FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
GATTEX® is indicated for the treatment of adults and pediatric patients 1 year of age and older with Short Bowel Syndrome (SBS) who are dependent on parenteral support.
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
GATTEX is for adult self-administration or caregiver administration. Self-administration in pediatric patients has not been tested.
Use of the GATTEX 5 mg kit is not recommended in pediatric patients weighing less than 10 kg.
Within 6 months prior to starting treatment with GATTEX:
- Adults: Perform a colonoscopy of the entire colon with removal of polyps [see Warnings and Precautions (5.1)].
- Pediatric patients: Perform fecal occult blood testing; if there is unexplained blood in the stool, perform colonoscopy/sigmoidoscopy [see Warnings and Precautions (5.1)].
- Obtain baseline laboratory assessments (bilirubin, alkaline phosphatase, lipase and amylase) [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage and Administration for Adults and Pediatric Patients 1 Year of Age and Older
GATTEX is for subcutaneous injection only. Not for intravenous or intramuscular administration.
The recommended dosage of GATTEX is 0.05 mg/kg once daily administered by subcutaneous injection.
If a dose is missed, that dose should be taken as soon as possible on that day. Do not take 2 doses on the same day.
Alternation of sites for subcutaneous injection is recommended, and can include the thighs, upper arms, and the abdomen.
2.3 Dosage Adjustment for Renal Impairment
The recommended dosage in adult and pediatric patients with moderate and severe renal impairment and end-stage renal disease (estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2) is 0.025 mg/kg once daily [see Use in Specific Populations (8.6)].
2.4 Monitoring to Assess Safety
Colonoscopy Schedule in Adults
A follow-up colonoscopy (or alternate imaging) is recommended at the end of 1 year of GATTEX. If no polyp is found, subsequent colonoscopies should be done no less frequently than every 5 years. If a polyp is found, adherence to current polyp follow-up guidelines is recommended [see Warnings and Precautions (5.1)].
Colonoscopy Schedule in Pediatric Patients
Perform subsequent fecal occult blood testing annually in children and adolescents while they are receiving GATTEX.
Colonoscopy/sigmoidoscopy is recommended for all children and adolescents after 1 year of treatment, every 5 years thereafter while on continuous treatment with GATTEX, and if they have new or unexplained gastrointestinal bleeding [see Warnings and Precautions (5.1)].
Laboratory Testing
Laboratory assessments are recommended every 6 months. If any clinically meaningful elevation is seen, further diagnostic workup is recommended as clinically indicated (i.e., imaging of the biliary tract, liver, or pancreas) [see Warnings and Precautions (5.1), (5.3)].
2.5 Discontinuation of Treatment
Discontinuation of treatment with GATTEX may result in fluid and electrolyte imbalance. Monitor fluid and electrolyte status in patients who discontinue GATTEX treatment [see Warnings and Precautions (5.4)].
2.6 Preparation Instructions
- Reconstitute each vial of GATTEX by slowly injecting the 0.5 mL of preservative-free Sterile Water for Injection provided in the prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution.
- Allow the vial containing GATTEX and water to stand for approximately 30 seconds and then gently roll the vial between the palms for about 15 seconds. Do not shake the vial.
- Allow the mixed contents to stand for about 2 minutes. Inspect the vial for any undissolved powder. If undissolved powder is observed, gently roll the vial again until all material is dissolved. Do not shake the vial.
- Reconstituted GATTEX is a sterile, clear, colorless to light straw-colored solution, which should be free from particulates. If there is any discoloration or particulates, discard the solution.
- A maximum of 0.38 mL of the reconstituted solution, containing 3.8 mg of teduglutide, can be withdrawn from the vial for dosing.
- If the product remains undissolved after the second attempt, do not use.
3 DOSAGE FORMS AND STRENGTHS
For Injection: 5 mg teduglutide as a white lyophilized powder for reconstitution in a single-dose vial supplied with 0.5 mL Sterile Water for Injection in a single-dose prefilled syringe.
5 WARNINGS AND PRECAUTIONS
5.1 Acceleration of Neoplastic Growth
Based on the pharmacologic activity and findings in animals, GATTEX has the potential to cause hyperplastic changes including neoplasia [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. In patients at increased risk for malignancy, the clinical decision to use GATTEX should be considered only if the benefits outweigh the risks. In patients who develop active gastrointestinal malignancy (GI tract, hepatobiliary, pancreatic) while on GATTEX, discontinue GATTEX treatment. In patients who develop active non-gastrointestinal malignancy while on GATTEX, the clinical decision to continue GATTEX should be made based on benefit-risk considerations.
