FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Monotherapy
Finasteride tablets are indicated for the treatment of symptomatic benign prostatic hyperplasia (BPH) in men with an enlarged prostate to:
-Improve symptoms
-Reduce the risk of acute urinary retention
-Reduce the risk of the need for surgery including transurethral resection of the prostate (TURP) and prostatectomy.
2 DOSAGE AND ADMINISTRATION
Finasteride tablets may be administered with or without meals.
2.1 Monotherapy
The recommended dose of finasteride is one tablet (5 mg) taken once a day [see Clinical Studies (14.1)].
2.2 Combination with Alpha-Blocker
The recommended dose of finasteride is one tablet (5 mg) taken once a day in combination with the
alpha-blocker doxazosin [see Clinical Studies (14.2)].
3 DOSAGE FORMS AND STRENGTHS
5 mg blue, film-coated, capsule-shaped, unscored tablets, debossed “93” on one side and “7355” on the other side.
4 CONTRAINDICATIONS
Finasteride is contraindicated in the following:
- Hypersensitivity to any component of this medication.
- Pregnancy. Finasteride use is contraindicated in females when they are or may potentially be pregnant. Because of the ability of Type II 5α-reductase inhibitors to inhibit the conversion of testosterone to 5α-dihydrotestosterone (DHT), finasteride may cause abnormalities of the external genitalia of a male fetus of a pregnant female who receives finasteride. If this drug is used during pregnancy, or if pregnancy occurs while taking this drug, the pregnant female should be apprised of the potential hazard to the male fetus [see also Warnings and Precautions (5.3), Use in Specific Populations (8.1), and How Supplied/Storage and Handling (16)]. In female rats, low doses of finasteride administered during pregnancy have produced abnormalities of the external genitalia in male offspring.
5 WARNINGS AND PRECAUTIONS
5.1 Effects on Prostate Specific Antigen (PSA) and the Use of PSA in Prostate Cancer Detection
In clinical studies, finasteride reduced serum PSA concentration by approximately 50% within six months of treatment. This decrease is predictable over the entire range of PSA values in patients with symptomatic BPH, although it may vary in individuals.
For interpretation of serial PSAs in men taking finasteride, a new PSA baseline should be established at least six months after starting treatment and PSA monitored periodically thereafter. Any confirmed increase from the lowest PSA value while on finasteride may signal the presence of prostate cancer and should be evaluated, even if PSA levels are still within the normal range for men not taking a 5α-reductase inhibitor. Non-compliance with finasteride therapy may also affect PSA test results. To interpret an isolated PSA value in patients treated with finasteride for six months or more, PSA values should be doubled for comparison with normal ranges in untreated men. These adjustments preserve the utility of PSA to detect prostate cancer in men treated with finasteride.
Finasteride may also cause decreases in serum PSA in the presence of prostate cancer.
The ratio of free to total PSA (percent free PSA) remains constant even under the influence of finasteride. If clinicians elect to use percent free PSA as an aid in the detection of prostate cancer in men undergoing finasteride therapy, no adjustment to its value appears necessary.
5.2 Increased Risk of High-Grade Prostate Cancer
Men aged 55 and over with a normal digital rectal examination and PSA ≤3.0 ng/mL at baseline taking finasteride 5 mg/day in the 7-year Prostate Cancer Prevention Trial (PCPT) had an increased risk of Gleason score 8 to 10 prostate cancer (finasteride 1.8% vs placebo 1.1%) [see Indications and Usage (1.3) and Adverse Reactions (6.1)]. Similar results were observed in a 4-year placebo-controlled clinical trial with another 5α-reductase inhibitor (dutasteride, AVODART) (1% dutasteride vs 0.5% placebo). 5α-reductase inhibitors may increase the risk of development of high-grade prostate cancer. Whether the effect of 5α-reductase inhibitors to reduce prostate volume, or study-related factors, impacted the results of these studies has not been established.
5.3 Exposure of Females — Risk to Male Fetus
Finasteride is contraindicated in pregnant females and in females who may potentially be pregnant and not indicated for use in females. Based on animal studies and the mechanism of action, finasteride may cause abnormal development of external genitalia in a male fetus if administered to a pregnant female. Females who are pregnant or may potentially be pregnant should not handle crushed or broken finasteride tablets. Finasteride tablets are coated and will prevent contact with the active ingredient during normal handling, provided that the tablets have not been broken or crushed. If a pregnant female comes in contact with crushed or broken finasteride tablets, the contact area should be washed immediately with soap and water [see Contraindications (4), Use in Specific Populations (8.1), Clinical Pharmacology (12.1 and 12.3), and How Supplied/Storage and Handling (16)].
5.4 Pediatric Patients and Females
Finasteride is not indicated for use in pediatric patients [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)] or females [see also Warnings and Precautions (5.3), Use in Specific Populations (8.1), Clinical Pharmacology (12.3), and How Supplied/Storage and Handling (16)].
5.5 Effect on Semen Characteristics
Treatment with finasteride for 24 weeks to evaluate semen parameters in healthy male volunteers revealed no clinically meaningful effects on sperm concentration, mobility, morphology, or pH. A 0.6 mL (22.1%) median decrease in ejaculate volume with a concomitant reduction in total sperm per ejaculate was observed. These parameters remained within the normal range and were reversible upon discontinuation of therapy with an average time to return to baseline of 84 weeks.
5.6 Consideration of Other Urological Conditions
Prior to initiating treatment with finasteride, consideration should be given to other urological conditions that may cause similar symptoms. In addition, prostate cancer and BPH may coexist.
Patients with large residual urinary volume and/or severely diminished urinary flow should be carefully monitored for obstructive uropathy. These patients may not be candidates for finasteride therapy.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
4-Year Placebo-Controlled Study (Long-term Efficacy and Safety Study)
In a long-term efficacy and safety study, 1524 patients treated with finasteride and 1516 patients treated with placebo were evaluated for safety over a period of 4 years. The most frequently reported adverse reactions were related to sexual function. 3.7% (57 patients) treated with finasteride and 2.1% (32 patients) treated with placebo discontinued therapy as a result of adverse reactions related to sexual function, which are the most frequently reported adverse reactions.
Table 1 presents the only clinical adverse reactions considered possibly, probably or definitely drug related by the investigator, for which the incidence on finasteride was ≥1% and greater than placebo over the 4 years of the study. In years 2 to 4 of the study, there was no significant difference between treatment groups in the incidences of impotence, decreased libido and ejaculation disorder.
|
Year 1 |
Years 2, 3 and 4* |
|||
|
Finasteride |
Placebo |
Finasteride |
Placebo |
|
|
Impotence |
8.1 |
3.7 |
5.1 |
5.1 |
|
Decreased Libido |
6.4 |
3.4 |
2.6 |
2.6 |
|
Decreased Volume of Ejaculate |
3.7 |
0.8 |
1.5 |
0.5 |
|
Ejaculation Disorder |
0.8 |
0.1 |
0.2 |
0.1 |
|
Breast Enlargement |
0.5 |
0.1 |
1.8 |
1.1 |
|
Breast Tenderness |
0.4 |
0.1 |
0.7 |
0.3 |
|
Rash |
0.5 |
0.2 |
0.5 |
0.1 |
| *Combined Years 2 to 4 | ||||
| N = 1524 and 1516, finasteride vs placebo, respectively | ||||
Phase III Studies and 5-Year Open Extensions
The adverse experience profile in the 1-year, placebo-controlled, Phase III studies, the 5-year open extensions, and the long-term efficacy and safety study were similar.
