DAUNORUBICIN HYDROCHLORIDE- daunorubicin hydrochloride injection, powder, for solution
Sanofi-Aventis U.S. LLC
Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here.
----------
Subject: Temporary importation of daunorubicin hydrochloride injection (Daunorubicin® 20 mg, Powder for I.V. Injection
IMPORTANT DRUG INFORMATION
June 17, 2014
Dear Healthcare Professional,
Due to the current shortage of daunorubicin hydrochloride solution for injection in the United States, Sanofi is coordinating with the U. S. Food and Drug Administration (FDA) to increase the availability of daunorubicin hydrochloride. In cooperation with the FDA, Sanofi has initiated temporary importation of Daunorubicin® 20 mg, Powder for I.V. Injection (Lot# 27498, 02/2017 expiration date) which is manufactured by Cenexi – Laboratoires Thissen SA, Belgium, into the US market. At this time, no other entity except Sanofi is authorized by the FDA to import or distribute Daunorubicin® 20 mg, Powder for I.V. Injection in the US. Any sales of this product from any entity other than Sanofi is considered a violation of the Federal Food, Drug, and Cosmetic Act and is subject to enforcement by the FDA.
While Daunorubicin® 20 mg, Powder for I.V. Injection is not an FDA approved drug in the United States, this importation involved a review of the product-associated information by FDA and has undergone review within Sanofi to ensure that the product meets the necessary quality and safety standards. Sanofi will continue to work with the FDA to potentially seek approval for additional importation of Daunorubicin® 20 mg, Powder for I.V. Injection as the drug shortage of daunorubicin hydrochloride continues.
Effective immediately, Sanofi will offer the following version of the product.
| Product name | Daunorubicin®
Daunorubicin Hydrochloride |
| Lot Number | 27498 |
| Expiration date | 02/2017 |
| Vial size | 20 mg vials |
| Quantity of vials in carton | 10 single vials in cartons |
It is important to note that there are several key differences between the current U.S. marketed daunorubicin and the imported product that you need to be aware of which appear in the following table. Please note the differences and follow the imported product handling and preparation instructions.
| Property | Daunorubicin Hydrochloride (U.S. product) | Daunorubicin Hydrochloride (Imported product) |
|---|---|---|
| Description | Sterile reddish lyophilized powder in vials | Microcrystalline orange-red hygroscopic sterile powder in vials |
| Dosage and Administration; Reconstitution | The contents of a vial should be reconstituted with 4 mL of Sterile Water for Injection and agitated gently until the material has completely dissolved. The sterile vial contents provide 20 mg of daunorubicin, with 5 mg of daunorubicin per mL. The desired dose is withdrawn into a syringe containing 10 mL to 15 mL of 0.9% Sodium Chloride Injection, USP and then injected into the tubing or sidearm in a rapidly flowing IV infusion of 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP. Cerubidine should not be administered mixed