VALPROIC ACID- valproic acid capsule
Upsher-Smith Laboratories, Inc
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use Valproic Acid Capsules, USP safely and effectively. See full prescribing information for Valproic Acid Capsules, USP.
Valproic Acid Capsules, USP for Oral use Initial U.S. Approval: 1978 WARNINGS: LIFE THREATENING ADVERSE REACTIONSSee full prescribing information for complete boxed warning
RECENT MAJOR CHANGESBoxed Warning, Hepatotoxicity 05/2013 Boxed Warning, Fetal Risk 05/2013 Indications and Usage, Important Limitations (1.2) 05/2013 Contraindications, Known or Suspected Mitochondrial Disorders (4) 05/2013 Warnings and Precautions, Hepatotoxicity (5.1) 05/2013 Warnings and Precautions, Birth Defects (5.2) 05/2013 Warnings and Precautions, Decreased IQ (5.3) 05/2013 Warnings and Precautions, Use in Women of Childbearing Potential (5.4) 05/2013 Warnings and Precautions, Brain Atrophy (5.7) 05/2013 INDICATIONS AND USAGEValproic Acid Capsules, USP are an anti-epileptic drug indicated for:
DOSAGE FORMS AND STRENGTHSCapsules: 250 mg valproic acid (3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Upsher-Smith Laboratories, Inc. at 1-855-899-9180 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 3/2014 |
FULL PRESCRIBING INFORMATION
WARNING: LIFE THREATENING ADVERSE REACTIONS
Hepatotoxicity
General Population: Hepatic failure resulting in fatalities has occurred in patients receiving valproate. These incidents usually have occurred during the first six months of treatment. Serious or fatal hepatotoxicity may be preceded by non-specific symptoms such as malaise, weakness, lethargy, facial edema, anorexia, and vomiting. In patients with epilepsy, a loss of seizure control may also occur. Patients should be monitored closely for appearance of these symptoms. Serum liver tests should be performed prior to therapy and at frequent intervals thereafter, especially during the first six months [see Warnings and Precautions (5.1)].
Children under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those on multiple anticonvulsants, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease. When valproic acid products are used in this patient group, they should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. The incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups.
Patients with Mitochondrial Disease: There is an increased risk of valproate-induced acute liver failure and resultant deaths in patients with hereditary neurometabolic syndromes caused by DNA mutations of the mitochondrial DNA Polymerase γ (POLG) gene (e.g. Alpers Huttenlocher Syndrome). Valproic acid is contraindicated in patients known to have mitochondrial disorders caused by POLG mutations and children under two years of age who are clinically suspected of having a mitochondrial disorder [see Contraindications (4)]. In patients over two years of age who are clinically suspected of having a hereditary mitochondrial disease, valproic acid should only be used after other anticonvulsants have failed. This older group of patients should be closely monitored during treatment with valproic acid for the development of acute liver injury with regular clinical assessments and serum liver testing. POLG mutation screening should be performed in accordance with current clinical practice [see Warnings and Precautions (5.1)].
Valproate can cause major congenital malformations, particularly neural tube defects (e.g., spina bifida). In addition, valproate can cause decreased IQ scores following in utero exposure.
Valproate should only be used to treat pregnant women with epilepsy if other medications have failed to control their symptoms or are otherwise unacceptable.
Valproate should not be administered to a woman of childbearing potential unless the drug is essential to the management of her medical condition. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death (e.g., migraine). Women should use effective contraception while using valproate [see Warnings and Precautions (5.2, 5.3, 5.4)].
A Medication Guide describing the risks of valproate is available for patients [see Patient Counseling Information (17)].
Pancreatitis
Cases of life-threatening pancreatitis have been reported in both children and adults receiving valproate. Some of the cases have been described as hemorrhagic with a rapid progression from initial symptoms to death. Cases have been reported shortly after initial use as well as after several years of use. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, valproate should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated [see Warnings and Precautions (5.5)].
1 INDICATIONS AND USAGE
1.1 Epilepsy
Valproic Acid Capsules, USP are indicated as monotherapy and adjunctive therapy in the treatment of patients with complex partial seizures that occur either in isolation or in association with other types of seizures. Valproic Acid Capsules, USP are indicated for use as sole and adjunctive therapy in the treatment of simple and complex absence seizures, and adjunctively in patients with multiple seizure types which include absence seizures.
Simple absence is defined as very brief clouding of the sensorium or loss of consciousness accompanied by certain generalized epileptic discharges without other detectable clinical signs. Complex absence is the term used when other signs are also present.
See Warnings and Precaution (5.1) for statement regarding fatal hepatic dysfunction.
1.2 Important Limitations
Because of the risk to the fetus of decreased IQ, neural tube defects, and other major congenital malformations, which may occur very early in pregnancy, valproate should not be administered to a woman of childbearing potential unless the drug is essential to the management of her medical condition [see Warnings and Precautions (5.2, 5.3, 5.4), Use in Specific Populations (8.1), and Patient Counseling Information (17.3)].
2 DOSAGE AND ADMINISTRATION
2.1 Epilepsy
Valproic acid capsules are intended for oral administration. Valproic acid capsules should be swallowed whole without chewing to avoid local irritation of the mouth and throat.
Patients should be informed to take valproic acid capsules every day as prescribed. If a dose is missed it should be taken as soon as possible, unless it is almost time for the next dose. If a dose is skipped, the patient should not double the next dose.
Valproic acid capsules are indicated as monotherapy and adjunctive therapy in complex partial seizures in adults and pediatric patients down to the age of 10 years, and in simple and complex absence seizures. As the valproic acid capsules dosage is titrated upward, concentrations of clonazepam, diazepam, ethosuximide, lamotrigine, tolbutamide, phenobarbital, carbamazepine, and/or phenytoin may be affected [see Drug Interactions (7.2)].
Complex Partial Seizures
For adults and children 10 years of age or older.
Monotherapy (Initial Therapy)
Valproic acid capsules have not been systematically studied as initial therapy. Patients should initiate therapy at 10 to 15 mg/kg/day. The dosage should be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made.
The probability of thrombocytopenia increases significantly at total trough valproate plasma concentrations above 110 mcg/mL in females and 135 mcg/mL in males. The benefit of improved seizure control with higher doses should be weighed against the possibility of a greater incidence of adverse reactions.
Conversion to Monotherapy
Patients should initiate therapy at 10 to 15 mg/kg/day. The dosage should be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50-100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made. Concomitant antiepilepsy drug (AED) dosage can ordinarily be reduced by approximately 25% every 2 weeks. This reduction may be started at initiation of valproic acid capsules therapy, or delayed by 1 to 2 weeks if there is a concern that seizures are likely to occur with a reduction. The speed and duration of withdrawal of the concomitant AED can be highly variable, and patients should be monitored closely during this period for increased seizure frequency.
Adjunctive Therapy
Valproic acid capsules may be added to the patient's regimen at a dosage of 10 to 15 mg/kg/day. The dosage may be increased by 5 to 10 mg/kg/week to achieve optimal clinical response. Ordinarily, optimal clinical response is achieved at daily doses below 60 mg/kg/day. If satisfactory clinical response has not been achieved, plasma levels should be measured to determine whether or not they are in the usually accepted therapeutic range (50 to 100 mcg/mL). No recommendation regarding the safety of valproate for use at doses above 60 mg/kg/day can be made. If the total daily dose exceeds 250 mg, it should be given in divided doses.
In a study of adjunctive therapy for complex partial seizures in which patients were receiving either carbamazepine or phenytoin in addition to divalproex sodium tablets, no adjustment of carbamazepine or phenytoin dosage was needed [see Clinical Studies (14)]. However, since valproate may interact with these or other concurrently administered AEDs as well as other drugs, periodic plasma concentration determinations of concomitant AEDs are recommended during the early course of therapy [see Drug Interactions (7)].
Simple and Complex Absence Seizures
The recommended initial dose is 15 mg/kg/day, increasing at one week intervals by 5 to 10 mg/kg/day until seizures are controlled or side effects preclude further increases. The maximum recommended dosage is 60 mg/kg/day. If the total daily dose exceeds 250 mg, it should be given in divided doses.
A good correlation has not been established between daily dose, serum concentrations, and therapeutic effect. However, therapeutic valproate serum concentration for most patients with absence seizures is considered to range from 50 to 100 mcg/mL. Some patients may be controlled with lower or higher serum concentrations [see Clinical Pharmacology (12.3)].
As the valproic acid capsules dosage is titrated upward, blood concentrations of phenobarbital and/or phenytoin may be affected [see Drug Interactions (7.2)].
Antiepilepsy drugs should not be abruptly discontinued in patients in whom the drug is administered to prevent major seizures because of the strong possibility of precipitating status epilepticus with attendant hypoxia and threat to life.