Medical Therapy of Prostatic Symptoms (MTOPS) Study
In the MTOPS study, 3047 men with symptomatic BPH were randomized to receive finasteride 5 mg/day (n=768), doxazosin 4 or 8 mg/day (n=756), the combination of finasteride 5 mg/day and doxazosin 4 or 8 mg/day (n=786), or placebo (n=737) for 4 to 6 years [see Clinical Studies (14.2)].
The incidence rates of drug-related adverse experiences reported by ≥2% of patients in any treatment group in the MTOPS Study are listed in Table 2.
The individual adverse effects which occurred more frequently in the combination group compared to either drug alone were: asthenia, postural hypotension, peripheral edema, dizziness, decreased libido, rhinitis, abnormal ejaculation, impotence and abnormal sexual function (see Table 2). Of these, the incidence of abnormal ejaculation in patients receiving combination therapy was comparable to the sum of the incidences of this adverse experience reported for the two monotherapies.
Combination therapy with finasteride and doxazosin was associated with no new clinical adverse experience.
Four patients in MTOPS reported the adverse experience breast cancer. Three of these patients were on finasteride only and one was on combination therapy [see Long-Term Data].
The MTOPS Study was not specifically designed to make statistical comparisons between groups for reported adverse experiences. In addition, direct comparisons of safety data between the MTOPS study and previous studies of the single agents may not be appropriate based upon differences in patient population, dosage or dose regimen, and other procedural and study design elements.
|
Adverse Experience |
Placebo |
Doxazosin 4 mg or 8 mg* |
Finasteride |
Combination |
|
(N=737) |
(N=756) |
(N=768) |
(N=786) |
|
|
(%) |
(%) |
(%) |
(%) |
|
|
Body as a whole | ||||
|
Asthenia |
7.1 |
15.7 |
5.3 |
16.8 |
|
Headache |
2.3 |
4.1 |
2.0 |
2.3 |
|
Cardiovascular | ||||
|
Hypotension |
0.7 |
3.4 |
1.2 |
1.5 |
|
Postural Hypotension | 8.0 |
16.7 |
9.1 |
17.8 |
|
Metabolic and Nutritional | ||||
|
Peripheral Edema |
0.9 |
2.6 |
1.3 |
3.3 |
|
Nervous | ||||
|
Dizziness |
8.1 |
17.7 |
7.4 |
23.2 |
|
Libido Decreased |
5.7 |
7.0 |
10.0 |
11.6 |
|
Somnolence |
1.5 |
3.7 |
1.7 |
3.1 |
|
Respiratory | ||||
|
Dyspnea |
0.7 |
2.1 |
0.7 |
1.9 |
|
Rhinitis |
0.5 |
1.3 |
1.0 |
2.4 |
|
Urogenital | ||||
|
Abnormal Ejaculation |
2.3 |
4.5 |
7.2 |
14.1 |
|
Gynecomastia |
0.7 |
1.1 |
2.2 |
1.5 |
|
Impotence |
12.2 |
14.4 |
18.5 |
22.6 |
|
Sexual Function Abnormal |
0.9 |
2.0 |
2.5 |
3.1 |
| *Doxazosin dose was achieved by weekly titration (1 mg to 2 mg to 4 mg to 8 mg). The final tolerated dose (4 mg or 8 mg) was administered at end-Week 4. Only those patients tolerating at least 4 mg were kept on doxazosin. The majority of patients received the 8-mg dose over the duration of the study. | ||||
Long-Term Data
High-Grade Prostate Cancer
The PCPT trial was a 7-year randomized, double-blind, placebo-controlled trial that enrolled 18,882 men ≥55 years of age with a normal digital rectal examination and a PSA
≤3.0 ng/mL. Men received either finasteride 5 mg or placebo daily. Patients were evaluated annually with PSA and digital rectal exams. Biopsies were performed for elevated PSA, an abnormal digital rectal exam, or the end of study. The incidence of Gleason score 8 to 10 prostate cancer was higher in men treated with finasteride (1.8%) than in those treated with placebo (1.1%) [see Indications and Usage (1.3) and Warnings and Precautions (5.2)]. In a 4-year placebo-controlled clinical trial with another 5α-reductase inhibitor (dutasteride, AVODART), similar results for Gleason score 8 to 10 prostate cancer were observed (1% dutasteride vs 0.5% placebo).
No clinical benefit has been demonstrated in patients with prostate cancer treated with finasteride.
Breast Cancer
During the 4- to 6-year placebo- and comparator-controlled MTOPS study that enrolled 3047 men, there were 4 cases of breast cancer in men treated with finasteride but no cases in men not treated with finasteride. During the 4-year, placebo-controlled long-term efficacy and safety study that enrolled 3040 men, there were 2 cases of breast cancer in placebo-treated men but no cases in men treated with finasteride. During the 7-year placebo-controlled Prostate Cancer Prevention Trial (PCPT) that enrolled 18,882 men, there was 1 case of breast cancer in men treated with finasteride, and 1 case of breast cancer in men treated with placebo. The relationship between long-term use of finasteride and male breast neoplasia is currently unknown.
Sexual Function
There is no evidence of increased sexual adverse experiences with increased duration of treatment with finasteride. New reports of drug-related sexual adverse experiences decreased with duration of therapy.
6.2 Postmarketing Experience
The following additional adverse events have been reported in postmarketing experience with finasteride. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- hypersensitivity reactions, such as pruritus, urticaria, and angioedema (including swelling of the lips, tongue, throat, and face)
- testicular pain
- hematospermia
- sexual dysfunction that continued after discontinuation of treatment, including erectile dysfunction, decreased libido and ejaculation disorders (e.g. reduced ejaculate volume). These events were reported rarely in men taking finasteride for the treatment of BPH. Most men were older and were taking concomitant medications and/or had co-morbid conditions. The independent role of finasteride in these events is unknown.
- male infertility and/or poor seminal quality were reported rarely in men taking finasteride for the treatment of BPH. Normalization or improvement of poor seminal quality has been reported after discontinuation of finasteride. The independent role of finasteride in these events is unknown.
- depression
- male breast cancer.
The following additional adverse event related to sexual dysfunction that continued after discontinuation of treatment has been reported in postmarketing experience with finasteride at lower doses used to treat male pattern baldness. Because the event is reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate its frequency or establish a causal relationship to drug exposure:
- orgasm disorders
7 DRUG INTERACTIONS
7.1 Cytochrome P450-Linked Drug Metabolizing Enzyme System
No drug interactions of clinical importance have been identified. Finasteride does not appear to affect the cytochrome P450-linked drug metabolizing enzyme system. Compounds that have been tested in man have included antipyrine, digoxin, propranolol, theophylline, and warfarin and no clinically meaningful interactions were found.
7.2 Other Concomitant Therapy
Although specific interaction studies were not performed, finasteride was concomitantly used in clinical studies with acetaminophen, acetylsalicylic acid, α-blockers, angiotensin-converting enzyme (ACE) inhibitors, analgesics, anti-convulsants, beta-adrenergic blocking agents, diuretics, calcium channel blockers, cardiac nitrates, HMG-CoA reductase inhibitors, nonsteroidal anti-inflammatory drugs (NSAIDs), benzodiazepines, H2 antagonists and quinolone anti-infectives without evidence of clinically significant adverse interactions.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Finasteride is contraindicated in pregnant females and not indicated for use in females. Based on animal studies and the mechanism of action, finasteride may cause abnormal development of external genitalia in a male fetus if administered to a pregnant female [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.1)].