with other drugs or heparin. | The contents of a vial should be reconstituted with 4ml Water for Injections giving a concentration of 5mg per ml. The calculated dose of Daunorubicin should be further diluted with normal saline to give a final concentration of 1mg per ml. This solution should be injected over a 20 minute period into the tubing or a side-arm of a well placed, rapidly flowing i.v. infusion of normal saline (to minimize extravasation and possible tissue necrosis). Alternatively, the Daunorubicin may be added to a minibag of sodium chloride injection 0.9%w/v and this solution infused into the side-arm of a rapidly flowing infusion of normal saline. Daunorubicin injection is compatible with sodium chloride injection 0.9%. However, a precipitate may form immediately if the solution is mixed with heparin sodium injection or dexamethasone sodium phosphate injection. |
| Storage Conditions | Store unreconstituted powder at controlled room temperature, 15° to 30° C (59° to 86° F). The reconstituted solution is stable for 24 hours at room temperature and 48 hours under refrigeration. It should be protected from exposure to sunlight. Protect from light. Retain in carton until time of use. | The Vials containing Daunorubicin should be stored below 25°C and protected from light. Solutions of Daunorubicin should be used immediately after preparation or stored at 2–8°C and used within 24 hours. The solutions should be protected from light. The reconstituted solution does not contain an antimicrobial preservative. |
| Handling | Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published. | Daunorubicin injections may be neutralized with sodium hydrochloride prior to disposal of unused drug if a vial is accidentally broken. The neutralised drug can be disposed of in the sink. |
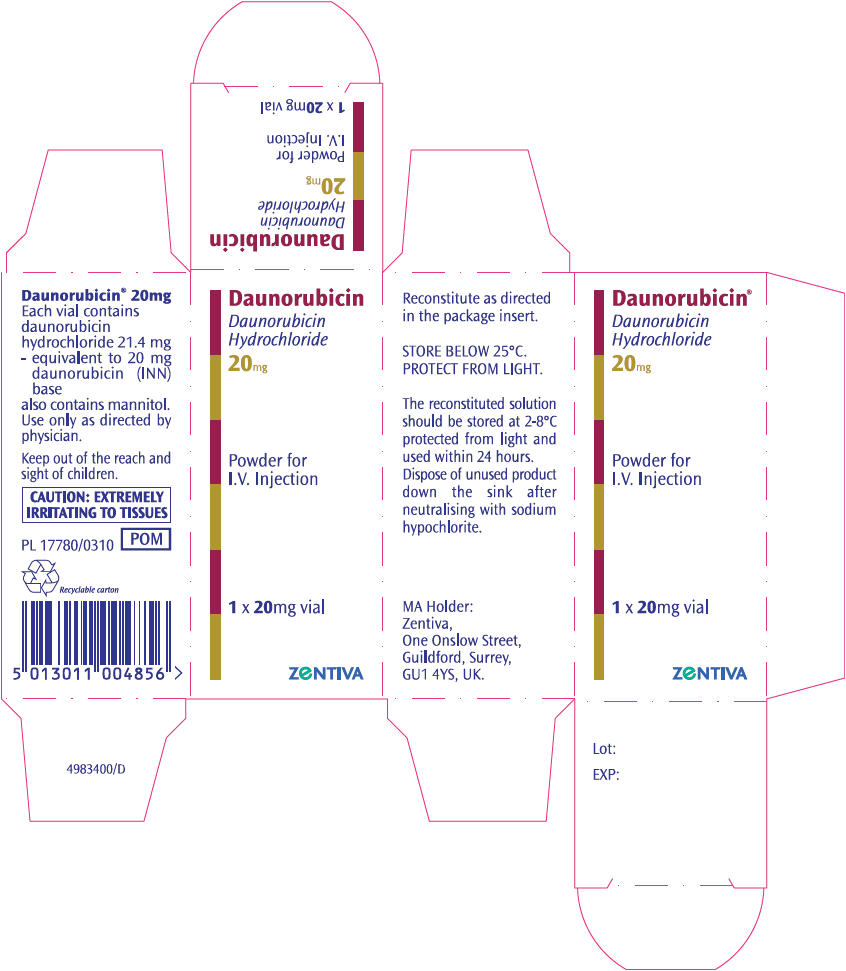
Daunorubicin hydrochloride (Imported product) vial label:

Daunorubicin hydrochloride (Imported product) carton:

Please note that the barcode used for the Cenexi – Laboratories Thissen SA's product may not be appropriately recognized by scanning systems used in the United States. Institutions should confirm the barcode systems do not provide incorrect information when the product is scanned. Alternate procedures should be followed to assure that the correct drug product is being used and administered to individual patients.
To report adverse events, product quality complaints, or to request product specific information related to these imported lots of DAUNORUBICIN HYDROCHLORIDE Injection, please contact Sanofi Medical Information Services at 1-800-633-1610.
Adverse events may also be reported to the FDA's MedWatch Adverse Event Reporting Program online, by regular mail or by fax:
- Online: www.fda.gov/medwatch/report.htm
- Regular mail: use postage paid FDA form 3500 available at www.fda.gov/MedWatch/getforms.htm
- Telephone: 1-800-332-1088
- Fax: 1-800-FDA-0178
For ongoing updates on daunorubicin injection supply issues, please visit the FDA's drug shortage website at www.fda.gov/Drugs/DrugSafety/DrugShortages/ucm050792.htm.
