FLECAINIDE ACETATE- flecainide acetate tablet
Apotex Corp.
----------
Flecainide Acetate Tablets
DESCRIPTION
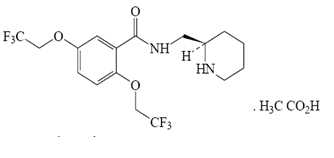
Flecainide acetate is an antiarrhythmic drug available in tablets of 50, 100 or 150 mg for oral administration. Flecainide acetate is benzamide, N-(2-piperidinylmethyl)-2,5-bis(2,2,2-trifluoroethoxy)-monoacetate. The structural formula is given below.
Flecainide acetate is a white crystalline substance with a pKa of 9.3. It has an aqueous solubility of 48.4 mg/mL at 37°C.
Flecainide acetate tablets also contain: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, methylcellulose and stearic acid.
CLINICAL PHARMACOLOGY
Flecainide acetate has local anesthetic activity and belongs to the membrane stabilizing (Class 1) group of antiarrhythmic agents; it has electrophysiologic effects characteristic of the IC class of antiarrhythmics.
Electrophysiology. In man, flecainide acetate produces a dose-related decrease in intracardiac conduction in all parts of the heart with the greatest effect on the His-Purkinje system (H-V conduction). Effects upon atrioventricular (AV) nodal conduction time and intra-atrial conduction times, although present, are less pronounced than those on ventricular conduction velocity. Significant effects on refractory periods were observed only in the ventricle.
Sinus node recovery times (corrected) following pacing and spontaneous cycle lengths are somewhat increased. This latter effect may become significant in patients with sinus node dysfunction. (See WARNINGS.)
Flecainide acetate causes a dose-related and plasma-level related decrease in single and multiple PVCs and can suppress recurrence of ventricular tachycardia. In limited studies of patients with a history of ventricular tachycardia, flecainide acetate has been successful 30-40% of the time in fully suppressing the inducibility of arrhythmias by programmed electrical stimulation. Based on PVC suppression, it appears that plasma levels of 0.2 to 1 mcg/mL may be needed to obtain the maximal therapeutic effect. It is more difficult to assess the dose needed to suppress serious arrhythmias, but trough plasma levels in patients successfully treated for recurrent ventricular tachycardia were between 0.2 and 1 mcg/mL. Plasma levels above 0.7 to 1 mcg/mL are associated with a higher rate of cardiac adverse experiences such as conduction defects or bradycardia. The relation of plasma levels to proarrhythmic events is not established, but dose reduction in clinical trials of patients with ventricular tachycardia appears to have led to a reduced frequency and severity of such events.
Hemodynamics. Flecainide acetate does not usually alter heart rate, although bradycardia and tachycardia have been reported occasionally.
In animals and isolated myocardium, a negative inotropic effect of flecainide has been demonstrated. Decreases in ejection fraction, consistent with a negative inotropic effect, have been observed after single administration of 200 to 250 mg of the drug in man; both increases and decreases in ejection fraction have been encountered during multidose therapy in patients at usual therapeutic doses. (See WARNINGS.)
Metabolism in Humans. Following oral administration, the absorption of flecainide acetate is nearly complete. Peak plasma levels are attained at about three hours in most individuals (range, 1 to 6 hours). Flecainide does not undergo any consequential presystemic biotransformation (first-pass effect). Food or antacid do not affect absorption. Milk, however, may inhibit absorption in infants. A reduction in flecainide acetate dosage should be considered when milk is removed from the diet of infants.
The apparent plasma half-life averages about 20 hours and is quite variable (range, 12 to 27 hours) after multiple oral doses in patients with premature ventricular contractions (PVCs). With multiple dosing, plasma levels increase because of its long half-life with steady-state levels approached in 3 to 5 days; once at steady-state, no additional (or unexpected) accumulation of drug in plasma occurs during chronic therapy. Over the usual therapeutic range, data suggest that plasma levels in an individual are approximately proportional to dose, deviating upwards from linearity only slightly (about 10 to 15% per 100 mg on average).
In healthy subjects, about 30% of a single oral dose (range, 10 to 50%) is excreted in urine as unchanged drug. The two major urinary metabolites are meta-O-dealkylated flecainide (active, but about one-fifth as potent) and the meta-O-dealkylated lactam of flecainide (non-active metabolite). These two metabolites (primarily conjugated) account for most of the remaining portion of the dose. Several minor metabolites (3% of the dose or less) are also found in urine; only 5% of an oral dose is excreted in feces. In patients, free (unconjugated) plasma levels of the two major metabolites are very low (less than 0.05 mcg/mL).
In vitro metabolic studies have confirmed that cytochrome P450IID6 is involved in the metabolism of flecainide.
When urinary pH is very alkaline (8 or higher), as may occur in rare conditions (e.g., renal tubular acidosis, strict vegetarian diet), flecainide elimination from plasma is much slower.
The elimination of flecainide from the body depends on renal function (i.e., 10 to 50% appears in urine as unchanged drug). With increasing renal impairment, the extent of unchanged drug excretion in urine is reduced and the plasma half-life of flecainide is prolonged. Since flecainide is also extensively metabolized, there is no simple relationship between creatinine clearance and the rate of flecainide elimination from plasma. (See DOSAGE AND ADMINISTRATION.)
