OLYSIO- simeprevir capsule
Janssen Products LP
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use OLYSIO® safely and effectively. See full prescribing information for OLYSIO.
OLYSIO (simeprevir) capsules, for oral use Initial U.S. Approval: 2013 WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBVSee full prescribing information for complete boxed warning.Hepatitis B virus (HBV) reactivation has been reported, in some cases resulting in fulminant hepatitis, hepatic failure, and death. (5.1) RECENT MAJOR CHANGES
INDICATIONS AND USAGEOLYSIO is a hepatitis C virus (HCV) NS3/4A protease inhibitor indicated for the treatment of adults with chronic hepatitis C virus (HCV) infection:
Limitations of Use:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCapsules: 150 mg (3) CONTRAINDICATIONSBecause OLYSIO is used only in combination with other antiviral drugs (including Peg-IFN-alfa and RBV) for the treatment of chronic HCV infection, the contraindications to other drugs also apply to the combination regimen. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 11/2017 |
||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
Test all patients for evidence of current or prior hepatitis B virus (HBV) infection before initiating treatment with OLYSIO. HBV reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals and were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Monitor HCV/HBV coinfected patients for hepatitis flare or HBV reactivation during HCV treatment and post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
OLYSIO® is indicated for the treatment of adults with chronic hepatitis C virus (HCV) infection [see Dosage and Administration (2.2) and Clinical Studies (14)]:
- in combination with sofosbuvir in patients with HCV genotype 1 without cirrhosis or with compensated cirrhosis
- in combination with peginterferon alfa (Peg-IFN-alfa) and ribavirin (RBV) in patients with HCV genotype 1 or 4 without cirrhosis or with compensated cirrhosis.
Limitations of Use:
- Efficacy of OLYSIO in combination with Peg-IFN-alfa and RBV is substantially reduced in patients infected with HCV genotype 1a with an NS3 Q80K polymorphism at baseline compared to patients infected with hepatitis C virus (HCV) genotype 1a without the Q80K polymorphism [see Dosage and Administration (2.1) and Microbiology (12.4)].
- OLYSIO is not recommended in patients who have previously failed therapy with a treatment regimen that included OLYSIO or other HCV protease inhibitors [see Microbiology (12.4)].
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to the Initiation of Therapy
Testing for HBV infection
Test all patients for evidence of current or prior HBV infection by measuring hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) before initiating HCV treatment with OLYSIO [see Warnings and Precautions (5.1)].
Q80K Testing in HCV Genotype 1a-Infected Patients
OLYSIO in Combination with Sofosbuvir
In HCV genotype 1a-infected patients with compensated cirrhosis, screening for the presence of virus with the NS3 Q80K polymorphism may be considered prior to initiation of treatment with OLYSIO with sofosbuvir [see Clinical Studies (14.2)].
OLYSIO in Combination with Peg-IFN-alfa and RBV
Prior to initiation of treatment with OLYSIO in combination with Peg-IFN-alfa and RBV, screening patients with HCV genotype 1a infection for the presence of virus with the NS3 Q80K polymorphism is strongly recommended and alternative therapy should be considered for patients infected with HCV genotype 1a containing the Q80K polymorphism [see Indications and Usage (1) and Microbiology (12.4)].
Hepatic Laboratory Testing
Monitor liver chemistry tests before and during OLYSIO combination therapy [see Warnings and Precautions (5.3)].
2.2 OLYSIO Combination Treatment
Administer OLYSIO in combination with other antiviral drugs for the treatment of chronic HCV infection. OLYSIO monotherapy is not recommended. The recommended dosage of OLYSIO is one 150 mg capsule taken orally once daily with food [see Clinical Pharmacology (12.3)]. The capsule should be swallowed as a whole. For specific dosing recommendations for the antiviral drugs used in combination with OLYSIO, refer to their respective prescribing information.
OLYSIO can be taken in combination with sofosbuvir or in combination with Peg-IFN-alfa and RBV.
OLYSIO in Combination with Sofosbuvir
Table 1 displays the recommended treatment regimen and duration of OLYSIO in combination with sofosbuvir in patients with chronic HCV genotype 1 infection.
| Patient Population (HCV Genotype 1) | Treatment Regimen and Duration |
|---|---|
|
|
| Treatment-naïve and treatment-experienced* patients: | |
| without cirrhosis | 12 weeks of OLYSIO + sofosbuvir |
| with compensated cirrhosis (Child-Pugh A) | 24 weeks of OLYSIO + sofosbuvir |
OLYSIO in Combination with Peg-IFN-alfa and RBV
Table 2 displays the recommended treatment regimen and duration of OLYSIO in combination with Peg-IFN-alfa and RBV in mono-infected and HCV/HIV-1 co-infected patients with HCV genotype 1 or 4 infection. Refer to Table 3 for treatment stopping rules for OLYSIO combination therapy with Peg-IFN-alfa and RBV.
| Patient Population (HCV Genotype 1 or 4) | Treatment Regimen and Duration |
|---|---|
| HIV = human immunodeficiency virus. | |
|
|
| Treatment-naïve patients and prior relapsers*: | |
| HCV mono-infected patients without cirrhosis or with compensated cirrhosis (Child-Pugh A) | 12 weeks of OLYSIO + Peg-IFN-alfa + RBV followed by additional 12 weeks of Peg-IFN-alfa + RBV (total treatment duration of 24 weeks)† |
| HCV/HIV-1 co-infected patients without cirrhosis | |
| HCV/HIV-1 co-infected patients with compensated cirrhosis (Child-Pugh A) | 12 weeks of OLYSIO + Peg-IFN-alfa + RBV followed by additional 36 weeks of Peg-IFN-alfa + RBV (total treatment duration of 48 weeks)† |
| Prior non-responders (including partial‡ and null responders§): | |
| HCV/HIV-1 co-infected or HCV mono-infected patients without cirrhosis or with compensated cirrhosis (Child-Pugh A) | 12 weeks of OLYSIO + Peg-IFN-alfa + RBV followed by additional 36 weeks of Peg-IFN-alfa + RBV (total treatment duration of 48 weeks)† |
2.3 Discontinuation of Dosing
OLYSIO in Combination with Sofosbuvir
No treatment stopping rules apply to the combination of OLYSIO with sofosbuvir [see Clinical Studies (14.2)].
OLYSIO in Combination with Peg-IFN-alfa and RBV
During treatment, HCV RNA levels should be monitored as clinically indicated using a sensitive assay with a lower limit of quantification of at least 25 IU/mL. Because patients with an inadequate on-treatment virologic response (i.e., HCV RNA greater or equal to 25 IU/mL) are not likely to achieve a sustained virologic response (SVR), discontinuation of treatment is recommended in these patients. Table 3 presents treatment stopping rules for patients who experience an inadequate on-treatment virologic response at Weeks 4, 12, and 24.
| Treatment Week | HCV RNA | Action |
|---|---|---|
| Week 4 | ≥ 25 IU/mL | Discontinue OLYSIO, Peg-IFN-alfa, and RBV |
| Week 12 | Discontinue Peg-IFN-alfa, and RBV (treatment with OLYSIO is complete at Week 12) | |
| Week 24 | Discontinue Peg-IFN-alfa, and RBV (treatment with OLYSIO is complete at Week 12) |
2.4 Dosage Adjustment or Interruption
To prevent treatment failure, avoid reducing the dosage of OLYSIO or interrupting treatment. If treatment with OLYSIO is discontinued because of adverse reactions or inadequate on-treatment virologic response, OLYSIO treatment must not be reinitiated [see Warnings and Precautions (5.3)].
If adverse reactions potentially related to the antiviral drug(s) used in combination with OLYSIO occur, refer to the instructions outlined in their respective prescribing information for recommendations on dosage adjustment or interruption.
If any of the other antiviral drug(s) used in combination with OLYSIO for the treatment of chronic HCV infection are permanently discontinued for any reason, OLYSIO should also be discontinued.
2.5 Not Recommended in Patients with Moderate or Severe Hepatic Impairment
OLYSIO is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C) [see Warnings and Precautions (5.3), Adverse Reactions (6.1), Use in Specific Populations (8.8), and Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
OLYSIO is available as a white gelatin capsule marked with "TMC435 150" in black ink. Each capsule contains 150 mg simeprevir.
4 CONTRAINDICATIONS
Because OLYSIO is used only in combination with other antiviral drugs (including Peg-IFN-alfa and RBV) for the treatment of chronic HCV infection, the contraindications to other drugs also apply to the combination regimen. Refer to the respective prescribing information for a list of contraindications.
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Cases have been reported in patients who are HBsAg positive and also in patients with serologic evidence of resolved HBV infection (i.e., HBsAg negative and anti-HBc positive). HBV reactivation has also been reported in patients receiving certain immunosuppressant or chemotherapeutic agents; the risk of HBV reactivation associated with treatment with HCV direct-acting antivirals may be increased in these patients.
HBV reactivation is characterized as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level. In patients with resolved HBV infection, reappearance of HBsAg can occur. Reactivation of HBV replication may be accompanied by hepatitis, i.e., increases in aminotransferase levels and, in severe cases, increases in bilirubin levels, liver failure, and death can occur.
Test all patients for evidence of current or prior HBV infection by measuring HBsAg and anti-HBc before initiating HCV treatment with OLYSIO. In patients with serologic evidence of HBV infection, monitor for clinical and laboratory signs of hepatitis flare or HBV reactivation during HCV treatment with OLYSIO and during post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated.
5.2 Serious Symptomatic Bradycardia When Coadministered with Sofosbuvir and Amiodarone
Postmarketing cases of symptomatic bradycardia and cases requiring pacemaker intervention have been reported when amiodarone was coadministered with a sofosbuvir-containing regimen. A fatal cardiac arrest was reported in a patient taking amiodarone who was coadministered a sofosbuvir-containing regimen (ledipasvir/sofosbuvir). Bradycardia has generally occurred within hours to days, but cases have been observed up to 2 weeks after initiating HCV treatment. Patients also taking beta blockers, or those with underlying cardiac comorbidities and/or advanced liver disease may be at increased risk for symptomatic bradycardia with coadministration of amiodarone. Bradycardia generally resolved after discontinuation of HCV treatment. The mechanism for this effect is unknown.
Coadministration of amiodarone with OLYSIO in combination with sofosbuvir is not recommended. For patients taking amiodarone who have no other alternative treatment options, and who will be coadministered OLYSIO and sofosbuvir:
- Counsel patients about the risk of serious symptomatic bradycardia.
- Cardiac monitoring in an in-patient setting for the first 48 hours of coadministration is recommended, after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.
Patients who are taking sofosbuvir in combination with OLYSIO who need to start amiodarone therapy due to no other alternative treatment options should undergo similar cardiac monitoring as outlined above.
Due to amiodarone's long elimination half-life, patients discontinuing amiodarone just prior to starting sofosbuvir in combination with OLYSIO should also undergo similar cardiac monitoring as outlined above.
Patients who develop signs or symptoms of bradycardia should seek medical evaluation immediately. Symptoms may include near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion or memory problems [see Adverse Reactions (6.2) and Drug Interactions (7.3)].
5.3 Hepatic Decompensation and Hepatic Failure
Hepatic decompensation and hepatic failure, including fatal cases, have been reported postmarketing in patients treated with OLYSIO in combination with Peg-IFN-alfa and RBV or in combination with sofosbuvir. Most cases were reported in patients with advanced and/or decompensated cirrhosis who are at increased risk for hepatic decompensation or hepatic failure. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made; and a causal relationship between treatment with OLYSIO and these events has not been established [see Adverse Reactions (6.2)].
OLYSIO is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C) [see Dosage and Administration (2.5) and Use in Specific Populations (8.8)].
In clinical trials of OLYSIO, modest increases in bilirubin levels were observed without impacting hepatic function [see Adverse Reactions (6.1)]. Postmarketing cases of hepatic decompensation with markedly elevated bilirubin levels have been reported. Monitor liver chemistry tests before and as clinically indicated during OLYSIO combination therapy. Patients who experience an increase in total bilirubin to greater than 2.5 times the upper limit of normal should be closely monitored:
- Patients should be instructed to contact their healthcare provider if they have onset of fatigue, weakness, lack of appetite, nausea and vomiting, jaundice or discolored feces.
- Discontinue OLYSIO if elevation in bilirubin is accompanied by liver transaminase increases or clinical signs and symptoms of hepatic decompensation.
5.4 Risk of Serious Adverse Reactions Associated with Combination Treatment
Because OLYSIO is used in combination with other antiviral drugs for the treatment of chronic HCV infection, consult the prescribing information for these drugs before starting therapy with OLYSIO. Warnings and Precautions related to these drugs also apply to their use in OLYSIO combination treatment.
5.5 Photosensitivity
Photosensitivity reactions have been observed with OLYSIO combination therapy. Serious photosensitivity reactions resulting in hospitalization have been observed with OLYSIO in combination with Peg-IFN-alfa and RBV [see Adverse Reactions (6.1)]. Photosensitivity reactions occurred most frequently in the first 4 weeks of treatment, but can occur at any time during treatment. Photosensitivity may present as an exaggerated sunburn reaction, usually affecting areas exposed to light (typically the face, "V" area of the neck, extensor surfaces of the forearms, and dorsa of the hands). Manifestations may include burning, erythema, exudation, blistering, and edema.
Use sun protective measures and limit sun exposure during treatment with OLYSIO. Avoid use of tanning devices during treatment with OLYSIO. Discontinuation of OLYSIO should be considered if a photosensitivity reaction occurs and patients should be monitored until the reaction has resolved. If a decision is made to continue OLYSIO in the setting of a photosensitivity reaction, expert consultation is advised.
5.6 Rash
Rash has been observed with OLYSIO combination therapy [see Adverse Reactions (6.1)]. Rash occurred most frequently in the first 4 weeks of treatment, but can occur at any time during treatment. Severe rash and rash requiring discontinuation of OLYSIO have been reported in subjects receiving OLYSIO in combination with Peg-IFN-alfa and RBV. Most of the rash events in OLYSIO-treated patients were of mild or moderate severity [see Adverse Reactions (6.1)]. Patients with mild to moderate rashes should be followed for possible progression of rash, including the development of mucosal signs (e.g., oral lesions, conjunctivitis) or systemic symptoms. If the rash becomes severe, OLYSIO should be discontinued. Patients should be monitored until the rash has resolved.