Sincerely,
Charles Hugh-Jones, MD
Chief Medical Officer
Sanofi North America
sanofi-aventis U.S.
| FDA Approved Cerubidine (daunorubicin hydrochloride) Injection, Powder, Lyophilized | Daunorubicin Hydrochloride Injection Powder Imported Product | |
|---|---|---|
| Composition | ||
| Active ingredient | Daunorubicin hydrochloride | Daunorubicin hydrochloride |
| Strength | Each vial contains 21.4 mg daunorubicin hydrochloride (equivalent to 20 mg of daunorubicin) and 100 mg of mannitol. When reconstituted with 4 mL of Sterile Water for Injection USP, each mL contains 5 mg daunorubicin activity. | Each vial contains the equivalent of 20 mg of daunorubicin (as hydrochloride); the contents of a vial should be reconstituted with 4ml Water for Injections giving a concentration of 5mg per ml. |
| Inactive ingredients | Mannitol | Mannitol |
| Physiochemical parameters | ||
| pH | pH of a 5 mg/mL aqueous solution is 4.5 to 6.5 | Daunorubicin solutions are unstable above pH8, decomposition being indicated by a change from red to blue-purple colour. |
| Description | Sterile reddish lyophilized powder in vials | Microcrystalline orange-red hygroscopic sterile powder in vials |
| How Supplied | 20 mg, single dose vials, carton of 10 | 20 mg vials, carton of 10 |
| Storage conditions | Store unreconstituted powder at controlled room temperature, 15° to 30° C (59° to 86° F). The reconstituted solution is stable for 24 hours at room temperature and 48 hours under refrigeration. It should be protected from exposure to sunlight. Protect from light. Retain in carton until time of use. | Vials containing Daunorubicin should be stored below 25°C and protected from light. Solutions of Daunorubicin should be used immediately after preparation or stored at 2–8°C and used within 24 hours. The solutions should be protected from light. The reconstituted solution does not contain an antimicrobial preservative. |
| Country-specific information | ||
| Expiration date format | MMM YY | MM/YYYY |
| Authorization holder | Bedford Laboratories, Bedford OH | Aventis Pharma Ltd., Kent, UK |
| FDA Approved Cerubidine (daunorubicin hydrochloride) Injection, Powder, Lyophilized | Daunorubicin Hydrochloride Injection Imported Product | |||||
|---|---|---|---|---|---|---|
| Clinical pharmacology | Mechanism of Action | |||||
| Cerubidine has antimitotic and cytotoxic activity through a number of proposed mechanisms of action. Cerubidine forms complexes with DNA by intercalation between base pairs. It inhibits topoisomerase II activity by stabilizing the DNA-topoisomerase II complex, preventing the religation portion of the ligation-religation reaction that topoisomerase II catalyzes. Single strand and double strand DNA breaks result. Cerubidine may also inhibit polymerase activity, affect regulation of gene expression, and produce free radical damage to DNA. Cerubidine possesses an antitumor effect against a wide spectrum of animal tumors, either grafted or spontaneous. | ||||||
| Pharmacokinetics | Pharmacokinetics | |||||
| General Following intravenous injection of Cerubidine, plasma levels of daunorubicin decline rapidly, indicating rapid tissue uptake and concentration. Thereafter, plasma levels decline slowly with a half-life of 45 minutes in the initial phase and 18.5 hours in the terminal phase. By 1 hour after drug administration, the predominant plasma species is daunorubicinol, an active metabolite, which disappears with a half-life of 26.7 hours. Distribution Cerubidine is rapidly and widely distributed in tissues, with highest levels in the spleen, kidneys, liver, lungs, and heart. The drug binds to many cellular components, particularly nucleic acids. There is no evidence that Cerubidine crosses the blood-brain barrier, but the drug apparently crosses the placenta. Metabolism and Elimination Cerubidine is extensively metabolized in the liver and other tissues, mainly by cytoplasmic aldo-keto reductases, producing daunorubicinol, the major metabolite which has antineoplastic activity. Approximately 40% of the drug in the plasma is present as daunorubicinol within 30 minutes and 60% in 4 hours after a dose of daunorubicin. Further metabolism via reduction cleavage of the glycosidic bond, 4-O demethylation, and conjugation with both sulfate and glucuronide have been demonstrated. Simple glycosidic cleavage of daunorubicin or daunorubicinol is not a significant metabolic pathway in man. Twenty-five percent of an administered dose of Cerubidine is eliminated in an active form by urinary excretion and an estimated 40% by biliary excretion. | Daunorubicin is rapidly taken up by the tissues, especially by the kidneys, spleen, liver and heart. It does not cross the blood-brain barrier, subsequent release of the drug and its metabolites by the tissue is slow (t1/2 = 55 hours). Daunorubicin is rapidly metabolized in the liver. The major metabolite daunorubicin is also active. Daunorubicin is excreted slowly in the urine, mainly as metabolites with 25% excreted in the first 5 days. Biliary excretion also makes a significant (40%) contribution to elimination. | |||||
| Special Populations | ||||||
|
