PANTOPRAZOLE SODIUM- pantoprazole sodium tablet, delayed release
REMEDYREPACK INC.
----------
These highlights do not include all the information needed to use pantoprazole sodium delayed-release tablets, USP safely and effectively. See full prescribing information for pantoprazole sodium delayed-release tablets, USP. Pantoprazole Sodium Delayed-Release Tablets, USP Initial U.S. approval: 2000
DRUG INTERACTIONS
Concomitant use of atazanavir or nelfinavir with proton pump inhibitors is not recommended. Coadministration of atazanavir or nelfinavir with proton pump inhibitors is expected to substantially decrease atazanavir or nelfinavir plasma concentrations and may result in a loss of therapeutic effect and development of drug resistance.
There have been postmarketing reports of increased INR and prothrombin time in patients receiving proton pump inhibitors, including pantoprazole sodium delayed-release tablets, and warfarin concomitantly. Increases in INR and prothrombin time may lead to abnormal bleeding and even death. Patients treated with proton pump inhibitors and warfarin concomitantly should be monitored for increases in INR and prothrombin time.
Pantoprazole causes long-lasting inhibition of gastric acid secretion. Therefore, pantoprazole may interfere with absorption of drugs where gastric pH is an important determinant of their bioavailability (e.g., ketoconazole, ampicillin esters, and iron salts).
DESCRIPTION
The active ingredient in pantoprazole sodium delayed-release tablets, USP is a substituted benzimidazole, sodium 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl] sulfinyl]1
H-benzimidazole sesquihydrate, a compound that inhibits gastric acid secretion. Its empirical formula is C
16H
14F
2N
3NaO
4S x 1.5 H
2O, with a molecular weight of 432.4. The structural formula is:
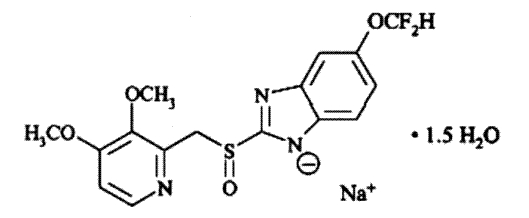
Pantoprazole sodium sesquihydrate, USP is a white to off-white powder and is racemic. Pantoprazole has weakly basic and acidic properties. Pantoprazole sodium sesquihydrate, USP is freely soluble in water and practically insoluble in hexane.
The stability of the compound in aqueous solution is pH-dependent. The rate of degradation increases with decreasing pH. At ambient temperature, the degradation half-life is approximately 2.8 hours at pH 5 and approximately 220 hours at pH 7.8.
Pantoprazole sodium is supplied as a delayed-release tablet for oral administration, available in 2 strengths (40 mg and 20 mg).
Each delayed-release tablet contains 45.1 mg or 22.56 mg of pantoprazole sodium sesquihydrate USP (equivalent to 40 mg or 20 mg pantoprazole, respectively) with the following inactive ingredients: aerosil 200, calcium stearate, colloidal silicon dioxide, mannitol, pregelatinized starch, shellac glaze, sodium carbonate anhydrous, sodium starch glycolate and talc. Each delayed-release tablet contains ammonium hydroxide, eudragit L 100-55, FD and C Blue #2, hypromellose, iron oxide black, iron oxide red, iron oxide yellow, polyethylene glycol 400, polyethylene glycol 4000, polyethylene glycol 6000, propylene glycol, sodium hydroxide, titanium dioxide and triethyl citrate as the coating ingredients.
Pantoprazole sodium delayed-release tablets USP, 20 mg and 40 mg, meet USP Dissolution Test 3.
HOW SUPPLIED
How Supplied
Pantoprazole sodium delayed-release tablets, USP are supplied as 20 mg yellow colored, biconvex oval shaped tablets plain on one side and imprinted with “W433” (brown ink) on other side.
HDPE bottle of 30 tablets.............................NDC 64679-433-01
HDPE bottle of 90 tablets.............................NDC 64679-433-04
HDPE bottle of 1000 tablets.........................NDC 64679-433-02
Unit dose packages of 100 tablets…..........NDC 64679-433-03
HDPE bottle of 100 tablets...........................NDC 64679-433-05
Pantoprazole sodium delayed-release tablets, USP are supplied as 40 mg yellow colored, biconvex oval shaped tablets plain on one side and imprinted with “W434” (brown ink) on other side.
HDPE bottle of 30 tablets............................NDC 64679-434-01
HDPE bottle of 90 tablets............................NDC 64679-434-04
HDPE bottle of 1000 tablets........................NDC 64679-434-02
Unit dose packages of 100 tablets.............NDC 64679-434-03
HDPE bottle of 100 tablets..........................NDC 64679-434-05
Storage
Store at 20°-25°C (68°-77°F); [See USP Controlled Room Temperature].

| PANTOPRAZOLE SODIUM
pantoprazole sodium tablet, delayed release |
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||
| Labeler - REMEDYREPACK INC. (829572556) |