ketorolac Tromethamine Injection
Ketorolac tromethamine injection should not be mixed in a small volume (e.g., in a syringe) with morphine sulfate, meperidine hydrochloride, promethazine hydrochloride or hydroxyzine hydrochloride; this will result in precipitation of ketorolac from solution.
NOTE: Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED
Ketorolac Tromethamine Injection, USP is supplied as follows:
Unit of Sale Each Concentration (mg/mL) Fill Volume/
Container Size
Total Ketorolac
Tromethamine
(Per Container)
NDC 0409-3793-01
Tray of 25
NDC 0409-3793-19
2mL Single-Dose Glass Fliptop Vial
15 mg/mL (1 mL/2 mL) 15 mg
NDC 0409-3795-01
Tray of 25
NDC 0409-3795-19
2mL Single-Dose Glass Fliptop Vial
30 mg/mL (1 mL/2 mL) 30 mg NDC 0409-3796-01*
Tray of 25
NDC 0409-3796-19*
2mL Single-Dose Glass Fliptop Vial
30 mg/mL (2 mL/2 mL) 60 mg - *
- FOR INTRAMUSCULAR USE ONLY.
Store at 20 to 25°C (68 to 77°F). [See USP Controlled Room Temperature.]
Protect from light.
Retain in carton until time of use.Revised: 7/2015
EN-3996
Hospira, Inc., Lake Forest, IL 60045 USA
-
Medication Guide for Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
What is the most important information I should know about medicines called Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
NSAIDs can cause serious side effects, including:
- Increased risk of a heart attack or stroke that can lead to death. This risk may happen early in treatment and may increase:
- with increasing doses of NSAIDs
- with longer use of NSAIDs
Do not take NSAIDs right before or after a heart surgery called a “coronary artery bypass graft (CABG).”
Avoid taking NSAIDs after a recent heart attack, unless your healthcare provider tells you to. You may have an increased risk of another heart attack if you take NSAIDs after a recent heart attack.
- Increased risk of bleeding, ulcers, and tears (perforation) of the esophagus (tube leading from the mouth to the stomach), stomach and intestines:
- anytime during use
- without warning symptoms
- that may cause death
The risk of getting an ulcer or bleeding increases with:
• past history of stomach ulcers, • increasing doses of NSAIDs
or stomach or intestinal • longer use of NSAIDS
bleeding with the use of • smoking
NSAIDs • drinking alcohol
• taking medicines called • older age
“corticosteroids”, • poor health
“anticoagulants”, “SSRIs”, or • advanced liver disease
“SNRIs” • bleeding problemsNSAIDs should only be used:
- exactly as prescribed
- at the lowest dose possible for your treatment
- for the shortest time needed
What are NSAIDs?
NSAIDs are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as different types of arthritis, menstrual cramps, and other types of short-term pain.
Who should not take NSAIDs?
Do not take NSAIDs:
- if you had an asthma attack, hives, or other allergic reaction with aspirin or any other NSAIDs.
- right before or after heart bypass surgery.
Before taking NSAIDs, tell your healthcare provider about all of your medical conditions, including if you:
- have liver or kidney problems
- have high blood pressure
- have asthma
- are pregnant or plan to become pregnant. Talk to your healthcare provider if you are considering taking NSAIDs during pregnancy. You should not take NSAIDs after 29 weeks of pregnancy.
- are breastfeeding or plan to breast feed
Tell your healthcare provider about all of the medicines you take, including prescription or over-the-counter medicines, vitamins or herbal supplements. NSAIDs and some other medicines can interact with each other and cause serious side effects. Do not start taking any new medicine without talking to your healthcare provider first.
What are the possible side effects of NSAIDs?
NSAIDs can cause serious side effects, including:See “What is the most important information I should know about medicines called NonSteroidal Anti-inflammatory Drugs (NSAIDs)?”