Although appropriate studies with Cerubidine have not been performed in the pediatric population, cardiotoxicity may be more frequent and occur at lower cumulative doses in children. Although appropriate studies with Cerubidine have not been performed in the geriatric population, cardiotoxicity may be more frequent in the elderly. Caution should also be used in patients who have inadequate bone marrow reserves due to old age. In addition, elderly patients are more likely to have age-related renal function impairment, which may require reduction of dosage in patients receiving Cerubidine. Renal and Hepatic Impairment Doses of Cerubidine should be reduced in patients with hepatic and renal impairment. Patients with serum bilirubin concentrations of 1.2 to 3 mg/dL should receive 75% of the usual daily dose and patients with serum bilirubin concentrations greater than 3 mg/dL should receive 50% of the usual daily dose. Patients with serum creatinine concentrations of greater than 3 mg/dL should receive 50% of the usual daily dose. (See WARNINGS, Evaluation of Hepatic and Renal Function). | ||||||
| Clinical Studies | ||||||
| In the treatment of adult acute nonlymphocytic leukemia, Cerubidine, used as a single agent, has produced complete remission rates of 40 to 50%, and in combination with cytarabine, has produced complete remission rates of 53 to 65%. The addition of Cerubidine to the two-drug induction regimen of vincristine-prednisone in the treatment of childhood acute lymphocytic leukemia does not increase the rate of complete remission. In children receiving identical CNS prophylaxis and maintenance therapy (without consolidation), there is prolongation of complete remission duration (statistically significant, p<0.02) in those children induced with the three drug (Cerubidine-vincristine-prednisone) regimen as compared to two drugs. There is no evidence of any impact of Cerubidine on the duration of complete remission when a consolidation (intensification) phase is employed as part of a total treatment program. In adult acute lymphocytic leukemia, in contrast to childhood acute lymphocytic leukemia, Cerubidine during induction significantly increases the rate of complete remission, but not remission duration, compared to that obtained with vincristine, prednisone, and L-asparaginase alone. The use of Cerubidine in combination with vincristine, prednisone, and L-asparaginase has produced complete remission rates of 83% in contrast to a 47% remission in patients not receiving Cerubidine. | ||||||
| Indications and usage | Cerubidine in combination with other approved anticancer drugs is indicated for remission induction in acute nonlymphocytic leukemia (myelogenous, monocytic, erythroid) of adults and for remission induction in acute lymphocytic leukemia of children and adults. | Daunorubicin is an anthracycline glycoside antibiotic and a potent antileukaemic agent. It also has immune suppressant effects. The exact mechanism of antineoplastic action of daunorubicin is uncertain but may involve binding to DNA by intercalation between base pairs and inhibition of DNA and RNA synthesis by template disordering and steric obstruction. Daunorubicin is most active in the S-phase of cell division but is not cycle phase-specific. Tumour cell cross resistance has been observed between daunorubicin and doxorubicin. No controlled paediatric have been conducted. The literature mentions the use of daunorubicin in treatment regimens for ALL and AML, including paediatric age groups. However, due to the ongoing search for a balance in gain or maintenance of efficacy and a decrease in toxicity the use of daunorubicin in the treatment of paediatric ALL and AML is fluctuating in clinical practice, mainly depending on risk stratification and specific subgroups. Published studies suggest no differences in safety profile between paediatric patients and adults. |
||||
| Boxed Warnings | WARNINGS | |||||
| ||||||
| Contraindications | Cerubidine is contraindicated in patients who have shown a hypersensitivity to it. | Do not use in patients recently exposed to, or with existing chicken pox or herpes zoster. | ||||
| Warnings/Precautions | Precautions and Warnings | |||||
| Bone Marrow
Cerubidine is a potent bone marrow suppressant. Suppression will occur in all patients given a therapeutic dose of this drug. Therapy with Cerubidine should not be started in patients with pre-existing drug-induced bone marrow suppression unless the benefit from such treatment warrants the risk. Persistent, severe myelosuppression may result in superinfection or hemorrhage. Special attention must be given to the potential cardiac toxicity of Cerubidine, particularly in infants and children. Pre-existing heart disease and previous therapy with doxorubicin are co-factors of increased risk of Cerubidine-induced cardiac toxicity and the benefit-to-risk ratio of Cerubidine therapy in such patients should be weighed before starting Cerubidine. In adults, at total cumulative doses less than 550 mg/m2, acute congestive heart failure is seldom encountered. However, rare instances of pericarditis-myocarditis, not dose-related, have been