In patients with NYHA class III congestive heart failure (CHF), the rate of flecainide elimination from plasma (mean half-life, 19 hours) is moderately slower than for healthy subjects (mean half-life, 14 hours), but similar to the rate for patients with PVCs without CHF. The extent of excretion of unchanged drug in urine is also similar. (See DOSAGE AND ADMINISTRATION.)
Under one year of age, currently available data are limited but suggest that the half-life at birth may be as long as 29 hours, decreasing to 11 to 12 hours by three months of age and 6 hours by one year of age. The pharmacokinetics in hydropic infants have not been studied, but case reports suggest prolonged elimination. In children aged 1 year to 12 years the half-life is approximately 8 hours. In adolescents (age 12 to 15) the plasma elimination half-life is approximately 11-12 hours. Since milk may inhibit absorption in infants, a reduction in flecainide acetate dosage should be considered when milk is removed from the diet (e.g., gastroenteritis, weaning). Plasma trough flecainide levels should be monitored during major changes in dietary milk intake.
From age 20 to 80, plasma levels are only slightly higher with advancing age; flecainide elimination from plasma is somewhat slower in elderly subjects than in younger subjects. Patients up to age 80+ have been safely treated with usual dosages.
The extent of flecainide binding to human plasma proteins is about 40% and is independent of plasma drug level over the range of 0.015 to about 3.4 mcg/mL. Thus, clinically significant drug interactions based on protein binding effects would not be expected.
Hemodialysis removes only about 1% of an oral dose as unchanged flecainide.
Small increases in plasma digoxin levels are seen during coadministration of flecainide acetate with digoxin. Small increases in both flecainide and propranolol plasma levels are seen during coadministration of these two drugs. (See PRECAUTIONS, DRUG INTERACTIONS.)
Clinical Trials. In two randomized, crossover, placebo-controlled clinical trials of 16 weeks double-blind duration, 79% of patients with paroxysmal supraventricular tachycardia (PSVT) receiving flecainide were attack free, whereas 15% of patients receiving placebo remained attack free. The median time-before-recurrence of PSVT in patients receiving placebo was 11 to 12 days, whereas over 85% of patients receiving flecainide had no recurrence at 60 days.
In two randomized, crossover, placebo-controlled clinical trials of 16 weeks double-blind duration, 31% of patients with paroxysmal atrial fibrillation/flutter (PAF) receiving flecainide were attack free, whereas 8% receiving placebo remained attack free. The median time-before-recurrence of PAF in patients receiving placebo was about 2 to 3 days, whereas for those receiving flecainide the median time-before-recurrence was 15 days.
INDICATIONS AND USAGE
In patients without structural heart disease, flecainide acetate is indicated for the prevention of
– paroxysmal supraventricular tachycardias (PSVT), including atrioventricular nodal reentrant tachycardia, atrioventricular reentrant tachycardia and other supraventricular tachycardias of unspecified mechanism associated with disabling symptoms
– paroxysmal atrial fibrillation/flutter (PAF) associated with disabling symptoms
Flecainide acetate is also indicated for the prevention of
– documented ventricular arrhythmias, such as sustained ventricular tachycardia (sustained VT), that in the judgment of the physician are life-threatening.
Use of flecainide acetate for the treatment of sustained VT, like other antiarrhythmics, should be initiated in the hospital. The use of flecainide acetate is not recommended in patients with less severe ventricular arrhythmias even if the patients are symptomatic.
Because of the proarrhythmic effects of flecainide acetate, its use should be reserved for patients in whom, in the opinion of the physician, the benefits of treatment outweigh the risks.
Flecainide acetate should not be used in patients with recent myocardial infarction. (See Boxed WARNINGS.)
Use of flecainide acetate in chronic atrial fibrillation has not been adequately studied and is not recommended. (See Boxed WARNINGS.)
As is the case for other antiarrhythmic agents, there is no evidence from controlled trials that the use of flecainide acetate favorably affects survival or the incidence of sudden death.
CONTRAINDICATIONS
Flecainide acetate is contraindicated in patients with pre-existing second- or third-degree AV block, or with right bundle branch block when associated with a left hemiblock (bifascicular block), unless a pacemaker is present to sustain the cardiac rhythm should complete heart block occur. Flecainide acetate is also contraindicated in the presence of cardiogenic shock or known hypersensitivity to the drug.
WARNINGS
Mortality. Flecainide acetate tablets were included in the National Heart Lung and Blood Institute's Cardiac Arrhythmia Suppression Trial (CAST), a longterm, multicenter, randomized, double-blind study in patients with asymptomatic non-life-threatening ventricular arrhythmias who had a myocardial infarction more than six days but less than two years previously. An excessive mortality or non-fatal cardiac arrest rate was seen in patients treated with flecainide acetate tablets compared with that seen in patients assigned to a carefully matched placebo-treated group. This rate was 16/315 (5.1%) for flecainide acetate tablets and 7/309 (2.3%) for the matched placebo. The average duration of treatment with flecainide acetate tablets in this study was ten months.