5.7 Sulfa Allergy
OLYSIO contains a sulfonamide moiety. In subjects with a history of sulfa allergy (n=16), no increased incidence of rash or photosensitivity reactions has been observed. However, there are insufficient data to exclude an association between sulfa allergy and the frequency or severity of adverse reactions observed with the use of OLYSIO.
5.8 Risk of Adverse Reactions or Reduced Therapeutic Effect Due to Drug Interactions
Coadministration of OLYSIO with substances that are moderate or strong inducers or inhibitors of cytochrome P450 3A (CYP3A) is not recommended as this may lead to significantly lower or higher exposure of simeprevir, respectively, which may result in reduced therapeutic effect or adverse reactions [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
6 ADVERSE REACTIONS
Because OLYSIO is administered in combination with other antiviral drugs, refer to the prescribing information of the antiviral drugs used in combination with OLYSIO for a description of adverse reactions associated with their use.
The following serious and otherwise important adverse reactions are described below and in other sections of the labeling:
- Serious Symptomatic Bradycardia When Coadministered with Sofosbuvir and Amiodarone [see Warnings and Precautions (5.2) and Drug Interactions (7.3)]
- Hepatic Decompensation and Hepatic Failure [see Warnings and Precautions (5.3)]
- Photosensitivity [see Warnings and Precautions (5.5)]
- Rash [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
OLYSIO in Combination with Sofosbuvir
The safety profile of OLYSIO in combination with sofosbuvir in patients with HCV genotype 1 infection with compensated cirrhosis (Child-Pugh A) or without cirrhosis is based on pooled data from the Phase 2 COSMOS trial and the Phase 3 OPTIMIST-1 and OPTIMIST-2 trials which included 317 subjects who received OLYSIO with sofosbuvir (without RBV) for 12 or 24 weeks [see Clinical Studies (14.2)].
Table 4 lists adverse events (all grades) that occurred with at least 10% frequency among subjects receiving 12 or 24 weeks of treatment with OLYSIO 150 mg once daily in combination with sofosbuvir 400 mg once daily without RBV. The overall safety profile appeared similar among cirrhotic and non-cirrhotic subjects [see Dosage and Administration (2.2)].
The majority of the adverse events reported were Grade 1 or 2 in severity. Grade 3 or 4 adverse events were reported in 4% and 13% of subjects receiving 12 or 24 weeks of OLYSIO with sofosbuvir, respectively. Serious adverse events were reported in 2% and 3% of subjects receiving 12 or 24 weeks of OLYSIO with sofosbuvir, respectively. One percent and 6% of subjects receiving 12 or 24 weeks of OLYSIO with sofosbuvir, respectively, discontinued treatment due to adverse events.
| Adverse Events | 12 Weeks OLYSIO + Sofosbuvir N=286 % (n) | 24 Weeks OLYSIO + Sofosbuvir N=31 % (n) |
|---|---|---|
|
||
| Headache | 17 (49) | 23 (7) |
| Fatigue | 16 (47) | 32 (10) |
| Nausea | 14 (40) | 13 (4) |
| Rash (including photosensitivity) | 12 (34) | 16 (5) |
| Diarrhea | 6 (18) | 16 (5) |
| Dizziness | 3 (10) | 16 (5) |
Rash and Photosensitivity
In trials of OLYSIO in combination with sofosbuvir, rash (including photosensitivity reactions) was observed in 12% of OLYSIO-treated subjects receiving 12 weeks of treatment compared to 16% of OLYSIO-treated subjects receiving 24 weeks of treatment.
Most of the rash events in OLYSIO-treated subjects were of mild or moderate severity (Grade 1 or 2). Among 317 subjects, Grade 3 rash was reported in one subject (<1%), leading to treatment discontinuation; none of the subjects experienced Grade 4 rash.
Most photosensitivity reactions were of mild severity (Grade 1); Grade 2 photosensitivity reactions were reported in 2 of 317 subjects (<1%). No Grade 3 or 4 photosensitivity reactions were reported and none of the subjects discontinued treatment due to photosensitivity reactions.
Laboratory Abnormalities
Among subjects who received OLYSIO in combination with sofosbuvir, the most common Grade 3 and 4 laboratory abnormalities were amylase and lipase elevations (Table 5). Most elevations in amylase and lipase were transient and of mild or moderate severity. Amylase and lipase elevations were not associated with pancreatitis.
| Laboratory Parameter | WHO Toxicity Range | 12 Weeks OLYSIO + Sofosbuvir N=286 % | 24 Weeks OLYSIO + Sofosbuvir N=31 % |
|---|---|---|---|
| Chemistry | |||
| Amylase† | |||
| Grade 1 | ≥ 1.1 to ≤ 1.5 × ULN‡ | 12 | 26 |
| Grade 2 | > 1.5 to ≤ 2.0 × ULN | 5 | 6 |
| Grade 3 | > 2.0 to ≤ 5.0 × ULN | 5 | 10 |
| Hyperbilirubinemia | |||
| Grade 1 | ≥ 1.1 to ≤ 1.5 × ULN | 12 | 16 |
| Grade 2 | > 1.5 to ≤ 3.0 × ULN | 3 | 3 |
| Grade 3 | > 3.0 to ≤ 5.0 × ULN | < 1 | 0 |
| Grade 4 | > 5.0 × ULN | 0 | 3 |
| Lipase | |||
| Grade 1 | ≥ 1.1 to ≤ 1.5 × ULN | 5 | 3 |
| Grade 2 | > 1.5 to ≤ 3.0 × ULN | 8 | 10 |
| Grade 3 | > 3.0 to ≤ 5.0 × ULN | < 1 | 3 |
| Grade 4 | > 5.0 × ULN | < 1 | 3 |
OLYSIO in Combination with Peg-IFN-alfa and RBV
The safety profile of OLYSIO in combination with Peg-IFN-alfa and RBV in patients with HCV genotype 1 infection is based on pooled data from three Phase 3 trials (QUEST-1, QUEST-2 and PROMISE) [see Clinical Studies (14.3)]. These trials included a total of 1178 subjects who received OLYSIO or placebo in combination with 24 or 48 weeks of Peg-IFN-alfa and RBV. Of the 1178 subjects, 781 subjects were randomized to receive OLYSIO 150 mg once daily for 12 weeks and 397 subjects were randomized to receive placebo once daily for 12 weeks.
In the pooled Phase 3 safety data, the majority of the adverse reactions reported during 12 weeks treatment with OLYSIO in combination with Peg-IFN-alfa and RBV were Grade 1 to 2 in severity. Grade 3 or 4 adverse reactions were reported in 23% of subjects receiving OLYSIO in combination with Peg-IFN-alfa and RBV versus 25% of subjects receiving placebo in combination with Peg-IFN-alfa and RBV. Serious adverse reactions were reported in 2% of subjects receiving OLYSIO in combination with Peg-IFN-alfa and RBV and in 3% of subjects receiving placebo in combination with Peg-IFN-alfa and RBV. Discontinuation of OLYSIO or placebo due to adverse reactions occurred in 2% and 1% of subjects receiving OLYSIO with Peg-IFN-alfa and RBV and subjects receiving placebo with Peg-IFN-alfa and RBV, respectively.
Table 6 lists adverse reactions (all Grades) that occurred with at least 3% higher frequency among subjects with HCV genotype 1 infection receiving OLYSIO 150 mg once daily in combination with Peg-IFN-alfa and RBV, compared to subjects receiving placebo in combination with Peg-IFN-alfa and RBV, during the first 12 weeks of treatment in the pooled Phase 3 trials in subjects who were treatment-naïve or who had previously relapsed after Peg-IFN-alfa and RBV therapy.
| Adverse Reaction‡ | OLYSIO 150 mg + Peg-IFN-alfa+ RBV First 12 Weeks N=781 % (n) | Placebo + Peg-IFN-alfa+ RBV First 12 Weeks N=397 % (n) |
|---|---|---|
| Rash (including photosensitivity) | 28 (218) | 20 (79) |
| Pruritus | 22 (168) | 15 (58) |
| Nausea | 22 (173) | 18 (70) |
| Myalgia | 16 (126) | 13 (53) |
| Dyspnea | 12 (92) | 8 (30) |
Rash and Photosensitivity
In the Phase 3 clinical trials of OLYSIO or placebo in combination with Peg-IFN-alfa and RBV, rash (including photosensitivity reactions) was observed in 28% of OLYSIO-treated subjects compared to 20% of placebo-treated subjects during the 12 weeks of treatment with OLYSIO or placebo in combination with Peg-IFN-alfa and RBV. Fifty-six percent (56%) of rash events in the OLYSIO group occurred in the first 4 weeks, with 42% of cases occurring in the first 2 weeks. Most of the rash events in OLYSIO-treated subjects were of mild or moderate severity (Grade 1 or 2). Severe (Grade 3) rash occurred in 1% of OLYSIO-treated subjects and in none of the placebo-treated subjects. There were no reports of life-threatening (Grade 4) rash. Discontinuation of OLYSIO or placebo due to rash occurred in 1% of OLYSIO-treated subjects, compared to less than 1% of placebo-treated subjects. The frequencies of rash and photosensitivity reactions were higher in subjects with higher simeprevir exposures.
All subjects enrolled in the Phase 3 trials were directed to use sun protection measures. In these trials, adverse reactions under the specific category of photosensitivity were reported in 5% of OLYSIO-treated subjects compared to 1% of placebo-treated subjects during the 12 weeks of treatment with OLYSIO or placebo in combination with Peg-IFN-alfa and RBV. Most photosensitivity reactions in OLYSIO-treated subjects were of mild or moderate severity (Grade 1 or 2). Two OLYSIO-treated subjects experienced photosensitivity reactions which resulted in hospitalization. No life-threatening photosensitivity reactions were reported.
Dyspnea
During the 12 weeks of treatment with OLYSIO or placebo in combination with Peg-IFN-alfa and RBV, dyspnea was reported in 12% of OLYSIO-treated subjects compared to 8% of placebo-treated subjects (all grades; pooled Phase 3 trials). All dyspnea events reported in OLYSIO-treated subjects were of mild or moderate severity (Grade 1 or 2). There were no Grade 3 or 4 dyspnea events reported and no subjects discontinued treatment with OLYSIO due to dyspnea. Sixty-one percent (61%) of dyspnea events occurred in the first 4 weeks of treatment with OLYSIO.
Laboratory Abnormalities
Among subjects who received OLYSIO or placebo plus Peg-IFN-alfa and RBV, there were no differences between treatment groups for the following laboratory parameters: hemoglobin, neutrophils, platelets, aspartate aminotransferase, alanine aminotransferase, amylase, or serum creatinine. Laboratory abnormalities that were observed at a higher incidence in OLYSIO-treated subjects than in placebo-treated subjects are listed in Table 7.
| Laboratory Parameter | WHO Toxicity Range | OLYSIO 150 mg + Peg-IFN-alfa + RBV N=781 % | Placebo + Peg-IFN-alfa + RBV N=397 % |
|---|---|---|---|
| Chemistry | |||
| Alkaline phosphatase† | |||
| Grade 1 | > 1.25 to ≤ 2.50 × ULN‡ | 3 | 1 |
| Grade 2 | > 2.50 to ≤ 5.00 × ULN | < 1 | 0 |
| Hyperbilirubinemia | |||
| Grade 1 | > 1.1 to ≤ 1.5 × ULN | 27 | 15 |
| Grade 2 | > 1.5 to ≤ 2.5 × ULN | 18 | 9 |
| Grade 3 | > 2.5 to ≤ 5.0 × ULN | 4 | 2 |
| Grade 4 | > 5.0 × ULN | < 1 | 0 |
Elevations in bilirubin were predominately mild to moderate (Grade 1 or 2) in severity, and included elevation of both direct and indirect bilirubin. Elevations in bilirubin occurred early after treatment initiation, peaking by study Week 2, and were rapidly reversible upon cessation of OLYSIO. Bilirubin elevations were generally not associated with elevations in liver transaminases. The frequency of elevated bilirubin was higher in subjects with higher simeprevir exposures.
Adverse Reactions in HCV/HIV-1 Co-infection
OLYSIO in combination with Peg-IFN-alfa and RBV was studied in 106 subjects with HCV genotype 1/HIV-1 co-infection (C212). The safety profile in HCV/HIV co-infected subjects was generally comparable to HCV mono-infected subjects.
Adverse Reactions in HCV Genotype 4 Infection
OLYSIO in combination with Peg-IFN-alfa and RBV was studied in 107 subjects with HCV genotype 4 infection (RESTORE). The safety profile of OLYSIO in subjects with HCV genotype 4 infection was comparable to subjects with HCV genotype 1 infection.
Adverse Reactions in East Asian Subjects
OLYSIO in combination with Peg-IFN-alfa and RBV was studied in a Phase 3 trial conducted in China and South Korea in treatment-naïve subjects with chronic HCV genotype 1 infection (TIGER). The safety profile of OLYSIO in East Asian subjects was similar to that of the pooled Phase 3 population from global trials; however, a higher incidence of the laboratory abnormality hyperbilirubinemia was observed in patients receiving 150 mg OLYSIO plus Peg-IFN-alfa and RBV compared to patients receiving placebo plus Peg-IFN-alfa and RBV. Elevation of total bilirubin (all grades) was observed in 66% (99/151) of subjects treated with 150 mg OLYSIO plus Peg-IFN-alfa and RBV and in 26% (40/152) of subjects treated with placebo plus Peg-IFN-alfa and RBV. Bilirubin elevations were mainly Grade 1 or Grade 2. Grade 3 elevations in bilirubin were observed in 9% (13/151) of subjects treated with 150 mg OLYSIO plus Peg-IFN-alfa and RBV and in 1% (2/152) of subjects treated with placebo plus Peg-IFN-alfa and RBV. There were no Grade 4 elevations in bilirubin. The bilirubin elevations were not associated with increases in liver transaminases and were reversible after the end of treatment [see Use in Specific Populations (8.6) and Clinical Studies (14.3)].
6.2 Postmarketing Experience
The following adverse reactions have been reported during post approval use of OLYSIO. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship between drug exposure and these adverse reactions.
Cardiac Disorders: Serious symptomatic bradycardia has been reported in patients taking amiodarone who initiated treatment with a sofosbuvir-containing regimen [see Warnings and Precautions (5.2) and Drug Interactions (7.3)].
Hepatobiliary Disorders: hepatic decompensation, hepatic failure [see Warnings and Precautions (5.3)].