reported. In adults, at cumulative doses exceeding 550 mg/m2, there is an increased incidence of drug-induced congestive heart failure. Based on prior clinical experience with doxorubicin, this limit appears lower, namely 400 mg/m2, in patients who received radiation therapy that encompassed the heart. In infants and children, there appears to be a greater susceptibility to anthracycline-induced cardiotoxicity compared to that in adults, which is more clearly dose-related. Anthracycline therapy (including daunorubicin) in pediatric patients has been reported to produce impaired left ventricular systolic performance, reduced contractility, congestive heart failure or death. These conditions may occur months to years following cessation of chemotherapy. This appears to be dose-dependent and aggravated by thoracic irradiation. Long-term periodic evaluation of cardiac function in such patients should, thus, be performed. In both children and adults, the total dose of Cerubidine administered should also take into account any previous or concomitant therapy with other potentially cardiotoxic agents or related compounds such as doxorubicin. There is no absolutely reliable method of predicting the patients in whom acute congestive heart failure will develop as a result of the cardiac toxic effect of Cerubidine. However, certain changes in the electrocardiogram and a decrease in the systolic ejection fraction from pre-treatment baseline may help to recognize those patients at greatest risk to develop congestive heart failure. On the basis of the electrocardiogram, a decrease equal to or greater than 30% in limb lead QRS voltage has been associated with a significant risk of drug-induced cardiomyopathy. Therefore, an electrocardiogram and/or determination of systolic ejection fraction should be performed before each course of Cerubidine. In the event that one or the other of these predictive parameters should occur, the benefit of continued therapy must be weighed against the risk of producing cardiac damage. Early clinical diagnosis of drug-induced congestive heart failure appears to be essential for successful treatment. Evaluation of Hepatic and Renal Function Significant hepatic or renal impairment can enhance the toxicity of the recommended doses of Cerubidine; therefore, prior to administration, evaluation of hepatic function and renal function using conventional clinical laboratory tests is recommended (see DOSAGE AND ADMINISTRATION). | Daunorubicin should be used under direction of a clinician conversant with the management of acute leukaemia and cytotoxic chemotherapy. The haematological status of patients should be monitored regularly. Daunorubicin might depress the bone marrow to the point where anti-infective agents would no longer be effective. If facilities are available, patients should be treated in a germ free environment or, where this is not possible, reverse barrier nursing and aseptic precautions should be employed. Anti-infective therapy should be employed in the presence of suspected or confirmed infection and during a phase of aplasia. It should be continued for some time after the marrow has regenerated. Care should also be used in patients at risk of infection. Rapid destruction of a large number of leukaemia cells may cause a rise in the blood uric acid or urea so it is a wise precaution to check these concentrations three or four times a week during the first week of treatment. Fluids should be administered and allopurinol used in severe cases to prevent the development of hyperuricaemia. Patients with heart disease should not be treated with this potentially cardiotoxic drug. Cardiotoxicity, if it occurs, is likely to be heralded by either a persistent tachycardia, shortness of breath and swelling of feet and lower limbs, or by minor changes in the electrocardiogram and for this reason an electrocardiographic examination should be made at regular intervals during treatment. Cardiotoxicity usually appears within 1 to 6 months after initiation of therapy. It may develop suddenly and nor be detected by routine ECG. It may be irreversible and fatal but responds to treatment if detected early. The risk of congestive heart failure increases significantly when the total cumulative dosage exceeds 600mg/m2 in adults, 300mg/m2 in children over 2 years or 10mg/kg in children under 2 years. Cardiotoxicity may be more frequent in children and the elderly. The dosage should be modified if previous or concomitant cardiotoxic drug therapy is used. Daunorubicin should be used with care in patients at risk of hyperuricaemia (e.g. in the presence of gout, urate and renal calculi), tumour cell infiltration of the bone marrow and in patients with inadequate bone marrow reserves due to previous cytotoxic drug or radiation therapy. The cumulative dose of Daunorubicin should be limited to 400mg/m2 when radiation therapy to the mediastinium has been previously administered. The dose of Daunorubicin should not be repeated in the presence of bone marrow depression or buccal ulceration. Care should be taken to avoid extravasation during intravenous administration. All steps should be taken