The following Table is a guide for the initial daily dose of valproic acid capsules (15 mg/kg/day):
| Weight | Total Daily Dose (mg) | Number of Capsules | |||
|---|---|---|---|---|---|
| (Kg) | (Lb) | Dose 1 | Dose 2 | Dose 3 | |
| 10 - 24.9 | 22 - 54.9 | 250 | 0 | 0 | 1 |
| 25 - 39.9 | 55 - 87.9 | 500 | 1 | 0 | 1 |
| 40 - 59.9 | 88 - 131.9 | 750 | 1 | 1 | 1 |
| 60 - 74.9 | 132 - 164.9 | 1,000 | 1 | 1 | 2 |
| 75 - 89.9 | 165 - 197.9 | 1,250 | 2 | 1 | 2 |
2.2 General Dosing Advice
Dosing in Elderly Patients
Due to a decrease in unbound clearance of valproate and possibly a greater sensitivity to somnolence in the elderly, the starting dose should be reduced in these patients. Dosage should be increased more slowly and with regular monitoring for fluid and nutritional intake, dehydration, somnolence, and other adverse reactions. Dose reductions or discontinuation of valproate should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence. The ultimate therapeutic dose should be achieved on the basis of both tolerability and clinical response [see Warnings and Precautions (5.15), Use in Specific Populations (8.5) and Clinical Pharmacology (12.3)].
Dose-Related Adverse Reactions
The frequency of adverse effects (particularly elevated liver enzymes and thrombocytopenia) may be dose-related. The probability of thrombocytopenia appears to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males) [see Warnings and Precautions (5.9)]. The benefit of improved therapeutic effect with higher doses should be weighed against the possibility of a greater incidence of adverse reactions.
3 DOSAGE FORMS AND STRENGTHS
Valproic acid capsules are supplied as 250 mg off-white colored soft gelatin capsules, imprinted with VALPROIC 250, packaged in bottles containing 100.
4 CONTRAINDICATIONS
- Valproic acid capsules should not be administered to patients with hepatic disease or significant hepatic dysfunction [see Warnings and Precautions (5.1)].
- Valproic acid capsules are contraindicated in patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder [see Warnings and Precautions (5.1)].
- Valproic acid capsules are contraindicated in patients with known hypersensitivity to the drug [see Warnings and Precautions (5.13)].
- Valproic acid capsules are contraindicated in patients with known urea cycle disorders [see Warnings and Precautions (5.6)].
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
General Information on Hepatotoxicity
Hepatic failure resulting in fatalities has occurred in patients receiving valproate. These incidents usually have occurred during the first six months of treatment. Serious or fatal hepatotoxicity may be preceded by non-specific symptoms such as malaise, weakness, lethargy, facial edema, anorexia, and vomiting. In patients with epilepsy, a loss of seizure control may also occur. Patients should be monitored closely for appearance of these symptoms. Serum liver tests should be performed prior to therapy and at frequent intervals thereafter, especially during the first six months. However, healthcare providers should not rely totally on serum biochemistry since these tests may not be abnormal in all instances, but should also consider the results of careful interim medical history and physical examination.
Caution should be observed when administering valproate products to patients with a prior history of hepatic disease. Patients on multiple anticonvulsants, children, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease may be at particular risk. See below, "Patients with Known or Suspected Mitochondrial Disease."
Experience has indicated that children under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those with the aforementioned conditions. When valproic acid capsules products are used in this patient group, they should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. In progressively older patient groups experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably.
Patients with Known or Suspected Mitochondrial Disease
Valproic acid capsules are contraindicated in patients known to have mitochondrial disorders caused by POLG mutations and children under two years of age who are clinically suspected of having a mitochondrial disorder [see Contraindications (4)]. Valproate-induced acute liver failure and liver-related deaths have been reported in patients with hereditary neurometabolic syndromes caused by mutations in the gene for mitochondrial DNA polymerase γ (POLG) (e.g., Alpers-Huttenlocher Syndrome) at a higher rate than those without these syndromes. Most of the reported cases of liver failure in patients with these syndromes have been identified in children and adolescents.
POLG-related disorders should be suspected in patients with a family history or suggestive symptoms of a POLG-related disorder, including but not limited to unexplained encephalopathy, refractory epilepsy (focal, myoclonic), status epilepticus at presentation, developmental delays, psychomotor regression, axonal sensorimotor neuropathy, myopathy cerebellar ataxia, opthalmoplegia, or complicated migraine with occipital aura. POLG mutation testing should be performed in accordance with current clinical practice for the diagnostic evaluation of such disorders. The A467T and W748S mutations are present in approximately 2/3 of patients with autosomal recessive POLG-related disorders.
In patients over two years of age who are clinically suspected of having a hereditary mitochondrial disease, valproic acid capsules should only be used after other anticonvulsants have failed. This older group of patients should be closely monitored during treatment with valproic acid capsules for the development of acute liver injury with regular clinical assessments and serum liver test monitoring.
The drug should be discontinued immediately in the presence of significant hepatic dysfunction, suspected or apparent. In some cases, hepatic dysfunction has progressed in spite of discontinuation of drug [see Boxed Warning and Contraindications (4)].
5.2 Birth Defects
Valproate can cause fetal harm when administered to a pregnant woman. Pregnancy registry data show that maternal valproate use can cause neural tube defects and other structural abnormalities (e.g., craniofacial defects, cardiovascular malformations and malformations involving various body systems). The rate of congenital malformations among babies born to mothers using valproate is about four times higher than the rate among babies born to epileptic mothers using other anti-seizure monotherapies. Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population.
5.3 Decreased IQ Following in utero Exposure
Valproate can cause decreased IQ scores following in utero exposure. Published epidemiological studies have indicated that children exposed to valproate in utero have lower cognitive test scores than children exposed in utero to either another antiepileptic drug or to no antiepileptic drugs. The largest of these studies1 is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94-101]) than children with prenatal exposure to the other antiepileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105-110]), carbamazepine (105 [95% C.I. 102-108]), and phenytoin (108 [95% C.I. 104-112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to antiepileptic drugs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed.
Although all of the available studies have methodological limitations, the weight of the evidence supports the conclusion that valproate exposure in utero can cause decreased IQ in children.
In animal studies, offspring with prenatal exposure to valproate had malformations similar to those seen in humans and demonstrated neurobehavioral deficits [see Use in Specific Populations (8.1)].
Women with epilepsy who are pregnant or who plan to become pregnant should not be treated with valproate unless other treatments have failed to provide adequate symptom control or are otherwise unacceptable. In such women, the benefits of treatment with valproate during pregnancy may still outweigh the risks.
5.4 Use in Women of Childbearing Potential
Because of the risk to the fetus of decreased IQ and major congenital malformations (including neural tube defects), which may occur very early in pregnancy, valproate should not be administered to a woman of childbearing potential unless the drug is essential to the management of her medical condition. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death (e.g., migraine). Women should use effective contraception while using valproate. Women who are planning a pregnancy should be counseled regarding the relative risks and benefits of valproate use during pregnancy, and alternative therapeutic options should be considered for these patients [see Boxed Warning and Use in Specific Populations (8.1)].
To prevent major seizures, valproate should not be discontinued abruptly, as this can precipitate status epilepticus with resulting maternal and fetal hypoxia and threat to life.
Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population. It is not known whether the risk of neural tube defects or decreased IQ in the offspring of women receiving valproate is reduced by folic acid supplementation. Dietary folic acid supplementation both prior to conception and during pregnancy should be routinely recommended for patients using valproate.
5.5 Pancreatitis
Cases of life-threatening pancreatitis have been reported in both children and adults receiving valproate. Some of the cases have been described as hemorrhagic with rapid progression from initial symptoms to death. Some cases have occurred shortly after initial use as well as after several years of use. The rate based upon the reported cases exceeds that expected in the general population and there have been cases in which pancreatitis recurred after rechallenge with valproate. In clinical trials, there were 2 cases of pancreatitis without alternative etiology in 2416 patients, representing 1044 patient-years experience. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, valproate should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated [see Boxed Warning].
5.6 Urea Cycle Disorders (UCD)
Valproic acid is contraindicated in patients with known urea cycle disorders.
Hyperammonemic encephalopathy, sometimes fatal, has been reported following initiation of valproate therapy in patients with urea cycle disorders, a group of uncommon genetic abnormalities, particularly ornithine transcarbamylase deficiency. Prior to the initiation of valproate therapy, evaluation for UCD should be considered in the following patients: 1) those with a history of unexplained encephalopathy or coma, encephalopathy associated with a protein load, pregnancy-related or postpartum encephalopathy, unexplained mental retardation, or history of elevated plasma ammonia or glutamine; 2) those with cyclical vomiting and lethargy, episodic extreme irritability, ataxia, low BUN, or protein avoidance; 3) those with a family history of UCD or a family history of unexplained infant deaths (particularly males); 4) those with other signs or symptoms of UCD. Patients who develop symptoms of unexplained hyperammonemic encephalopathy while receiving valproate therapy should receive prompt treatment (including discontinuation of valproate therapy) and be evaluated for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.11)].
5.7 Brain Atrophy
There have been postmarketing reports of reversible and irreversible cerebral and cerebellar atrophy temporally associated with the use valproate products; in some cases, patients recovered with permanent sequelae [see Adverse Reactions (6.4)]. The motor and cognitive functions of patients on valproate should be routinely monitored and drug should be evaluated for continued use in the presence of suspected or apparent signs of brain atrophy.
Reports of cerebral atrophy have also been reported in children who were exposed in utero to valproate products [see Use in Specific Populations (8.1)].
5.8 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including valproic acid capsules, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed.