7 DRUG INTERACTIONS
7.1 Potential for OLYSIO to Affect Other Drugs
Simeprevir mildly inhibits CYP1A2 activity and intestinal CYP3A4 activity, but does not affect hepatic CYP3A4 activity. Coadministration of OLYSIO with drugs that are primarily metabolized by CYP3A4 may result in increased plasma concentrations of such drugs (see Table 8).
Simeprevir inhibits OATP1B1/3, P-glycoprotein (P-gp) and BCRP transporters, and does not inhibit OCT2 in vitro. Coadministration of OLYSIO with drugs that are substrates for OATP1B1/3, and P-gp and BCRP transport may result in increased plasma concentrations of such drugs (see Table 8).
Fluctuations in INR values may occur in patients receiving warfarin concomitant with HCV treatment, including treatment with OLYSIO. Close monitoring of INR values is recommended during treatment and post-treatment follow-up.
7.2 Potential for Other Drugs to Affect OLYSIO
The primary enzyme involved in the biotransformation of simeprevir is CYP3A [see Clinical Pharmacology (12.3)]. Clinically relevant effects of other drugs on simeprevir pharmacokinetics via CYP3A may occur. Coadministration of OLYSIO with moderate or strong inhibitors of CYP3A may significantly increase the plasma exposure of simeprevir. Coadministration with moderate or strong inducers of CYP3A may significantly reduce the plasma exposure of simeprevir and lead to loss of efficacy (see Table 8). Therefore, coadministration of OLYSIO with substances that are moderate or strong inducers or inhibitors of CYP3A is not recommended [see Warnings and Precautions (5.8) and Clinical Pharmacology (12.3)].
7.3 Established and Other Potentially Significant Drug Interactions
Table 8 shows the established and other potentially significant drug interactions based on which alterations in dose or regimen of OLYSIO and/or coadministered drug may be recommended. Drugs that are not recommended for coadministration with OLYSIO are also included in Table 8. For information regarding the magnitude of interaction, see Tables 9 and 10 [see Clinical Pharmacology (12.3)].
| Concomitant Drug Class Drug Name | Effect on Concentration of Simeprevir or Concomitant Drug | Clinical Comment |
|---|---|---|
| The direction of the arrow (↑ = increase, ↓ = decrease, ↔ = no change) indicates the direction of the change in PK. | ||
|
||
| Antiarrhythmics | ||
| Amiodarone | Effect on amiodarone, simeprevir, and sofosbuvir concentrations unknown | Coadministration of amiodarone with OLYSIO in combination with sofosbuvir is not recommended because it may result in serious symptomatic bradycardia. If coadministration is required, cardiac monitoring is recommended [see Warnings and Precautions (5.2), Adverse Reactions (6.2)]. |
| ↑ amiodarone | Caution is warranted and therapeutic drug monitoring of amiodarone, if available, is recommended for concomitant use of amiodarone with an OLYSIO-containing regimen that does not contain sofosbuvir. | |
| Digoxin* | ↑ digoxin | Routine therapeutic drug monitoring of digoxin concentrations is recommended. |
| Oral administration
Disopyramide, Flecainide, Mexiletine, Propafenone, Quinidine | ↑ antiarrhythmics | Therapeutic drug monitoring for these antiarrhythmics, if available, is recommended when coadministered with OLYSIO. |
| Anticonvulsants | ||
| Carbamazepine, Oxcarbazepine, Phenobarbital, Phenytoin | ↓ simeprevir | Coadministration is not recommended. |
| Anti-infectives | ||
| Antibiotics (systemic administration):
Erythromycin* | ↑ simeprevir ↑ erythromycin | Coadministration is not recommended. |
| Antibiotics (systemic administration):
Clarithromycin, Telithromycin | ↑ simeprevir | Coadministration is not recommended. |
| Antifungals (systemic administration):
Itraconazole, Ketoconazole, Posaconazole | ↑ simeprevir | Coadministration is not recommended. |
| Antifungals (systemic administration):
Fluconazole, Voriconazole | ↑ simeprevir | Coadministration is not recommended. |
| Antimycobacterials:
Rifampin*†, Rifabutin, Rifapentine | ↓ simeprevir ↔ rifampin, rifabutin, rifapentine | Coadministration is not recommended. |
| Calcium Channel Blockers (oral administration) | ||
| Amlodipine, Diltiazem, Felodipine, Nicardipine, Nifedipine, Nisoldipine, Verapamil | ↑ calcium channel blockers | Clinical monitoring of patients is recommended when OLYSIO is coadministered with calcium channel blockers. |
| Corticosteroids | ||
| Systemic
Dexamethasone | ↓ simeprevir | Coadministration is not recommended. |
| Gastrointestinal Products | ||
| Propulsive:
Cisapride | ↑ cisapride | Coadministration is not recommended. |
| HCV Products | ||
| Antiviral:
Ledipasvir‡ | ↑ ledipasvir ↑ simeprevir | Coadministration of OLYSIO with products containing ledipasvir is not recommended. |
| Herbal Products | ||
| Milk thistle (Silybum marianum) | ↑ simeprevir | Coadministration is not recommended. |
| St. John's wort (Hypericum perforatum) | ↓ simeprevir | Coadministration of OLYSIO with products containing St. John's wort is not recommended. |
| HIV Products | ||
| Cobicistat-containing products | ↑ simeprevir | Coadministration is not recommended. |
| Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs):
Efavirenz* | ↓ simeprevir ↔ efavirenz | Coadministration is not recommended. |
| Other NNRTIs
Delavirdine Etravirine, Nevirapine |
↑ simeprevir ↓ simeprevir | Coadministration is not recommended. |
| Protease Inhibitors (PIs):
Darunavir/ritonavir*,§ | ↑ simeprevir ↑ darunavir | Coadministration is not recommended. |
| Protease Inhibitors (PIs):
Ritonavir*,¶ | ↑ simeprevir | Coadministration is not recommended. |
| Other ritonavir-boosted or unboosted HIV PIs (Atazanavir, Fosamprenavir, Lopinavir, Indinavir, Nelfinavir, Saquinavir, Tipranavir) | ↑ or ↓ simeprevir | Coadministration of OLYSIO with any HIV PI, with or without ritonavir is not recommended. |
| HMG CO-A Reductase Inhibitors | ||
| Atorvastatin, Rosuvastatin, Simvastatin* | ↑ statin | Coadministration of OLYSIO with statins is expected to increase statin concentrations, which is associated with increased risk of myopathy, including rhabdomyolysis. Use the lowest necessary statin dose, titrate the statin dose carefully, and monitor closely for statin-associated adverse reactions, such as myopathy or rhabdomyolysis. |
| Pitavastatin, Pravastatin, Lovastatin, Fluvastatin | ↑statin | |
| Immunosuppressants | ||
| Cyclosporine* | ↑ cyclosporine ↑ simeprevir# | Coadministration is not recommended. |
| Sirolimus | ↑ or ↓ sirolimus | Routine monitoring of blood concentrations of sirolimus is recommended. |
| Phosphodiesterase Type 5 (PDE-5) Inhibitors | ||
| Sildenafil, Tadalafil, Vardenafil | ↑ PDE-5 inhibitors | Dose adjustment of the PDE-5 inhibitor may be required when OLYSIO is coadministered with sildenafil or tadalafil administered chronically at doses used for the treatment of pulmonary arterial hypertension. Consider starting with the lowest dose of the PDE-5 inhibitor and increase as needed, with clinical monitoring as appropriate. No dose adjustment is required when OLYSIO is coadministered with doses of sildenafil, tadalafil or vardenafil indicated for the treatment of erectile dysfunction. |
| Sedatives/Anxiolytics | ||
| Midazolam* (oral administration) | ↑ midazolam | Caution is warranted when midazolam, which has a narrow therapeutic index, is coadministered with OLYSIO. |
| Triazolam (oral administration) | ↑ triazolam | Caution is warranted when triazolam, which has a narrow therapeutic index, is coadministered with OLYSIO. |
7.4 Drugs Without Clinically Significant Interactions with OLYSIO
In addition to the drugs included in Table 8, the interaction between OLYSIO and the following drugs were evaluated in clinical studies and no dose adjustments are needed for either drug [see Clinical Pharmacology (12.3)]: caffeine, daclatasvir, dextromethorphan, escitalopram, ethinyl estradiol/norethindrone, methadone, midazolam (intravenous administration), omeprazole, raltegravir, rilpivirine, sofosbuvir, tacrolimus, and tenofovir disoproxil fumarate.
No clinically relevant drug-drug interaction is expected when OLYSIO is coadministered with antacids, azithromycin, bedaquiline, corticosteroids (budesonide, fluticasone, methylprednisolone, and prednisone), dolutegravir, H2-receptor antagonists, the narcotic analgesics buprenorphine and naloxone, NRTIs (such as abacavir, didanosine, emtricitabine, lamivudine, stavudine, zidovudine), maraviroc, methylphenidate, and proton pump inhibitors.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
If OLYSIO is administered with RBV, the combination regimen is contraindicated in pregnant women and in men whose female partners are pregnant. Refer to prescribing information for RBV and for other drugs used in combination with OLYSIO for information on use in pregnancy.
No adequate human data are available to establish whether or not OLYSIO poses a risk to pregnancy outcomes. In animal reproduction studies with simeprevir, embryofetal developmental toxicity (including fetal loss) was observed in mice at simeprevir exposures greater than or equal to 1.9 times higher than exposure in humans at the recommended clinical dose while no adverse embryofetal developmental outcomes were observed in mice and rats at exposures similar to the exposure in humans at the recommended clinical dose [see Data]. Given these findings, pregnant women should be advised of potential risk to the fetus. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Animal Data
In embryofetal development studies in rats and mice, pregnant animals were administered simeprevir at doses up to 500 mg/kg/day (rats) and at 150, 500 and 1000 mg/kg/day (mice) on gestation days 6 to 17 (rats) and gestation days 6 to 15 (mice), resulting in late in utero fetal losses in mice at an exposure greater than or equal to 1.9 times higher than the exposure in humans at the recommended clinical dose. In addition, decreased fetal weights and an increase in fetal skeletal variations were observed in mice at exposures greater than or equal to 1.2 times higher than the exposure in humans at the recommended clinical dose. No adverse embryofetal developmental effects were observed in mice (at the lowest dose tested) or in rats (at up to the highest dose tested) at exposures similar to the exposure in humans at the recommended clinical dose.
In a rat pre- and post-natal development study, maternal animals were exposed to simeprevir from gestation day 6 to lactation/post-partum day 20 at doses up to 1000 mg/kg/day. At maternally toxic doses, the developing rat offspring exhibited significantly decreased body weight and negative effects on physical growth (delay and small size) and development (decreased motor activity) following simeprevir exposure in utero (via maternal dosing) and during lactation (via maternal milk to nursing pups) at maternal exposures similar to the exposure in humans at the recommended clinical dose. Subsequent survival, behavior and reproductive capacity of the offspring were not affected.
8.2 Lactation
Risk Summary
It is not known whether OLYSIO and its metabolites are present in human breast milk, affect human milk production, or have effects on the breastfed infant. When administered to lactating rats, simeprevir was detected in plasma of nursing pups, likely due to the presence of simeprevir in milk [see Data].
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OLYSIO and any potential adverse effects on the breastfed child from OLYSIO or from the underlying maternal condition.
If OLYSIO is administered with RBV, the nursing mother's information for RBV also applies to this combination regimen. Refer to prescribing information for RBV and for other drugs used in combination with OLYSIO for more information on use during lactation.
Animal Data
Although not measured directly, simeprevir was likely present in the milk of lactating rats in the pre- and post-natal development study, because systemic exposures (AUC) of simeprevir were observed in nursing pups on lactation/post-partum day 6 at concentrations approximately 10% of maternal simeprevir exposures [see Use in Specific Populations (8.1)].
8.3 Females and Males of Reproductive Potential
If OLYSIO is administered with RBV, follow the recommendations for pregnancy testing and contraception within RBV's prescribing information. Refer to prescribing information for other drugs used in combination with OLYSIO for additional information on use in females and males of reproductive potential.
Infertility
There are no data on the effect of simeprevir on human fertility. Limited effects on male fertility were observed in animal studies [see Nonclinical Toxicology (13.1)]. If OLYSIO is administered with RBV, the information for RBV with regard to infertility also applies to this combination regimen. In addition, refer to prescribing information for other drugs used in combination with OLYSIO for information on effects on fertility.
8.4 Pediatric Use
The safety and efficacy of OLYSIO in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of OLYSIO did not include sufficient numbers of patients older than 65 years to determine whether they respond differently from younger patients. No dosage adjustment of OLYSIO is required in geriatric patients [see Clinical Pharmacology (12.3)].
8.6 Race
Patients of East Asian ancestry exhibit higher simeprevir plasma exposures, but no dosage adjustment is required based on race [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.3)].
8.7 Renal Impairment
No dosage adjustment of OLYSIO is required in patients with mild, moderate or severe renal impairment [see Clinical Pharmacology (12.3)]. The safety and efficacy of OLYSIO have not been studied in HCV-infected patients with severe renal impairment (creatinine clearance below 30 mL/min) or end-stage renal disease, including patients requiring dialysis. Simeprevir is highly protein-bound; therefore, dialysis is unlikely to result in significant removal of simeprevir [see Clinical Pharmacology (12.3)].
Refer to the prescribing information for the other antiviral drug(s) used in combination with OLYSIO regarding their use in patients with renal impairment.
8.8 Hepatic Impairment
No dosage adjustment of OLYSIO is required in patients with mild hepatic impairment (Child-Pugh A) [see Clinical Pharmacology (12.3)].
OLYSIO is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C). Simeprevir exposures are increased in patients with moderate or severe hepatic impairment (Child-Pugh B or C). In clinical trials of OLYSIO in combination with Peg-IFN-alfa and RBV, higher simeprevir exposures were associated with increased frequency of adverse reactions, including increased bilirubin, rash and photosensitivity. There have been postmarketing reports of hepatic decompensation, hepatic failure, and death in patients with advanced or decompensated cirrhosis receiving OLYSIO combination therapy [see Dosage and Administration (2.5), Warnings and Precautions (5.3), Adverse Reactions (6.1, 6.2), and Clinical Pharmacology (12.3)].
The safety and efficacy of OLYSIO have not been established in liver transplant patients.
See the Peg-IFN-alfa prescribing information regarding its contraindication in patients with hepatic decompensation.
10 OVERDOSAGE
Human experience of overdose with OLYSIO is limited. There is no specific antidote for overdose with OLYSIO. In the event of an overdose, the patient's clinical status should be observed and the usual supportive measures employed.