to avoid tissuing and bandages should not be used. Facial flushing or erythematous streaking along the vein indicates too rapid injection. If tissue necrosis is suspected, the infusion should be stopped immediately and resumed in another vein. Where extravasation has occurred, and attempt should be made to aspirate the fluid back through the needle. The effected area may be injected with hydrocortisone. Sodium bicarbonate (5ml of 8–4% w/v solution) may also be injected in the hope that through pH changes the drug will hydrolyse. The opinion of the plastic surgeon should be sought as skin grafting may be required. Application of ice packs may help to decrease local discomfort and also prevent extension. Liberal application of corticosteroid cream and dressing the area with sterile gauze should then be carried out. |
|||||
| Pregnancy | ||||||
| Cerubidine may cause fetal harm when administered to a pregnant woman. An increased incidence of fetal abnormalities (parieto-occipital cranioschisis, umbilical hernias, or rachischisis) and abortions was reported in rabbits at doses of 0.05 mg/kg/day or approximately 1/100th of the highest recommended human dose on a body surface area basis. Rats showed an increased incidence of esophageal, cardiovascular and urogenital abnormalities as well as rib fusions at doses of 4 mg/kg/day or approximately 1/2 the human dose on a body surface area basis. Decreases in fetal birth weight and post-delivery growth rate were observed in mice. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant. Secondary Leukemias There have been reports of secondary leukemias in patients exposed to topoisomerase II inhibitors when used in combination with other antineoplastic agents or radiation therapy. Extravasation at Injection Site Extravasation of Cerubidine at the site of intravenous administration can cause severe local tissue necrosis. (See ADVERSE REACTIONS) | Daunorubicin crosses the placenta and experiments in animals have shown it to be mutagenic, carcinogenic and teratogenic. There is also the possibility that treatment during pregnancy may produce delayed effects in the offspring. If appropriate, the mother should be offered the opportunity of a therapeutic abortion. Owing to potential toxic risks to infants, breastfeeding should be discontinued during treatment. |
|||||
| Precautions
General Therapy with Cerubidine requires close patient observation and frequent complete blood-count determinations. Cardiac, renal, and hepatic function should be evaluated prior to each course of treatment. Appropriate measures must be taken to control any systemic infection before beginning therapy with Cerubidine. Cerubidine may transiently impart a red coloration to the urine after administration, and patients should be advised to expect this. Laboratory Tests Cerubidine may induce hyperuricemia secondary to rapid lysis of leukemic renal cells. As a precaution, allopurinol administration is usually begun prior to initiating antileukemic therapy. Blood uric acid levels should be monitored and appropriate therapy initiated in the event that hyperuricemia develops. Carcinogenesis, Mutagenesis, Impairment of Fertility Cerubidine, when injected subcutaneously into mice, causes fibrosarcomas to develop at the injection site. When administered to mice thrice weekly intraperitoneally, no carcinogenic effect was noted after 18 months of observation. In male rats administered Cerubidine thrice weekly for 6 months, at 1/70th the recommended human dose on a body surface area basis, peritoneal sarcomas were found at 18 months. A single IV dose of Cerubidine administered to rats at 1.6 fold the recommended human dose on a body surface area basis caused mammary adenocarcinomas to appear at 1 year. Cerubidine was mutagenic in vitro (Ames assay, V79 hamster cell assay), and clastogenic in vitro (CCRFCEM human lymphoblasts) and in vivo (SCE assay in mouse bone marrow) tests. In male dogs at a daily dose of 0.25 mg/kg administered intravenously, testicular atrophy was noted at autopsy. Histologic examination revealed total aplasia of the spermatocyte series in the seminiferous tubules with complete aspermatogenesis. Pregnancy Category D (See WARNINGS) Nursing Mothers It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Cerubidine, mothers should be advised to discontinue nursing during Cerubidine therapy. Elderly See CLINICAL PHARMACOLOGY, Special Populations, Geriatric Patients. Pediatric Use See CLINICAL PHARMACOLOGY, Special Populations, Pediatric Patients section and WARNINGS, Cardiac Effects. Drug Interactions Use of Cerubidine in a patient who has previously received doxorubicin increases the risk of cardiotoxicity. Cerubidine should not be used in patients who have previously received the recommended maximum cumulative doses of doxorubicin or Cerubidine. Cyclophosphamide used concurrently with Cerubidine may also result in increased cardiotoxicity. Dosage reduction of Cerubidine may be required when used concurrently with other myelosuppressive agents. Hepatotoxic medications, such as high-dose methotrexate, may impair liver function and increase the risk of toxicity. | ||||||