Table 2 shows absolute and relative risk by indication for all evaluated AEDs.
| Indication | Placebo Patients with Events Per 1000 Patients | Drug Patients with Events Per 1000 Patients | Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients | Risk Difference: Additional Drug Patients with Events Per 1000 Patients |
|---|---|---|---|---|
| Epilepsy | 1.0 | 3.4 | 3.5 | 2.4 |
| Psychiatric | 5.7 | 8.5 | 1.5 | 2.9 |
| Other | 1.0 | 1.8 | 1.9 | 0.9 |
| Total | 2.4 | 4.3 | 1.8 | 1.9 |
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing valproic acid capsules or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
5.9 Thrombocytopenia
The frequency of adverse effects (particularly elevated liver enzymes and thrombocytopenia may be dose-related. In a clinical trial of divalproex sodium as monotherapy in patients with epilepsy, 34/126 patients (27%) receiving approximately 50 mg/kg/day on average, had at least one value of platelets ≤ 75 × 109/L. Approximately half of these patients had treatment discontinued, with return of platelet counts to normal. In the remaining patients, platelet counts normalized with continued treatment. In this study, the probability of thrombocytopenia appeared to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males). The therapeutic benefit which may accompany the higher doses should therefore be weighed against the possibility of a greater incidence of adverse effects.
Because of reports of thrombocytopenia, inhibition of the secondary phase of platelet aggregation, and abnormal coagulation parameters, (e.g., low fibrinogen), platelet counts and coagulation tests are recommended before initiating therapy and at periodic intervals. It is recommended that patients receiving valproic acid capsules be monitored for platelet count and coagulation parameters prior to planned surgery. Evidence of hemorrhage, bruising, or a disorder of hemostasis/coagulation is an indication for reduction of the dosage or withdrawal of therapy.
5.10 Hyperammonemia
Hyperammonemia has been reported in association with valproate therapy and may be present despite normal liver function tests. In patients who develop unexplained lethargy and vomiting or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured. Hyperammonemia should also be considered in patients who present with hypothermia [see Warnings and Precautions (5.12)]. If ammonia is increased, valproate therapy should be discontinued. Appropriate interventions for treatment of hyperammonemia should be initiated, and such patients should undergo investigation for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.6, 5.11)].
Asymptomatic elevations of ammonia are more common and when present, require close monitoring of plasma ammonia levels. If the elevation persists, discontinuation of valproate therapy should be considered.
5.11 Hyperammonemia and Encephalopathy Associated with Concomitant Topiramate Use
Concomitant administration of topiramate and valproate has been associated with hyperammonemia with or without encephalopathy in patients who have tolerated either drug alone. Clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. Hypothermia can also be a manifestation of hyperammonemia [see Warnings and Precautions (5.12)]. In most cases, symptoms and signs abated with discontinuation of either drug. This adverse reaction is not due to a pharmacokinetic interaction. It is not known if topiramate monotherapy is associated with hyperammonemia. Patients with inborn errors of metabolism or reduced hepatic mitochondrial activity may be at an increased risk for hyperammonemia with or without encephalopathy. Although not studied, an interaction of topiramate and valproate may exacerbate existing defects or unmask deficiencies in susceptible persons. In patients who develop unexplained lethargy, vomiting, or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured [see Contraindications (4) and Warnings and Precautions (5.6, 5.10)].
5.12 Hypothermia
Hypothermia, defined as an unintentional drop in body core temperature to <35°C (95°F), has been reported in association with valproate therapy both in conjunction with and in the absence of hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate with valproate after starting topiramate treatment or after increasing the daily dose of topiramate [see Drug Interactions (7.3)]. Consideration should be given to stopping valproate in patients who develop hypothermia, which may be manifested by a variety of clinical abnormalities including lethargy, confusion, coma, and significant alterations in other major organ systems such as the cardiovascular and respiratory systems. Clinical management and assessment should include examination of blood ammonia levels.
5.13 Multi-Organ Hypersensitivity Reactions
Multi-organ hypersensitivity reactions have been rarely reported in close temporal association to the initiation of valproate therapy in adult and pediatric patients (median time to detection 21 days: range 1 to 40 days). Although there have been a limited number of reports, many of these cases resulted in hospitalization and at least one death has been reported. Signs and symptoms of this disorder were diverse; however, patients typically, although not exclusively, presented with fever and rash associated with other organ system involvement. Other associated manifestations may include lymphadenopathy, hepatitis, liver function test abnormalities, hematological abnormalities (e.g., eosinophilia, thrombocytopenia, neutropenia), pruritus, nephritis, oliguria, hepato-renal syndrome, arthralgia, and asthenia. Because the disorder is variable in its expression, other organ system symptoms and signs, not noted here, may occur. If this reaction is suspected, valproate should be discontinued and an alternative treatment started. Although the existence of cross sensitivity with other drugs that produce this syndrome is unclear, the experience amongst drugs associated with multi-organ hypersensitivity would indicate this to be a possibility.
5.14 Interaction with Carbapenem Antibiotics
Carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) may reduce serum valproate concentrations to subtherapeutic levels, resulting in loss of seizure control. Serum valproate concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproate concentrations drop significantly or seizure control deteriorates [see Drug Interactions (7.1)].
5.15 Somnolence in the Elderly
In a double-blind, multicenter trial of valproate in elderly patients with dementia (mean age = 83 years), doses were increased by 125 mg/day to a target dose of 20 mg/kg/day. A significantly higher proportion of valproate patients had somnolence compared to placebo, and although not statistically significant, there was a higher proportion of patients with dehydration. Discontinuations for somnolence were also significantly higher than with placebo. In some patients with somnolence (approximately one-half), there was associated reduced nutritional intake and weight loss. There was a trend for the patients who experienced these events to have a lower baseline albumin concentration, lower valproate clearance, and a higher BUN. In elderly patients, dosage should be increased more slowly and with regular monitoring for fluid and nutritional intake, dehydration, somnolence, and other adverse reactions. Dose reductions or discontinuation of valproate should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence [see Dosage and Administration (2.2)].
5.16 Monitoring: Drug Plasma Concentration
Since valproate may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy [see Drug Interactions (7)].
5.17 Effect on Ketone and Thyroid Function Tests
Valproate is partially eliminated in the urine as a keto-metabolite which may lead to a false interpretation of the urine ketone test.
There have been reports of altered thyroid function tests associated with valproate. The clinical significance of these is unknown.
5.18 Effect on HIV and CMV Viruses Replication
There are in vitro studies that suggest valproate stimulates the replication of the HIV and CMV viruses under certain experimental conditions. The clinical consequence, if any, is not known. Additionally, the relevance of these in vitro findings is uncertain for patients receiving maximally suppressive antiretroviral therapy. Nevertheless, these data should be borne in mind when interpreting the results from regular monitoring of the viral load in HIV infected patients receiving valproate or when following CMV infected patients clinically.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
Hepatic failure (5.1)
Birth defects (5.2)
Decreased IQ following in utero exposure (5.3)
Pancreatitis (5.5)
Thrombocytopenia (5.9)
Hyperammonemic encephalopathy (5.10, 5.11)
Multi-organ hypersensitivity reactions (5.13)
Somnolence in the elderly (5.15)
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
6.1 Epilepsy
The data described in the following section were obtained using divalproex sodium tablets.
Based on a placebo-controlled trial of adjunctive therapy for treatment of complex partial seizures, divalproex sodium tablets were generally well tolerated with most adverse reactions rated as mild to moderate in severity. Intolerance was the primary reason for discontinuation in the divalproex sodium tablets-treated patients (6%), compared to 1% of placebo-treated patients.
Table 3 lists treatment-emergent adverse reactions which were reported by ≥ 5% of divalproex sodium-treated patients and for which the incidence was greater than in the placebo group, in a placebo-controlled trial of adjunctive therapy for the treatment of complex partial seizures. Since patients were also treated with other antiepilepsy drugs, it is not possible, in most cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium tablets alone, or the combination of divalproex sodium tablets and other antiepilepsy drugs.
| Body System/Reaction | Divalproex Sodium Tablets (%) (n = 77) | Placebo (%) (n = 70) |
|---|---|---|
| Body as a Whole | ||
| Headache | 31 | 21 |
| Asthenia | 27 | 7 |
| Fever | 6 | 4 |
| Gastrointestinal System | ||
| Nausea | 48 | 14 |
| Vomiting | 27 | 7 |
| Abdominal Pain | 23 | 6 |
| Diarrhea | 13 | 6 |
| Anorexia | 12 | 0 |
| Dyspepsia | 8 | 4 |
| Constipation | 5 | 1 |
| Nervous System | ||
| Somnolence | 27 | 11 |
| Tremor | 25 | 6 |
| Dizziness | 25 | 13 |
| Diplopia | 16 | 9 |
| Amblyopia/Blurred Vision | 12 | 9 |
| Ataxia | 8 | 1 |
| Nystagmus | 8 | 1 |
| Emotional Lability | 6 | 4 |
| Thinking Abnormal | 6 | 0 |
| Amnesia | 5 | 1 |
| Respiratory System | ||
| Flu Syndrome | 12 | 9 |
| Infection | 12 | 6 |
| Bronchitis | 5 | 1 |
| Rhinitis | 5 | 4 |
| Other | ||
| Alopecia | 6 | 1 |
| Weight Loss | 6 | 0 |
Table 4 lists treatment-emergent adverse reactions which were reported by ≥ 5% of patients in the high dose divalproex sodium group, and for which the incidence was greater than in the low dose group, in a controlled trial of divalproex sodium tablets monotherapy treatment of complex partial seizures. Since patients were being titrated off another antiepilepsy drug during the first portion of the trial, it is not possible, in many cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium tablets alone, or the combination of divalproex sodium tablets and other antiepilepsy drugs.