Simeprevir is highly protein-bound; therefore, dialysis is unlikely to result in significant removal of simeprevir [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
OLYSIO (simeprevir) is an inhibitor of the HCV NS3/4A protease.
The chemical name for simeprevir is (2R,3aR,10Z,11aS,12aR,14aR)-N-(cyclopropylsulfonyl)-2-[[2-(4-isopropyl-1,3-thiazol-2-yl)-7-methoxy-8-methyl-4-quinolinyl]oxy]-5-methyl-4,14-dioxo-2,3,3a,4,5,6,7,8,9,11a,12,13,14,14a-tetradecahydrocyclopenta[c]cyclopropa[g][1,6]diazacyclotetradecine-12a(1H)-carboxamide. Its molecular formula is C38H47N5O7S2 and its molecular weight is 749.94. Simeprevir has the following structural formula:
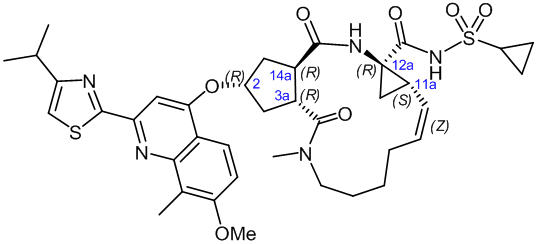
Simeprevir drug substance is a white to almost white powder. Simeprevir is practically insoluble in water over a wide pH range. It is practically insoluble in propylene glycol, very slightly soluble in ethanol, and slightly soluble in acetone. It is soluble in dichloromethane and freely soluble in some organic solvents (e.g., tetrahydrofuran and N,N-dimethylformamide).
OLYSIO (simeprevir) for oral administration is available as 150 mg strength hard gelatin capsules. Each capsule contains 154.4 mg of simeprevir sodium salt, which is equivalent to 150 mg of simeprevir. OLYSIO (simeprevir) capsules contain the following inactive ingredients: colloidal anhydrous silica, croscarmellose sodium, lactose monohydrate, magnesium stearate and sodium lauryl sulphate. The white capsule contains gelatin and titanium dioxide (E171) and is printed with ink containing iron oxide black (E172) and shellac (E904).
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Simeprevir is a direct-acting antiviral (DAA) agent against the hepatitis C virus [see Microbiology (12.4)].
12.3 Pharmacokinetics
The pharmacokinetic properties of simeprevir have been evaluated in healthy adult subjects and in adult HCV-infected subjects. Plasma Cmax and AUC increased more than dose-proportionally after multiple doses between 75 mg and 200 mg once daily, with accumulation occurring following repeated dosing. Steady-state was reached after 7 days of once-daily dosing. Plasma exposure (AUC) of simeprevir in HCV-infected subjects was about 2- to 3-fold higher compared to that observed in HCV-uninfected subjects. Plasma Cmax and AUC of simeprevir were similar during coadministration of simeprevir with Peg-IFN-alfa and RBV compared with administration of simeprevir alone. In Phase 3 trials with Peg-IFN-alfa and RBV in HCV-infected subjects, the geometric mean steady-state pre-dose plasma concentration was 1009 ng/mL (geometric coefficient of variation [gCV] = 162%) and the geometric mean steady-state AUC24 was 39140 ng.h/mL (gCV = 98%).
Absorption
The mean absolute bioavailability of simeprevir following a single oral 150 mg dose of OLYSIO in fed conditions is 62%. Maximum plasma concentrations (Cmax) are typically achieved between 4 to 6 hours post-dose.
In vitro studies with human Caco-2 cells indicated that simeprevir is a substrate of P-gp.
Effects of Food on Oral Absorption
Compared to intake without food, administration of simeprevir with food to healthy subjects increased the AUC by 61% after a high-fat, high-caloric breakfast (928 kcal) and by 69% after a normal-caloric breakfast (533 kcal), and delayed the absorption by 1 hour and 1.5 hours, respectively.
Distribution
Simeprevir is extensively bound to plasma proteins (greater than 99.9%), primarily to albumin and, to a lesser extent, alfa 1-acid glycoprotein. Plasma protein binding is not meaningfully altered in patients with renal or hepatic impairment.
In animals, simeprevir is extensively distributed to gut and liver (liver:blood ratio of 29:1 in rat) tissues. In vitro data and physiologically-based pharmacokinetic modeling and simulations indicate that hepatic uptake in humans is mediated by OATP1B1/3.
Metabolism
Simeprevir is metabolized in the liver. In vitro experiments with human liver microsomes indicated that simeprevir primarily undergoes oxidative metabolism by the hepatic CYP3A system. Involvement of CYP2C8 and CYP2C19 cannot be excluded. Coadministration of OLYSIO with moderate or strong inhibitors of CYP3A may significantly increase the plasma exposure of simeprevir, and coadministration with moderate or strong inducers of CYP3A may significantly reduce the plasma exposure of simeprevir [see Drug Interactions (7)].
Following a single oral administration of 200 mg (1.3 times the recommended dosage) 14C-simeprevir to healthy subjects, the majority of the radioactivity in plasma (mean: 83%) was accounted for by unchanged drug and a small part of the radioactivity in plasma was related to metabolites (none being major metabolites). Metabolites identified in feces were formed via oxidation at the macrocyclic moiety or aromatic moiety or both and by O-demethylation followed by oxidation.
Elimination
Elimination of simeprevir occurs via biliary excretion. Renal clearance plays an insignificant role in its elimination. Following a single oral administration of 200 mg 14C-simeprevir to healthy subjects, on average 91% of the total radioactivity was recovered in feces. Less than 1% of the administered dose was recovered in urine. Unchanged simeprevir in feces accounted for on average 31% of the administered dose.
The terminal elimination half-life of simeprevir was 10 to 13 hours in HCV-uninfected subjects and 41 hours in HCV-infected subjects receiving 200 mg (1.3 times the recommended dosage) of simeprevir.
Specific Populations
Geriatric Use
There is limited data on the use of OLYSIO in patients aged 65 years and older. Age (18–73 years) had no clinically meaningful effect on the pharmacokinetics of simeprevir based on a population pharmacokinetic analysis of HCV-infected subjects treated with OLYSIO [see Use in Specific Populations (8.5)].
Renal Impairment
Compared to HCV-uninfected subjects with normal renal function (classified using the Modification of Diet in Renal Disease [MDRD] eGFR formula; eGFR greater than or equal to 80 mL/min) the mean steady-state AUC of simeprevir was 62% higher in HCV-uninfected subjects with severe renal impairment (eGFR below 30 mL/min).
In a population pharmacokinetic analysis of mild or moderate renally impaired HCV-infected subjects treated with OLYSIO 150 mg once daily, creatinine clearance was not found to influence the pharmacokinetic parameters of simeprevir. It is therefore not expected that renal impairment will have a clinically relevant effect on the exposure to simeprevir [see Use in Specific Populations (8.7)].
As simeprevir is highly bound to plasma proteins, it is unlikely that it will be significantly removed by dialysis.
Hepatic Impairment
Compared to HCV-uninfected subjects with normal hepatic function, the mean steady-state AUC of simeprevir was 2.4-fold higher in HCV-uninfected subjects with moderate hepatic impairment (Child-Pugh B) and 5.2-fold higher in HCV-uninfected subjects with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.8)].
Based on a population pharmacokinetic analysis of HCV-infected subjects with mild hepatic impairment (Child-Pugh A) treated with OLYSIO, liver fibrosis stage did not have a clinically relevant effect on the pharmacokinetics of simeprevir.
Gender, Body Weight, Body Mass Index
Gender, body weight or body mass index have no clinically meaningful relevant effect on the pharmacokinetics of simeprevir based on a population pharmacokinetic analysis of HCV-infected subjects treated with OLYSIO.
Race
Population pharmacokinetic estimates of exposure of simeprevir were comparable between Caucasian and Black/African American HCV-infected subjects.
In a Phase 3 trial conducted in China and South Korea, the mean plasma exposure of simeprevir in East Asian HCV-infected subjects was 2.1-fold higher compared to non-Asian HCV-infected subjects in a pooled Phase 3 population from global trials [see Use in Specific Populations (8.6)].
Drug Interactions
[See also Warnings and Precautions (5.8) and Drug Interactions (7)]
In vitro studies indicated that simeprevir is a substrate and mild inhibitor of CYP3A. Simeprevir does not affect CYP2C9, CYP2C19 or CYP2D6 in vivo. Simeprevir does not induce CYP1A2 or CYP3A4 in vitro. In vivo, simeprevir mildly inhibits the CYP1A2 activity and intestinal CYP3A4 activity, while it does not affect hepatic CYP3A4 activity. Simeprevir is not a clinically relevant inhibitor of cathepsin A enzyme activity.
In vitro, simeprevir is a substrate for P-gp, MRP2, BCRP, OATP1B1/3 and OATP2B1; simeprevir inhibits the uptake transporters OATP1B1/3 and NTCP and the efflux transporters P-gp/MDR1, MRP2, BCRP and BSEP and does not inhibit OCT2. The inhibitory effects of simeprevir on the bilirubin transporters OATP1B1/3 and MRP2 likely contribute to clinical observations of elevated bilirubin [see Adverse Reactions (6.1)].
Simeprevir is transported into the liver by OATP1B1/3 where it undergoes metabolism by CYP3A. Based on results from in vivo studies, coadministration of OLYSIO with moderate or strong inhibitors of CYP3A may significantly increase the plasma exposure of simeprevir and coadministration with moderate or strong inducers of CYP3A may significantly reduce the plasma exposure of simeprevir, which may lead to loss of efficacy.
Drug interaction studies were performed in healthy adults with simeprevir (at the recommended dose of 150 mg once daily unless otherwise noted) and drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interactions. The effects of coadministration of other drugs on the Cmax, AUC, and Cmin values of simeprevir are summarized in Table 9 (effect of other drugs on OLYSIO). The effect of coadministration of OLYSIO on the Cmax, AUC, and Cmin values of other drugs are summarized in Table 10 (effect of OLYSIO on other drugs). For information regarding clinical recommendations, see Drug Interactions (7).