The applicability of the CAST results to other populations (e.g., those without recent myocardial infarction) is uncertain, but at present, it is prudent to consider the risks of Class IC agents (including flecainide acetate), coupled with the lack of any evidence of improved survival, generally unacceptable in patients without life-threatening ventricular arrhythmias, even if the patients are experiencing unpleasant, but not life-threatening, symptoms or signs.
Ventricular Pro-arrhythmic Effects in Patients with Atrial Fibrillation/Flutter.A review of the world literature revealed reports of 568 patients treated with oral flecainide acetate tablets for paroxysmal atrial fibrillation/flutter (PAF). Ventricular tachycardia was experienced in 0.4% (2/568) of these patients. Of 19 patients in the literature with chronic atrial fibrillation (CAF), 10.5% (2) experienced VT or VF. FLECAINIDE IS NOT RECOMMENDED FOR USE IN PATIENTS WITH CHRONIC ATRIAL FIBRILLATION. Case reports of ventricular proarrhythmic effects in patients treated with flecainide acetate tablets for atrial fibrillation/flutter have included increased PVCs, VT, ventricular fibrillation (VF), and death.
As with other Class I agents, patients treated with flecainide acetate for atrial flutter have been reported with 1:1 atrioventricular conduction due to slowing the atrial rate. A paradoxical increase in the ventricular rate also may occur in patients with atrial fibrillation who receive flecainide acetate. Concomitant negative chronotropic therapy such as digoxin or beta-blockers may lower the risk of this complication.
PROARRHYTHMIC EFFECTS
Flecainide acetate, like other antiarrhythmic agents, can cause new or worsened supraventricular or ventricular arrhythmias. Ventricular proarrhythmic effects range from an increase in frequency of PVCs to the development of more severe ventricular tachycardia, e.g., tachycardia that is more sustained or more resistant to conversion to sinus rhythm, with potentially fatal consequences. In studies of ventricular arrhythmia patients treated with flecainide acetate, three-fourths of proarrhythmic events were new or worsened ventricular tachyarrhythmias, the remainder being increased frequency of PVCs or new supraventricular arrhythmias. In patients treated with flecainide for sustained ventricular tachycardia, 80% (51/64) of proarrhythmic events occurred within 14 days of the onset of therapy. In studies of 225 patients with supraventricular arrhythmia (108 with paroxysmal supraventricular tachycardia and 117 with paroxysmal atrial fibrillation), there were 9 (4%) proarrhythmic events, 8 of them in patients with paroxysmal atrial fibrillation. Of the 9, 7 (including the one in a PSVT patient) were exacerbations of supraventricular arrhythmias (longer duration, more rapid rate, harder to reverse) while 2 were ventricular arrhythmias, including one fatal case of VT/VF and one wide complex VT (the patient showed inducible VT, however, after withdrawal of flecainide), both in patients with paroxysmal atrial fibrillation and known coronary artery disease.
It is uncertain if flecainide acetate's risk of proarrhythmia is exaggerated in patients with chronic atrial fibrillation (CAF), high ventricular rate, and/or exercise. Wide complex tachycardia and ventricular fibrillation have been reported in two of 12 CAF patients undergoing maximal exercise tolerance testing.
In patients with complex ventricular arrhythmias, it is often difficult to distinguish a spontaneous variation in the patient's underlying rhythm disorder from drug-induced worsening, so that the following occurrence rates must be considered approximations. Their frequency appears to be related to dose and to the underlying cardiac disease.
Among patients treated for sustained VT (who frequently also had CHF, a low ejection fraction, a history of myocardial infarction and/or an episode of cardiac arrest), the incidence of proarrhythmic events was 13% when dosage was initiated at 200 mg/day with slow upward titration, and did not exceed 300 mg/day in most patients. In early studies in patients with sustained VT utilizing a higher initial dose (400 mg/day) the incidence of proarrhythmic events was 26%; moreover, in about 10% of the patients treated proarrhythmic events resulted in death, despite prompt medical attention. With lower initial doses, the incidence of proarrhythmic events resulting in death decreased to 0.5% of these patients. Accordingly, it is extremely important to follow the recommended dosage schedule. (See DOSAGE AND ADMINISTRATION.)
The relatively high frequency of proarrhythmic events in patients with sustained VT and serious underlying heart disease, and the need for careful titration and monitoring, requires that therapy of patients with sustained VT be started in the hospital. (See DOSAGE AND ADMINISTRATION.)