| Dose-limiting toxicity includes myelosuppression and cardiotoxicity (see WARNINGS). Other reactions include: Cutaneous Reversible alopecia occurs in most patients. Rash, contact dermatitis and urticaria have occurred rarely. Gastrointestinal Acute nausea and vomiting occur but are usually mild. Antiemetic therapy may be of some help. Mucositis may occur 3 to 7 days after administration. Diarrhea and abdominal pain have occasionally been reported. Local If extravasation occurs during administration, severe local tissue necrosis, severe cellulitis, thrombophlebitis, or painful induration can result. Acute Reactions Rarely, anaphylactoid reaction, fever, and chills can occur. Hyperuricemia may occur, especially in patients with leukemia, and serum uric acid levels should be monitored. | Bone marrow depression: in every patient bone marrow function will be depressed by treatment with Daunorubicin and in a variable proportion of cases, severe aplasia will develop. The consequence may include severe infection and opportunistic infection. Leucopenia is usually more significant than thrombocytopenia. The nadir for leucopenia usually occurs between 10–14 days and recovery occurs gradually over the next 1–2 weeks. Bone marrow depression must be anticipated in every case by eliminating infection before the treatment, by isolating the patient from infection during the treatment and by means of supportive therapy. This includes the continuous administration of anti-infective agents, the administration of platelet-rich plasma or fresh whole blood transfusion and, under some circumstances, the transfusion of white cell concentrates. Other less serious adverse reactions that have been reported (in order of reducing frequency) are: stomatitis, alopecia, phlebitis, fever, anaemia, nausea, vomiting, mucositis, diarrhoea and rash. The urine may be temporarily coloured red after treatment. |
|||||
| Overdose | Although no cases have been reported to our knowledge, overdosage may result in drastic myelosuppression and severe cardiotoxicity with or without transient reversible ECG changes leading to congestive heart failure. Treatment should be supportive and symptomatic. | |||||
| Parenteral drug products should be inspected visually for particulate matter prior to administration, whenever solution and container permit. Principles In order to eradicate the leukemic cells and induce a complete remission, a profound suppression of the bone marrow is usually required. Evaluation of both the peripheral blood and bone marrow is mandatory in the formulation of appropriate treatment plans. It is recommended that the dosage of Cerubidine be reduced in instances of hepatic or renal impairment. For example, using serum bilirubin and serum creatinine as indicators of liver and kidney function, the following dose modifications are recommended: | Daunorubicin is extremely irritating to tissues and may only be administered intravenously. Do not administer by the intramuscular or subcutaneous route.
The contents of a vial should be reconstituted with 4ml Water for Injections giving a concentration of 5mg per ml. The calculated dose of Daunorubicin should be further diluted with normal saline to give a final concentration of 1mg per ml. This solution should be injected over a 20 minute period into the tubing or a side-arm of a well placed, rapidly flowing i.v. infusion of normal saline (to minimize extravasation and possible tissue necrosis). Alternatively, the Daunorubicin may be added to a minibag of sodium chloride injection 0.9%w/v and this solution infused into the side-arm of a rapidly flowing infusion of normal saline. |
|||||
| Serum Bilirubin | Serum Creatinine | Dose Reduction | ||||
| 1.2 to 3.0 mg% | - | 25% | ||||
| >3 mg% | - | 50% | ||||
| - | >3 mg% | 50% | ||||
|
Representative Dose Schedules and Combination for the Approved Indication of Remission Induction in Adult Acute Nonlymphocytic Leukemia In Combination For patients under age 60, Cerubidine 45 mg/m2/day IV on days 1, 2, and 3 of the first course and on days 1, 2 of subsequent courses AND cytosine arabinoside 100 mg/m2/day IV infusion daily for 7 days for the first course and for 5 days for subsequent courses. For patients 60 years of age and above, Cerubidine 30 mg/m2/day IV on days 1, 2, and 3 of the first course and on days 1, 2 of subsequent courses AND cytosine arabinoside 100 mg/m2/day IV infusion daily for 7 days for the first course and for 5 days for subsequent courses. This Cerubidine dose-reduction is based on a single study and may not be appropriate if optimal supportive care is available. The attainment of a normal-appearing bone marrow may require up to three courses of induction therapy. Evaluation of the bone marrow following recovery from the previous course of induction therapy determines whether a further course of induction treatment is required. Representative Dose Schedule and Combination for the Approved Indication of Remission Induction in Pediatric Acute Lymphocytic Leukemia In Combination Cerubidine 25 mg/m2 IV on day 1 every week, vincristine 1.5 mg/m2 IV on day 1 every week, prednisone 40 mg/m2 PO