In an embryo-fetal development study in rats, there was a dose-dependent increase in hypospadias that occurred in 3.6% to 100% of male offspring of pregnant rats administered oral finasteride during the period of major organogenesis at doses approximately 0.1 to 86 times the maximum recommended human dose (MRHD) of 5 mg/day (based on AUC at animal doses of 0.1 mg/kg/day to 100 mg/kg/day). Decreased prostatic and seminal vesicular weights, delayed preputial separation and transient nipple development were also observed in male offspring at oral maternal doses approximately 0.03 times the MRHD (based on AUC at animal dose of 0.03 mg/kg/day), along with decreased anogenital distance in male offspring at oral maternal doses approximately 0.003 times the MRHD (based on AUC at animal dose of 0.003 mg/kg/day).
Finasteride is a Type II 5α-reductase inhibitor that prevents conversion of testosterone to 5α-dihydrotestosterone (DHT), a hormone necessary for normal development of male genitalia. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the male fetus.
Abnormal male genital development is an expected consequence when conversion of testosterone to
5α-dihydrotestosterone (DHT) is inhibited by 5α-reductase inhibitors. These outcomes are similar to those reported in male infants with genetic 5α-reductase deficiency. Females could be exposed to finasteride through contact with crushed or broken finasteride tablets or semen from a male partner taking finasteride. With regard to finasteride exposure through the skin, finasteride tablets are coated and will prevent skin contact with finasteride during normal handling if the tablets have not been crushed or broken. Females who are pregnant or may potentially be pregnant should not handle crushed or broken finasteride tablets because of possible exposure of a male fetus. With regard to potential finasteride exposure through semen, three studies have been conducted that measured finasteride concentrations in semen in men receiving finasteride 5 mg/day. In these studies the highest amount of finasteride in semen was estimated to be 50- to 100-fold less than the dose of finasteride (5 mcg) that had no effect on circulating DHT levels in men [see Data and Clinical Pharmacology (12.3)].
Data
Human Data
In 2 studies of healthy subjects (n=69) receiving finasteride 5 mg/day for 6 to 24 weeks, finasteride concentrations in semen ranged from undetectable (<0.1 ng/mL) to 10.54 ng/mL. In an earlier study using a less sensitive assay, finasteride concentrations in semen of 16 subjects receiving finasteride 5 mg/day ranged from undetectable (<1.0 ng/mL) to 21 ng/mL. Using the highest semen level measured and assuming 100% absorption would be up to 105 ng per day, which is 50- to 100-fold less than the dose of finasteride (5 mcg) that had no effect on circulating DHT levels in men [see Clinical Pharmacology (12.3)].
Animal Data
In an embryo-fetal development study, pregnant rats received finasteride during the period of major organogenesis (gestation days 6 to 17). At maternal doses of oral finasteride approximately 0.1 to 86 times the maximum recommended human dose (MRHD) of 5 mg/day (based on AUC at animal doses of 0.1 mg/kg/day to 100 mg/kg/day) there was a dose-dependent increase in hypospadias that occurred in 3.6%to 100% of male offspring. Exposure multiples were estimated using data from nonpregnant rats. Days 16 to 17 of gestation is a critical period in male fetal rats for differentiation of the external genitalia. At oral maternal doses approximately 0.03 times the MRHD (based on AUC at animal dose of 0.03 mg/kg/day), male offspring had decreased prostatic and seminal vesicular weights, delayed preputial separation and transient nipple development. Decreased anogenital distance occurred in male offspring of pregnant rats that received approximately 0.003 times the MRHD (based on AUC at animal dose of 0.003 mg/kg/day). No abnormalities were observed in female offspring at any maternal dose of finasteride.
No developmental abnormalities were observed in the offspring of untreated females mated with finasteride treated male rats that received approximately 61 times the MRHD (based on AUC at animal dose of 80 mg/kg/day). Slightly decreased fertility was observed in male offspring after administration of about 3 times the MRHD (based on AUC at animal dose of 3 mg/kg/day) to female rats during late gestation and lactation. No effects on fertility were seen in female offspring under these conditions.
No evidence of male external genital malformations or other abnormalities were observed in rabbit fetuses exposed to finasteride during the period of major organogenesis (gestation days 6 to 18) at maternal oral doses up to 100 mg/kg/day, (finasteride exposure levels were not measured in rabbits). However, this study may not have included the critical period for finasteride effects on development of male external genitalia in the rabbit.
The fetal effects of maternal finasteride exposure during the period of embryonic and fetal development were evaluated in the rhesus monkey (gestation days 20 to 100), in a species and development period more predictive of specific effects in humans than the studies in rats and rabbits. Intravenous administration of finasteride to pregnant monkeys at doses as high as 800 ng/day (estimated maximal blood concentration of 1.86 ng/mL or about 143 times the highest estimated exposure of pregnant females to finasteride from semen of men taking 5 mg/day) resulted in no abnormalities in male fetuses. In confirmation of the relevance of the rhesus model for human fetal development, oral administration of a dose of finasteride (2 mg/kg/day or approximately 18,000 times the highest estimated blood levels of finasteride from semen of men taking 5 mg/day) to pregnant monkeys resulted in external genital abnormalities in male fetuses. No other abnormalities were observed in male fetuses and no finasteride-related abnormalities were observed in female fetuses at any dose.
8.3 Females and Males of Reproductive Potential
Infertility
Females
Finasteride is not indicated for use in females.
Males
Treatment with finasteride for 24 weeks to evaluate semen parameters in healthy male volunteers revealed no clinically meaningful effects on sperm concentration, mobility, morphology, or pH. A 0.6 mL (22.1%) median decrease in ejaculate volume with a concomitant reduction in total sperm per ejaculate was observed. These parameters remained within the normal range and were reversible upon discontinuation of therapy with an average time to return to baseline of 84 weeks [see Warnings and Precautions (5.5)].
There have been postmarketing reports of male infertility and/or poor seminal quality; normalization or improvement of seminal quality has been reported after discontinuation of finasteride [see Adverse Reactions (6.2)].
8.4 Pediatric Use
Finasteride is not indicated for use in pediatric patients. Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of subjects included in the long-term efficacy and safety study, 1480 and 105 subjects were 65 and over and 75 and over, respectively. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients. No dosage adjustment is necessary in the elderly [see Clinical Pharmacology (12.3) and Clinical Studies (14)].
8.6 Hepatic Impairment
Caution should be exercised in the administration of finasteride in those patients with liver function abnormalities, as finasteride is metabolized extensively in the liver [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is necessary in patients with renal impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Patients have received single doses of finasteride up to 400 mg and multiple doses of finasteride up to 80 mg/day for three months without adverse effects. Until further experience is obtained, no specific treatment for an overdose with finasteride can be recommended.
Significant lethality was observed in male and female mice at single oral doses of 1500 mg/m2 (500 mg/kg) and in female and male rats at single oral doses of 2360 mg/m2 (400 mg/kg) and 5900 mg/m2 (1000 mg/kg), respectively.
11 DESCRIPTION
Finasteride USP, a synthetic 4-azasteroid compound, is a specific inhibitor of steroid Type II 5α-reductase, an intracellular enzyme that converts the androgen testosterone into 5α-dihydrotestosterone (DHT).