Colorectal Polyps
Colorectal polyps were identified during the clinical studies [see Adverse Reactions (6.1)]. In adults, within 6 months prior to starting treatment with GATTEX, perform colonoscopy of the entire colon with removal of polyps. A follow-up colonoscopy (or alternate imaging) is recommended at the end of 1 year of GATTEX. Perform subsequent colonoscopies every 5 years or more often as needed. If a polyp is found, adherence to current polyp follow-up guidelines is recommended. If colorectal cancer is diagnosed, discontinue GATTEX therapy.
In children and adolescents, perform fecal occult blood testing prior to initiating treatment with GATTEX. Colonoscopy/sigmoidoscopy is required if there is unexplained blood in the stool. Perform subsequent fecal occult blood testing annually in children and adolescents while they are receiving GATTEX. Colonoscopy/sigmoidoscopy is recommended for all children and adolescents after 1 year of treatment, every 5 years thereafter while on continuous treatment with GATTEX, and if they have new or unexplained gastrointestinal bleeding [see Dosage and Administration (2.1)].
Small Bowel Neoplasia
Based on tumor findings in the rat and mouse carcinogenicity studies, monitor patients clinically for small bowel neoplasia [see Nonclinical Toxicology (13.1)]. If a benign neoplasm is found, it should be removed. In case of small bowel cancer, discontinue GATTEX therapy.
5.2 Intestinal Obstruction
Intestinal obstruction has been reported in clinical studies [see Adverse Reactions (6.1)] and postmarketing. In patients who develop intestinal or stomal obstruction, temporarily discontinue GATTEX while the patient is clinically managed. GATTEX may be restarted when the obstructive presentation resolves, if clinically indicated.
5.3 Biliary and Pancreatic Disease
Gallbladder and Biliary Tract Disease
Cholecystitis, cholangitis, and cholelithiasis have been reported in clinical studies [see Adverse Reactions (6.1)] and postmarketing. For identification of the onset or worsening of gallbladder/biliary disease, obtain laboratory assessment of bilirubin and alkaline phosphatase within 6 months prior to starting GATTEX, and at least every 6 months while on GATTEX; or more frequently if needed. If clinically meaningful changes are seen, further evaluation including imaging of the gallbladder and/or biliary tract is recommended; and reassess the need for continued GATTEX treatment.
Pancreatic Disease
Pancreatitis has been reported in clinical studies [see Adverse Reactions (6.1)]. For identification of onset or worsening of pancreatic disease, obtain laboratory assessments of lipase and amylase within 6 months prior to starting GATTEX, and at least every 6 months while on GATTEX; or more frequently if needed. If clinically meaningful changes are seen, further evaluation such as imaging of the pancreas is recommended; and reassess the need for continued GATTEX treatment.
5.4 Fluid Imbalance and Fluid Overload
Fluid Overload
Fluid overload and congestive heart failure have been observed in clinical studies, which were deemed to be related to enhanced fluid absorption associated with GATTEX [see Adverse Reactions (6.1)]. If fluid overload occurs, adjust parenteral support and reassess GATTEX treatment, especially in patients with underlying cardiovascular disease. If significant cardiac deterioration develops while on GATTEX, reassess the need for continued GATTEX treatment.
Fluid and Electrolyte Imbalance
Discontinuation of treatment with GATTEX may also result in fluid and electrolyte imbalance. Monitor fluid and electrolyte status in patients who discontinue treatment with GATTEX [see Dosage and Administration (2.5)].
5.5 Increased Absorption of Concomitant Oral Medication
In the adult placebo-controlled studies, an analysis of episodes of cognition and attention disturbances was performed for patients on benzodiazepines. One patient receiving prazepam concomitantly with GATTEX 0.05 mg/kg once daily experienced a dramatic deterioration in mental status progressing to coma during the first week of GATTEX therapy. The patient was admitted to the ICU and the prazepam blood concentration was >300 mcg/L. GATTEX and prazepam were discontinued, and coma resolved 5 days later.
Monitor patients receiving concomitant oral drugs requiring titration or with a narrow therapeutic index, for adverse reactions due to potential increased absorption of the concomitant drug. The concomitant drug may require a reduction in dosage [see Drug Interactions (7.1)].
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Acceleration of Neoplastic Growth [see Warnings and Precautions (5.1)]
- Intestinal Obstruction [see Warnings and Precautions (5.2)]
- Biliary and Pancreatic Disease [see Warnings and Precautions (5.3)]
- Fluid Imbalance and Fluid Overload [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Adults
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
The rates of adverse reactions in 136 adult patients with SBS participating in two randomized, placebo-controlled, 24-week, double-blind clinical studies (Study 1 and Study 3) are summarized in Table 1. Only those reactions with a rate of at least 5% in the GATTEX group, and greater than placebo group, are summarized in Table 1.