daily. Generally, a complete remission will be obtained within four such courses of therapy; however; if after four courses the patient is in partial remission, an additional one or, if necessary, two courses may be given in an effort to obtain a complete remission. In children less than 2 years of age or below 0.5 m2 body surface area, it has been recommended that the Cerubidine dosage calculation should be based on weight (1 mg/kg) instead of body surface area. Representative Dose Schedules and Combination for the Approved Indication of Remission Induction in Adult Acute Lymphocytic Leukemia In Combination Cerubidine 45 mg/m2/day IV on days 1, 2, and 3 AND vincristine 2 mg IV on days 1, 8, and 15; prednisone 40 mg/m2/day PO on days 1 through 22, then tapered between days 22 to 29; L-asparaginase 500 IU/kg/day × 10 days IV on days 22 through 32. The contents of a vial should be reconstituted with 4 mL of Sterile Water for Injection and agitated gently until the material has completely dissolved. The sterile vial contents provide 20 mg of daunorubicin with 5 mg of daunorubicin per mL. The desired dose is withdrawn into a syringe containing 10 mL to 15 mL of 0.9% Sodium Chloride Injection, USP and then injected into the tubing or sidearm in a rapidly flowing IV infusion of 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP. Cerubidine should not be administered mixed with other drugs or heparin. Storage and Handling Store reconstituted powder at controlled room temperature, 15° to 30°C (59° to 86°F). The reconstituted solution is stable for 24 hours at room temperature and 48 hours under refrigeration. It should be protected from exposure to sunlight. Protect from light. Retain in carton until time of use. If Cerubidine contacts the skin or mucosae, the area should be washed thoroughly with soap and water. Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published. There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate. |
Adults: 40–60mg/m2 on alternate day for a course of up to three injections for the induction of remissions. Acute myelogenous leukaemia: The recommended dose is 45mg/m2. Acute lymphocytic leukaemia: The recommended dose is 45mg/m2. Paediatric patients: Daunorubicin dosage for children (over 2 years) is usually calculated by based on body surface area and adjusted to meet the individual requirements of each patient, on the basis of clinical response and the patients' haematological status. Courses may be repeated after 3 to 6 weeks. Current specialised protocols and guidelines should be consulted for appropriate treatment regimen. For children over 2 years the maximum cumulative dose os 300mg/m2. For children under 2 years (or below 0.5m2 body surface area), the maximum cumulative dose is 10mg/kg. Elderly: Daunorubicin should be used with care in patients with inadequate bone marrow reserves due to old age. A reduction of up to 50% in dosage is recommended. The number of injections required varies widely from patient to patient and must be determined in each case according to response and tolerance. Daunorubicin should be administered through a large vein and the infusion should be kept free flowing. When second or subsequent injections are given, the doses and the time intervals depend on the effect of the previous doses and must be the subject of careful deliberation, examination of the peripheral blood and, under some circumstances, of the bone marrow. The effect of Daunorubicin on the disease process and on the normal blood precursors cannot be exactly predicted for any particular use. The difference between incomplete treatment, a satisfactory remission, and overdosage with possible irreversible aplasia of the bone marrow depends on the correct choice of dosage, time intervals and total number of doses. The dosage should be reduced in patients with impaired hepatic or renal function. A 25% reduction is recommended in patients with serum bilirubin concentrations of 20–50 umol/l or creatinine of 105–265 umol/l. A 50% reduction is recommended in cases with serum bilirubin concentrations of above 50 umol/l or creatinine of above 265 umol/l. Pharmaceutical Precautions Daunorubicin injection is compatible with sodium chloride injection 0.9%. However a precipitate may form immediately if the solution is mixed with heparin sodium injection or dexamethasone sodium phosphate injection. Further Information Spill or leak procedures : Daunorubicin injection may be neutralised with sodium hypochlorite prior to disposal of unused drug or if vial is accidently broken. The neutralised drug can be disposed of in the sink. Special Protection Information Daunorubicin should only be handled by staff experienced with cytotoxic drugs. Further information is available from the Marketing Autorisation holder. |
|||||
| February 2008 | Text issued June 2013. | |||||

| DAUNORUBICIN HYDROCHLORIDE
daunorubicin hydrochloride injection, powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Sanofi-Aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Cenexi - Laboratoires Thissen Sa | 370088959 | MANUFACTURE(0024-5858), ANALYSIS(0024-5858), LABEL(0024-5858), PACK(0024-5858) | |