| Coadministered Drug | Dose (mg) and Schedule | N | Effect on PK* | LS Mean Ratio (90% CI) of Simeprevir PK Parameters with/without Drug | |||
|---|---|---|---|---|---|---|---|
| Drug | Simeprevir | Cmax | AUC | Cmin | |||
| CI = Confidence Interval; N = number of subjects with data; NA = not available; PK = pharmacokinetics; LS = least square; q.d. = once daily; b.i.d. = twice daily; t.i.d. = three times a day | |||||||
|
|||||||
| Cyclosporine† | individualized dose‡ | 150 mg q.d. for 14 days | 10 | ↑ | 4.53 (3.05–6.74) | 5.68 (3.58–9.00) | NA |
| Erythromycin | 500 mg t.i.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↑ | 4.53 (3.91–5.25) | 7.47 (6.41–8.70) | 12.74 (10.19–15.93) |
| Escitalopram | 10 mg q.d. for 7 days | 150 mg q.d. for 7 days | 18 | ↓ | 0.80 (0.71–0.89) | 0.75 (0.68–0.83) | 0.68 (0.59–0.79) |
| Rifampin | 600 mg q.d. for 7 days | 200 mg q.d. for 7 days | 18 | ↓ | 1.31 (1.03–1.66) | 0.52 (0.41–0.67) | 0.08 (0.06–0.11) |
| Tacrolimus† | individualized dose‡ | 150 mg q.d. for 14 days | 25 | ↑ | 1.85 (1.40–2.46) | 1.90 (1.37–2.63) | NA |
| Anti-HCV Drug | |||||||
| Daclatasvir | 60 mg q.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↑ | 1.39 (1.27–1.52) | 1.44 (1.32–1.56) | 1.49 (1.33–1.67) |
| Ledipasvir§ | 90 mg q.d. for 14 days | 150 mg q.d. for 14 days | 20 | ↑ | 2.34 (1.95–2.81) | 3.05 (2.34–3.84) | 4.69 (3.40–6.47) |
| Sofosbuvir¶ | 400 mg q.d. | 150 mg q.d. | 21 | ↔ | 0.96 (0.71–1.30) | 0.94 (0.67–1.33) | NA |
| Anti-HIV Drugs | |||||||
| Darunavir/Ritonavir# | 800/100 mg q.d. for 7 days | 50 mg and 150 mg q.d. for 7 days | 25 | ↑ | 1.79 (1.55–2.06) | 2.59 (2.15–3.11) | 4.58 (3.54–5.92) |
| Efavirenz | 600 mg q.d. for 14 days | 150 mg q.d. for 14 days | 23 | ↓ | 0.49 (0.44–0.54) | 0.29 (0.26–0.33) | 0.09 (0.08–0.12) |
| Raltegravir | 400 mg b.i.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↔ | 0.93 (0.85–1.02) | 0.89 (0.81–0.98) | 0.86 (0.75–0.98) |
| Rilpivirine | 25 mg q.d. for 11 days | 150 mg q.d. for 11 days | 21 | ↔ | 1.10 (0.97–1.26) | 1.06 (0.94–1.19) | 0.96 (0.83–1.11) |
| Ritonavir | 100 mg b.i.d. for 15 days | 200 mg q.d. for 7 days | 12 | ↑ | 4.70 (3.84–5.76) | 7.18 (5.63–9.15) | 14.35 (10.29–20.01) |
| Tenofovir disoproxil fumarate | 300 mg q.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↓ | 0.85 (0.73–0.99) | 0.86 (0.76–0.98) | 0.93 (0.78–1.11) |
| Coadministered Drug | Dose (mg) and Schedule | N | Effect on PK* | LS Mean Ratio (90% CI) of Coadministered Drug PK Parameters with/without OLYSIO | |||
|---|---|---|---|---|---|---|---|
| Drug | Simeprevir | Cmax | AUC | Cmin | |||
| CI = Confidence Interval; i.v.= intravenous; N = number of subjects with data; NA = not available; PK = pharmacokinetics; LS = least square; q.d. = once daily; b.i.d. = twice daily; t.i.d. = three times a day | |||||||
|
|||||||
| Atorvastatin | 40 mg single dose | 150 mg q.d. for 10 days | 18 | ↑ | 1.70 (1.42–2.04) | 2.12 (1.72–2.62) | NA |
| 2-hydroxy-atorvastatin | ↑ | 1.98 (1.70–2.31) | 2.29 (2.08–2.52) | NA | |||
| Caffeine | 150 mg | 150 mg q.d. for 11 days | 16 | ↑ | 1.12 (1.06–1.19) | 1.26 (1.21–1.32) | NA |
| Cyclosporine | 100 mg single dose | 150 mg q.d. for 7 days | 14 | ↑ | 1.16 (1.07–1.26) | 1.19 (1.13–1.26) | NA |
| Dextromethorphan | 30 mg | 150 mg q.d. for 11 days | 16 | ↑ | 1.21 (0.93–1.57) | 1.08 (0.87–1.35) | NA |
| Dextrorphan | ↔ | 1.03 (0.93–1.15) | 1.09 (1.03–1.15) | NA | |||
| Digoxin | 0.25 mg single dose | 150 mg q.d. for 7 days | 16 | ↑ | 1.31 (1.14–1.51) | 1.39 (1.16–1.67) | NA |
| Erythromycin | 500 mg t.i.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↑ | 1.59 (1.23–2.05) | 1.90 (1.53–2.36) | 3.08 (2.54–3.73) |
| Escitalopram | 10 mg q.d. for 7 days | 150 mg q.d. for 7 days | 17 | ↔ | 1.03 (0.99–1.07) | 1.00 (0.97–1.03) | 1.00 (0.95–1.05) |
| Ethinyl estradiol (EE), coadministered with norethindrone (NE) | 0.035 mg q.d. EE + 1 mg q.d. NE for 21 days | 150 mg q.d. for 10 days | 18 | ↔ | 1.18 (1.09–1.27) | 1.12 (1.05–1.20) | 1.00 (0.89–1.13) |
| Midazolam (oral) | 0.075 mg/kg | 150 mg q.d. for 10 days | 16 | ↑ | 1.31 (1.19–1.45) | 1.45 (1.35–1.57) | NA |
| Midazolam (i.v.) | 0.025 mg/kg | 150 mg q.d. for 11 days | 16 | ↑ | 0.78 (0.52–1.17) | 1.10 (0.95–1.26) | NA |
| R(-) methadone† | 30–150 mg q.d., individualized dose | 150 mg q.d. for 7 days | 12 | ↔ | 1.03 (0.97–1.09) | 0.99 (0.91–1.09) | 1.02 (0.93–1.12) |
| Norethindrone (NE), coadministered with EE | 0.035 mg q.d. EE + 1 mg q.d. NE for 21 days | 150 mg q.d. for 10 days | 18 | ↔ | 1.06 (0.99–1.14) | 1.15 (1.08–1.22) | 1.24 (1.13–1.35) |
| Omeprazole | 40 mg single dose | 150 mg q.d. for 11 days | 16 | ↑ | 1.14 (0.93–1.39) | 1.21 (1.00–1.46) | NA |
| Rifampin | 600 mg q.d. for 7 days | 200 mg q.d. for 7 days | 18 | ↔ | 0.92 (0.80–1.07) | 1.00 (0.93–1.08) | NA |
| 25-desacetyl-rifampin | 17 | ↑ | 1.08 (0.98–1.19) | 1.24 (1.13–1.36) | NA | ||
| Rosuvastatin | 10 mg single dose | 150 mg q.d. for 7 days | 16 | ↑ | 3.17 (2.57–3.91) | 2.81 (2.34–3.37) | NA |
| Simvastatin | 40 mg single dose | 150 mg q.d. for 10 days | 18 | ↑ | 1.46 (1.17–1.82) | 1.51 (1.32–1.73) | NA |
| Simvastatin acid | ↑ | 3.03 (2.49–3.69) | 1.88 (1.63–2.17) | NA | |||
| Tacrolimus | 2 mg single dose | 150 mg q.d. for 7 days | 14 | ↓ | 0.76 (0.65–0.90) | 0.83 (0.59–1.16) | NA |
| S-Warfarin | 10 mg single dose | 150 mg q.d. for 11 days | 16 | ↔ | 1.00 (0.94–1.06) | 1.04 (1.00–1.07) | NA |
| Anti-HCV Drug | |||||||
| Daclatasvir | 60 mg q.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↑ | 1.50 (1.39–1.62) | 1.96 (1.84–2.10) | 2.68 (2.42–2.98) |
| Ledipasvir‡ | 90 mg q.d. for 14 days | 150 mg q.d. for 14 days | 20 | ↑ | 1.64 (1.45–1.86) | 1.75 (1.56–1.96) | 1.74 (1.55–1.97) |
| Sofosbuvir§ | 400 mg q.d. | 150 mg q.d. | 22 | ↑ | 1.91 (1.26–2.90) | 3.16 (2.25–4.44) | NA |
| GS-331007¶ | ↔ | 0.69 (0.52–0.93) | 1.09 (0.87–1.37) | NA | |||
| Anti-HIV Drugs | |||||||
| Darunavir# | 800 mg q.d. for 7 days | 50 mg q.d. for 7 days | 25 | ↑ | 1.04 (0.99–1.10) | 1.18 (1.11–1.25) | 1.31 (1.13–1.52) |
| Ritonavir# | 100 mg q.d. for 7 days | ↑ | 1.23 (1.14–1.32) | 1.32 (1.25–1.40) | 1.44 (1.30–1.61) |
||
| Efavirenz | 600 mg q.d. for 14 days | 150 mg q.d. for 14 days | 23 | ↔ | 0.97 (0.89–1.06) | 0.90 (0.85–0.95) | 0.87 (0.81–0.93) |
| Raltegravir | 400 mg b.i.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↑ | 1.03 (0.78–1.36) | 1.08 (0.85–1.38) | 1.14 (0.97–1.36) |
| Rilpivirine | 25 mg q.d. for 11 days | 150 mg q.d. for 11 days | 23 | ↔ | 1.04 (0.95–1.13) | 1.12 (1.05–1.19) | 1.25 (1.16–1.35) |
| Tenofovir disoproxil fumarate | 300 mg q.d. for 7 days | 150 mg q.d. for 7 days | 24 | ↔ | 1.19 (1.10–1.30) | 1.18 (1.13–1.24) | 1.24 (1.15–1.33) |
12.4 Microbiology
Mechanism of Action
Simeprevir is an inhibitor of the HCV NS3/4A protease which is essential for viral replication. In a biochemical assay simeprevir inhibited the proteolytic activity of recombinant genotype 1a and 1b HCV NS3/4A proteases, with median Ki values of 0.5 nM and 1.4 nM, respectively.
Antiviral Activity
The median simeprevir EC50 and EC90 values against a HCV genotype 1b replicon were 9.4 nM (7.05 ng/mL) and 19 nM (14.25 ng/mL), respectively. Chimeric replicons carrying NS3 sequences derived from HCV protease inhibitor treatment-naïve genotype 1a- or genotype 1b-infected patients displayed median fold change (FC) in EC50 values of 1.4 (interquartile range, IQR: 0.8 to 11; N=78) and 0.4 (IQR: 0.3 to 0.7; N=59) compared to reference genotype 1b replicon, respectively. Genotype 1a (N=33) and 1b (N=2) isolates with a baseline Q80K polymorphism resulted in median FC in simeprevir EC50 value of 11 (IQR: 7.4 to 13) and 8.4, respectively. Chimeric replicons carrying NS3 sequences derived from HCV protease inhibitor treatment-naïve genotype 4a-, 4d-, or 4r-infected patients displayed median FC in EC50 values of 0.5 (IQR: 0.4 to 0.6; N=38), 0.4 (IQR: 0.2 to 0.5; N=24), and 1.6 (IQR: 0.7 to 4.5; N=8), compared to reference genotype 1b replicon, respectively. A pooled analysis of chimeric replicons carrying the NS3 sequences from HCV protease inhibitor-naïve patients infected with other HCV genotype 4 subtypes, including 4c (N=1), 4e (N=2), 4f (N=3), 4h (N=3), 4k (N=1), 4o (N=2), 4q (N=2), or unidentified subtype (N=7) displayed a median FC in EC50 value of 0.7 (IQR: 0.5 to 1.1; N=21) compared to reference genotype 1b replicon. The presence of 50% human serum reduced simeprevir replicon activity by 2.4-fold. Combination of simeprevir with IFN, RBV, NS5A inhibitors, nucleoside analog NS5B polymerase inhibitors or non-nucleoside analog NS5B polymerase inhibitors, including NS5B thumb 1-, thumb 2-, and palm-domain targeting drugs, was not antagonistic.
Resistance in Cell Culture
Resistance to simeprevir was characterized in HCV genotype 1a and 1b replicon-containing cells. Ninety-six percent (96%) of simeprevir-selected genotype 1 replicons carried one or multiple amino acid substitutions at NS3 protease positions F43, Q80, R155, A156, and/or D168, with substitutions at NS3 position D168 being most frequently observed (78%). Additionally, resistance to simeprevir was evaluated in HCV genotype 1a and 1b replicon assays using site-directed mutants and chimeric replicons carrying NS3 sequences derived from clinical isolates. Amino acid substitutions at NS3 positions F43, Q80, S122, R155, A156, and D168 reduced susceptibility to simeprevir. Replicons with D168V or A, and R155K substitutions displayed large reductions in susceptibility to simeprevir (FC in EC50 value greater than 50), whereas other substitutions such as Q80K or R, S122R, and D168E displayed lower reductions in susceptibility (FC in EC50 value between 2 and 50). Other substitutions such as Q80G or L, S122G, N or T did not reduce susceptibility to simeprevir in the replicon assay (FC in EC50 value lower than 2). Amino acid substitutions at NS3 positions Q80, S122, R155, and/or D168 that were associated with lower reductions in susceptibility to simeprevir when occurring alone, reduced susceptibility to simeprevir by more than 50-fold when present in combination.
Resistance in Clinical Studies
In a pooled analysis of subjects treated with 150 mg OLYSIO in combination with Peg-IFN-alfa and RBV who did not achieve SVR in the controlled Phase 2 and Phase 3 clinical trials (PILLAR, ASPIRE, QUEST 1 and QUEST 2, PROMISE), emerging virus with amino acid substitutions at NS3 positions Q80, S122, R155 and/or D168 were observed in 180 out of 197 (91%) subjects. Substitutions D168V and R155K alone or in combination with other substitutions at these positions emerged most frequently (Table 11). Most of these emerging substitutions have been shown to reduce susceptibility to simeprevir in cell culture replicon assays.
HCV genotype 1 subtype-specific patterns of simeprevir treatment-emergent amino acid substitutions were observed. HCV genotype 1a predominately had emerging R155K alone or in combination with amino acid substitutions at NS3 positions Q80, S122 and/or D168, while HCV genotype 1b had most often an emerging D168V substitution (Table 11). In HCV genotype 1a with a baseline Q80K amino acid polymorphism, an emerging R155K substitution was observed most frequently at failure.
| Emerging Amino Acid Substitutions in NS3 | Genotype 1a*
N=116 % (n) | Genotype 1b N=81 % (n) |
|---|---|---|
| Note: substitutions at NS3 position F43 and A156 were selected in cell culture and associated with reduced simeprevir activity in the replicon assay but were not observed at time of failure. | ||
|
||
| Any substitution at NS3 position F43, Q80, S122, R155, A156, or D168† | 95 (110) | 86 (70) |
| D168E | 15 (17) | 17 (14) |
| D168V | 10 (12) | 60 (49) |
| Q80R‡ | 4 (5) | 12 (10) |
| R155K | 77 (89) | 0 (0) |
| Q80X+D168X§ | 4 (5) | 14 (11) |
| R155X+D168X§ | 13 (15) | 4 (3) |
| Q80K‡, S122A/G/I/T‡, S122R, R155Q‡, D168A, D168F‡, D168H, D168T, I170T¶ | Less than 10% | Less than 10% |
The majority of HCV genotype 1-infected subjects treated with OLYSIO in combination with sofosbuvir (with or without RBV) for 12 or 24 weeks who did not achieve SVR due to virologic reasons and with sequencing data available had emerging NS3 amino acid substitutions at position 168 and/or R155K: 5 out of 6 subjects in COSMOS and 1 out of 3 subjects in OPTIMIST-1. The emerging NS3 amino acid substitutions were similar to those observed in subjects who did not achieve SVR following treatment with OLYSIO in combination with Peg-IFN-alfa and RBV. No emerging NS5B amino acid substitutions associated with sofosbuvir resistance were observed in subjects who did not achieve SVR following treatment of OLYSIO in combination with sofosbuvir (with or without RBV) for 12 or 24 weeks.
In the RESTORE trial in genotype 4-infected subjects, 30 out of 34 (88%) subjects who did not achieve SVR had emerging amino acid substitutions at NS3 positions Q80, T122, R155, A156 and/or D168 (mainly substitutions at position D168; 26 out of 34 [76%] subjects), similar to the emerging amino acid substitutions observed in genotype 1-infected subjects.
Persistence of Resistance–Associated Substitutions
The persistence of simeprevir-resistant virus was assessed following treatment failure in the pooled analysis of subjects receiving 150 mg OLYSIO in combination with Peg-IFN-alfa and RBV in the controlled Phase 2 and Phase 3 trials. The proportion of subjects with detectable levels of treatment-emergent, resistance-associated variants was followed post-treatment for a median time of 28 weeks (range 0 to 70 weeks). Resistant variants remained at detectable levels in 32 out of 66 subjects (48%) with single emerging R155K and in 16 out of 48 subjects (33%) with single emerging D168V.
The lack of detection of virus containing a resistance-associated substitution does not necessarily indicate that the resistant virus is no longer present at clinically significant levels. The long-term clinical impact of the emergence or persistence of virus containing OLYSIO-resistance-associated substitutions is unknown.
Effect of Baseline HCV Polymorphisms on Treatment Response
Analyses were conducted to explore the association between naturally-occurring baseline NS3/4A amino acid substitutions (polymorphisms) and treatment outcome. In the pooled analysis of the Phase 3 trials QUEST 1 and QUEST 2, and in the PROMISE trial, the efficacy of OLYSIO in combination with Peg-IFN-alfa and RBV was substantially reduced in subjects infected with HCV genotype 1a virus with the NS3 Q80K polymorphism at baseline [see Clinical Studies (14.3)].