HEART FAILURE
Flecainide acetate has a negative inotropic effect and may cause or worsen CHF, particularly in patients with cardiomyopathy, preexisting severe heart failure (NYHA functional class III or IV) or low ejection fractions (less than 30%). In patients with supraventricular arrhythmias new or worsened CHF developed in 0.4% (1/225) of patients. In patients withsustainedventricular tachycardia during a mean duration of 7.9 months of flecainide acetate therapy, 6.3% (20/317) developed new CHF. In patients withsustainedventricular tachycardia and a history of CHF, during a mean duration of 5.4 months of flecainide acetate therapy, 25.7% (78/304) developed worsened CHF. Exacerbation of preexisting CHF occurred more commonly in studies which included patients with class III or IV failure than in studies which excluded such patients. Flecainide acetate should be used cautiously in patients who are known to have a history of CHF or myocardial dysfunction. The initial dosage in such patients should be no more than 100 mg bid (see DOSAGE AND ADMINISTRATION) and patients should be monitored carefully. Close attention must be given to maintenance of cardiac function, including optimization of digitalis, diuretic, or other therapy. In cases where CHF has developed or worsened during treatment with flecainide acetate, the time of onset has ranged from a few hours to several months after starting therapy. Some patients who develop evidence of reduced myocardial function while on flecainide acetate can continue on flecainide acetate with adjustment of digitalis or diuretics, others may require dosage reduction or discontinuation of flecainide acetate. When feasible, it is recommended that plasma flecainide levels be monitored. Attempts should be made to keep trough plasma levels below 0.7 to 1.0 mcg/mL.
Effects on Cardiac Conduction. Flecainide acetate slows cardiac conduction in most patients to produce dose-related increases in PR, QRS, and QT intervals. PR interval increases on average about 25% (0.04 seconds) and as much as 118% in some patients. Approximately one-third of patients may develop new first-degree AV heart block (PR interval ≥0.20 seconds). The QRS complex increases on average about 25% (0.02 seconds) and as much as 150% in some patients. Many patients develop QRS complexes with a duration of 0.12 seconds or more. In one study, 4% of patients developed new bundle branch block while on flecainide acetate. The degree of lengthening of PR and QRS intervals does not predict either efficacy or the development of cardiac adverse effects. In clinical trials, it was unusual for PR intervals to increase to 0.30 seconds or more, or for QRS intervals to increase to 0.18 seconds or more. Thus, caution should be used when such intervals occur, and dose reductions may be considered. The QT interval widens about 8%, but most of this widening (about 60% to 90%) is due to widening of the QRS duration. The JT interval (QT minus QRS) only widens about 4% on the average. Significant JT prolongation occurs in less than 2% of patients. There have been rare cases of Torsade de Pointes-type arrhythmia associated with flecainide acetate therapy.
Clinically significant conduction changes have been observed at these rates: sinus node dysfunction such as sinus pause, sinus arrest and symptomatic bradycardia (1.2%), second-degree AV block (0.5%) and third-degree AV block (0.4%). An attempt should be made to manage the patient on the lowest effective dose in an effort to minimize these effects. (See DOSAGE AND ADMINISTRATION) If second- or third-degree AV block, or right bundle branch block associated with a left hemiblock occur, flecainide acetate therapy should be discontinued unless a temporary or implanted ventricular pacemaker is in place to ensure an adequate ventricular rate.
Sick Sinus Syndrome (Bradycardia-Tachycardia Syndrome). Flecainide acetateshould be used only with extreme caution in patients with sick sinus syndrome because it may cause sinus bradycardia, sinus pause, or sinus arrest.
Effects on Pacemaker Thresholds. Flecainide acetateis known to increase endocardial pacing thresholds and may suppress ventricular escape rhythms. These effects are reversible if flecainide is discontinued. It should be used with caution in patients with permanent pacemakers or temporary pacing electrodes and should not be administered to patients with existing poor thresholds or nonprogrammable pacemakers unless suitable pacing rescue is available.
The pacing threshold in patients with pacemakers should be determined prior to instituting therapy with flecainide acetate, again after one week of administration and at regular intervals thereafter. Generally threshold changes are within the range of multiprogrammable pacemakers and, when these occur, a doubling of either voltage or pulse width is usually sufficient to regain capture.
Electrolyte Disturbances. Hypokalemia or hyperkalemia may alter the effects of Class I antiarrhythmic drugs. Preexisting hypokalemia or hyperkalemia should be corrected before administration of flecainide acetate.
Pediatric Use. The safety and efficacy of flecainide acetate in the fetus, infant, or child have not been established in double-blind, randomized, placebo-controlled trials. The proarrhythmic effects of flecainide acetate, as described previously, apply also to children. In pediatric patients with structural heart disease, flecainide acetate has been associated with cardiac arrest and sudden death. Flecainide acetate should be started in the hospital with rhythm monitoring. Any use of flecainide acetate in children should be directly supervised by a cardiologist skilled in the treatment of arrhythmias in children.
PRECAUTIONS
Drug Interactions. Flecainide acetatehas been administered to patients receiving digitalis preparations or beta-adrenergic blocking agents without adverse effects. During administration of multiple oral doses of flecainide acetate to healthy subjects stabilized on a maintenance dose of digoxin, a 13% to 19% increase in plasma digoxin levels occurred at six hours postdose.
In a study involving healthy subjects receiving flecainide acetate and propranolol concurrently, plasma flecainide levels were increased about 20% and propranolol levels were increased about 30% compared to control values. In this formal interaction study, flecainide acetate and propranolol were each found to have negative inotropic effects; when the drugs were administered together, the effects were additive. The effects of concomitant administration of flecainide acetate and propranolol on the PR interval were less than additive. In flecainide acetate clinical trials, patients who were receiving beta-blockers concurrently did not experience an increased incidence of side effects. Nevertheless, the possibility of additive negative inotropic effects of beta- blockers and flecainide should be recognized.