| Body System/Reaction | High Dose (%) (n = 131) | Low Dose (%) (n = 134) |
|---|---|---|
|
||
| Body as a Whole | ||
| Asthenia | 21 | 10 |
| Digestive System | ||
| Nausea | 34 | 26 |
| Diarrhea | 23 | 19 |
| Vomiting | 23 | 15 |
| Abdominal Pain | 12 | 9 |
| Anorexia | 11 | 4 |
| Dyspepsia | 11 | 10 |
| Hemic/Lymphatic System | ||
| Thrombocytopenia | 24 | 1 |
| Ecchymosis | 5 | 4 |
| Metabolic/Nutritional | ||
| Weight Gain | 9 | 4 |
| Peripheral Edema | 8 | 3 |
| Nervous System | ||
| Tremor | 57 | 19 |
| Somnolence | 30 | 18 |
| Dizziness | 18 | 13 |
| Insomnia | 15 | 9 |
| Nervousness | 11 | 7 |
| Amnesia | 7 | 4 |
| Nystagmus | 7 | 1 |
| Depression | 5 | 4 |
| Respiratory System | ||
| Infection | 20 | 13 |
| Pharyngitis | 8 | 2 |
| Dyspnea | 5 | 1 |
| Skin and Appendages | ||
| Alopecia | 24 | 13 |
| Special Senses | ||
| Amblyopia/Blurred Vision | 8 | 4 |
| Tinnitus | 7 | 1 |
The following additional adverse reactions were reported by greater than 1% but less than 5% of the 358 patients treated with divalproex sodium tablets in the controlled trials of complex partial seizures:
Body as a Whole: Back pain, chest pain, malaise.
Cardiovascular System: Tachycardia, hypertension, palpitation.
Digestive System: Increased appetite, flatulence, hematemesis, eructation, pancreatitis, periodontal abscess.
Hemic and Lymphatic System: Petechia.
Metabolic and Nutritional Disorders: SGOT increased, SGPT increased.
Musculoskeletal System: Myalgia, twitching, arthralgia, leg cramps, myasthenia.
Nervous System: Anxiety, confusion, abnormal gait, paresthesia, hypertonia, incoordination, abnormal dreams, personality disorder.
Respiratory System: Sinusitis, cough increased, pneumonia, epistaxis.
Skin and Appendages: Rash, pruritus, dry skin.
Special Senses: Taste perversion, abnormal vision, deafness, otitis media.
Urogenital System: Urinary incontinence, vaginitis, dysmenorrhea, amenorrhea, urinary frequency.
6.2 Mania
Although valproic acid capsules have not been evaluated for safety and efficacy in the treatment of manic episodes associated with bipolar disorder, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Chills, neck pain, neck rigidity.
Cardiovascular System: Hypotension, postural hypotension, vasodilation.
Digestive System: Fecal incontinence, gastroenteritis, glossitis.
Musculoskeletal System: Arthrosis.
Nervous System: Agitation, catatonic reaction, hypokinesia, reflexes increased, tardive dyskinesia, vertigo.
Skin and Appendages: Furunculosis, maculopapular rash, seborrhea.
Special Senses: Conjunctivitis, dry eyes, eye pain.
Urogenital System: Dysuria.
6.3 Migraine
Although valproic acid capsules have not been evaluated for safety and efficacy in the treatment of prophylaxis of migraine headaches, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Face edema.
Digestive System: Dry mouth, stomatitis.
Urogenital System: Cystitis, metrorrhagia, and vaginal hemorrhage.
6.4 Other Patient Populations
Adverse reactions that have been reported with all dosage forms of valproate from epilepsy trials, spontaneous reports, and other sources are listed below by body system.
Gastrointestinal: The most commonly reported side effects at the initiation of therapy are nausea, vomiting, and indigestion. These effects are usually transient and rarely require discontinuation of therapy. Diarrhea, abdominal cramps, and constipation have been reported. Both anorexia with some weight loss and increased appetite with weight gain have also been reported. The administration of delayed-release divalproex sodium may result in reduction of gastrointestinal side effects in some patients.
CNS Effects: Sedative effects have occurred in patients receiving valproate alone but occur most often in patients receiving combination therapy. Sedation usually abates upon reduction of other antiepileptic medication. Tremor (may be dose-related), hallucinations, ataxia, headache, nystagmus, diplopia, asterixis, "spots before eyes", dysarthria, dizziness, confusion, hypesthesia, vertigo, incoordination, and Parkinsonism have been reported with the use of valproate. Rare cases of coma have occurred in patients receiving valproate alone or in conjunction with phenobarbital. In rare instances encephalopathy with or without fever has developed shortly after the introduction of valproate monotherapy without evidence of hepatic dysfunction or inappropriately high plasma valproate levels. Although recovery has been described following drug withdrawal, there have been fatalities in patients with hyperammonemic encephalopathy, particularly in patients with underlying urea cycle disorders [see Warnings and Precautions (5.6)].
There have been postmarketing reports of reversible and irreversible cerebral and cerebellar atrophy temporally associated with the use of valproate products. In some cases the patients recovered with permanent sequelae [see Warnings and Precautions (5.7)].
Cerebral atrophy has been reported in children exposed to valproate in utero [see Use in Specific Populations (8.1)].
Dermatologic: Transient hair loss, skin rash, photosensitivity, generalized pruritus, erythema multiforme, and Stevens-Johnson syndrome. Rare cases of toxic epidermal necrolysis have been reported including a fatal case in a 6 month old infant taking valproate and several other concomitant medications. An additional case of toxic epidermal necrosis resulting in death was reported in a 35 year old patient with AIDS taking several concomitant medications and with a history of multiple cutaneous drug reactions. Serious skin reactions have been reported with concomitant administration of lamotrigine and valproate [see Drug Interactions (7)].
Psychiatric: Emotional upset, depression, psychosis, aggression, hyperactivity, hostility, and behavioral deterioration.
Hematologic: Thrombocytopenia and inhibition of the secondary phase of platelet aggregation may be reflected in altered bleeding time, petechiae, bruising, hematoma formation, epistaxis, and frank hemorrhage [see Warnings and Precautions (5.9) and Drug Interactions (7)]. Relative lymphocytosis, macrocytosis, hypofibrinogenemia, leucopenia, eosinophilia, anemia including macrocytic with or without folate deficiency, bone marrow suppression, pancytopenia, aplastic anemia, agranulocytosis, and acute intermittent porphyria.
Hepatic: Minor elevations of transaminases (e.g., SGOT and SGPT) and LDH are frequent and appear to be dose-related. Occasionally, laboratory test results include increases in serum bilirubin and abnormal changes in other liver function tests. These results may reflect potentially serious hepatotoxicity [see Warnings and Precautions (5.1)].
Endocrine: Irregular menses, secondary amenorrhea, breast enlargement, galactorrhea, and parotid gland swelling. Abnormal thyroid function tests [see Warnings and Precautions (5.17)].
There have been rare spontaneous reports of polycystic ovary disease. A cause and effect relationship has not been established.
Pancreatic: Acute pancreatitis, including fatalities [see Warnings and Precautions (5.5)].
Metabolic: Hyperammonemia [see Warnings and Precautions (5.10)], hyponatremia, and inappropriate ADH secretion.
There have been rare reports of Fanconi's syndrome occurring chiefly in children.
Decreased carnitine concentrations have been reported although the clinical relevance is undetermined.
Hyperglycinemia has occurred and was associated with a fatal outcome in a patient with preexistent nonketotic hyperglycinemia.
Special Senses: Hearing loss, either reversible or irreversible, has been reported; however, a cause and effect relationship has not been established. Ear pain has also been reported.
Other: Allergic reaction, anaphylaxis, edema of the extremities, lupus erythematosus, bone pain, cough increased, pneumonia, otitis media, bradycardia, cutaneous vasculitis, fever, and hypothermia.
There have been reports of developmental delay, autism and/or autism spectrum disorder in the offspring of women exposed to valproate during pregnancy.
7 DRUG INTERACTIONS
7.1 Effects of Co-Administered Drugs on Valproate Clearance
Drugs that affect the level of expression of hepatic enzymes, particularly those that elevate levels of glucuronosyltransferases, may increase the clearance of valproate. For example, phenytoin, carbamazepine, and phenobarbital (or primidone) can double the clearance of valproate. Thus, patients on monotherapy will generally have longer half-lives and higher concentrations than patients receiving polytherapy with antiepilepsy drugs.
In contrast, drugs that are inhibitors of cytochrome P450 isozymes, e.g., antidepressants, may be expected to have little effect on valproate clearance because cytochrome P450 microsomal mediated oxidation is a relatively minor secondary metabolic pathway compared to glucuronidation and beta-oxidation.
Because of these changes in valproate clearance, monitoring of valproate and concomitant drug concentrations should be increased whenever enzyme inducing drugs are introduced or withdrawn.
The following list provides information about the potential for an influence of several commonly prescribed medications on valproate pharmacokinetics. The list is not exhaustive nor could it be, since new interactions are continuously being reported.
Drugs for which a potentially important interaction has been observed
Aspirin
A study involving the co-administration of aspirin at antipyretic doses (11 to 16 mg/kg) with valproate to pediatric patients (n = 6) revealed a decrease in protein binding and an inhibition of metabolism of valproate. Valproate free fraction was increased 4-fold in the presence of aspirin compared to valproate alone. The β-oxidation pathway consisting of 2-E-valproic acid, 3-OH-valproic acid, and 3-keto valproic acid was decreased from 25% of total metabolites excreted on valproate alone to 8.3% in the presence of aspirin. Caution should be observed if valproate and aspirin are to be co-administered.