The observed prevalence of NS3 Q80K polymorphic variants at baseline in the overall population of the Phase 2 and Phase 3 trials (PILLAR, ASPIRE, PROMISE, QUEST 1 and QUEST 2) was 14%; while the observed prevalence of the Q80K polymorphism was 30% in subjects infected with HCV genotype 1a and 0.5% in subjects infected with HCV genotype 1b. The observed prevalence of Q80K polymorphic variants at baseline in the U.S. population of these Phase 2 and Phase 3 trials was 35% overall, 48% in subjects infected with HCV genotype 1a and 0% in subjects infected with HCV genotype 1b. With the exception of the NS3 Q80K polymorphism, baseline HCV variants with polymorphisms at NS3 positions F43, Q80, S122, R155, A156, and/or D168 that were associated with reduced simeprevir activity in replicon assays were generally uncommon (1.3%) in subjects with HCV genotype 1 infection in these Phase 2 and Phase 3 trials (n=2007).
The Q80K polymorphic variant was not observed in subjects infected with HCV genotype 4.
Cross-Resistance
Based on resistance patterns observed in cell culture replicon studies and HCV-infected subjects, cross-resistance between OLYSIO and other NS3/4A protease inhibitors is expected. No cross-resistance is expected between direct-acting antiviral agents with different mechanisms of action. OLYSIO remained fully active against substitutions associated with resistance to NS5A inhibitors, NS5B nucleoside and non-nucleoside polymerase inhibitors.
12.5 Pharmacogenomics
A genetic variant near the gene encoding interferon-lambda-3 (IL28B rs12979860, a C [cytosine] to T [thymine] substitution) is a strong predictor of response to Peg-IFN-alfa and RBV (PR). In the Phase 3 trials, IL28B genotype was a stratification factor.
Overall, SVR rates were lower in subjects with the CT and TT genotypes compared to those with the CC genotype (Tables 12 and 13). Among both treatment-naïve subjects and those who experienced previous treatment failures, subjects of all IL28B genotypes had the highest SVR rates with OLYSIO-containing regimens (Table 12).
| Trial (Population) | IL28B rs12979860 Genotype | OLYSIO + PR % (n/N) | Placebo + PR % (n/N) |
|---|---|---|---|
| SVR12: sustained virologic response 12 weeks after planned end of treatment (EOT). | |||
| QUEST 1 and QUEST 2 (treatment-naïve subjects) | C/C | 95 (144/152) | 80 (63/79) |
| C/T | 78 (228/292) | 41 (61/147) | |
| T/T | 61 (47/77) | 21 (8/38) | |
| PROMISE (prior relapsers) | C/C | 89 (55/62) | 53 (18/34) |
| C/T | 78 (131/167) | 34 (28/83) | |
| T/T | 65 (20/31) | 19 (3/16) | |
| Trial (Population) | IL28B rs12979860 Genotype | Treatment-Naïve Subjects % (n/N) | Prior Relapsers % (n/N) | Prior Partial Responders % (n/N) | Prior Null Responders % (n/N) |
|---|---|---|---|---|---|
| SVR12: sustained virologic response 12 weeks after planned EOT. | |||||
| C212 (HIV-1 co-infection) | C/C | 100 (15/15) | 100 (7/7) | 100 (1/1) | 80 (4/5) |
| C/T | 70 (19/27) | 100 (6/6) | 71 (5/7) | 53 (10/19) | |
| T/T | 80 (8/10) | 0 (0/2) | 50 (1/2) | 50 (2/4) | |
| RESTORE (HCV genotype 4) | C/C | 100 (7/7) | 100 (1/1) | - | - |
| C/T | 82 (14/17) | 82 (14/17) | 60 (3/5) | 41 (9/22) | |
| T/T | 80 (8/10) | 100 (4/4) | 60 (3/5) | 39 (7/18) | |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Simeprevir was not genotoxic in a series of in vitro and in vivo tests including the Ames test, the mammalian forward mutation assay in mouse lymphoma cells or the in vivo mammalian micronucleus test. Carcinogenicity studies with simeprevir have not been conducted.
If OLYSIO is administered in a combination regimen containing RBV, refer to the prescribing information for RBV for information on carcinogenesis and mutagenesis.
Impairment of Fertility
In a rat fertility study at doses up to 500 mg/kg/day, 3 male rats treated with simeprevir (2/24 rats at 50 mg/kg/day and 1/24 rats at 500 mg/kg/day) showed no motile sperm, small testes and epididymides, and resulted in infertility in 2 out of 3 of the male rats at exposures less than the exposure in humans at the recommended clinical dose.
If OLYSIO is administered with Peg-IFN-alfa and RBV, refer to the prescribing information for Peg-IFN-alfa and RBV for information on impairment of fertility.
13.2 Animal Toxicology and/or Pharmacology
Cardiovascular toxicity consisting of acute endocardial and myocardial necrosis restricted to the left ventricular subendocardial area was seen in 2 out of 6 animals in a 2-week oral dog toxicity study at an exposure approximately 28 times the mean AUC in humans at the recommended daily dose of 150 mg. No cardiac findings were observed in a 6-month and a 9-month oral toxicity study at exposures, respectively, of 11 and 4 times the mean AUC in humans at the recommended daily dose of 150 mg.
If OLYSIO is administered with Peg-IFN-alfa and RBV, refer to the prescribing information for Peg-IFN-alfa and RBV for information on animal toxicology.
14 CLINICAL STUDIES
14.1 Overview of Clinical Trials
The efficacy of OLYSIO in combination with sofosbuvir in subjects with HCV genotype 1 infection was evaluated in one Phase 2 trial (COSMOS) in prior null responders and treatment-naïve subjects with compensated cirrhosis (Child-Pugh A) or without cirrhosis, and in two Phase 3 trials in subjects with compensated cirrhosis (Child-Pugh A) or without cirrhosis (OPTIMIST-2 and OPTIMIST-1, respectively) who were HCV treatment-naïve or treatment-experienced (following prior treatment with IFN [pegylated or non-pegylated], with or without RBV) (see Table 14). Efficacy data from OPTIMIST-2, which evaluated OLYSIO in combination with sofosbuvir in subjects with compensated cirrhosis, are not shown because subjects in this trial received a shorter than recommended duration of therapy.
| Trial | Population | Relevant Study Arms (Number of Subjects Treated) |
|---|---|---|
| GT: genotype; TN: treatment-naïve; TE: treatment-experienced. | ||
| COSMOS (open-label) | GT 1, TN or TE*, with compensated cirrhosis or without cirrhosis |
|
| OPTIMIST-1 (open-label) | GT 1, TN or TE†, without cirrhosis |
|
| OPTIMIST-2 (open-label) | GT 1, TN or TE†, with compensated cirrhosis |
|
The efficacy of OLYSIO in combination with Peg-IFN-alfa and RBV in patients with HCV genotype 1 infection was evaluated in three Phase 3 trials in treatment-naïve subjects (QUEST 1, QUEST 2 and TIGER), one Phase 3 trial in subjects who relapsed after prior interferon-based therapy (PROMISE), one Phase 2 trial in subjects who failed prior therapy with Peg-IFN and RBV (including prior relapsers, partial and null responders) (ASPIRE), and one Phase 3 trial in subjects with HCV genotype 1 and HIV-1 co-infection who were HCV treatment-naïve or failed previous HCV therapy with Peg-IFN and RBV (C212), as summarized in Table 15.
The efficacy of OLYSIO in combination with Peg-IFN-alfa and RBV in patients with HCV genotype 4 infection was evaluated in one Phase 3 trial in treatment-naïve subjects or subjects who failed previous therapy with Peg-IFN and RBV (RESTORE) (see Table 15).
| Trial | Population | Relevant Study Arms (Number of Subjects Treated) |
|---|---|---|
| GT: genotype; TN: treatment-naïve; TE: treatment-experienced, includes prior relapsers, partial responders and null responders following prior treatment with Peg-IFN and RBV. | ||
|
||
| QUEST-1 (double-blind) | GT 1, TN, with compensated cirrhosis or without cirrhosis |
|
| QUEST-2 (double-blind) | GT 1, TN, with compensated cirrhosis or without cirrhosis |
|
| TIGER (double-blind) | GT 1, TN, with compensated cirrhosis or without cirrhosis |
|
| PROMISE (double-blind) | GT 1, TE*, with compensated cirrhosis or without cirrhosis |
|
| ASPIRE (double-blind) | GT 1, TE, with compensated cirrhosis or without cirrhosis |
|
| C212 (open-label) | GT 1, TN or TE, with compensated cirrhosis or without cirrhosis, HCV/HIV-1 co-infected |
|
| RESTORE (open-label) | GT 4, TN or TE, with compensated cirrhosis or without cirrhosis |
|
Prior relapsers were subjects who had HCV RNA not detected at the end of prior IFN-based therapy and HCV RNA detected during follow-up; prior partial responders were subjects with prior on-treatment greater than or equal to 2 log10 reduction in HCV RNA from baseline at Week 12 and HCV RNA detected at the end of prior therapy with Peg-IFN and RBV; and null responders were subjects with prior on-treatment less than 2 log10 reduction in HCV RNA from baseline at Week 12 during prior therapy with Peg-IFN and RBV. These trials included subjects with compensated cirrhosis (Child-Pugh A) or without cirrhosis, HCV RNA of at least 10000 IU/mL, and liver histopathology consistent with chronic HCV infection. In subjects who were treatment-naïve and prior relapsers, the overall duration of treatment with Peg-IFN-alfa and RBV in the Phase 3 trials was response-guided. In these subjects, the planned total duration of HCV treatment was 24 weeks if the following on-treatment protocol-defined response-guided therapy (RGT) criteria were met: HCV RNA lower than 25 IU/mL (detected or not detected) at Week 4 AND HCV RNA not detected at Week 12. Plasma HCV RNA levels were measured using the Roche COBAS® TaqMan® HCV test (version 2.0), for use with the High Pure System (25 IU/mL lower limit of quantification and 15 IU/mL limit of detection). Treatment stopping rules for HCV therapy were used to ensure that subjects with inadequate on-treatment virologic response discontinued treatment in a timely manner. In the Phase 3 trial C212 in HCV/HIV-1 co-infected subjects, the total duration of treatment with Peg-IFN-alfa and RBV in treatment-naïve and prior relapser subjects with compensated cirrhosis was not response-guided; these subjects received a fixed total duration of HCV treatment of 48 weeks. The total duration of treatment with Peg-IFN-alfa and RBV in non-cirrhotic HCV/HIV-1 co-infected treatment-naïve or prior relapser subjects was response-guided using the same criteria.
14.2 OLYSIO in Combination with Sofosbuvir
Adult Subjects with HCV Genotype 1 Infection
The efficacy of OLYSIO (150 mg once daily) in combination with sofosbuvir (400 mg once daily) in HCV genotype 1-infected treatment-naïve or treatment-experienced subjects with compensated cirrhosis (Child-Pugh A) or without cirrhosis was demonstrated in one Phase 2 trial (COSMOS) and one Phase 3 trial (OPTIMIST-1).
The COSMOS trial was an open-label, randomized Phase 2 trial to investigate the efficacy and safety of 12 or 24 weeks of OLYSIO (150 mg once daily) in combination with sofosbuvir (400 mg once daily) without or with RBV in HCV genotype 1-infected prior null responders with METAVIR fibrosis score F0–F2, or treatment-naïve subjects and prior null responders with METAVIR fibrosis score F3–F4 and compensated liver disease.
Results from treatment arms containing RBV in addition to OLYSIO and sofosbuvir in the COSMOS trial are not shown because efficacy was similar with or without RBV, and thus addition of RBV to OLYSIO and sofosbuvir is not recommended. In this trial, 28 subjects received 12 weeks of OLYSIO in combination with sofosbuvir and 31 subjects received 24 weeks of OLYSIO in combination with sofosbuvir. These 59 subjects had a median age of 57 years (range 27 to 68 years; with 2% above 65 years); 53% were male; 76% were White, and 24% Black or African American; 46% had a BMI greater than or equal to 30 kg/m2; the median baseline HCV RNA level was 6.75 log10 IU/mL; 19%, 31% and 22% had METAVIR fibrosis scores F0–F1, F2 and F3, respectively, and 29% had METAVIR fibrosis score F4 (cirrhosis); 75% had HCV genotype 1a of which 41% carried Q80K at baseline, and 25% had HCV genotype 1b; 14% had IL28B CC genotype, 64% IL28B CT genotype, and 22% IL28B TT genotype; 75% were prior null responders to Peg-IFN-alfa and RBV, and 25% were treatment-naïve.
OPTIMIST-1 was an open-label, randomized Phase 3 trial in HCV genotype 1-infected subjects without cirrhosis who were treatment-naïve or treatment-experienced (including prior relapsers, non-responders and IFN-intolerant subjects). Subjects were randomized to treatment arms of different durations. One hundred fifty-five subjects received 12 weeks of OLYSIO with sofosbuvir. The 155 subjects without cirrhosis receiving 12 weeks of OLYSIO with sofosbuvir had a median age of 56 years (range 19 to 70 years; with 7% above 65 years); 53% were male; 78% were White, 20% Black or African American, and 16% Hispanic; 37% had a BMI ≥ 30 kg/m2; the median baseline HCV RNA level was 6.83 log10 IU/mL; 75% had HCV genotype 1a of which 40% had Q80K polymorphism at baseline, and 25% had HCV genotype 1b; 28% had IL28B CC genotype, 55% IL28B CT genotype, and 17% IL28B TT genotype; 74% were treatment-naïve and 26% were treatment-experienced.
In the COSMOS and OPTIMIST-1 trials, SVR12 was achieved in 170/176 (97%) subjects without cirrhosis treated with 12 weeks OLYSIO in combination with sofosbuvir, as shown in Table 16. In the COSMOS trial, 10/10 (100%) subjects with compensated cirrhosis (Child-Pugh A) who received 24 weeks of OLYSIO with sofosbuvir achieved SVR12.
| Response Rates | OLYSIO+sofosbuvir*
12 weeks N=176 % (n/N) |
|---|---|
| SVR12: sustained virologic response 12 weeks after actual (OPTIMIST-1) or planned (COSMOS) EOT. | |
|
|
| Overall SVR12 | 97 (170/176) |
| Outcome for subjects without SVR12 | |
| Viral relapse† | 3 (5/175) |
Among subjects without cirrhosis in OPTIMIST-1 who received 12 weeks of OLYSIO in combination with sofosbuvir, similar SVR12 rates were observed among subgroups, including: treatment-naïve and treatment-experienced subjects (112/115 [97%] and 38/40 [95%] respectively), subjects with HCV genotype 1a with and without NS3 Q80K polymorphism (44/46 [96%] and 68/70 [97%], respectively), genotype 1b (38/39 [97%]), and subjects with IL28B CC and non-CC genotypes (43/43 [100%] and 107/112 [96%], respectively).