- new or worse high blood pressure
- heart failure
- liver problems including liver failure
- kidney problems including kidney failure
- low red blood cells (anemia)
- life-threatening skin reactions
- life-threatening allergic reactions
- Other side effects of NSAIDs include: stomach pain, constipation, diarrhea, gas, heartburn, nausea, vomiting, and dizziness.
Get emergency help right away if you get any of the following symptoms:
- shortness of breath or trouble breathing
- chest pain
- weakness in one part or side of your body
- slurred speech
- swelling of the face or throat
Stop taking your NSAID and call your healthcare provider right away if you get any of the following symptoms:
• nausea • vomit blood
• more tired or weaker than usual • there is blood in your bowel movement
• diarrhea or it is black and sticky like tar
• itching • unusual weight gain
• your skin or eyes look yellow • skin rash or blisters with fever
• indigestion or stomach pain • swelling of the arms and legs, hands and
• flu-like symptoms feetIf you take too much of your NSAID, call your healthcare provider or get medical help right away.
These are not all the possible side effects of NSAIDs. For more information, ask your pharmacist about NSAIDs.
Call your doctor for medical advice about side effects. You may report side effects to FDA at
1-800-FDA-1088.Other information about NSAIDs
- Aspirin is an NSAID medicine but it does not increase the chance of a heart attack. Aspirin can cause bleeding in the brain, stomach, and intestines. Aspirin can also cause ulcers in the stomach and intestines.
- Some NSAIDs are sold in lower doses without a prescription (over-the-counter). Talk to your healthcare provider before using over-the-counter NSAIDs for more than 10 days.
General information about the safe and effective use of NSAIDs
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use NSAIDs for a condition for which it was not prescribed. Do not give NSAIDs to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information about NSAIDs, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about NSAIDs that is written for health professionals.
For more information go to www.hospira.com or call 1-800-615-0187.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 7/2015
Hospira, Inc., Lake Forest, IL 60045 USA
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
READYSHARP ANESTHETICS PLUS KETOROLAC
lidocaine hydrochloride, bupivacaine hydrochloride, and ketorolac tromethamine kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53225-4000 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53225-4000-1 1 in 1 KIT; Type 1: Convenience Kit of Co-Package 12/09/2016 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 AMPULE 2 mL Part 2 1 VIAL, SINGLE-DOSE 10 mL Part 3 1 VIAL, SINGLE-DOSE 1 mL Part 1 of 3 LIDOCAINE HYDROCHLORIDE
lidocaine hydrochloride injection, solutionProduct Information Item Code (Source) NDC:0409-4713 Route of Administration INFILTRATION Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LIDOCAINE HYDROCHLORIDE (UNII: V13007Z41A) (LIDOCAINE - UNII:98PI200987) LIDOCAINE HYDROCHLORIDE ANHYDROUS 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0409-4713-42 2 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA080408 05/16/1984 Part 2 of 3 MARCAINE
bupivacaine hydrochloride injection, solutionProduct Information Item Code (Source) NDC:0409-1560 Route of Administration EPIDURAL, INFILTRATION Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUPIVACAINE HYDROCHLORIDE (UNII: 7TQO7W3VT8) (BUPIVACAINE - UNII:Y8335394RO) BUPIVACAINE HYDROCHLORIDE ANHYDROUS 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0409-1560-10 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA016964 05/04/2010 Part 3 of 3 KETOROLAC TROMETHAMINE
ketorolac tromethamine injection, solutionProduct Information Item Code (Source) NDC:0409-3793 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength KETOROLAC TROMETHAMINE (UNII: 4EVE5946BQ) (KETOROLAC - UNII:YZI5105V0L) KETOROLAC TROMETHAMINE 15 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM HYDROXIDE (UNII: 55X04QC32I) SODIUM CHLORIDE (UNII: 451W47IQ8X) 6.68 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0409-3793-19 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074802 06/05/1997 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA016964 12/29/2015 Labeler - Terrain Pharmaceuticals (078358750)