Flecainide is not extensively bound to plasma proteins. In vitro studies with several drugs which may be administered concomitantly showed that the extent of flecainide binding to human plasma proteins is either unchanged or only slightly less. Consequently, interactions with other drugs which are highly protein bound (e.g., anticoagulants) would not be expected. Flecainide acetatehas been used in a large number of patients receiving diuretics without apparent interaction. Limited data in patients receiving known enzyme inducers (phenytoin, phenobarbital, carbamazepine) indicate only a 30% increase in the rate of flecainide elimination. In healthy subjects receiving cimetidine (1 gram daily) for one week, plasma flecainide levels increased by about 30% and half-life increased by about 10%.
When amiodarone is added to flecainide therapy, plasma flecainide levels may increase two-fold or more in some patients, if flecainide dosage is not reduced. (See DOSAGE AND ADMINISTRATION)
Drugs that inhibit cytochrome P450IID6, such as quinidine, might increase the plasma concentrations of flecainide in patients that are on chronic flecainide therapy; especially if these patients are extensive metabolizers.
There has been little experience with the coadministration of flecainide acetate and either disopyramide or verapamil. Because both of these drugs have negative inotropic properties and the effects of coadministration with flecainide acetate are unknown, neither disopyramide nor verapamil should be administered concurrently with flecainide acetate unless, in the judgment of the physician, the benefits of this combination outweigh the risks. There has been too little experience with the coadministration of flecainide acetate with nifedipine or diltiazem to recommend concomitant use.
Carcinogenesis, Mutagenesis, Impairment of Fertility. Long-term studies with flecainide in rats and mice at doses up to 60 mg/kg/day have not revealed any compound-related carcinogenic effects. Mutagenicity studies (Ames test, mouse lymphoma and in vivo cytogenetics) did not reveal any mutagenic effects. A rat reproduction study at doses up to 50 mg/kg/day (seven times the usual human dose) did not reveal any adverse effect on male or female fertility.
Pregnancy. Pregnancy Category C. Flecainide has been shown to have teratogenic effects (club paws, sternebrae and vertebrae abnormalities, pale hearts with contracted ventricular septum) and an embryotoxic effect (increased resorptions) in one breed of rabbit (New Zealand White) when given doses of 30 and 35 mg/kg/day, but not in another breed of rabbit (Dutch Belted) when given doses up to 30 mg/kg/day. No teratogenic effects were observed in rats and mice given doses up to 50 and 80 mg/kg/day, respectively; however, delayed sternebral and vertebral ossification was observed at the high dose in rats. Because there are no adequate and well-controlled studies in pregnant women, flecainide acetate should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Labor and Delivery. It is not known whether the use of flecainide acetate during labor or delivery has immediate or delayed adverse effects on the mother or fetus, affects the duration of labor or delivery, or increases the possibility of forceps delivery or other obstetrical intervention.
Nursing Mothers. Results from a multiple dose study conducted in mothers soon after delivery indicates that flecainide is excreted in human breast milk in concentrations as high as 4 times (with average levels about 2.5 times) corresponding plasma levels; assuming a maternal plasma level at the top of the therapeutic range (1 mcg/mL), the calculated daily dose to a nursing infant (assuming about 700 mL breast milk over 24 hours) would be less than 3 mg.
Pediatric Use. The safety and efficacy of flecainide acetate in the fetus, infant, or child have not been established in double-blind, randomized, placebo-controlled trials (see CLINICAL PHARMACOLOGY,WARNINGS, and DOSAGE AND ADMINISTRATION).
Hepatic Impairment. Since flecainide elimination from plasma can be markedly slower in patients with significant hepatic impairment, flecainide acetate should not be used in such patients unless the potential benefits clearly outweigh the risks. If used, frequent and early plasma level monitoring is required to guide dosage (see DOSAGE AND ADMINISTRATION, Plasma Level Monitoring); dosage increases should be made very cautiously when plasma levels have plateaued (after more than four days).
ADVERSE REACTIONS
In post-myocardial infarction patients with asymptomatic PVCs and non-sustained ventricular tachycardia, flecainide acetate therapy was found to be associated with a 5.1% rate of death and non-fatal cardiac arrest, compared with a 2.3% rate in a matched placebo group. (See WARNINGS.)
Adverse effects reported for flecainide acetate tablets, described in detail in the Warnings section, were new or worsened arrhythmias which occurred in 1% of 108 patients with PSVT and in 7% of 117 patients with PAF; and new or exacerbated ventricular arrhythmias which occurred in 7% of 1330 patients with PVCs, non-sustained or sustained VT. In patients treated with flecainide for sustained VT, 80% (51/64) of proarrhythmic events occurred within 14 days of the onset of therapy. 198 patients with sustained VT experienced a 13% incidence of new or exacerbated ventricular arrhythmias when dosage was initiated at 200 mg/day with slow upward titration, and did not exceed 300 mg/day in most patients. In some patients, flecainide acetate tablets treatment has been associated with episodes of unresuscitatable VT or ventricular fibrillation (cardiac arrest). (See WARNINGS.) New or worsened CHF occurred in 6.3% of 1046 patients with PVCs, non-sustained or sustained VT. Of 297 patients with sustained VT, 9.1% experienced new or worsened CHF. New or worsened CHF was reported in 0.4% of 225 patients with supraventricular arrhythmias. There have also been instances of second- (0.5%) or third-degree (0.4%) AV block. Patients have developed sinus bradycardia, sinus pause, or sinus arrest, about 1.2% altogether (see WARNINGS). The frequency of most of these serious adverse events probably increases with higher trough plasma levels, especially when these trough levels exceed 1 mcg/mL.