Finasteride, USP is 4-azaandrost-1-ene-17-carboxamide, N-(1,1-dimethylethyl)-3-oxo-,(5α,17β)-. Its structural formula is:
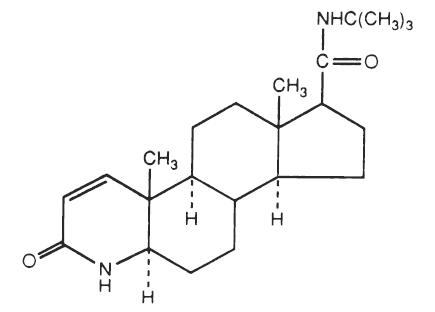
C23H36N2O2 M.W. 372.55
Finasteride, USP is a white to off-white crystalline powder with a melting point near 250°C. It is freely soluble in chloroform and in lower alcohol solvents, but is practically insoluble in water.
Finasteride Tablets, USP for oral administration are film-coated tablets that contain 5 mg of finasteride, USP and the following inactive ingredients: FD&C Blue No. 2 aluminum lake, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized corn starch, sodium lauryl sulfate, sodium starch glycolate type A potato, and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The development and enlargement of the prostate gland is dependent on the potent androgen, 5α-dihydrotestosterone (DHT). Type II 5α-reductase metabolizes testosterone to DHT in the prostate gland, liver and skin. DHT induces androgenic effects by binding to androgen receptors in the cell nuclei of these organs.
Finasteride is a competitive and specific inhibitor of Type II 5α-reductase with which it slowly forms a stable enzyme complex. Turnover from this complex is extremely slow (t½ ~ 30 days). This has been demonstrated both in vivo and in vitro. Finasteride has no affinity for the androgen receptor. In man, the 5α-reduced steroid metabolites in blood and urine are decreased after administration of finasteride.
12.2 Pharmacodynamics
In man, a single 5-mg oral dose of finasteride produces a rapid reduction in serum DHT concentration, with the maximum effect observed 8 hours after the first dose. The suppression of DHT is maintained throughout the 24-hour dosing interval and with continued treatment. Daily dosing of finasteride at 5 mg/day for up to 4 years has been shown to reduce the serum DHT concentration by approximately 70%. The median circulating level of testosterone increased by approximately 10% to 20% but remained within the physiologic range. In a separate study in healthy men treated with finasteride 1 mg per day (n=82) or placebo (n=69), mean circulating levels of testosterone and estradiol were increased by approximately 15% as compared to baseline, but these remained within the physiologic range.
In patients receiving finasteride 5 mg/day, increases of about 10% were observed in luteinizing hormone (LH) and follicle-stimulating hormone (FSH), but levels remained within the normal range. In healthy volunteers, treatment with finasteride did not alter the response of LH and FSH to gonadotropin-releasing hormone indicating that the hypothalamic-pituitary-testicular axis was not affected.
In patients with BPH, finasteride has no effect on circulating levels of cortisol, prolactin, thyroid-stimulating hormone, or thyroxine. No clinically meaningful effect was observed on the plasma lipid profile (i.e., total cholesterol, low density lipoproteins, high density lipoproteins and triglycerides) or bone mineral density.
Adult males with genetically inherited Type II 5α-reductase deficiency also have decreased levels of DHT. Except for the associated urogenital defects present at birth, no other clinical abnormalities related to Type II 5α-reductase deficiency have been observed in these individuals. These individuals have a small prostate gland throughout life and do not develop BPH.
In patients with BPH treated with finasteride (1 mg/day to 100 mg/day) for 7 to 10 days prior to prostatectomy, an approximate 80% lower DHT content was measured in prostatic tissue removed at surgery, compared to placebo; testosterone tissue concentration was increased up to 10 times over pretreatment levels, relative to placebo. Intraprostatic content of PSA was also decreased.
In healthy male volunteers treated with finasteride for 14 days, discontinuation of therapy resulted in a return of DHT levels to pretreatment levels in approximately 2 weeks. In patients treated for three months, prostate volume, which declined by approximately 20%, returned to close to baseline value after approximately three months of discontinuation of therapy.
12.3 Pharmacokinetics
Absorption
In a study of 15 healthy young subjects, the mean bioavailability of finasteride 5-mg tablets was 63% (range 34 to 108%), based on the ratio of area under the curve (AUC) relative to an intravenous (IV) reference dose. Maximum finasteride plasma concentration averaged 37 ng/mL (range, 27 ng/mL to 49 ng/mL) and was reached 1 to 2 hours postdose. Bioavailability of finasteride was not affected by food.
Distribution
Mean steady-state volume of distribution was 76 liters (range, 44 to 96 liters). Approximately 90% of circulating finasteride is bound to plasma proteins. There is a slow accumulation phase for finasteride after multiple dosing. After dosing with 5 mg/day of finasteride for 17 days, plasma concentrations of finasteride were 47 and 54% higher than after the first dose in men 45 to 60 years old (n=12) and ≥70 years old (n=12), respectively. Mean trough concentrations after 17 days of dosing were 6.2 ng/mL (range, 2.4 ng/mL to 9.8 ng/mL) and 8.1 ng/mL (range, 1.8 ng/mL to 19.7 ng/mL), respectively, in the two age groups. Although steady state was not reached in this study, mean trough plasma concentration in another study in patients with BPH (mean age, 65 years) receiving 5 mg/day was 9.4 ng/mL (range, 7.1 ng/mL to 13.3 ng/mL; n=22) after over a year of dosing.
Finasteride has been shown to cross the blood brain barrier but does not appear to distribute preferentially to the CSF.
In 2 studies of healthy subjects (n=69) receiving finasteride 5 mg/day for 6 to 24 weeks, finasteride concentrations in semen ranged from undetectable (<0.1 ng/mL) to 10.54 ng/mL. In an earlier study using a less sensitive assay, finasteride concentrations in the semen of 16 subjects receiving finasteride 5 mg/day ranged from undetectable (<1.0 ng/mL) to 21 ng/mL. Thus, based on a 5-mL ejaculate volume, the amount of finasteride in semen was estimated to be 50- to 100-fold less than the dose of finasteride (5 mcg) that had no effect on circulating DHT levels in men [see also Use in Specific Populations (8.1)].
Metabolism
Finasteride is extensively metabolized in the liver, primarily via the cytochrome P450 3A4 enzyme subfamily. Two metabolites, the t-butyl side chain monohydroxylated and monocarboxylic acid metabolites, have been identified that possess no more than 20% of the 5α-reductase inhibitory activity of finasteride.
Excretion
In healthy young subjects (n=15), mean plasma clearance of finasteride was 165 mL/min (range, 70 mL/min to 279 mL/min) and mean elimination half-life in plasma was 6 hours (range, 3 to 16 hours). Following an oral dose of 14C-finasteride in man (n=6), a mean of 39% (range, 32% to 46%) of the dose was excreted in the urine in the form of metabolites; 57% (range, 51% to 64%) was excreted in the feces. The mean terminal half-life of finasteride in subjects ≥70 years of age was approximately 8 hours (range, 6 to 15 hours; n=12), compared with 6 hours (range, 4 to 12 hours; n=12) in subjects 45 to 60 years of age. As a result, mean AUC(0-24 hr) after 17 days of dosing was 15% higher in subjects ≥70 years of age than in subjects 45 to 60 years of age (p=0.02).
|
Mean (± SD) |
|
|
Bioavailability |
63% (34% to 108%)* |
|
Clearance (mL/min) |
165 (55) |
|
Volume of Distribution (L) |
76 (14) |
|
Half-Life (hours) |
6.2 (2.1) |
| *Range | |
Pediatric
Finasteride pharmacokinetics have not been investigated in patients <18 years of age. Finasteride is not indicated for use in pediatric patients [see Warnings and Precautions (5.4), Use in Specific Populations (8.4)].