| Adverse Reaction | Placebo (N=59) (%) | GATTEX 0.05 mg/kg Once Daily (N=77) (%) |
|---|---|---|
|
||
| Abdominal pain† | 22 | 30 |
| Nausea | 20 | 23 |
| Upper respiratory tract infection‡ | 12 | 21 |
| Abdominal distension | 2 | 20 |
| Injection site reaction§ | 12 | 13 |
| Vomiting | 10 | 12 |
| Fluid Overload¶ | 7 | 12 |
| Hypersensitivity# | 7 | 10 |
| Flatulence | 7 | 9 |
| Decreased appetite | 3 | 7 |
| InfluenzaÞ | 2 | 7 |
| Skin hemorrhageß | 2 | 5 |
| Cough | 0 | 5 |
| Sleep disturbancesà | 0 | 5 |
Adverse Reactions in the Subset of Patients with a Stoma
Among the 53 patients with a stoma in the placebo-controlled studies (Study 1 and Study 3), the percentage of patients with gastrointestinal stoma complication was 42% (13/31) for patients receiving GATTEX 0.05 mg/kg/day and 14% (3/22) for patients receiving placebo.
Pediatric Patients 1 Year to Less Than 17 Years of Age
In two clinical studies of 24-week and 12-week duration, 41 pediatric patients aged 1 year to less than 17 years were treated with GATTEX 0.05 mg/kg/day [see Use in Specific Populations (8.4), Clinical Studies (14.2)]. Overall, the safety profile of GATTEX was similar to that in adults. In the long-term extension studies with mean exposure of 41 weeks, no new safety signals were identified.
Less Common Adverse Reactions
Adverse Reactions of Special Interest
Malignancy
Three patients were diagnosed with malignancy in the SBS clinical studies in adults, all of whom were male and had received GATTEX 0.05 mg/kg/day in Study 2. One patient had a history of abdominal radiation for Hodgkin's disease two decades prior to receiving GATTEX and prior liver lesion on CT scan, and was diagnosed with metastatic adenocarcinoma of unconfirmed origin after 11 months of exposure to GATTEX. Two patients had extensive smoking histories and were diagnosed with lung cancers (squamous and non-small cell) after 12 months and 3 months of GATTEX exposure, respectively [see Warnings and Precautions (5.1)].
Intestinal Polyps
In the adult clinical studies, 14 patients with SBS were diagnosed with polyps of the GI tract after initiation of study treatment. In the SBS placebo-controlled studies, 1/59 (2%) of patients on placebo and 1/109 (1%) of patients on GATTEX 0.05 mg/kg/day were diagnosed with intestinal polyps (inflammatory stomal and hyperplastic sigmoidal after 3 and 5 months, respectively). The remaining 12 polyp cases occurred in the extension studies – 2 colorectal villous adenomas (onset at 6 and 7 months in GATTEX 0.1 mg/kg/day (twice the recommended dose) and 0.05 mg/kg/day dose groups, respectively), 2 hyperplastic polyps (onset 6 months in GATTEX 0.1 mg/kg/day dose group and 24 months in GATTEX 0.05 mg/kg/day dose group), 4 colorectal tubular adenomas (onset between 24 and 29 months in GATTEX 0.05 mg/kg/day dose group), 1 serrated adenoma (onset at 24 months in GATTEX 0.05 mg/kg/day dose group), 1 colorectal polyp biopsy not done (onset at 24 months in GATTEX 0.05 mg/kg/day dose group), 1 rectal inflammatory polyp (onset at 10 months in the GATTEX 0.05 mg/kg/day dose group, and 1 small duodenal polyp (onset at 3 months in GATTEX 0.05 mg/kg/day dose group) [see Warnings and Precautions (5.1)].
In the pediatric clinical studies (up to 69 weeks of exposure), there was one case of cecal polyp that was not biopsied and was not seen on repeat colonoscopy.
Gastrointestinal Obstruction
Overall, in the adult clinical studies, 12 patients with SBS experienced one or more episodes of intestinal obstruction/stenosis: 6 in SBS placebo-controlled studies and 6 in the extension studies. The 6 patients in the placebo-controlled studies were all on GATTEX: 3/77 (4%) on GATTEX 0.05 mg/kg/day and 3/32 (9%) on GATTEX 0.1 mg/kg/day (twice the recommended dose). No cases of intestinal obstruction occurred in the placebo group. Onset ranged from 1 day to 6 months. In the adult extension studies, 6 additional patients (all on GATTEX 0.05 mg/kg/day) were diagnosed with intestinal obstruction/stenosis with onsets ranging from 6 days to 19 months. Two of the 6 patients from the placebo-controlled studies experienced recurrence of obstruction in the extension studies. Of all 8 patients with an episode of intestinal obstruction/stenosis in these extension studies, 2 patients required endoscopic dilation and 1 required surgical intervention) [see Warnings and Precautions (5.2)].
In the pediatric clinical studies (up to 69 weeks of exposure), there was 1 serious adverse reaction of obstruction. Teduglutide was temporarily withheld, obstruction resolved without additional intervention, and there was no recurrence once teduglutide was restarted.