Carbapenem Antibiotics
A clinically significant reduction in serum valproic acid concentration has been reported in patients receiving carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) and may result in loss of seizure control. The mechanism of this interaction is not well understood. Serum valproic acid concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop significantly or seizure control deteriorates [see Warnings and Precautions (5.14)].
Felbamate
A study involving the co-administration of 1200 mg/day of felbamate with valproate to patients with epilepsy (n = 10) revealed an increase in mean valproate peak concentration by 35% (from 86 to 115 mcg/mL) compared to valproate alone. Increasing the felbamate dose to 2400 mg/day increased the mean valproate peak concentration to 133 mcg/mL (another 16% increase). A decrease in valproate dosage may be necessary when felbamate therapy is initiated.
Drugs for which either no interaction or a likely clinically unimportant interaction has been observed
Antacids
A study involving the co-administration of valproate 500 mg with commonly administered antacids (Maalox, Trisogel, and Titralac - 160 mEq doses) did not reveal any effect on the extent of absorption of valproate.
Chlorpromazine
A study involving the administration of 100 to 300 mg/day of chlorpromazine to schizophrenic patients already receiving valproate (200 mg BID) revealed a 15% increase in trough plasma levels of valproate.
7.2 Effects of Valproate on Other Drugs
Valproate has been found to be a weak inhibitor of some P450 isozymes, epoxide hydrase, and glucuronyltransferases.
The following list provides information about the potential for an influence of valproate co-administration on the pharmacokinetics or pharmacodynamics of several commonly prescribed medications. The list is not exhaustive, since new interactions are continuously being reported.
Drugs for which a potentially important valproate interaction has been observed
Amitriptyline/Nortriptyline
Administration of a single oral 50 mg dose of amitriptyline to 15 normal volunteers (10 males and 5 females) who received valproate (500 mg BID) resulted in a 21% decrease in plasma clearance of amitriptyline and a 34% decrease in the net clearance of nortriptyline. Rare postmarketing reports of concurrent use of valproate and amitriptyline resulting in an increased amitriptyline level have been received. Concurrent use of valproate and amitriptyline has rarely been associated with toxicity. Monitoring of amitriptyline levels should be considered for patients taking valproate concomitantly with amitriptyline. Consideration should be given to lowering the dose of amitriptyline/nortriptyline in the presence of valproate.
Carbamazepine/carbamazepine-10,11-Epoxide
Serum levels of carbamazepine (CBZ) decreased 17% while that of carbamazepine-10,11-epoxide (CBZ-E) increased by 45% upon co-administration of valproate and CBZ to epileptic patients.
Clonazepam
The concomitant use of valproate and clonazepam may induce absence status in patients with a history of absence type seizures.
Diazepam
Valproate displaces diazepam from its plasma albumin binding sites and inhibits its metabolism. Co-administration of valproate (1500 mg daily) increased the free fraction of diazepam (10 mg) by 90% in healthy volunteers (n = 6). Plasma clearance and volume of distribution for free diazepam were reduced by 25% and 20%, respectively, in the presence of valproate. The elimination half-life of diazepam remained unchanged upon addition of valproate.
Ethosuximide
Valproate inhibits the metabolism of ethosuximide. Administration of a single ethosuximide dose of 500 mg with valproate (800 to 1600 mg/day) to healthy volunteers (n=6) was accompanied by a 25% increase in elimination half-life of ethosuximide and a 15% decrease in its total clearance as compared to ethosuximide alone. Patients receiving valproate and ethosuximide, especially along with other anticonvulsants, should be monitored for alterations in serum concentrations of both drugs.
Lamotrigine
In a steady-state study involving 10 healthy volunteers, the elimination half-life of lamotrigine increased from 26 to 70 hours with valproate co-administration (a 165% increase). The dose of lamotrigine should be reduced when co-administered with valproate. Serious skin reactions (such as Stevens-Johnson Syndrome and toxic epidermal necrolysis) have been reported with concomitant lamotrigine and valproate administration. See lamotrigine package insert for details on lamotrigine dosing with concomitant valproate administration.
Phenobarbital
Valproate was found to inhibit the metabolism of phenobarbital. Co-administration of valproate (250 mg BID for 14 days) with phenobarbital to normal subjects (n = 6) resulted in a 50% increase in half-life and a 30% decrease in plasma clearance of phenobarbital (60 mg single-dose). The fraction of phenobarbital dose excreted unchanged increased by 50% in presence of valproate.
There is evidence for severe CNS depression, with or without significant elevations of barbiturate or valproate serum concentrations. All patients receiving concomitant barbiturate therapy should be closely monitored for neurological toxicity. Serum barbiturate concentrations should be obtained, if possible, and the barbiturate dosage decreased, if appropriate.
Primidone, which is metabolized to a barbiturate, may be involved in a similar interaction with valproate.
Phenytoin
Valproate displaces phenytoin from its plasma albumin binding sites and inhibits its hepatic metabolism. Co-administration of valproate (400 mg TID) with phenytoin (250 mg) in normal volunteers (n = 7) was associated with a 60% increase in the free fraction of phenytoin. Total plasma clearance and apparent volume of distribution of phenytoin increased 30% in the presence of valproate. Both the clearance and apparent volume of distribution of free phenytoin were reduced by 25%.
In patients with epilepsy, there have been reports of breakthrough seizures occurring with the combination of valproate and phenytoin. The dosage of phenytoin should be adjusted as required by the clinical situation.
Tolbutamide
From in vitro experiments, the unbound fraction of tolbutamide was increased from 20% to 50% when added to plasma samples taken from patients treated with valproate. The clinical relevance of this displacement is unknown.
Drugs for which either no interaction or a likely clinically unimportant interaction has been observed
Acetaminophen
Valproate had no effect on any of the pharmacokinetic parameters of acetaminophen when it was concurrently administered to three epileptic patients.
Clozapine
In psychotic patients (n = 11), no interaction was observed when valproate was co-administered with clozapine.
Lithium
Co-administration of valproate (500 mg BID) and lithium carbonate (300 mg TID) to normal male volunteers (n = 16) had no effect on the steady-state kinetics of lithium.
7.3 Topiramate
Concomitant administration of valproate and topiramate has been associated with hyperammonemia with and without encephalopathy [see Contraindications (4) and Warnings and Precautions (5.10, 5.11)]. Concomitant administration of topiramate with valproate has also been associated with hypothermia in patients who have tolerated either drug alone. It may be prudent to examine blood ammonia levels in patients in whom the onset of hypothermia has been reported [see Warnings and Precautions (5.10, 5.12)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D for epilepsy [see Warnings and Precautions (5.2, 5.3)].
Pregnancy Registry
To collect information on the effects of in utero exposure to valproic acid, physicians should encourage pregnant patients taking valproic acid capsules to enroll in the NAAED Pregnancy Registry. This can be done by calling toll free 1-888-233-2334, and must be done by the patients themselves. Information on the registry can be found at the website, http://www.aedpregnancyregistry.org/.
Fetal Risk Summary
All pregnancies have a background risk of birth defects (about 3%), pregnancy loss (about 15%), or other adverse outcomes regardless of drug exposure. Maternal valproate use during pregnancy for any indication increases the risk of congenital malformations, particularly neural tube defects, but also malformations involving other body systems (e.g., craniofacial defects, cardiovascular malformations). The risk of major structural abnormalities is greatest during the first trimester; however, other serious developmental effects can occur with valproate use throughout pregnancy. The rate of congenital malformations among babies born to epileptic mothers who used valproate during pregnancy has been shown to be about four times higher than the rate among babies born to epileptic mothers who used other anti-seizure monotherapies [see Warnings and Precautions (5.3)].
Exposure in utero to valproate products has been associated with cerebral atrophy [see Warnings and Precautions (5.7) and Adverse Reactions (6.4)].
Several published epidemiological studies have indicated that children exposed to valproate in utero have lower IQ scores than children exposed to either another antiepileptic drug in utero or to no antiepileptic drugs in utero [see Warnings and Precautions (5.3)].
In animal studies, offspring with prenatal exposure to valproate had structural malformations similar to those seen in humans and demonstrated neurobehavioral deficits.
Clinical Considerations
- Neural tube defects are the congenital malformation most strongly associated with maternal valproate use. The risk of spina bifida following in utero valproate exposure is generally estimated as 1-2%, compared to an estimated general population risk for spina bifida of about 0.06 to 0.07% (6 to 7 in 10,000 births).
- Valproate can cause decreased IQ scores in children whose mothers were treated with valproate during pregnancy.
- Because of the risks of decreased IQ, neural tube defects, and other fetal adverse events, which may occur very early in pregnancy:
- Valproate should not be administered to a woman of childbearing potential unless the drug is essential to the management of her medical condition. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death (e.g., migraine).
- Valproic acid capsules should not be used to treat women with epilepsy who are pregnant or who plan to become pregnant unless other treatments have failed to provide adequate symptom control or are otherwise unacceptable. In such women, the benefits of treatment with valproate during pregnancy may still outweigh the risks. When treating a pregnant woman or a woman of childbearing potential, carefully consider both the potential risks and benefits of treatment and provide appropriate counseling.