Gender
Finasteride is not indicated for use in females [see Contraindications (4), Warnings and Precautions (5.3 and 5.4), Use in Specific Populations (8.1), and How Supplied/Storage and Handling (16)].
Geriatric
No dosage adjustment is necessary in the elderly. Although the elimination rate of finasteride is decreased in the elderly, these findings are of no clinical significance [see Clinical Pharmacology (12.3) and Use in Specific Populations (8.5)].
|
Mean (± SD) |
||
|
45 to 60 years old (n=12) |
≥70 years old (n=12) |
|
|
AUC (ng•hr/mL) |
389 (98) |
463 (186) |
|
Peak Concentration (ng/mL) |
46.2 (8.7) |
48.4 (14.7) |
|
Time to Peak (hours) |
1.8 (0.7) |
1.8 (0.6) |
|
Half-Life (hours)* |
6.0 (1.5) |
8.2 (2.5) |
| *First-dose values; all other parameters are last-dose values | ||
Race
The effect of race on finasteride pharmacokinetics has not been studied.
Hepatic Impairment
The effect of hepatic impairment on finasteride pharmacokinetics has not been studied. Caution should be exercised in the administration of finasteride in those patients with liver function abnormalities, as finasteride is metabolized extensively in the liver.
Renal Impairment
No dosage adjustment is necessary in patients with renal impairment. In patients with chronic renal impairment, with creatinine clearances ranging from 9.0 mL/min to 55 mL/min, AUC, maximum plasma concentration, half-life, and protein binding after a single dose of 14C-finasteride were similar to values obtained in healthy volunteers. Urinary excretion of metabolites was decreased in patients with renal impairment. This decrease was associated with an increase in fecal excretion of metabolites. Plasma concentrations of metabolites were significantly higher in patients with renal impairment (based on a 60% increase in total radioactivity AUC). However, finasteride has been well tolerated in BPH patients with normal renal function receiving up to 80 mg/day for 12 weeks, where exposure of these patients to metabolites would presumably be much greater.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of a tumorigenic effect was observed in a 24-month study in Sprague-Dawley rats receiving doses of finasteride up to 160 mg/kg/day in males and 320 mg/kg/day in females. These doses produced respective systemic exposure in rats of 111 and 274 times those observed in man receiving the recommended human dose of 5 mg/day. All exposure calculations were based on calculated AUC(0-24 hr) for animals and mean AUC(0-24 hr) for man (0.4 mcg•hr/mL).
In a 19-month carcinogenicity study in CD-1 mice, a statistically significant (p≤0.05) increase in the incidence of testicular Leydig cell adenomas was observed at 228 times the human exposure (250 mg/kg/day). In mice at 23 times the human exposure, estimated (25 mg/kg/day) and in rats at 39 times the human exposure (40 mg/kg/day) an increase in the incidence of Leydig cell hyperplasia was observed. A positive correlation between the proliferative changes in the Leydig cells and an increase in serum LH levels (2- to 3-fold above control) has been demonstrated in both rodent species treated with high doses of finasteride. No drug-related Leydig cell changes were seen in either rats or dogs treated with finasteride for 1 year at 30 and 350 times (20 mg/kg/day and 45 mg/kg/day, respectively) or in mice treated for 19 months at 2.3 times the human exposure, estimated (2.5 mg/kg/day).
Mutagenesis
No evidence of mutagenicity was observed in an in vitro bacterial mutagenesis assay, a mammalian cell mutagenesis assay, or in an in vitro alkaline elution assay. In an in vitro chromosome aberration assay, using Chinese hamster ovary cells, there was a slight increase in chromosome aberrations. These concentrations correspond to 4000 to 5000 times the peak plasma levels in man given a total dose of 5 mg. In an in vivo chromosome aberration assay in mice, no treatment-related increase in chromosome aberration was observed with finasteride at the maximum tolerated dose of 250 mg/kg/day (228 times the human exposure) as determined in the carcinogenicity studies.
Impairment of Fertility
In sexually mature male rabbits treated with finasteride at 543 times the human exposure (80 mg/kg/day) for up to 12 weeks, no effect on fertility, sperm count, or ejaculate volume was seen. In sexually mature male rats treated with 61 times the human exposure (80 mg/kg/day), there were no significant effects on fertility after 6 or 12 weeks of treatment; however, when treatment was continued for up to 24 or 30 weeks, there was an apparent decrease in fertility, fecundity and an associated significant decrease in the weights of the seminal vesicles and prostate. All these effects were reversible within 6 weeks of discontinuation of treatment. No drug-related effect on testes or on mating performance has been seen in rats or rabbits. This decrease in fertility in finasteride-treated rats is secondary to its effect on accessory sex organs (prostate and seminal vesicles) resulting in failure to form a seminal plug. The seminal plug is essential for normal fertility in rats and is not relevant in man.
14 CLINICAL STUDIES
14.1 Monotherapy
Finasteride 5 mg/day was initially evaluated in patients with symptoms of BPH and enlarged prostates by digital rectal examination in two 1-year, placebo-controlled, randomized, double-blind studies and their 5-year open extensions.
Finasteride was further evaluated in a long-term efficacy and safety study, a double-blind, randomized, placebo-controlled, 4-year, multicenter study. 3040 patients between the ages of 45 and 78, with moderate to severe symptoms of BPH and an enlarged prostate upon digital rectal examination, were randomized into the study (1524 to finasteride, 1516 to placebo) and 3016 patients were evaluable for efficacy. 1883 patients completed the 4-year study (1000 in the finasteride group, 883 in the placebo group).
Effect on Symptom Score
Symptoms were quantified using a score similar to the American Urological Association Symptom Score, which evaluated both obstructive symptoms (impairment of size and force of stream, sensation of incomplete bladder emptying, delayed or interrupted urination) and irritative symptoms (nocturia, daytime frequency, need to strain or push the flow of urine) by rating on a 0 to 5 scale for six symptoms and a 0 to 4 scale for one symptom, for a total possible score of 34.
Patients in a long-term efficacy and safety study had moderate to severe symptoms at baseline (mean of approximately 15 points on a 0 to 34 point scale). Patients randomized to finasteride who remained on therapy for 4 years had a mean (± 1 SD) decrease in symptom score of 3.3 (± 5.8) points compared with 1.3 (± 5.6) points in the placebo group. (See Figure 1.) A statistically significant improvement in symptom score was evident at 1 year in patients treated with finasteride vs placebo (–2.3 vs –1.6), and this improvement continued through Year 4.
Figure 1
Symptom Score in Long-Term Efficacy and Safety Study
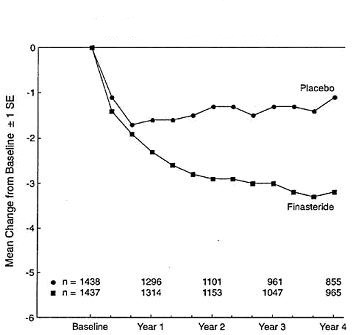
Results seen in earlier studies were comparable to those seen in a long-term efficacy and safety study. Although an early improvement in urinary symptoms was seen in some patients, a therapeutic trial of at least 6 months was generally necessary to assess whether a beneficial response in symptom relief had been achieved. The improvement in BPH symptoms was seen during the first year and maintained throughout an additional 5 years of open extension studies.