Gallbladder, Biliary and Pancreatic Disease
For gallbladder and biliary disease in the adult placebo-controlled studies, 3 patients with SBS were diagnosed with cholecystitis, all of whom had a prior history of gallbladder disease and were in the GATTEX 0.05 mg/kg/day dose group. No cases were reported in the placebo group. One of these 3 cases had gallbladder perforation and underwent cholecystectomy the next day. The remaining 2 cases underwent elective cholecystectomy at a later date. In the adult extension studies, 4 patients had an episode of acute cholecystitis; 3 patients had new-onset cholelithiasis; and 1 patient experienced cholestasis secondary to an obstructed biliary stent. For pancreatic disease in the adult placebo-controlled studies, 1 patient (GATTEX 0.05 mg/kg/day dose group) had a pancreatic pseudocyst diagnosed after 4 months of GATTEX. In the adult extension studies, 1 patient was diagnosed with chronic pancreatitis; and 1 patient was diagnosed with acute pancreatitis) [see Warnings and Precautions (5.3)].
Fluid Overload
In the adult placebo-controlled studies, peripheral edema was reported in 2/59 (3%) of patients on placebo and 8/77 (10%) patients on GATTEX; fluid overload was reported in 1/77 (1%) patient in the GATTEX group; no cases of fluid overload were seen in the placebo arm. There were 2 cases of congestive heart failure (CHF, 3%) in the GATTEX arm, 1 of which was reported as a serious adverse event and the other as non-serious. The serious case had onset at 6 months and was possibly associated with previously undiagnosed hypothyroidism and/or cardiac dysfunction [see Warnings and Precautions (5.4)].
6.2 Immunogenicity
As with all peptides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to teduglutide in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Adults
Based on integrated data from two studies in adults with SBS (a 6-month randomized placebo-controlled study, followed by a 24-month open-label study), the development of anti-teduglutide antibodies in patients who received subcutaneous administration of 0.05 mg/kg GATTEX once daily was 3% (2/60) at Month 3, 17% (13/77) at Month 6, 24% (16/67) at Month 12, 33% (11/33) at Month 24, and 48% (14/29) at Month 30. Anti-teduglutide antibodies were cross-reactive to native glucagon-like peptide (GLP-2) in 5 of the 6 patients (83%) who had anti-teduglutide antibodies and were tested for cross-reactivity. In the same two studies, a total of 36 patients were tested for neutralizing antibodies: one patient developed borderline positive neutralizing antibody responses at month 24 of the extension study. The antibody formation has not been associated with clinically relevant safety findings, reduced efficacy or changed pharmacokinetics of GATTEX.
Pediatric Patients 1 Year to Less than 17 Years of Age
In pediatric patients who received subcutaneous administration of 0.05 mg/kg GATTEX once daily for 24 weeks, the rate of anti-teduglutide antibody formation at Month 6 was 19% (5/26) and was similar to the rate of antibody formation in adult patients (17%). Of the 5 pediatric patients who had developed antibodies to teduglutide at Month 6, 2 patients had neutralizing antibodies. However, with the longer duration of treatment, the rate of anti-teduglutide formation at Month 12 was higher in pediatric patients with 54% (14/26), compared to that of adults (24%). Of the 14 pediatric patients who had developed antibodies to teduglutide at Month 12, 1 patient had neutralizing antibodies.
Among the small number of pediatric patients who developed anti-teduglutide antibodies, no association with adverse events or lack of efficacy was observed.
7 DRUG INTERACTIONS
7.1 Potential for Increased Absorption of Oral Medications
Based upon the pharmacodynamic effect of GATTEX, there is a potential for increased absorption of concomitant oral medications. Altered mental status has been observed in patients taking GATTEX and benzodiazepines in the adult clinical studies [see Warnings and Precautions (5.5)].
Monitor patients on concomitant oral drugs requiring titration or with a narrow therapeutic index for adverse reactions related to the concomitant drug while on GATTEX. The concomitant drug may require a reduction in dosage.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports with GATTEX use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Pregnant women with short bowel syndrome are at risk for malnutrition, which is associated with adverse maternal and fetal outcomes (see Clinical Considerations). In animal reproduction studies, no effects on embryo-fetal development were observed with the subcutaneous administration of teduglutide to pregnant rats and rabbits during organogenesis at exposures up to 686 and 657 times, respectively, the clinical exposure at the recommended human dose (based on AUC) (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Reproduction studies have been performed in pregnant rats at subcutaneous doses of teduglutide up to 25 mg/kg twice daily (50 mg/kg/day) (about 686 times the clinical exposure (AUC) at the recommended daily human dose of 0.05 mg/kg) and in pregnant rabbits at subcutaneous doses up to 25 mg/kg twice daily (50 mg/kg/day) (about 657 times the clinical exposure (AUC) at the recommended daily human dose of 0.05 mg/kg) during the period of organogenesis. These studies did not reveal any evidence of impaired fertility or harm to the fetus due to teduglutide. In a pre- and postnatal development study in rats (gestation day 7 to lactation day 20), teduglutide did not show any significant adverse effects on pre- and postnatal development at doses up to 25 mg/kg twice daily (50 mg/kg/day) (about 343 times the clinical exposure (AUC) at the recommended daily human dose of 0.05 mg/kg).