14.3 OLYSIO in Combination with Peg-IFN-alfa and RBV
Treatment-Naïve Adult Subjects with HCV Genotype 1 Infection
The efficacy of OLYSIO in treatment-naïve patients with HCV genotype 1 infection was demonstrated in two randomized, double-blind, placebo-controlled, 2-arm, multicenter, Phase 3 trials (QUEST 1 and QUEST 2). The designs of both trials were similar. All subjects received 12 weeks of once daily treatment with 150 mg OLYSIO or placebo, plus Peg-IFN-alfa-2a (QUEST 1 and QUEST 2) or Peg-IFN-alfa-2b (QUEST 2) and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa and RBV in accordance with the on-treatment protocol-defined RGT criteria. Subjects in the control groups received 48 weeks of Peg-IFN-alfa-2a or -2b and RBV.
In the pooled analysis for QUEST 1 and QUEST 2, demographics and baseline characteristics were balanced between both trials and between the OLYSIO and placebo treatment groups. In the pooled analysis of trials (QUEST 1 and QUEST 2), the 785 enrolled subjects had a median age of 47 years (range: 18 to 73 years; with 2% above 65 years); 56% were male; 91% were White, 7% Black or African American, 1% Asian, and 17% Hispanic; 23% had a body mass index (BMI) greater than or equal to 30 kg/m2; 78% had baseline HCV RNA levels greater than 800000 IU/mL; 74% had METAVIR fibrosis score F0, F1 or F2, 16% METAVIR fibrosis score F3, and 10% METAVIR fibrosis score F4 (cirrhosis); 48% had HCV genotype 1a, and 51% HCV genotype 1b; 29% had IL28B CC genotype, 56% IL28B CT genotype, and 15% IL28B TT genotype; 17% of the overall population and 34% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. In QUEST 1, all subjects received Peg-IFN-alfa-2a; in QUEST 2, 69% of the subjects received Peg-IFN-alfa-2a and 31% received Peg-IFN-alfa-2b.
Table 17 shows the response rates in treatment-naïve adult subjects with HCV genotype 1 infection. In the OLYSIO treatment group, SVR12 rates were lower in subjects with genotype 1a virus with the NS3 Q80K polymorphism at baseline compared to subjects infected with genotype 1a virus without the Q80K polymorphism.
| Response Rate | OLYSIO + PR N=521 % (n/N) | Placebo + PR N=264 % (n/N) |
|---|---|---|
| OLYSIO: 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a or -2b and RBV for 24 or 48 weeks; Placebo: placebo for 12 weeks with Peg-IFN-alfa-2a or -2b and RBV for 48 weeks. SVR12: sustained virologic response 12 weeks after planned EOT. | ||
|
||
| Overall SVR12 (genotype 1a and 1b) | 80 (419/521) | 50 (132/264) |
| Genotype 1a | 75 (191/254) | 47 (62/131) |
| Without Q80K | 84 (138/165) | 43 (36/83) |
| With Q80K | 58 (49/84) | 52 (23/44) |
| Genotype 1b | 85 (228/267) | 53 (70/133) |
| Outcome for subjects without SVR12 | ||
| On-treatment failure* | 8 (42/521) | 33 (87/264) |
| Viral relapse† | 11 (51/470) | 23 (39/172) |
In the pooled analysis of QUEST 1 and QUEST 2, 88% (459/521) of OLYSIO-treated subjects were eligible for a total treatment duration of 24 weeks. In these subjects, the SVR12 rate was 88% (405/459).
Seventy-nine percent (79%; 404/509) of OLYSIO-treated subjects had HCV RNA not detected at Week 4 (RVR); in these subjects the SVR12 rate was 90% (362/404).
SVR12 rates were higher for the OLYSIO treatment group compared to the placebo treatment group by sex, age, race, BMI, HCV genotype/subtype, baseline HCV RNA load (less than or equal to 800000 IU/mL, greater than 800000 IU/mL), METAVIR fibrosis score, and IL28B genotype. Table 18 shows the SVR rates by METAVIR fibrosis score.
| Subgroup | OLYSIO + PR % (n/N) | Placebo + PR % (n/N) |
|---|---|---|
| OLYSIO: 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a or -2b and RBV for 24 or 48 weeks; Placebo: placebo for 12 weeks with Peg-IFN-alfa-2a or -2b and RBV for 48 weeks. SVR12: sustained virologic response 12 weeks after planned EOT. | ||
| F0–2 | 84 (317/378) | 55 (106/192) |
| F3–4 | 68 (89/130) | 36 (26/72) |
SVR12 rates were higher for subjects receiving OLYSIO with Peg-IFN-alfa-2a or Peg-IFN-alfa-2b and RBV (88% and 78%, respectively) compared to subjects receiving placebo with Peg-IFN-alfa-2a or Peg-IFN-alfa-2b and RBV (62% and 42%, respectively) (QUEST 2).
Treatment-Naïve East Asian Subjects with HCV Genotype 1 Infection
TIGER was a Phase 3, randomized, double-blind, placebo-controlled trial in HCV genotype 1-infected treatment-naïve adult subjects from China and South Korea.
In this trial, 152 subjects received 12 weeks of once-daily treatment with 150 mg OLYSIO plus Peg-IFN-alfa-2a and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa-2a and RBV in accordance with protocol-defined RGT criteria; and 152 subjects received 12 weeks of placebo plus Peg-IFN-alfa-2a and RBV, followed by 36 weeks therapy with Peg-IFN-alfa-2a and RBV. These 304 subjects had a median age of 45 years (range: 18 to 68 years; with 2% above 65 years); 49% were male; all were East Asians (81% were enrolled in China, and 19% in South Korea); 3% had a body mass index (BMI) greater or equal to 30 kg/m2; 84% had baseline HCV RNA levels greater than 800000 IU/mL; 82% had METAVIR fibrosis score F0, F1 or F2, 12% METAVIR fibrosis score F3, and 6% METAVIR fibrosis score F4 (cirrhosis); 1% had HCV genotype 1a, and 99% HCV genotype 1b; less than 1% of the overall population had Q80K polymorphism at baseline; 79% had IL28B CC genotype, 20% IL28B CT genotype, and 1% IL28B TT genotype. Demographics and baseline characteristics were balanced across the OLYSIO 150 mg and placebo treatment groups.
SVR12 rates were 91% (138/152) in the OLYSIO 150 mg treatment group and 76% (115/152) in the placebo treatment group [see Adverse Reactions (6.1), Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Adult Subjects with HCV Genotype 1 Infection who Failed Prior Peg-IFN-alfa and RBV Therapy
The PROMISE trial was a randomized, double-blind, placebo-controlled, 2-arm, multicenter, Phase 3 trial in subjects with HCV genotype 1 infection who relapsed after prior IFN-based therapy. All subjects received 12 weeks of once daily treatment with 150 mg OLYSIO or placebo, plus Peg-IFN-alfa-2a and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa-2a and RBV in accordance with the protocol-defined RGT criteria. Subjects in the control group received 48 weeks of Peg-IFN-alfa-2a and RBV.
Demographics and baseline characteristics were balanced between the OLYSIO and placebo treatment groups. The 393 subjects enrolled in the PROMISE trial had a median age of 52 years (range: 20 to 71 years; with 3% above 65 years); 66% were male; 94% were White, 3% Black or African American, 2% Asian, and 7% Hispanic; 26% had a BMI greater than or equal to 30 kg/m2; 84% had baseline HCV RNA levels greater than 800000 IU/mL; 69% had METAVIR fibrosis score F0, F1 or F2, 15% METAVIR fibrosis score F3, and 15% METAVIR fibrosis score F4 (cirrhosis); 42% had HCV genotype 1a, and 58% HCV genotype 1b; 24% had IL28B CC genotype, 64% IL28B CT genotype, and 12% IL28B TT genotype; 13% of the overall population and 31% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. The prior IFN-based HCV therapy was Peg-IFN-alfa-2a/RBV (68%) or Peg-IFN-alfa-2b/RBV (27%).
Table 19 shows the response rates for the OLYSIO and placebo treatment groups in adult subjects with HCV genotype 1 infection who relapsed after prior interferon-based therapy. In the OLYSIO treatment group, SVR12 rates were lower in subjects infected with genotype 1a virus with the NS3 Q80K polymorphism at baseline compared to subjects infected with genotype 1a virus without the Q80K polymorphism.
| Response Rates | OLYSIO + PR N=260 % (n/N) | Placebo + PR N=133 % (n/N) |
|---|---|---|
| OLYSIO: 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks; Placebo: placebo for 12 weeks with Peg-IFN-alfa-2a and RBV for 48 weeks. SVR12: sustained virologic response 12 weeks after planned EOT. | ||
|
||
| Overall SVR12 (genotype 1a and 1b) | 79 (206/260) | 37 (49/133) |
| Genotype 1a | 70 (78/111) | 28 (15/54) |
| Without Q80K | 78 (62/79) | 26 (9/34) |
| With Q80K | 47 (14/30) | 30 (6/20) |
| Genotype 1b | 86 (128/149) | 43 (34/79) |
| Outcome for subjects without SVR12 | ||
| On-treatment failure* | 3 (8/260) | 27 (36/133) |
| Viral relapse† | 18 (46/249) | 48 (45/93) |
In PROMISE, 93% (241/260) of OLYSIO-treated subjects were eligible for a total treatment duration of 24 weeks. In these subjects, the SVR12 rate was 83% (200/241).
Seventy-seven percent (77%; 200/259) of OLYSIO-treated subjects had HCV RNA not detected at Week 4 (RVR); in these subjects the SVR12 rate was 87% (173/200).
SVR12 rates were higher for the OLYSIO treatment group compared to the placebo treatment group by sex, age, race, BMI, HCV genotype/subtype, baseline HCV RNA load (less than or equal to 800000 IU/mL, greater than 800000 IU/mL), prior HCV therapy, METAVIR fibrosis score, and IL28B genotype. Table 20 shows the SVR rates by METAVIR fibrosis score.
| Subgroup | OLYSIO + PR % (n/N) | Placebo + PR % (n/N) |
|---|---|---|
| OLYSIO: 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks; Placebo: placebo for 12 weeks with Peg-IFN-alfa-2a and RBV for 48 weeks. SVR12: sustained virologic response 12 weeks after planned EOT. | ||
| F0–2 | 82 (137/167) | 41 (40/98) |
| F3–4 | 73 (61/83) | 24 (8/34) |
The ASPIRE trial was a randomized, double-blind, placebo-controlled, Phase 2 trial in subjects with HCV genotype 1 infection, who failed prior therapy with Peg-IFN-alfa and RBV (including prior relapsers, partial responders or null responders).
In this trial, 66 subjects received 12 weeks of 150 mg OLYSIO in combination with Peg-IFN-alfa-2a and RBV for 48 weeks, and 66 subjects received placebo in combination with Peg-IFN-alfa-2a and RBV for 48 weeks. These 132 subjects had a median age of 49 years (range: 20 to 66 years; with 1% above 65 years); 66% were male; 93% were White, 3% Black or African American, and 2% Asian; 27% had a BMI greater than or equal to 30 kg/m2; 85% had baseline HCV RNA levels greater than 800000 IU/mL; 64% had METAVIR fibrosis score F0, F1, or F2, 18% METAVIR fibrosis score F3, and 18% METAVIR fibrosis score F4 (cirrhosis); 43% had HCV genotype 1a, and 57% HCV genotype 1b; 17% had IL28B CC genotype, 67% IL28B CT genotype, and 16% IL28B TT genotype (information available for 93 subjects); 27% of the overall population and 23% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. Forty percent (40%) of subjects were prior relapsers, 35% prior partial responders, and 25% prior null responders following prior therapy with Peg-IFN-alfa and RBV. Demographics and baseline characteristics were balanced between the 12 weeks 150 mg OLYSIO and placebo treatment groups.
Table 21 shows the response rates for the 12 weeks of 150 mg OLYSIO and placebo treatment groups in prior relapsers, prior partial responders and prior null responders.
| Response Rates | OLYSIO + PR N=66 % (n/N) | Placebo + PR N=66 % (n/N) |
|---|---|---|
| 150 mg OLYSIO: 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 48 weeks; Placebo: placebo with Peg-IFN-alfa-2a and RBV for 48 weeks. | ||
| SVR24: sustained virologic response defined as undetectable HCV RNA 24 weeks after planned EOT. | ||
|
||
| SVR24 | ||
| Prior relapsers | 77 (20/26) | 37 (10/27) |
| Prior partial responders | 65 (15/23) | 9 (2/23) |
| Prior null responders | 53 (9/17) | 19 (3/16) |
| Outcome for subjects without SVR24 | ||
| On-treatment virologic failure* | ||
| Prior relapsers | 8 (2/26) | 22 (6/27) |
| Prior partial responders | 22 (5/23) | 78 (18/23) |
| Prior null responders | 35 (6/17) | 75 (12/16) |
| Viral relapse† | ||
| Prior relapsers | 13 (3/23) | 47 (9/19) |
| Prior partial responders | 6 (1/17) | 50 (2/4) |
| Prior null responders | 18 (2/11) | 25 (1/4) |
SVR24 rates were higher in the OLYSIO-treated subjects compared to subjects receiving placebo in combination with Peg-IFN-alfa and RBV, regardless of HCV geno/subtype, METAVIR fibrosis score, and IL28B genotype.
Subjects with HCV/HIV-1 Co-Infection
C212 was an open-label, single-arm Phase 3 trial in HIV-1 subjects co-infected with HCV genotype 1 who were treatment-naïve or failed prior HCV therapy with Peg-IFN-alfa and RBV (including prior relapsers, partial responders or null responders). Non-cirrhotic treatment-naïve subjects or prior relapsers received 12 weeks of once-daily treatment with 150 mg OLYSIO plus Peg-IFN-alfa-2a and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa-2a and RBV in accordance with the protocol-defined RGT criteria. Prior non-responder subjects (partial and null response) and all cirrhotic subjects (METAVIR fibrosis score F4) received 36 weeks of Peg-IFN-alfa-2a and RBV after the initial 12 weeks of OLYSIO in combination with Peg-IFN-alfa-2a and RBV.