Effect on Acute Urinary Retention and the Need for Surgery
In a long-term efficacy and safety study, efficacy was also assessed by evaluating treatment failures. Treatment failure was prospectively defined as BPH-related urological events or clinical deterioration, lack of improvement and/or the need for alternative therapy. BPH-related urological events were defined as urological surgical intervention and acute urinary retention requiring catheterization. Complete event information was available for 92% of the patients. The following table (Table 5) summarizes the results.
|
Patients (%)* | |||||
|
Event |
Placebo |
Finasteride |
Relative |
95% CI |
P |
|
All Treatment Failures |
37.1 |
26.2 |
0.68 |
(0.57 to 0.79) |
<0.001 |
|
Surgical Interventions for BPH |
10.1 |
4.6 |
0.45 |
(0.32 to 0.63) |
<0.001 |
|
Acute Urinary Retention Requiring Catheterization |
6.6 |
2.8 |
0.43 |
(0.28 to 0.66) |
<0.001 |
|
Two consecutive symptom scores ≥20 |
9.2 |
6.7 | |||
|
Bladder Stone |
0.4 |
0.5 | |||
|
Incontinence |
2.1 |
1.7 | |||
|
Renal Failure |
0.5 |
0.6 | |||
|
UTI |
5.7 |
4.9 | |||
|
Discontinuation due to worsening of BPH, lack of improvement, or to receive other medical treatment |
21.8 |
13.3 | |||
|
*patients with multiple events may be counted more than once for each type of event †Hazard ratio based on log rank test |
|||||
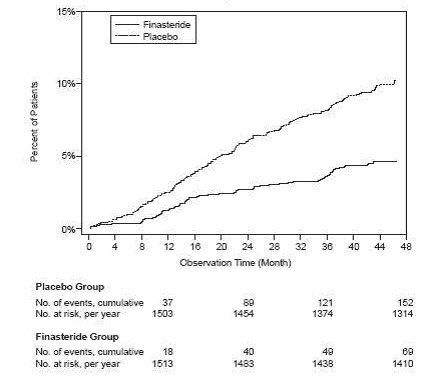
Compared with placebo, finasteride was associated with a significantly lower risk for acute urinary retention or the need for BPH-related surgery [13.2% for placebo vs 6.6% for finasteride; 51% reduction in risk, 95% CI: (34% to 63%)]. Compared with placebo, finasteride was associated with a significantly lower risk for surgery [10.1% for placebo vs 4.6% for finasteride; 55% reduction in risk, 95% CI: (37% to 68%)] and with a significantly lower risk of acute urinary retention [6.6% for placebo vs 2.8% for finasteride; 57% reduction in risk, 95% CI: (34% to 72%)]; see Figures 2 and 3.
Figure 2
Percent of Patients Having Surgery for BPH, Including TURP
Effect on Maximum Urinary Flow Rate
In the patients in a long-term efficacy and safety study who remained on therapy for the duration of the study and had evaluable urinary flow data, finasteride increased maximum urinary flow rate by 1.9 mL/sec compared with 0.2 mL/sec in the placebo group.
There was a clear difference between treatment groups in maximum urinary flow rate in favor of finasteride by month 4 (1.0 vs 0.3 mL/sec) which was maintained throughout the study. In the earlier 1-year studies, increase in maximum urinary flow rate was comparable to a long-term efficacy and safety study and was maintained through the first year and throughout an additional 5 years of open extension studies.
Effect on Prostate Volume
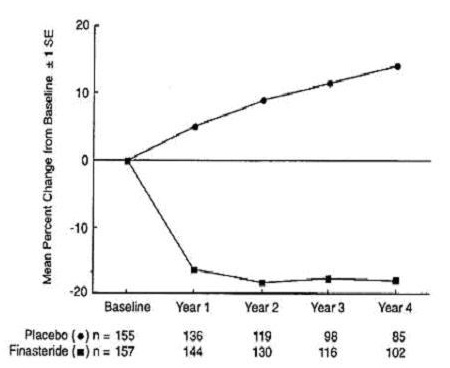
In a long-term efficacy and safety study, prostate volume was assessed yearly by magnetic resonance imaging (MRI) in a subset of patients. In patients treated with finasteride who remained on therapy, prostate volume was reduced compared with both baseline and placebo throughout the 4-year study. Finasteride decreased prostate volume by 17.9% (from 55.9 cc at baseline to 45.8 cc at 4 years) compared with an increase of 14.1% (from 51.3 cc to 58.5 cc) in the placebo group (p<0.001) (see Figure 4).
Results seen in earlier studies were comparable to those seen in a long-term efficacy and safety study. Mean prostate volume at baseline ranged between 40 to 50 cc. The reduction in prostate volume was seen during the first year and maintained throughout an additional five years of open extension studies.
Figure 4.
Prostate Volume in Long-Term Efficacy and Safety Study
Prostate Volume as a Predictor of Therapeutic Response
A meta-analysis combining 1-year data from seven double-blind, placebo-controlled studies of similar design, including 4491 patients with symptomatic BPH, demonstrated that, in patients treated with finasteride, the magnitude of symptom response and degree of improvement in maximum urinary flow rate were greater in patients with an enlarged prostate at baseline.
14.2 Combination with Alpha-Blocker Therapy
The Medical Therapy of Prostatic Symptoms (MTOPS) Trial was a double-blind, randomized, placebo-controlled, multicenter, 4- to 6-year study (average 5 years) in 3047 men with symptomatic BPH, who were randomized to receive finasteride 5 mg/day (n=768), doxazosin 4 or 8 mg/day (n=756), the combination of finasteride 5 mg/day and doxazosin 4 or 8 mg/day (n=786), or placebo (n=737). All participants underwent weekly titration of doxazosin (or its placebo) from 1 mg/day to 2 mg/day to 4 mg/day to 8 mg/day. Only those who tolerated the 4 or 8 mg dose level were kept on doxazosin (or its placebo) in the study. The participant’s final tolerated dose (either 4 mg or 8 mg) was administered beginning at end-Week 4. The final doxazosin dose was administered once per day, at bedtime.
The mean patient age at randomization was 62.6 years (±7.3 years). Patients were Caucasian (82%), African American (9%), Hispanic (7%), Asian (1%) or Native American (<1%). The mean duration of BPH symptoms was 4.7 years (±4.6 years). Patients had moderate to severe BPH symptoms at baseline with a mean AUA symptom score of approximately 17 out of 35 points. Mean maximum urinary flow rate was 10.5 mL/sec (±2.6 mL/sec). The mean prostate volume as measured by transrectal ultrasound was 36.3 mL (±20.1 mL). Prostate volume was ≤20 mL in 16% of patients, ≥50 mL in 18% of patients and between 21 and 49 mL in 66% of patients.