8.2 Lactation
Risk Summary
There is no information regarding the presence of GATTEX in human milk, the effects of GATTEX on the breastfed infant, or the effects of GATTEX on milk production. Teduglutide is present in the milk of lactating rats (see Data). Systemic exposure of teduglutide to a breastfed infant is expected to be low. However, because of the potential for serious adverse reactions in a breastfed infant, including tumorigenicity [see Nonclinical Toxicology (13.1)], advise patients that breastfeeding is not recommended during treatment with GATTEX.
Data
In a milk excretion study in the rat, a single subcutaneous dose of 25 mg/kg of teduglutide (81 times the recommended daily human dose of 0.05 mg/kg based on body surface area) was administered to lactating female rats at Day 12 postpartum. The maximum concentration of teduglutide in the milk corresponded to 0.9% and 2.9% of the plasma concentration at 1.5 and 4 hours after dosing, respectively.
8.4 Pediatric Use
The safety and effectiveness in pediatric patients less than 1 year of age have not been established.
The safety and effectiveness of GATTEX have been established in pediatric patients 1 year to less than 17 years of age who are dependent on parenteral support for the treatment of SBS. Use of GATTEX in this population is supported by evidence from adequate and well-controlled studies in adults, with additional efficacy, safety, pharmacokinetic and pharmacodynamic data in pediatric patients 1 year to less than 17 years of age [see Dosage and Administration (2), Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2)]. These data were derived from two studies of 24-week (Study 5) and 12-week (NCT01952080) duration in which 41 pediatric patients were treated with GATTEX in the following groups: 1 infant (1 year to less than 2 years), 37 children (2 years to less than 12 years) and 3 adolescents (12 years to less than 17 years).
In these 2 studies and the corresponding extension studies (Study 6 and NCT02949362), 29 pediatric patients were administered GATTEX prospectively for up to 94 weeks [see Clinical Studies (14.2)]. Adverse reactions in pediatric patients were similar to those seen in adults [see Adverse Reactions (6.1)].
Juvenile Animal Toxicity Data
In a juvenile toxicity study, teduglutide was administered to juvenile minipigs at subcutaneous doses of 0.5, 2.5 and 12.5 mg/kg twice daily (1, 5, and 25 mg/kg/day) from post-natal day 7 and continuing for 90 days). Exposures (AUC) at these doses were at least 12-, 25-, and 170-fold the pediatric clinical exposure for ages 1 year to 11 years at 0.05 mg/kg, respectively, and 10-, 21-, and 141-fold the pediatric clinical exposure for ages 12 years to 17 years at 0.05 mg/kg, respectively.
In juvenile minipigs, subcutaneous teduglutide caused intestinotrophic effects, gall bladder mucosal hyperplasia, bile duct mucosal hyperplasia, and injection site reactions, similar to those observed in adult animals.
8.5 Geriatric Use
Of the 134 patients with SBS that were treated with GATTEX at the recommended dosage of 0.05 mg/kg/day in the clinical studies, 19 patients were 65 years or older while 5 patients were 75 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
In adult subjects with moderate to severe renal impairment or end-stage renal disease (ESRD) (creatinine clearance <60 mL/min), the exposure to teduglutide increased with the degree of renal impairment [see Clinical Pharmacology (12.3)]. Reduce the dosage of GATTEX by half in both pediatric and adult patients with eGFR less than 60 mL/min/1.73 m2 [see Dosage and Administration (2.3)].
8.7 Hepatic Impairment
GATTEX has not been studied in patients with severe hepatic impairment (Child-Pugh grade C). No dosage adjustment is recommended for patients with mild and moderate hepatic impairment (Child-Pugh grade A and B) [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
The maximum dose of GATTEX studied during clinical development was 80 mg/day for 8 days. No unexpected systemic adverse reactions were seen. In the event of overdose, the patient should be carefully monitored by the medical professional.