There have been rare reports of isolated elevations of serum alkaline phosphatase and isolated elevations of serum transaminase levels. These elevations have been asymptomatic and no cause and effect relationship with flecainide acetate tablets has been established. In foreign postmarketing surveillance studies, there have been rare reports of hepatic dysfunction including reports of cholestasis and hepatic failure, and extremely rare reports of blood dyscrasias. Although no cause and effect relationship has been established, it is advisable to discontinue flecainide acetate tablets in patients who develop unexplained jaundice or signs of hepatic dysfunction or blood dyscrasias in order to eliminate flecainide acetate tablets as the possible causative agent.
Incidence figures for other adverse effects in patients with ventricular arrhythmias are based on a multicenter efficacy study, utilizing starting doses of 200 mg/day with gradual upward titration to 400 mg/day. Patients were treated for an average of 4.7 months, with some receiving up to 22 months of therapy. In this trial, 5.4% of patients discontinued due to non-cardiac adverse effects.
|
* Dizziness includes reports of dizziness, lightheadedness, faintness, unsteadiness, near syncope, etc. |
||||
|
† Visual disturbance includes reports of blurred vision, difficulty in focusing, spots before eyes, etc. |
||||
| Incidence by Dose During Upward Titration |
||||
|
Incidence |
||||
| All 429 | 200 | 300 | 400 | |
| Adverse | Patients at | mg/Day | mg/Day | mg/Day |
| Effect | Any Dose | (N=426) | (N=293) | (N=100) |
| Dizziness* | 18.9% | 11.0% | 10.6% | 13.0% |
| Visual Disturbances† | 15.9% | 5.4% | 12.3% | 18.0% |
| Dyspnea | 10.3% | 5.2% | 7.5% | 4.0% |
| Headache | 9.6% | 4.5% | 6.1% | 9.0% |
| Nausea | 8.9% | 4.9% | 4.8% | 6.0% |
| Fatigue | 7.7% | 4.5% | 4.4% | 3.0% |
| Palpitation | 6.1% | 3.5% | 2.4% | 7.0% |
| Chest Pain | 5.4% | 3.1% | 3.8% | 1.0% |
| Asthenia | 4.9% | 2.6% | 2.0% | 4.0% |
| Tremor | 4.7% | 2.4% | 3.4% | 2.0% |
| Constipation | 4.4% | 2.8% | 2.1% | 1.0% |
| Edema | 3.5% | 1.9% | 1.4% | 2.0% |
| Abdominal Pain | 3.3% | 1.9% | 2.4% | 1.0% |
The following additional adverse experiences, possibly related to flecainide acetate tablets therapy and occurring in 1% to less than 3% of patients, have been reported in acute and chronic studies: Body as a Whole– malaise, fever; Cardiovascular– tachycardia, sinus pause or arrest; Gastrointestinal– vomiting, diarrhea, dyspepsia, anorexia; Skin– rash; Visual– diplopia; Nervous System– hypoesthesia, paresthesia, paresis, ataxia, flushing, increased sweating, vertigo, syncope, somnolence, tinnitus; Psychiatric– anxiety, insomnia, depression.
The following additional adverse experiences, possibly related to flecainide acetate tablets, have been reported in less than 1% of patients: Body as a Whole– swollen lips, tongue and mouth; arthralgia, bronchospasm, myalgia; Cardiovascular– angina pectoris, second-degree and third-degree AV block, bradycardia, hypertension, hypotension; Gastrointestinal– flatulence; Urinary System– polyuria, urinary retention; Hematologic– leukopenia, granulocytopenia, thrombocytopenia; Skin– urticaria, exfoliative dermatitis, pruritus, alopecia; Visual– eye pain or irritation, photophobia, nystagmus; Nervous System– twitching, weakness, change in taste, dry mouth, convulsions, impotence, speech disorder, stupor, neuropathy; Respiratory– pneumonitis/pulmonary infiltration possibly due to chronic flecainide treatment; Psychiatric– amnesia, confusion, decreased libido, depersonalization, euphoria, morbid dreams, apathy.
For patients with supraventricular arrhythmias, the most commonly reported noncardiac adverse experiences remain consistent with those known for patients treated with flecainide acetate tablets for ventricular arrhythmias. Dizziness is possibly more frequent in PAF patients.