- To prevent major seizures, women with epilepsy should not discontinue valproate abruptly, as this can precipitate status epilepticus with resulting maternal and fetal hypoxia and threat to life. Even minor seizures may pose some hazard to the developing embryo or fetus. However, discontinuation of the drug may be considered prior to and during pregnancy in individual cases if the seizure disorder severity and frequency do not pose a serious threat to the patient.
- Available prenatal diagnostic testing to detect neural tube and other defects should be offered to pregnant women using valproate.
- Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population. It is not known whether the risk of neural tube defects or decreased IQ in the offspring of women receiving valproate is reduced by folic acid supplementation. Dietary folic acid supplementation both prior to conception and during pregnancy should be routinely recommended for patients using valproate.
- Patients taking valproate may develop clotting abnormalities [see Warnings and Precautions (5.9)]. A patient who had low fibrinogen when taking multiple anticonvulsants including valproate gave birth to an infant with afibrinogenemia who subsequently died of hemorrhage. If valproate is used in pregnancy, the clotting parameters should be monitored carefully.
- Patients taking valproate may develop hepatic failure [see Boxed Warning and Warnings and Precautions (5.1)]. Fatal cases of hepatic failure in infants exposed to valproate in utero have also been reported following maternal use of valproate during pregnancy.
Data
Human
There is an extensive body of evidence demonstrating that exposure to valproate in utero increases the risk of neural tube defects and other structural abnormalities. Based on published data from the CDC's National Birth Defects Prevention Network, the risk of spina bifida in the general population is about 0.06 to 0.07%. The risk of spina bifida following in utero valproate exposure has been estimated to be approximately 1 to 2%.
In one study using NAAED Pregnancy Registry data, 16 cases of major malformations following prenatal valproate exposure were reported among offspring of 149 enrolled women who used valproate during pregnancy. Three of the 16 cases were neural tube defects; the remaining cases included craniofacial defects, cardiovascular malformations and malformations of varying severity involving other body systems. The NAAED Pregnancy Registry has reported a major malformation rate of 10.7% (95% C.I. 6.3% – 16.9%) in the offspring of women exposed to an average of 1,000 mg/day of valproate monotherapy during pregnancy (dose range 500-2000 mg/day). The major malformation rate among the internal comparison group of 1,048 epileptic women who received any other antiepileptic drug monotherapy during pregnancy was 2.9% (95% CI 2.0% to 4.1%). These data show a four-fold increased risk for any major malformation (Odds Ratio 4.0; 95% CI 2.1 to 7.4) following valproate exposure in utero compared to the risk following exposure in utero to any other antiepileptic drug monotherapy.
Published epidemiological studies have indicated that children exposed to valproate in utero have lower IQ scores than children exposed to either another antiepileptic drug in utero or to no antiepileptic drugs in utero. The largest of these studies is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94-101]) than children with prenatal exposure to the other anti-epileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105-110]), carbamazepine (105 [95% C.I. 102-108]) and phenytoin (108 [95% C.I. 104-112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to antiepileptic drugs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed.
Although all of the available studies have methodological limitations, the weight of the evidence supports a causal association between valproate exposure in utero and subsequent adverse effects on cognitive development.
There are published case reports of fatal hepatic failure in offspring of women who used valproate during pregnancy.
Animal
In developmental toxicity studies conducted in mice, rats, rabbits, and monkeys, increased rates of fetal structural abnormalities, intrauterine growth retardation, and embryo-fetal death occurred following treatment of pregnant animals with valproate during organogenesis at clinically relevant doses (calculated on a body surface area basis). Valproate induced malformations of multiple organ systems, including skeletal, cardiac, and urogenital defects. In mice, in addition to other malformations, fetal neural tube defects have been reported following valproate administration during critical periods of organogenesis, and the teratogenic response correlated with peak maternal drug levels. Behavioral abnormalities (including cognitive, locomotor, and social interaction deficits) and brain histopathological changes have also been reported in mice and rat offspring exposed prenatally to clinically relevant doses of valproate.
8.3 Nursing Mothers
Valproate is excreted in human milk. Caution should be exercised when valproate is administered to a nursing woman.
8.4 Pediatric Use
Experience has indicated that pediatric patients under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those with the aforementioned conditions [see Boxed Warning]. When valproic acid capsules are used in this patient group, it should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. Above the age of 2 years, experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups.
Younger children, especially those receiving enzyme-inducing drugs, will require larger maintenance doses to attain targeted total and unbound valproic acid concentrations. Pediatric patients (i.e., between 3 months and 10 years) have 50% higher clearances expressed on weight (i.e., mL/min/kg) than do adults. Over the age of 10 years, children have pharmacokinetic parameters that approximate those of adults.
The variability in free fraction limits the clinical usefulness of monitoring total serum valproic acid concentrations. Interpretation of valproic acid concentrations in children should include consideration of factors that affect hepatic metabolism and protein binding.
Pediatric Clinical Trials
Divalproex sodium tablets were studied in seven pediatric clinical trials.
Two of the pediatric studies were double-blinded placebo-controlled trials to evaluate the efficacy of divalproex sodium tablets ER for the indications of mania (150 patients aged 10 to 17 years, 76 of whom were on divalproex sodium tablets ER) and migraine (304 patients aged 12 to 17 years, 231 of whom were on divalproex sodium tablets ER). Efficacy was not established for either the treatment of migraine or the treatment of mania. The most common drug-related adverse reactions (reported >5% and twice the rate of placebo) reported in the controlled pediatric mania study were nausea, upper abdominal pain, somnolence, increased ammonia, gastritis and rash.
The remaining five trials were long term safety studies. Two six-month pediatric studies were conducted to evaluate the long-term safety of divalproex sodium tablets ER for the indication of mania (292 patients aged 10 to 17 years). Two twelve-month pediatric studies were conducted to evaluate the long-term safety of divalproex sodium tablets ER for the indication of migraine (353 patients aged 12 to 17 years). One twelve-month study was conducted to evaluate the safety of divalproex sodium sprinkle capsules in the indication of partial seizures (169 patients aged 3 to 10 years).
In these seven trials, the safety and tolerability of divalproex sodium tablets in pediatric patients were shown to be comparable to those in adults [see Adverse Reactions (6)].
Juvenile Animal Toxicology
In studies of valproate in immature animals, toxic effects not observed in adult animals included retinal dysplasia in rats treated during the neonatal period (from postnatal day 4) and nephrotoxicity in rats treated during the neonatal and juvenile (from postnatal day 14) periods. The no-effect dose for these findings was less than the maximum recommended human dose on a mg/m2 basis.
8.5 Geriatric Use
No patients above the age of 65 years were enrolled in double-blind prospective clinical trials of mania associated with bipolar illness. In a case review study of 583 patients, 72 patients (12%) were greater than 65 years of age. A higher percentage of patients above 65 years of age reported accidental injury, infection, pain, somnolence, and tremor.
Discontinuation of valproate was occasionally associated with the latter two events. It is not clear whether these events indicate additional risk or whether they result from preexisting medical illness and concomitant medication use among these patients.
A study of elderly patients with dementia revealed drug related somnolence and discontinuation for somnolence [see Warnings and Precautions (5.15)]. The starting dose should be reduced in these patients, and dosage reductions or discontinuation should be considered in patients with excessive somnolence [see Dosage and Administration (2.2)].
10 OVERDOSAGE
Overdosage with valproate may result in somnolence, heart block, and deep coma. Fatalities have been reported; however, patients have recovered from valproate levels as high as 2120 mcg/mL.
In overdose situations, the fraction of drug not bound to protein is high and hemodialysis or tandem hemodialysis plus hemoperfusion may result in significant removal of drug. The benefit of gastric lavage or emesis will vary with the time since ingestion. General supportive measures should be applied with particular attention to the maintenance of adequate urinary output.
Naloxone has been reported to reverse the CNS depressant effects of valproate overdosage. Because naloxone could theoretically also reverse the antiepileptic effects of valproate, it should be used with caution in patients with epilepsy.
11 DESCRIPTION
Valproic acid is a carboxylic acid designated as 2-propylpentanoic acid. It is also known as dipropylacetic acid. Valproic acid has the following structure:
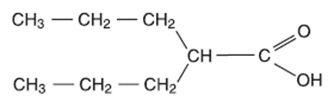
Valproic acid (pKa 4.8) has a molecular weight of 144 and occurs as a colorless liquid with a characteristic odor. It is slightly soluble in water (1.3 mg/mL) and very soluble in organic solvents.
Valproic Acid Capsules, USP are antiepileptics for oral administration. Each soft gelatin capsule contains 250 mg valproic acid.
Inactive Ingredients
Peanut oil, gelatin, glycerin and titanium dioxide
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Valproic acid dissociates to the valproate ion in the gastrointestinal tract. The mechanisms by which valproate exerts its antiepileptic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
12.2 Pharmacodynamics
The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate which affects the clearance of the drug. Thus, monitoring of total serum valproate cannot provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
12.3 Pharmacokinetics
Absorption/Bioavailability
Equivalent oral doses of divalproex sodium products and valproic acid capsules deliver equivalent quantities of valproate ion systemically. Although the rate of valproate ion absorption may vary with the formulation administered (liquid, solid, or sprinkle), conditions of use (e.g., fasting or postprandial) and the method of administration (e.g., whether the contents of the capsule are sprinkled on food or the capsule is taken intact), these differences should be of minor clinical importance under the steady state conditions achieved in chronic use in the treatment of epilepsy.