11 DESCRIPTION
The active ingredient in GATTEX (teduglutide) for injection is teduglutide, which is a 33 amino acid glucagon-like peptide-2 (GLP-2) analog manufactured using a strain of Escherichia coli modified by recombinant DNA technology. The chemical composition of teduglutide is L-histidyl-L-glycyl-L-aspartyl-L-glycyl-L-seryl-L-phenylalanyl-L-seryl-L-aspartyl-L-glutamyl-L-methionyl-L-asparaginyl-L-threonyl-L-isoleucyl-L-leucyl-L-aspartyl-L-asparaginyl-L-leucyl-L-alanyl-L-alanyl-L-arginyl-L-aspartyl-L-phenylalanyl-L-isoleucyl-L-asparaginyl-L-tryptophanyl-L-leucyl-L-isoleucyl-L-glutaminyl-L-threonyl-L-lysyl-L-isoleucyl-L-threonyl-L-aspartic acid. The structural formula is:
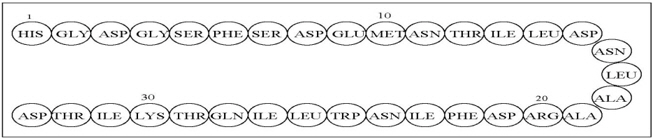
Figure 1: Structural formula of teduglutide
Teduglutide has a molecular weight of 3752 Daltons. Teduglutide drug substance is a clear, colorless to light-straw–colored liquid.
Each single-dose vial of GATTEX contains 5 mg of teduglutide as a white lyophilized powder for reconstitution and administration by subcutaneous injection. In addition to the active pharmaceutical ingredient (teduglutide), each vial of GATTEX contains 3.434 mg dibasic sodium phosphate heptahydrate, 3.88 mg L-histidine, 15 mg mannitol, and 0.644 mg monobasic sodium phosphate monohydrate as excipients. No preservatives are present.
At the time of administration, the lyophilized powder is reconstituted with 0.5 mL of Sterile Water for Injection, which is provided in a single-dose prefilled syringe. A 10 mg/mL sterile solution is obtained after reconstitution. Up to 0.38 mL of the reconstituted solution which contains 3.8 mg of teduglutide can be withdrawn for subcutaneous injection upon reconstitution.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Teduglutide is an analog of naturally occurring human glucagon-like peptide-2 (GLP-2), a peptide secreted by L-cells of the distal intestine. GLP-2 is known to increase intestinal and portal blood flow and inhibit gastric acid secretion. Teduglutide binds to the glucagon-like peptide-2 receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial myofibroblasts and enteric neurons of the submucosal and myenteric plexus. Activation of these receptors results in the local release of multiple mediators including insulin-like growth factor (IGF)-1, nitric oxide and keratinocyte growth factor (KGF).
12.2 Pharmacodynamics
Intestinal Fluid Absorption
The ability of GATTEX to improve intestinal absorption was studied in 17 adult subjects with Short Bowel Syndrome (N=2-3 per dose group) using daily doses of 0.03, 0.1, 0.15 mg/kg (doses ranging from 0.6 to 3 times the recommended dose) in a 21-day, open-label, multi-center, dose-ranging study. All subcutaneous (abdomen) doses studied, except 0.03 mg/kg once daily, resulted in enhanced gastrointestinal fluid (wet weight) absorption of approximately 750 to 1000 mL/day, and increased villus height and crypt depth of the intestinal mucosa.
12.3 Pharmacokinetics
Absorption
In healthy subjects, GATTEX administered subcutaneously had an absolute bioavailability of 88% and reached maximum plasma teduglutide concentrations at 3 to 5 hours after administration. Following a 0.05 mg/kg subcutaneous dose in SBS subjects, the median peak teduglutide concentration (Cmax) was 36 ng/mL and the median area under the curve at steady state (AUCtau) was 0.15 µg∙hr/mL. No accumulation of teduglutide was observed following repeated subcutaneous administrations.
The Cmax and AUC of teduglutide was dose proportional over the dose range of 0.05 to 0.4 mg/kg (up to 8 times the recommended dose of GATTEX).
Distribution
In healthy subjects, teduglutide has a volume of distribution (103 mL/kg) similar to blood volume.
Elimination
Metabolism
The metabolic pathway of teduglutide was not investigated in humans. However, teduglutide is expected to be degraded into small peptides and amino acids via catabolic pathways, similar to the catabolism of endogenous GLP-2.
Excretion
In healthy subjects, teduglutide plasma clearance was approximately 123 mL/hr/kg which is similar to the GFR suggesting that teduglutide is primarily cleared by the kidney. Teduglutide has a mean terminal half-life (t1/2) of approximately 2 hours in healthy subjects and 1.3 hours in SBS subjects.
Use in Specific Populations
Geriatric Patients
No differences were observed between healthy subjects younger than 65 years and those older than 65 years. Experience in subjects 75 years and above is limited.