The primary endpoint was a composite measure of the first occurrence of any of the following five outcomes: a ≥4 point confirmed increase from baseline in symptom score, acute urinary retention, BPH-related renal insufficiency (creatinine rise), recurrent urinary tract infections or urosepsis, or incontinence. Compared to placebo, treatment with finasteride, doxazosin, or combination therapy resulted in a reduction in the risk of experiencing one of these five outcome events by 34% (p=0.002), 39% (p<0.001), and 67% (p<0.001), respectively. Combination therapy resulted in a significant reduction in the risk of the primary endpoint compared to treatment with finasteride alone (49%; p≤0.001) or doxazosin alone (46%; p≤0.001). (See Table 6.)
|
Event |
Treatment Group |
||||
|
Placebo |
Doxazosin |
Finasteride |
Combination |
Total |
|
|
N=737 |
N=756 |
N=768 |
N=786 |
N=3047 |
|
|
N (%) |
N (%) |
N (%) |
N (%) |
N (%) |
|
|
AUA 4-point rise |
100 (13.6) |
59 (7.8) |
74 (9.6) |
41 (5.2) |
274 (9.0) |
|
Acute urinary retention |
18 (2.4) |
13 (1.7) |
6 (0.8) |
4 (0.5) |
41 (1.3) |
|
Incontinence |
8 (1.1) |
11 (1.5) |
9 (1.2) |
3 (0.4) |
31 (1.0) |
|
Recurrent UTI/urosepsis |
2 (0.3) |
2 (0.3) |
0 (0.0) |
1 (0.1) |
5 (0.2) |
|
Creatinine rise |
0 (0.0) |
0 (0.0) |
0 (0.0) |
0 (0.0) |
0 (0.0) |
|
Total Events |
128 (17.4) |
85 (11.2) |
89 (11.6) |
49 (6.2) |
351 (11.5) |
The majority of the events (274 out of 351; 78%) was a confirmed ≥4 point increase in symptom score, referred to as symptom score progression. The risk of symptom score progression was reduced by 30% (p=0.016), 46% (p<0.001), and 64% (p<0.001) in patients treated with finasteride, doxazosin, or the combination, respectively, compared to patients treated with placebo (see Figure 5). Combination therapy significantly reduced the risk of symptom score progression compared to the effect of finasteride alone (p<0.001) and compared to doxazosin alone (p=0.037).
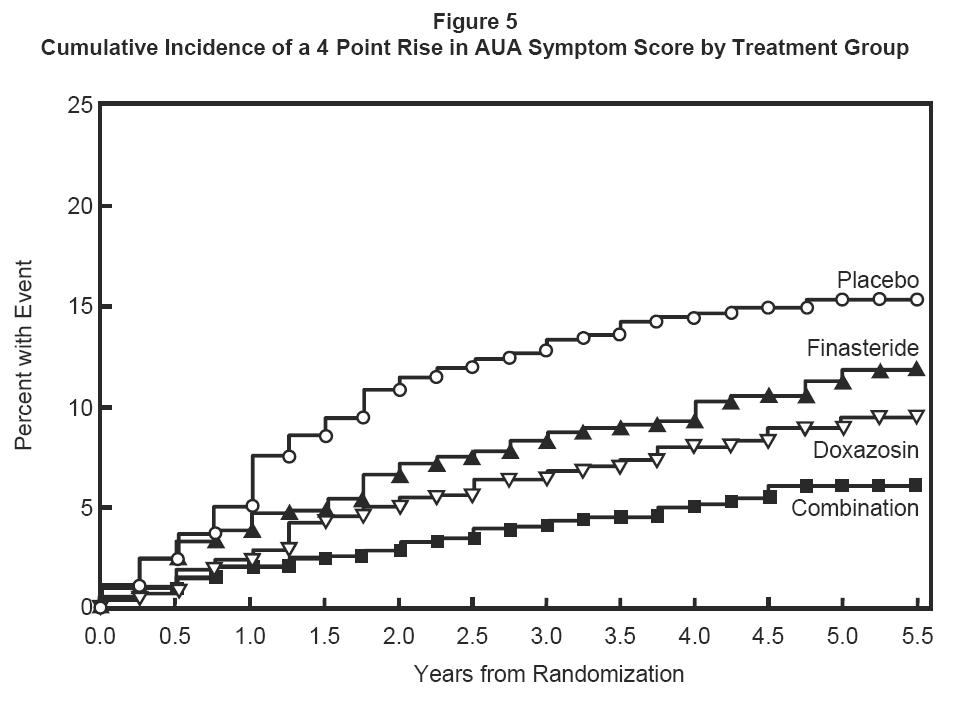
Treatment with finasteride, doxazosin or the combination of finasteride with doxazosin, reduced the mean symptom score from baseline at year 4. Table 7 provides the mean change from baseline for AUA symptom score by treatment group for patients who remained on therapy for four years.
|
Placebo |
Doxazosin |
Finasteride |
Combination |
|
|
N=534 |
N=582 |
N=565 |
N=598 |
|
|
Baseline Mean (SD) |
16.8 (6.0) |
17.0 (5.9) |
17.1 (6.0) |
16.8 (5.8) |
|
Mean Change AUA Symptom Score (SD) |
-4.9 (5.8) |
-6.6 (6.1) |
-5.6 (5.9) |
-7.4 (6.3) |
|
Comparison to Placebo (95% CI) |
-1.8 (-2.5, -1.1) |
-0.7 (-1.4, 0.0) |
-2.5 (-3.2, -1.8) |
|
|
Comparison to Doxazosin alone (95% CI) |
-0.7 (-1.4, 0.0) |
|||
|
Comparison to Finasteride alone (95% CI) |
-1.8 (-2.5, -1.1) |
The results of MTOPS are consistent with the findings of the 4-year, placebo-controlled study a long-term efficacy and safety study [see Clinical Studies (14.1)] in that treatment with finasteride reduces the risk of acute urinary retention and the need for BPH-related surgery. In MTOPS, the risk of developing acute urinary retention was reduced by 67% in patients treated with finasteride compared to patients treated with placebo (0.8% for finasteride and 2.4% for placebo). Also, the risk of requiring BPH-related invasive therapy was reduced by 64% in patients treated with finasteride compared to patients treated with placebo (2.0% for finasteride and 5.4% for placebo).
14.3 Summary of Clinical Studies
The data from these studies, showing improvement in BPH-related symptoms, reduction in treatment failure (BPH-related urological events), increased maximum urinary flow rates, and decreasing prostate volume, suggest that finasteride arrests the disease process of BPH in men with an enlarged prostate.
16 HOW SUPPLIED/STORAGE AND HANDLING
Finasteride Tablets USP, 5 mg are available as blue, film-coated, capsule-shaped, unscored tablets, debossed “93” on one side and “7355” on the other side, in bottles of 30, 90, and 500 tablets.
NDC 0093-7355-56 bottle of 30.
NDC 0093-7355-98 bottle of 90.
NDC 0093-7355-05 bottle of 500.
Storage and Handling
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Protect from light. Keep container tightly closed.
Females should not handle crushed or broken finasteride tablets, USP when they are pregnant or may potentially be pregnant because of the possibility of absorption of finasteride, USP and the subsequent potential risk to a male fetus [see Warnings and Precautions (5.3), and Use in Specific Populations (8.1)].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
17.1 Increased Risk of High-Grade Prostate Cancer
Patients should be informed that there was an increase in high-grade prostate cancer in men treated with 5α-reductase inhibitors indicated for BPH treatment, including finasteride, compared to those treated with placebo in studies looking at the use of these drugs to prevent prostate cancer [see Indications and Usage (1.3), Warnings and Precautions (5.2), and Adverse Reactions (6.1)].
17.2 Exposure of Females – Risk to Male Fetus
Physicians should inform patients that females who are pregnant or may potentially be pregnant should not handle crushed or broken finasteride tablets because of the possibility of absorption of finasteride and the subsequent potential risk to the male fetus. Finasteride tablets are coated and will prevent contact with the active ingredient during normal handling, provided that the tablets have not been broken or crushed. If a female who is pregnant or may potentially be pregnant comes in contact with crushed or broken finasteride tablets, the contact area should be washed immediately with soap and water [see Contraindications (4), Warnings and Precautions (5.3), Use in Specific Populations (8.1) and How Supplied/Storage and Handling (16)].