However, it is possible that differences among the various valproate products in Tmax and Cmax could be important upon initiation of treatment. For example, in single dose studies, the effect of feeding had a greater influence on the rate of absorption of the divalproex sodium tablet (increase in Tmax from 4 to 8 hours) than on the absorption of the divalproex sodium sprinkle capsules (increase in Tmax from 3.3 to 4.8 hours).
While the absorption rate from the G.I. tract and fluctuation in valproate plasma concentrations vary with dosing regimen and formulation, the efficacy of valproate as an anticonvulsant in chronic use is unlikely to be affected. Experience employing dosing regimens from once-a-day to four-times-a-day, as well as studies in primate epilepsy models involving constant rate infusion, indicate that total daily systemic bioavailability (extent of absorption) is the primary determinant of seizure control and that differences in the ratios of plasma peak to trough concentrations between valproate formulations are inconsequential from a practical clinical standpoint.
Co-administration of oral valproate products with food and substitution among the various divalproex sodium and valproic acid formulations should cause no clinical problems in the management of patients with epilepsy [see Dosage and Administration (2.1)]. Nonetheless, any changes in dosage administration, or the addition or discontinuance of concomitant drugs should ordinarily be accompanied by close monitoring of clinical status and valproate plasma concentrations.
Distribution
Protein Binding
The plasma protein binding of valproate is concentration dependent and the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Protein binding of valproate is reduced in the elderly, in patients with chronic hepatic diseases, in patients with renal impairment, and in the presence of other drugs (e.g., aspirin). Conversely, valproate may displace certain protein-bound drugs (e.g., phenytoin, carbamazepine, warfarin, and tolbutamide). (See Drug Interactions (7.2) for more detailed information on the pharmacokinetic interactions of valproate with other drugs.)
Metabolism
Valproate is metabolized almost entirely by the liver. In adult patients on monotherapy, 30-50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial β-oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine.
The relationship between dose and total valproate concentration is nonlinear; concentration does not increase proportionally with the dose, but rather, increases to a lesser extent due to saturable plasma protein binding. The kinetics of unbound drug are linear.
Elimination
Mean plasma clearance and volume of distribution for total valproate are 0.56 L/hr/1.73 m2 and 11 L/1.73 m2, respectively. Mean plasma clearance and volume of distribution for free valproate are 4.6 L/hr/1.73 m2 and 92 L/1.73 m2. Mean terminal half-life for valproate monotherapy ranged from 9 to 16 hours following oral dosing regimens of 250 to 1000 mg.
The estimates cited apply primarily to patients who are not taking drugs that affect hepatic metabolizing enzyme systems. For example, patients taking enzyme-inducing antiepileptic drugs (carbamazepine, phenytoin, and phenobarbital) will clear valproate more rapidly. Because of these changes in valproate clearance, monitoring of antiepileptic concentrations should be intensified whenever concomitant antiepileptics are introduced or withdrawn.
Special Populations
Effect of Age
Neonates
Children within the first two months of life have a markedly decreased ability to eliminate valproate compared to older children and adults. This is a result of reduced clearance (perhaps due to delay in development of glucuronosyltransferase and other enzyme systems involved in valproate elimination) as well as increased volume of distribution (in part due to decreased plasma protein binding). For example, in one study, the half-life in children under 10 days ranged from 10 to 67 hours compared to a range of 7 to 13 hours in children greater than 2 months.
Children
Pediatric patients (i.e., between 3 months and 10 years) have 50% higher clearances expressed on weight (i.e., mL/min/kg) than do adults. Over the age of 10 years, children have pharmacokinetic parameters that approximate those of adults.
Elderly
The capacity of elderly patients (age range: 68 to 89 years) to eliminate valproate has been shown to be reduced compared to younger adults (age range: 22 to 26). Intrinsic clearance is reduced by 39%; the free fraction is increased by 44%. Accordingly, the initial dosage should be reduced in the elderly [see Dosage and Administration (2.2)].
Effect of Sex
There are no differences in the body surface area adjusted unbound clearance between males and females (4.8 ± 0.17 and 4.7 ± 0.07 L/hr per 1.73 m2, respectively).
Effect of Disease
Liver Disease
[See Boxed Warning, Contraindications (4), and Warnings and Precautions (5.1)]. Liver disease impairs the capacity to eliminate valproate. In one study, the clearance of free valproate was decreased by 50% in 7 patients with cirrhosis and by 16% in 4 patients with acute hepatitis, compared with 6 healthy subjects. In that study, the half-life of valproate was increased from 12 to 18 hours. Liver disease is also associated with decreased albumin concentrations and larger unbound fractions (2 to 2.6 fold increase) of valproate. Accordingly, monitoring of total concentrations may be misleading since free concentrations may be substantially elevated in patients with hepatic disease whereas total concentrations may appear to be normal.
Renal Disease
A slight reduction (27%) in the unbound clearance of valproate has been reported in patients with renal failure (creatinine clearance < 10 mL/minute); however, hemodialysis typically reduces valproate concentrations by about 20%. Therefore, no dosage adjustment appears to be necessary in patients with renal failure. Protein binding in these patients is substantially reduced; thus, monitoring total concentrations may be misleading.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
Valproate was administered orally to rats and mice at doses of 80 and 170 mg/kg/day (less than the maximum recommended human dose on a mg/m2 basis) for two years. The primary findings were an increase in the incidence of subcutaneous fibrosarcomas in high-dose male rats receiving valproate and a dose-related trend for benign pulmonary adenomas in male mice receiving valproate. The significance of these findings for humans is unknown.
Mutagenesis
Valproate was not mutagenic in an in vitro bacterial assay (Ames test), did not produce dominant lethal effects in mice, and did not increase chromosome aberration frequency in an in vivo cytogenetic study in rats. Increased frequencies of sister chromatid exchange (SCE) have been reported in a study of epileptic children taking valproate, but this association was not observed in another study conducted in adults. There is some evidence that increased SCE frequencies may be associated with epilepsy. The biological significance of an increase in SCE frequency is not known.
Fertility
Chronic toxicity studies of valproate in juvenile and adult rats and dogs demonstrated reduced spermatogenesis and testicular atrophy at oral doses of 400 mg/kg/day or greater in rats (approximately equivalent to or greater than the maximum recommended human dose (MRHD) on a mg/m2 basis) and 150 mg/kg/day or greater in dogs (approximately 1.4 times the MRHD or greater on a mg/m2 basis). Fertility studies in rats have shown no effect on fertility at oral doses of valproate up to 350 mg/kg/day (approximately equal to the MRHD on a mg/m2 basis) for 60 days. The effect of valproate on testicular development and on sperm production and fertility in humans is unknown.
14 CLINICAL STUDIES
The studies described in the following section were conducted using divalproex sodium tablets.
14.1 Epilepsy
The efficacy of divalproex sodium tablets in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
In one, multi-clinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per 8 weeks during an 8 week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the "therapeutic range" were randomized to receive, in addition to their original antiepilepsy drug (AED), either divalproex sodium tablets or placebo. Randomized patients were to be followed for a total of 16 weeks. The following Table presents the findings.
| Add-on Treatment | Number of Patients | Baseline Incidence | Experimental Incidence |
|---|---|---|---|
|
|||
| Divalproex Sodium Tablets | 75 | 16.0 | 8.9* |
| Placebo | 69 | 14.5 | 11.5 |
Figure 1 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This Figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for divalproex sodium tablets than for placebo. For example, 45% of patients treated with divalproex sodium tablets had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo.
Figure 1
The second study assessed the capacity of divalproex sodium tablets to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if 1) they continued to experience 2 or more CPS per 4 weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone) and 2) they made a successful transition over a two week interval to divalproex sodium tablets. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to divalproex sodium tablets monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
The following Table presents the findings for all patients randomized who had at least one post-randomization assessment.
| Treatment | Number of Patients | Baseline Incidence | Randomized Phase Incidence |
|---|---|---|---|
|
|||
| High dose | 131 | 13.2 | 10.7* |
| Divalproex Sodium Tablets | |||
| Low dose | 134 | 14.2 | 13.8 |
| Divalproex Sodium Tablets | |||
Figure 2 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This Figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose divalproex sodium tablets than for low dose divalproex sodium tablets. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose divalproex sodium tablets monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose divalproex sodium tablets.
Figure 2
15 REFERENCES
- Meador KJ, Baker GA, Browning N, et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurology 2013; 12 (3):244-252.
16 HOW SUPPLIED/STORAGE AND HANDLING
Valproic Acid Capsules, USP 250 mg: Each off-white colored soft gelatin capsule, imprinted with VALPROIC 250, contains Valproic Acid, USP packaged in bottles containing 100 (NDC 0832-1008-00).
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Medication Guide
17.1 Hepatotoxicity
Warn patients and guardians that nausea, vomiting, abdominal pain, anorexia, diarrhea, asthenia, and/or jaundice can be symptoms of hepatotoxicity and, therefore, require further medical evaluation promptly [see Warnings and Precautions (5.1)].
17.2 Pancreatitis
Warn patients and guardians that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis and, therefore, require further medical evaluation promptly [see Warnings and Precautions (5.5)].
17.3 Birth Defects and Decreased IQ
Inform pregnant women and women of childbearing potential that use of valproate during pregnancy increases the risk of birth defects and decreased IQ in children who were exposed. Advise women to use effective contraception while using valproate. When appropriate, counsel these patients about alternative therapeutic options. This is particularly important when valproate use is considered for a condition not usually associated with permanent injury or death. Advise patients to read the Medication Guide, which appears as the last section of the labeling [see Warnings and Precautions (5.2, 5.3, 5.4) and Use in Specific Populations (8.1)].