Pediatric Patients
Following subcutaneous administration, similar steady state Cmax of teduglutide across age groups was demonstrated by population pharmacokinetics modeling (see Table 2). However, lower AUC was seen in pediatric patients 1 to 17 years of age as compared with adults and increases with increasing age.
| Age | Parameters (Mean ± SD) | |||
|---|---|---|---|---|
| Cmax, ss (ng/mL) | AUCss (ng∙h/mL) | CL/F (L/h) | t1/2 (h) | |
| 12 to 17 years (n=3) | 29.7 ± 8.4 | 154 ± 17.6 | 13.0 ± 2.3 | 1.0 ± 0.01 |
| 1 to 11 years (n=37) | 33.5 ± 11.5 | 128 ± 56.7 | 7.45 ± 2.1 | 0.7 ± 0.2 |
Patients with Renal Impairment
In adult subjects with moderate to severe renal impairment or end stage renal disease (ESRD) (creatinine clearance <60 mL/min), the Cmax and AUC0-inf of teduglutide increased with the degree of renal impairment following a single subcutaneous dose of 10 mg GATTEX. Teduglutide exposure increased by a factor of 1.6, 1.4, and 2.1 (Cmax) and 1.5, 1.7, and 2.6 (AUC0-inf) in subjects with moderate, severe renal impairment and ESRD, respectively, compared to healthy subjects [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Subjects with moderate hepatic impairment (Child-Pugh Class B) had an approximately 10 to 15% lower teduglutide Cmax and AUC compared to healthy matched control subjects after a single subcutaneous dose of 20 mg GATTEX. This reduction in teduglutide exposure is not thought to be clinically meaningful. GATTEX was not studied in subjects with severe hepatic impairment (Child-Pugh Class C).
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic potential of GATTEX was assessed in 2-year subcutaneous carcinogenicity studies in rats and mice. In a 2-year carcinogenicity study in Wistar Han rats at subcutaneous doses of 3, 10, and 35 mg/kg/day (about 15, 41, and 199 times the exposures [AUC] achieved at the recommended daily human dose of 0.05 mg/kg, respectively), teduglutide caused statistically significant increases in the incidences of adenomas in the bile duct and jejunum of male rats. In a 2-year carcinogenicity study in Crl:CD1(ICR) mice at subcutaneous doses of 1, 3.5, and 12.5 mg/kg/day (about 32, 66, and 244 times the exposures [AUC] achieved at the recommended daily human dose of 0.05 mg/kg, respectively), teduglutide caused a significant increase in papillary adenomas in the gall bladder; it also caused adenocarcinomas in the jejunum in male mice at the high dose of 12.5 mg/kg/day.
Teduglutide was negative in the Ames test, chromosomal aberration test in Chinese hamster ovary cells, and in vivo mouse micronucleus assay.
Teduglutide at subcutaneous doses up to 25 mg/kg twice daily (50 mg/kg/day or at least 202 times the clinical exposure (AUC) at the recommended daily human dose of 0.05 mg/kg) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
14 CLINICAL STUDIES
14.1 Treatment of SBS in Adults
Study 1 (Placebo-controlled) and Study 2 (Open-label Extension of Study 1)
Study 1 (CL0600-020, NCT00798967)
The efficacy, safety, and tolerability of GATTEX was evaluated in a randomized, double-blind, placebo-controlled, parallel-group, multi-national, multi-center clinical study (Study 1) in adults with SBS who were dependent on parenteral nutrition/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. For 8 weeks (or less) prior to randomization, investigators optimized the PN/I.V. volume of all patients. Optimization was followed by a 4-week to 8-week period of fluid stabilization. Patients then were randomized (1:1) to placebo (n=43) or GATTEX 0.05 mg/kg/day (n=43). Study treatment was administered subcutaneously once daily for 24 weeks. PN/I.V. volume adjustments (up to 30% decrease) and clinical assessments were made at 2, 4, 8, 12, 20, and 24 weeks.
The primary efficacy endpoint was based on a clinical response, defined as a patient achieving at least 20% reduction in weekly PN/I.V. volume from Baseline (immediately before randomization) to both Weeks 20 and 24.
The mean age of patients was 50 years. Mean duration of PN/I.V. dependency prior to enrollment was 6 years (range 1 to 26 years). The most common reasons for intestinal resection leading to SBS were vascular disease (34%, 29/85), Crohn's Disease (21%, 18/85), and "other" (21%, 18/85). Stoma was present in 45% (38/85) of patients, and the most common type was jejunostomy/ileostomy (82%, 31/38). The mean length of remaining small intestine was 77.3±64.4 cm (range: 5 to 343 cm). The colon was not in continuity in 44% (37/85) patients. At baseline, the mean (± SD) prescribed days per week for PN/I.V. infusion was 5.73 (±1.59) days.
The percentages of treatment group responders were compared in the intent-to-treat population of this study which was defined as all randomized patients. Sixty-three percent (27/43) of GATTEX-treated patients versus 30% (13/43) of placebo-treated patients were considered responders (p=0.002).
At Week 24, the mean reduction in weekly PN/I.V. volume was 4.4 Liters for GATTEX-treated patients (from pre-treatment baseline of 12.9 Liters) versus 2.3 Liters for placebo-treated patients (from pre-treatment baseline of 13.2 Liters/week) (p<0.001).