The 106 enrolled subjects in the C212 trial had a median age of 48 years (range: 27 to 67 years; with 2% above 65 years); 85% were male; 82% were White, 14% Black or African American, 1% Asian, and 6% Hispanic; 12% had a BMI greater than or equal to 30 kg/m2; 86% had baseline HCV RNA levels greater than 800,000 IU/mL; 68% had METAVIR fibrosis score F0, F1 or F2, 19% METAVIR fibrosis score F3, and 13% METAVIR fibrosis score F4; 82% had HCV genotype 1a, and 17% HCV genotype 1b; 28% of the overall population and 34% of the subjects with genotype 1a had Q80K polymorphism at baseline; 27% had IL28B CC genotype, 56% IL28B CT genotype, and 17% IL28B TT genotype; 50% (n=53) were HCV treatment-naïve subjects, 14% (n=15) prior relapsers, 9% (n=10) prior partial responders, and 26% (n=28) prior null responders. Eighty-eight percent (n=93) of the subjects were on highly active antiretroviral therapy (HAART), with nucleoside reverse transcriptase inhibitors and the integrase inhibitor raltegravir being the most commonly used HIV antiretroviral. HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors (except rilpivirine) were prohibited from use in this study.
The median baseline HIV-1 RNA levels and CD4+ cell count in subjects not on HAART were 4.18 log10 copies/mL (range: 1.3–4.9 log10 copies/mL) and 677 × 106 cells/L (range: 489–1076 × 106 cells/L), respectively. The median baseline CD4+ cell count in subjects on HAART was 561 × 106 cells/mL (range: 275–1407 × 106 cells/mL).
Table 22 shows the response rates in treatment-naïve, prior relapsers, prior partial responders and null responders.
| Response Rates | Treatment-Naïve Subjects N=53 % (n/N) | Prior Relapsers N=15 % (n/N) | Prior Partial Responders N=10 % (n/N) | Prior Null Responders N=28 % (n/N) |
|---|---|---|---|---|
| 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks. | ||||
| SVR12: sustained virologic response 12 weeks after planned EOT. | ||||
|
||||
| Overall SVR12 (genotype 1a and 1b) | 79 (42/53) | 87 (13/15) | 70 (7/10) | 57 (16/28) |
| Genotype 1a | 77 (33/43) | 83 (10/12) | 67 (6/9) | 54 (13/24) |
| Genotype 1b | 90 (9/10) | 100 (3/3) | 100 (1/1) | 75 (3/4) |
| Outcome for subjects without SVR12 | ||||
| On-treatment failure* | 9 (5/53) | 0 (0/15) | 20 (2/10) | 39 (11/28) |
| Viral relapse† | 10 (5/48) | 13 (2/15) | 0 (0/7) | 12 (2/17) |
Eighty-nine percent (n=54/61) of the OLYSIO-treated treatment-naïve subjects and prior relapsers without cirrhosis were eligible for a total treatment duration of 24 weeks. In these subjects, the SVR12 rate was 87%.
Seventy-one percent (n=37/52), 93% (n=14/15), 80% (n=8/10) and 36% (n=10/28) of OLYSIO-treated treatment-naïve subjects, prior relapsers, prior partial responders and prior null responders had undetectable HCV RNA at week 4 (RVR). In these subjects the SVR12 rates were 89%, 93%, 75% and 90%, respectively.
Table 23 shows the SVR rates by METAVIR fibrosis scores.
| Subgroup | Treatment-Naïve Subjects % (n/N) | Prior Relapsers % (n/N) | Prior Partial Responders % (n/N) | Prior Null Responders % (n/N) |
|---|---|---|---|---|
| 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks. | ||||
| SVR12: sustained virologic response 12 weeks after planned EOT. | ||||
| F0–2 | 89 (24/27) | 78 (7/9) | 50 (1/2) | 57 (4/7) |
| F3–4 | 57 (4/7) | 100 (2/2) | 67 (2/3) | 60 (6/10) |
Two subjects had HIV virologic failure defined as confirmed HIV-1 RNA at least 200 copies/mL after previous less than 50 copies/mL; these failures occurred 36 and 48 weeks after end of OLYSIO treatment.
Adult Subjects with HCV Genotype 4 Infection
RESTORE was an open-label, single-arm Phase 3 trial in subjects with HCV genotype 4 infection who were treatment-naïve or failed prior therapy with Peg-IFN-alfa and RBV (including prior relapsers, partial responders or null responders). Treatment-naïve subjects or prior relapsers received once-daily treatment with 150 mg OLYSIO plus Peg-IFN-alfa-2a and RBV for 12 weeks, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa-2a and RBV in accordance with the protocol-defined RGT criteria. Prior non-responder subjects (partial and null response) received once-daily treatment with 150 mg OLYSIO plus Peg-IFN-alfa-2a and RBV for 12 weeks, followed by 36 weeks of Peg-IFN-alfa-2a and RBV.
The 107 enrolled subjects in the RESTORE trial with HCV genotype 4 had a median age of 49 years (range: 27 to 69 years; with 5% above 65 years); 79% were male; 72% were White, 28% Black or African American, and 7% Hispanic; 14% had a BMI greater than or equal to 30 kg/m2; 60% had baseline HCV RNA levels greater than 800,000 IU/mL; 57% had METAVIR fibrosis score F0, F1 or F2, 14% METAVIR fibrosis score F3, and 29% METAVIR fibrosis score F4; 42% had HCV genotype 4a, and 24% had HCV genotype 4d; 8% had IL28B CC genotype, 58% IL28B CT genotype, and 35% IL28B TT genotype; 33% (n=35) were treatment-naïve HCV subjects, 21% (n=22) prior relapsers, 9% (n=10) prior partial responders, and 37% (n=40) prior null responders.
Table 24 shows the response rates in treatment-naïve, prior relapsers, prior partial responders and null responders. Table 25 shows the SVR rates by METAVIR fibrosis scores.
| Response Rates | Treatment-Naïve Subjects N=35 % (n/N) | Prior Relapsers N=22 % (n/N) | Prior Partial Responders N=10 % (n/N) | Prior Null Responders N=40 % (n/N) |
|---|---|---|---|---|
| 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks. | ||||
| SVR12: sustained virologic response 12 weeks after planned EOT. | ||||
|
||||
| Overall SVR12 | 83 (29/35) | 86 (19/22) | 60 (6/10) | 40 (16/40) |
| Outcome for subjects without SVR12 | ||||
| On-treatment failure* | 9 (3/35) | 9 (2/22) | 20 (2/10) | 45 (18/40) |
| Viral relapse† | 9 (3/35) | 5 (1/22) | 20 (2/10) | 15 (6/40) |
| Subgroup | Treatment-Naïve Subjects % (n/N) | Prior Relapsers % (n/N) | Prior Partial Responders % (n/N) | Prior Null Responders % (n/N) |
|---|---|---|---|---|
| 150 mg OLYSIO for 12 weeks with Peg-IFN-alfa-2a and RBV for 24 or 48 weeks. | ||||
| SVR12: sustained virologic response 12 weeks after planned EOT. | ||||
| F0–2 | 85 (22/26) | 91 (10/11) | 100 (5/5) | 47 (8/17) |
| F3–4 | 78 (7/9) | 82 (9/11) | 20 (1/5) | 35 (7/20) |
16 HOW SUPPLIED/STORAGE AND HANDLING
OLYSIO 150 mg capsules are white, marked with "TMC435 150" in black ink. The capsules are packaged into a bottle containing 28 capsules (NDC 59676-225-28).
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Inform patients that HBV reactivation can occur in patients coinfected with HBV during or after treatment of HCV infection. Advise patients to tell their healthcare provider if they have a history of HBV infection [see Warnings and Precautions (5.1)].
Symptomatic Bradycardia When Used in Combination with Sofosbuvir and Amiodarone
Advise patients to seek medical evaluation immediately for symptoms of bradycardia such as near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion or memory problems [see Warnings and Precautions (5.2), Adverse Reactions (6.2) and Drug Interactions (7.3)].
Pregnancy
Advise patients taking OLYSIO of the potential risk to the fetus. In addition, when OLYSIO is taken with RBV, advise patients to avoid pregnancy during treatment and within 6 months of stopping RBV and to notify their healthcare provider immediately in the event of a pregnancy [see Use in Specific Populations (8.1)].
Hepatic Decompensation and Failure
Inform patients to watch for early warning signs of liver inflammation, such as fatigue, weakness, lack of appetite, nausea and vomiting, as well as later signs such as jaundice and discolored feces, and to contact their healthcare provider immediately if such symptoms occur [see Warnings and Precautions (5.3)].
Photosensitivity
Advise patients of the risk of photosensitivity reactions related to OLYSIO combination treatment and that these reactions may be severe. Instruct patients to use effective sun protection measures to limit exposure to natural sunlight and to avoid artificial sunlight (tanning beds or phototherapy) during treatment with OLYSIO.
Advise patients to contact their healthcare provider immediately if they develop a photosensitivity reaction. Inform patients not to stop OLYSIO due to photosensitivity reactions unless instructed by their healthcare provider [see Warnings and Precautions (5.5)].
Rash
Advise patients of the risk of rash related to OLYSIO combination treatment and that rash may become severe. Advise patients to contact their healthcare provider immediately if they develop a rash. Inform patients not to stop OLYSIO due to rash unless instructed by their healthcare provider [see Warnings and Precautions (5.6)].
Administration
Advise patients to use OLYSIO only in combination with other antiviral drugs for the treatment of chronic HCV infection. Advise patients to discontinue OLYSIO if any of the other antiviral drugs used in combination with OLYSIO are permanently discontinued for any reason. Advise patients that the dose of OLYSIO must not be reduced or interrupted, as it may increase the possibility of treatment failure [see Dosage and Administration (2.1)].
Advise patients to take OLYSIO every day at the regularly scheduled time with food. Inform patients that it is important not to miss or skip doses and to take OLYSIO for the duration that is recommended by the healthcare provider. Inform patients not to take more or less than the prescribed dose of OLYSIO at any one time.
Product of Belgium
Manufactured by:
Janssen-Cilag SpA, Latina, Italy
Manufactured for:
Janssen Therapeutics, Division of Janssen Products, LP
Titusville NJ 08560
Licensed from Medivir AB
OLYSIO® is a registered trademark of Johnson & Johnson
© Janssen Products, LP 2017
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised February 2017 | |||
| PATIENT INFORMATION OLYSIO® (oh li see oh) (simeprevir) capsules |
||||
| Read this Patient Information before you start taking OLYSIO and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. | ||||
| Important: You should not take OLYSIO alone. OLYSIO should be used together with other antiviral medicines to treat chronic hepatitis C virus infection. When taking OLYSIO in combination with peginterferon alfa and ribavirin you should also read those Medication Guides. When taking OLYSIO in combination with sofosbuvir, you should also read its Patient Information leaflet. | ||||
| What is the most important information I should know about OLYSIO?
OLYSIO can cause serious side effects, including: Hepatitis B virus reactivation: Before starting treatment with OLYSIO, your healthcare provider will do blood tests to check for hepatitis B virus infection. If you have ever had hepatitis B virus infection, the hepatitis B virus could become active again during or after treatment of hepatitis C virus with OLYSIO. Hepatitis B virus becoming active again (called reactivation) may cause serious liver problems including liver failure and death. Your healthcare provider will monitor you if you are at risk for hepatitis B virus reactivation during treatment and after you stop taking OLYSIO. OLYSIO combination treatment with sofosbuvir (Sovaldi®) may result in slowing of the heart rate (pulse) along with other symptoms when taken with amiodarone (Cordarone®, Nexterone®, Pacerone®), a medicine used to treat certain heart problems.
|
||||
|
|
|||
|
OLYSIO may cause severe liver problems in some people. These severe liver problems may lead to liver failure or death.
|
||||
|
|
|||
|
OLYSIO combination treatment may cause rashes and skin reactions to sunlight. These rashes and skin reactions to sunlight can be severe and you may need to be treated in a hospital. Rashes and skin reactions to sunlight are most common during the first 4 weeks of treatment, but can happen at any time during combination treatment with OLYSIO.
|
||||
|
|
|||
| For more information about side effects, see the section "What are the possible side effects of OLYSIO?" | ||||
|---|---|---|---|---|
What is OLYSIO?
It is not known if OLYSIO is safe and effective in children under 18 years of age. |
||||
| What should I tell my healthcare provider before taking OLYSIO?
Before taking OLYSIO, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may interact with OLYSIO. This can cause you to have too much or not enough OLYSIO or other medicines in your body, which may affect the way OLYSIO or your other medicines work, or may cause side effects. Keep a list of your medicines and show it to your healthcare provider and pharmacist.
|
||||
How should I take OLYSIO?
|
||||
| What are the possible side effects of OLYSIO?
OLYSIO can cause serious side effects, including:
|
||||
|
The most common side effects of OLYSIO when used in combination with sofosbuvir include: |
||||
|
|
|
||
|
The most common side effects of OLYSIO when used in combination with peginterferon alfa and ribavirin include: |
||||
|
|
|
||
|
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of OLYSIO. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store OLYSIO?
Keep OLYSIO and all medicines out of the reach of children. |
||||
| General information about the safe and effective use of OLYSIO
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use OLYSIO for a condition for which it was not prescribed. Do not give your OLYSIO to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information about OLYSIO, talk with your pharmacist or healthcare provider. You can ask your pharmacist or healthcare provider for information about OLYSIO that is written for health professionals. For more information about OLYSIO, go to www.OLYSIO.com or call 1-800-526-7736. |
||||
| What are the ingredients in OLYSIO?
Active ingredient: simeprevir Inactive ingredients: colloidal anhydrous silica, croscarmellose sodium, lactose monohydrate, magnesium stearate, sodium lauryl sulphate. The white capsule contains gelatin and titanium dioxide (E171) and is printed with ink containing iron oxide black (E172) and shellac (E904). Product of Belgium Manufactured by: Janssen-Cilag SpA, Latina, Italy Manufactured for: Janssen Therapeutics, Division of Janssen Products, LP, Titusville NJ 08560 Licensed from Medivir AB OLYSIO® is a registered trademark of Johnson & Johnson © Janssen Products, LP 2017 |
||||

| OLYSIO
simeprevir capsule |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - Janssen Products LP (804684207) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceutica NV | 374747970 | API MANUFACTURE(59676-225) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Cilag SpA | 542797928 | MANUFACTURE(59676-225) , ANALYSIS(59676-225) , PACK(59676-225) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AndersonBrecon Inc. | 053217022 | LABEL(59676-225) | |