Advise women of childbearing potential to discuss pregnancy planning with their doctor and to contact their doctor immediately if they think they are pregnant.
Encourage patients to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll free number 1-888-233-2334 [see Use in Specific Populations (8.1)].
17.4 Suicidal Thinking and Behavior
Counsel patients, their caregivers, and families that AEDs, including valproic acid capsules, may increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Instruct patients, caregivers, and families to report behaviors of concern immediately to the healthcare providers [see Warnings and Precautions (5.8)].
17.5 Hyperammonemia
Inform patients of the signs and symptoms associated with hyperammonemic encephalopathy and be told to inform the prescriber if any of these symptoms occur [see Warnings and Precautions (5.10, 5.11)].
17.6 CNS Depression
Since valproate products may produce CNS depression, especially when combined with another CNS depressant (e.g., alcohol), advise patients not to engage in hazardous activities, such as driving an automobile or operating dangerous machinery, until it is known that they do not become drowsy from the drug.
17.7 Multi-Organ Hypersensitivity Reactions
Instruct patients that a fever associated with other organ system involvement (rash, lymphadenopathy, etc.) may be drug-related and should be reported to the physician immediately [see Warnings and Precautions (5.13)].
Manufactured for
UPSHER-SMITH LABORATORIES, INC.
Maple Grove, MN 55369
Manufactured by
Catalent Pharma Solutions
St. Petersburg, FL 33716-1016
Revised 1013
MEDICATION GUIDE
VALPROIC ACID (val•pro•ic acid) CAPSULES, USP
Read this Medication Guide before you start taking valproic acid capsules and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is the most important information I should know about valproic acid capsules?
Do not stop taking valproic acid capsules without first talking to your healthcare provider.
Stopping valproic acid capsules suddenly can cause serious problems.
Valproic acid capsules can cause serious side effects, including:
- 1.
-
Serious liver damage that can cause death, especially in children younger than 2 years old.
The risk of getting this serious liver damage is more likely to happen within the first 6 months of treatment.
Call your healthcare provider right away if you get any of the following symptoms:- nausea or vomiting that does not go away
- loss of appetite
- pain on the right side of your stomach (abdomen)
- dark urine
- swelling of your face
- yellowing of your skin or the whites of your eyes
- 2.
-
Valproic acid capsules may harm your unborn baby.
- If you take valproic acid capsules during pregnancy for any medical condition, your baby is at risk for serious birth defects. The most common birth defects with valproic acid capsules affect the brain and spinal cord and are called spina bifida or neural tube defects. These defects occur in 1 to 2 out of every 100 babies born to mothers who use this medicine during pregnancy. These defects can begin in the first month, even before you know you are pregnant. Other birth defects can happen.
- Birth defects may occur even in children born to women who are not taking any medicines and do not have other risk factors.
- Taking folic acid supplements before getting pregnant and during early pregnancy can lower the chance of having a baby with a neural tube defect.
If you take valproic acid capsules during pregnancy for any medical condition, your child is at risk for having a lower IQ. - There may be other medicines to treat your condition that have a lower chance of causing birth defects and decreased IQ in your child.
- Women who are pregnant must not take valproic acid capsules to prevent migraine headaches.
- All women of child-bearing age should talk to their healthcare provider about using other possible treatments instead of valproic acid capsules. If the decision is made to use valproic acid capsules, you should use effective birth control (contraception).
- Tell your healthcare provider right away if you become pregnant while taking valproic acid capsules. You and your healthcare provider should decide if you will continue to take valproic acid capsules while you are pregnant.
-
Pregnancy Registry: If you become pregnant while taking valproic acid capsules, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy.
- 3.
-
Inflammation of your pancreas that can cause death.
Call your healthcare provider right away if you have any of these symptoms:- severe stomach pain that you may also feel in your back
- nausea or vomiting that does not go away
- 4.
-
Like other antiepileptic drugs, valproic acid capsules may cause suicidal thoughts or actions in a very small number of people, about 1 in 500.
Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
How can I watch for early symptoms of suicidal thoughts and actions?
-
- Pay attention to any changes, especially sudden changes in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
Do not stop valproic acid capsules without first talking to a healthcare provider. Stopping valproic acid capsules suddenly can cause serious problems. Stopping a seizure medicine suddenly in a patient who has epilepsy can cause seizures that do not stop (status epilepticus).
Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
What are valproic acid capsules?
Valproic acid capsules are prescription medicines used alone or with other medicines, to treat:
- complex partial seizures in adults and children 10 years of age and older
- simple and complex absence seizures, with or without other seizure types
Who should not take valproic acid capsules?
Do not take valproic acid capsules if you:
- have liver problems
- have or think you have a genetic liver problem caused by a mitochondrial disorder (e.g. Alpers-Huttenlocher syndrome)
- are allergic to divalproex sodium, valproic acid, sodium valproate, or any of the ingredients in valproic acid capsules. See the end of this leaflet for a complete list of ingredients in valproic acid capsules.
- have a genetic problem called urea cycle disorder
- are pregnant for the prevention of migraine headaches
What should I tell my healthcare provider before taking valproic acid capsules?
Before you take valproic acid capsules, tell your healthcare provider if you:
- have a genetic liver problem caused by a mitochondrial disorder (e.g. Alpers-Huttenlocher syndrome)
- drink alcohol
- are pregnant or breastfeeding. Valproic acid capsules can pass into breast milk. Talk to your healthcare provider about the best way to feed your baby if you take valproic acid capsules.
- have or have had depression, mood problems, or suicidal thoughts or behavior
- have any other medical conditions
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, herbal supplements and medicines that you take for a short period of time.
Taking valproic acid capsules with certain other medicines can cause side effects or affect how well they work. Do not start or stop other medicines without talking to your healthcare provider.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist each time you get a new medicine.
How should I take valproic acid capsules?
- Take valproic acid capsules exactly as your healthcare provider tells you. Your healthcare provider will tell you how much valproic acid capsules to take and when to take it.
- Your healthcare provider may change your dose.
- Do not change your dose of valproic acid capsules without talking to your healthcare provider.
- Do not stop taking valproic acid capsules without first talking to your healthcare provider. Stopping valproic acid capsules suddenly can cause serious problems.
- Swallow valproic acid capsules whole. Do not crush or chew valproic acid capsules. Tell your healthcare provider if you can not swallow valproic acid capsules whole. You may need a different medicine.
- If you take too much valproic acid capsules, call your healthcare provider or local Poison Control Center right away.
What should I avoid while taking valproic acid capsules?
- Valproic acid capsules can cause drowsiness and dizziness. Do not drink alcohol or take other medicines that make you sleepy or dizzy while taking valproic acid capsules, until you talk with your doctor. Taking valproic acid capsules with alcohol or drugs that cause sleepiness or dizziness may make your sleepiness or dizziness worse.
- Do not drive a car or operate dangerous machinery until you know how valproic acid capsules affect you. Valproic acid capsules can slow your thinking and motor skills.
What are the possible side effects of valproic acid capsules?
- See "What is the most important information I should know about valproic acid capsules?"
Valproic acid capsules may cause other serious side effects including: - Low blood count: red or purple spots on your skin, bruising, bleeding from your mouth, teeth or nose.
- High ammonia levels in your blood: feeling tired, vomiting, changes in mental status.
- Low body temperature (hypothermia): drop in your body temperature to less than 95°F, feeling tired, confusion, coma.
- Allergic (hypersensitivity) reactions: fever, skin rash, hives, sores in your mouth, skin blistering and peeling of your skin, swelling of your lymph nodes, swelling of your face, eyes, lips, tongue, or throat, trouble swallowing or breathing.
- Drowsiness or sleepiness in the elderly. This extreme drowsiness may cause you to eat or drink less than you normally would. Tell your doctor if you are not able to eat or drink as you normally do. Your doctor may start you at a lower dose of valproic acid capsules.
Call your healthcare provider right away, if you have any of the symptoms listed above.
The common side effects of valproic acid capsules include:
- nausea
- headache
- sleepiness
- vomiting
- weakness
- tremor
- dizziness
- stomach pain
- blurry vision
- double vision
- diarrhea
- increased appetite
- weight gain
- hair loss
- loss of appetite
- problems with walking or coordination
These are not all of the possible side effects of valproic acid capsules. For more information, ask your healthcare provider or pharmacist.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store valproic acid capsules?
- Store valproic acid capsules between 59°F to 86°F (15°C to 30°C).
Keep valproic acid capsules and all medicines out of the reach of children.
General information about the safe and effective use of valproic acid capsules
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
Do not use valproic acid capsules for a condition for which it was not prescribed. Do not give valproic acid capsules to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about valproic acid capsules. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about valproic acid capsules that is written for health professionals.
For more information, go to www.upsher-smith.com or call 1-888-350-3789.
What are the ingredients in valproic acid capsules?
Valproic Acid Capsules, USP:
Active ingredient: valproic acid
Inactive ingredients: peanut oil, gelatin, glycerin, and titanium dioxide
This Medication Guide has been approved by the U.S. Food and Drug Administration.

| VALPROIC ACID
valproic acid capsule |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Upsher-Smith Laboratories, Inc (809088862) |