OVERDOSAGE
No specific antidote has been identified for the treatment of flecainide acetate tablets overdosage. Overdoses ranging up to 8000 mg have been survived, with peak plasma flecainide concentrations as high as 5.3 mcg/mL. Untoward effects in these cases included nausea and vomiting, convulsions, hypotension, bradycardia, syncope, extreme widening of the QRS complex, widening of the QT interval, widening of the PR interval, ventricular tachycardia, AV nodal block, asystole, bundle branch block, cardiac failure, and cardiac arrest. The spectrum of events observed in fatal cases was much the same as that seen in the non-fatal cases. Death has resulted following ingestion of as little as 1000 mg; concomitant overdose of other drugs and/or alcohol in many instances undoubtedly contributed to the fatal outcome. Treatment of overdosage should be supportive and may include the following: removal of unabsorbed drug from the gastrointestinal tract, administration of inotropic agents or cardiac stimulants such as dopamine, dobutamine or isoproterenol; mechanically assisted respiration; circulatory assists such as intra-aortic balloon pumping; and transvenous pacing in the event of conduction block. Because of the long plasma half-life of flecainide (12 to 27 hours in patients receiving usual doses), and the possibility of markedly non-linear elimination kinetics at very high doses, these supportive treatments may need to be continued for extended periods of time.
Hemodialysis is not an effective means of removing flecainide from the body. Since flecainide elimination is much slower when urine is very alkaline (pH 8 or higher), theoretically, acidification of urine to promote drug excretion may be beneficial in overdose cases with very alkaline urine. There is no evidence that acidification from normal urinary pH increases excretion.
DOSAGE AND ADMINISTRATION
For patients withsustainedVT, no matter what their cardiac status, flecainide acetate tablets, like other antiarrhythmics, should be initiated in-hospital with rhythm monitoring.
Flecainide has a long half-life (12 to 27 hours in patients). Steady-state plasma levels, in patients with normal renal and hepatic function, may not be achieved until the patient has received 3 to 5 days of therapy at a given dose. Therefore, increases in dosage should be made no more frequently than once every four days, since during the first 2 to 3 days of therapy the optimal effect of a given dose may not be achieved.
For patients with PSVT and patients with PAF the recommended starting dose is 50 mg every 12 hours. Flecainide acetate tablets doses may be increased in increments of 50 mg bid every four days until efficacy is achieved. For PAF patients, a substantial increase in efficacy without a substantial increase in discontinuations for adverse experiences may be achieved by increasing the flecainide acetate tablets dose from 50 to 100 mg bid. The maximum recommended dose for patients with paroxysmal supraventricular arrhythmias is 300 mg/day.
For sustained VT the recommended starting dose is 100 mg every 12 hours. This dose may be increased in increments of 50 mg bid every four days until efficacy is achieved. Most patients with sustained VT do not require more than 150 mg every 12 hours (300 mg/day) and the maximum dose recommended is 400 mg/day.
In patients with sustained VT, use of higher initial doses and more rapid dosage adjustments have resulted in an increased incidence of proarrhythmic events and CHF, particularly during the first few days of dosing (see WARNINGS). Therefore, a loading dose is not recommended.
Intravenous lidocaine has been used occasionally with flecainide acetate tablets while awaiting the therapeutic effect of flecainide acetate tablets. No adverse drug interactions were apparent. However, no formal studies have been performed to demonstrate the usefulness of this regimen.
An occasional patient not adequately controlled by (or intolerant to) a dose given at 12-hour intervals may be dosed at eight-hour intervals.
Once adequate control of the arrhythmia has been achieved, it may be possible in some patients to reduce the dose as necessary to minimize side effects or effects on conduction. In such patients, efficacy at the lower dose should be evaluated.
Flecainide acetate tablets should be used cautiously in patients with a history of CHF or myocardial dysfunction (see WARNINGS).
Any use of flecainide acetate tablets in children should be directly supervised by a cardiologist skilled in the treatment of arrhythmias in children. Because of the evolving nature of information in this area, specialized literature should be consulted. Under six months of age, the initial starting dose of flecainide acetate tablets in children is approximately 50 mg/M2 body surface area daily, divided into two or three equally spaced doses. Over six months of age, the initial starting dose may be increased to 100 mg/M2 per day. The maximum recommended dose is 200 mg/M2 per day. This dose should not be exceeded. In some children on higher doses, despite previously low plasma levels, the level has increased rapidly to far above therapeutic values while taking the same dose. Small changes in dose may also lead to disproportionate increases in plasma levels. Plasma trough (less than one hour pre-dose) flecainide levels and electrocardiograms should be obtained at presumed steady state (after at least five doses) either after initiation or change in flecainide acetate tablet dose, whether the dose was increased for lack of effectiveness, or increased growth of the patient. For the first year on therapy, whenever the patient is seen for reasons of clinical follow-up, it is suggested that a 12-lead electrocardiogram and plasma trough flecainide level are obtained. The usual therapeutic level of flecainide in children is 200 to 500 ng/mL. In some cases, levels as high as 800 ng/mL may be required for control.