17.3 Additional Instructions
Physicians should inform patients that the volume of ejaculate may be decreased in some patients during treatment with finasteride. This decrease does not appear to interfere with normal sexual function. However, impotence and decreased libido may occur in patients treated with finasteride [see Adverse Reactions (6.1)].
Physicians should instruct their patients to promptly report any changes in their breasts such as lumps, pain or nipple discharge. Breast changes including breast enlargement, tenderness and neoplasm have been reported [see Adverse Reactions (6.1)].
Physicians should instruct their patients to read the patient package insert before starting therapy with finasteride and to re-read it each time the prescription is renewed so that they are aware of current information for patients regarding finasteride.
Brands listed are the trademarks of their respective owners.
Manufactured In Croatia By:
Pliva Hrvatska d.o.o.
Zagreb, Croatia
Manufactured For:
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Rev. Q 1/2022
PATIENT INFORMATION
Finasteride (fin as' ter ide) Tablets
Finasteride tablets are for use by men only.
Please read this leaflet before you start taking finasteride. Also, read it each time you renew your prescription, just in case anything has changed. Remember, this leaflet does not take the place of careful discussions with your doctor. You and your doctor should discuss finasteride when you start taking your medication and at regular checkups.
What is Finasteride?
Finasteride is a medication used to treat symptoms of benign prostatic hyperplasia (BPH) in men with an enlarged prostate. Finasteride may also be used to reduce the risk of a sudden inability to pass urine and the need for surgery related to BPH in men with an enlarged prostate.
Finasteride may be prescribed along with another medicine, an alpha-blocker called doxazosin, to help you better manage your BPH symptoms.
Who should NOT take Finasteride?
Finasteride is for use by MEN only.
Do Not Take finasteride if you are:
- a woman who is pregnant or may potentially be pregnant. Finasteride may harm your unborn baby. Do not touch or handle crushed or broken finasteride tablets (see “A warning about finasteride and pregnancy”).
- allergic to finasteride or any of the ingredients in finasteride tablets. See the end of this leaflet for a complete list of ingredients in finasteride tablets.
A warning about finasteride and pregnancy:
Women who are or may potentially be pregnant must not use finasteride. They should also not handle crushed or broken tablets of finasteride. Finasteride tablets are coated and will prevent contact with the active ingredient during normal handling, provided that the tablets are not broken or crushed.
If a woman who is pregnant with a male baby absorbs the active ingredient in finasteride after oral use or through the skin, it may cause the male baby to be born with abnormalities of the sex organs. If a woman who is pregnant comes into contact with the active ingredient in finasteride, a doctor should be consulted.
How should I take finasteride?
Follow your doctor's instruction.
- Take one tablet by mouth each day. To avoid forgetting to take finasteride, you can take it at the same time every day.
- If you forget to take finasteride, do not take an extra tablet. Just take the next tablet as usual.
- You may take finasteride with or without food.
- Do not share finasteride with anyone else; it was prescribed only for you.
What are the possible side effects of finasteride?
Finasteride may increase the chance of a more serious form of prostate cancer.
The most common side effects of finasteride include:
- trouble getting or keeping an erection (impotence)
- decrease in sex drive
- decreased volume of ejaculate
- ejaculation disorders
- enlarged or painful breast. You should promptly report to your doctor any changes in your breasts such as lumps, pain or nipple discharge.
The following have been reported in general use with finasteride tablets 5 mg and/or finasteride at lower doses:
- allergic reactions, including rash, itching, hives, and swelling of the lips, tongue, throat, and face
- rarely, some men may have testicular pain
- blood in semen
- trouble getting or keeping an erection that continued after stopping the medication
- problems with ejaculation that continued after stopping the medication
- male infertility and/or poor quality of semen. Improvement in the quality of semen has been reported after stopping the medication.
- depression
- decrease in sex drive that continued after stopping the medication
- in rare cases, male breast cancer has been reported.
You should discuss side effects with your doctor before taking finasteride and anytime you think you are having a side effect. These are not all the possible side effects with finasteride. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at: 1-800-FDA-1088.
What you need to know while taking finasteride:
- You should see your doctor regularly while taking finasteride. Follow your doctor's advice about when to have these checkups.
- Checking for prostate cancer. Your doctor has prescribed finasteride for BPH and not for treatment of prostate cancer — but a man can have BPH and prostate cancer at the same time. Your doctor may continue checking for prostate cancer while you take finasteride.
- About Prostate-Specific Antigen (PSA). Your doctor may have done a blood test called PSA for the screening of prostate cancer. Because finasteride decreases PSA levels, you should tell your doctor(s) that you are taking finasteride. Changes in PSA levels will need to be evaluated by your doctor(s). Any increase in follow-up PSA levels from their lowest point may signal the presence of prostate cancer and should be evaluated, even if the test results are still within the normal range. You should also tell your doctor if you have not been taking finasteride as prescribed because this may affect the PSA test results. For more information, talk to your doctor.
How should I store finasteride?
- Store finasteride tablets in a dry place at room temperature.
- Keep finasteride in the original container and keep the container closed. Finasteride tablets are coated and will prevent contact with the active ingredient during normal handling, provided that the tablets are not broken or crushed.
Keep finasteride tablets and all medications out of the reach of children.
Do not give your finasteride tablets to anyone else. It has been prescribed only for you.
For more information call 1-888-838-2872.
What are the ingredients in finasteride?
Active ingredient: finasteride
Inactive ingredients: FD&C Blue No. 2 aluminum lake, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized corn starch, sodium lauryl sulfate, sodium starch glycolate type A potato, and titanium dioxide.
What is BPH?
BPH is an enlargement of the prostate gland. The prostate is located below the bladder. As the prostate enlarges, it may slowly restrict the flow of urine. This can lead to symptoms such as:
- a weak or interrupted urinary stream
- a feeling that you cannot empty your bladder completely
- a feeling of delay or hesitation when you start to urinate
- a need to urinate often, especially at night
- a feeling that you must urinate right away.
In some men, BPH can lead to serious problems, including urinary tract infections, a sudden inability to pass urine (acute urinary retention), as well as the need for surgery.
What finasteride does:
Finasteride lowers levels of a hormone called DHT (dihydrotestosterone), which is a cause of prostate growth. Lowering DHT leads to shrinkage of the enlarged prostate gland in most men. This can lead to gradual improvement in urine flow and symptoms over the next several months. Finasteride will help reduce the risk of developing a sudden inability to pass urine and the need for surgery related to an enlarged prostate. However, since each case of BPH is different, you should know that:
- Even though the prostate shrinks, you may NOT notice an improvement in urine flow or symptoms.
- You may need to take finasteride for six (6) months or more to see whether it improves your symptoms.
- Therapy with finasteride may reduce your risk for a sudden inability to pass urine and the need for surgery for an enlarged prostate.
Manufactured In Croatia By:
Pliva Hrvatska d.o.o.
Zagreb, Croatia
Manufactured For:
Teva Pharmaceuticals USA, Inc.
North Wales, PA 19454
Rev. N 1/2022
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 0093-7355-05
Finasteride Tablets USP
5 mg
WARNING: Finasteride should not be used by women or children. Women who are or may potentially be pregnant must not use finasteride.
They should also not handle crushed or broken tablets of finasteride. (See accompanying circular.)
Rx only
500 TABLETS