In patients with severe renal impairment (creatinine clearance of 35 mL/min/1.73 square meters or less), the initial dosage should be 100 mg once daily (or 50 mg bid); when used in such patients, frequent plasma level monitoring is required to guide dosage adjustments (see Plasma Level Monitoring). In patients with less severe renal disease, the initial dosage should be 100 mg every 12 hours; plasma level monitoring may also be useful in these patients during dosage adjustment. In both groups of patients, dosage increases should be made very cautiously when plasma levels have plateaued (after more than four days), observing the patient closely for signs of adverse cardiac effects or other toxicity. It should be borne in mind that in these patients it may take longer than four days before a new steady-state plasma level is reached following a dosage change.
Based on theoretical considerations, rather than experimental data, the following suggestion is made: when transferring patients from another antiarrhythmic drug to flecainide acetate tablets allow at least two to four plasma half-lives to elapse for the drug being discontinued before starting flecainide acetate tablets at the usual dosage. In patients where withdrawal of a previous antiarrhythmic agent is likely to produce life-threatening arrhythmias, the physician should consider hospitalizing the patient.
When flecainide is given in the presence of amiodarone, reduce the usual flecainide dose by 50% and monitor the patient closely for adverse effects.
Plasma level monitoring is strongly recommended to guide dosage with such combination therapy (see below).
Plasma Level Monitoring: The large majority of patients successfully treated with flecainide acetate tablets were found to have trough plasma levels between 0.2 and 1.0 mcg/mL. The probability of adverse experiences, especially cardiac, may increase with higher trough plasma levels, especially when these exceed 1.0 mcg/mL. Periodic monitoring of trough plasma levels may be useful in patient management. Plasma level monitoring is required in patients with severe renal failure or severe hepatic disease, since elimination of flecainide from plasma may be markedly slower. Monitoring of plasma levels is strongly recommended in patients on concurrent amiodarone therapy and may also be helpful in patients with CHF and in patients with moderate renal disease.
HOW SUPPLIED
Flecainide Acetate Tablets USP 50 mg are available for oral administration as white to off-white, round, biconvex tablets. Engraved “APO” on one side, “FLE” over “50” on the other side. They are supplied as follows:
Bottles of 30 NDC 60505-2660-3
Bottles of 100 NDC 60505-2660-1
Bottles of 500 NDC 60505-2660-5
Bottles of 1000 NDC 60505-2660-8
Unit Dose Cartons of 100 (10x10) NDC 60505-2660-0
Flecainide Acetate Tablets USP 100 mg are available for oral administration as white to off-white, round biconvex tablets. Engraved “APO” on one side, “FLE” over bisect “100” on the other side. They are supplied as follows:
Bottles of 30 NDC 60505-2661-3
Bottles of 100 NDC 60505-2661-1
Bottles of 500 NDC 60505-2661-5
Bottles of 1000 NDC 60505-2661-8
Unit Dose Cartons of 100 (10x10) NDC 60505-2661-0
Flecainide Acetate Tablets USP 150 mg are available for oral administration as white to off-white, oval, biconvex tablets. Engraved “APO” on one side, “FLE” over bisect “150” on the other side. They are supplied as follows:
Bottles of 30 NDC 60505-2662-3
Bottles of 100 NDC 60505-2662-1
Bottles of 500 NDC 60505-2662-5
Bottles of 1000 NDC 60505-2662-8
Unit Dose Cartons of 100 (10x10) NDC 60505-2662-0
Storage
Store at 20º to 25 ºC (68 º to 77 º F); excursions permitted to 15 º to 30 º C (59º to 86 º F) [see USP Controlled Room Temperature].
Dispense in a tight, light-resistant container [see USP].
APOTEX INC.
FLECAINIDE ACETATE TABLETS USP
50 mg, 100 mg, and 150 mg
Rx only
| Manufactured by: Apotex Inc. Toronto, Ontario Canada M9L 1T9 | Manufactured for: Apotex Corp. Weston, Florida 33326 |
Revised: June 2008
Revision: 2
PACKAGE LABEL PRINCIPAL DISPLAY PANEL SECTION
APOTEX INC. NDC 60505-2660-3
Flecainide Acetate Tablets USP
Rx Only
50 mg
30 bottle count

PACKAGE LABEL PRINCIPAL DISPLAY PANEL SECTION
APOTEX INC. NDC 60505-2661-3
Flecainide Acetate Tablets USP
Rx Only
100 mg
30 bottle count

PACKAGE LABEL PRINCIPAL DISPLAY PANEL SECTION
APOTEX INC. NDC 60505-2662-3
Flecainide Acetate Tablets USP
Rx Only
150 mg
30 bottle count

PACKAGE LABEL PRINCIPAL DISPLAY PANEL SECTION
APOTEX INC. NDC 60505-2660-0
Flecainide Acetate Tablets USP
Rx Only
50 mg
10 X 10 Unit dose



| FLECAINIDE ACETATE
flecainide acetate tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| FLECAINIDE ACETATE
flecainide acetate tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| FLECAINIDE ACETATE
flecainide acetate tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Apotex Corp. (845263701) |
| Registrant - Apotex Inc. (209429182) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Apotex Inc. | 209429182 | analysis(60505-2660, 60505-2661, 60505-2662) , manufacture(60505-2660, 60505-2661, 60505-2662) | |