Twenty-one patients on GATTEX (54%) versus 9 on placebo (23%) achieved at least a one-day reduction in PN/I.V. support.
The mean changes from Baseline in PN/I.V. volume by visit are shown in Figure 2.
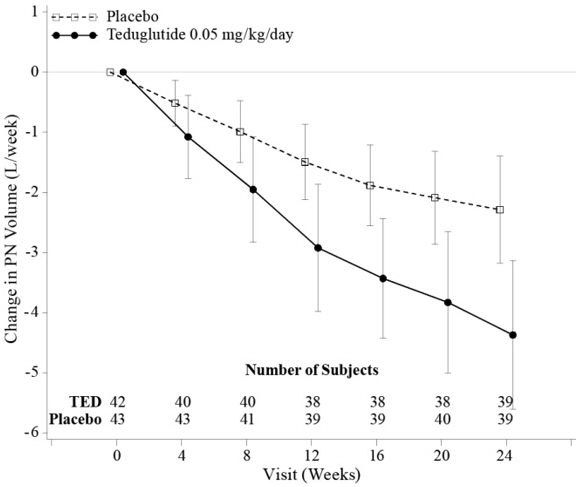
Figure 2: Change (±95% CI) in PN/I.V. volume (L/week)
Study 2 (CL0600-021, NCT00930644)
Study 2 was a 2-year open-label extension of Study 1 in which 88 patients received GATTEX 0.05 mg/kg/day. Ninety-seven percent (76/78) of patients who completed Study 1 elected to enroll in Study 2 (37 received GATTEX; 39 received Placebo). An additional 12 patients entered Study 2, who had been optimized and stabilized but not randomized in Study 1 because of closed enrollment.
30 months exposure
Thirty GATTEX patients completed a total duration of 30 months (Study 1 followed by Study 2 treatment). Of these, 28 patients (93%) achieved a 20% or greater reduction of parenteral support (PS). Of responders in Study 1 who had completed 2 additional years of continuous treatment with GATTEX, 96% (21/22) sustained their response to GATTEX. The mean reduction in PN/I.V. (n=30) was 7.55 L/week (a 66% reduction from baseline). Ten patients were weaned off their PN/I.V. support while on GATTEX treatment for 30 months. Patients were maintained on GATTEX even if no longer requiring PN/I.V. support. These 10 patients had required PN/I.V. support for 1.2 to 15.5 years, and prior to GATTEX had required between 3.5 L/week and 13.4 L/week of PN/I.V. support. At the end of study, 21 (70%), 18 (60%) and 18 (60%) of the 30 completers achieved a reduction of 1, 2, or 3 days per week in PN/I.V. support, respectively.
24 months exposure
Of the 39 placebo-treated patients from Study 1 entering Study 2, 29 completed 24 months of treatment with GATTEX. The mean reduction in PN/I.V. was 3.11 L/week (an additional 28.3% reduction) from the start of Study 2. Sixteen (55%) of the 29 completers achieved a 20% or greater reduction of PS. At the end of the study, 14 (48%), 7 (24%) and 5 (17%) achieved a reduction of 1, 2, or 3 days per week in PN/I.V. support, respectively. Two patients were weaned off their PN/I.V. support while on GATTEX. Of the 12 patients entering Study 2 directly, 6 completed 24 months of treatment with GATTEX. Similar effects were seen. One of the six patients was weaned off their PN/I.V. support while on GATTEX.
Study 3 (Placebo-controlled) and Study 4 (Blinded Uncontrolled Extension of Study 3)
Study 3 (CL0600-004, NCT00081458)
Study 3 was a randomized, double-blind, placebo-controlled, three parallel-group, multinational study in adults with SBS who were dependent on parenteral nutrition/intravenous (PN/I.V.) support for at least 12 months and required PN at least 3 times per week. After a period of optimization and stabilization similar to Study 1, patients were randomized to receive 24 weeks of one of the following treatment regimens: GATTEX 0.05 mg/kg/day (n=35), GATTEX 0.1 mg/kg/day (twice the recommended dose) (n=33), or placebo (n=16). GATTEX 0.1 mg/kg/day is not a recommended dosage [see Dosage and Administration (2.2)]. The treatment groups were compared using the intent-to-treat population of this study which was defined as all randomized patients who were administered at least one dose of study drug. This population contained one less patient in the 0.1 mg/kg/day dose group hence n=32 in this group for all analyses. The primary efficacy endpoint was a graded categorical score that did not achieve statistical significance for the high dose. Further evaluation of PN/I.V. volume reduction using the endpoint of response (defined as at least 20% reduction in PN/I.V. fluid from Baseline to Weeks 20 and 24) showed that 46% of patients on GATTEX 0.05 mg/kg/day responded versus 6% on placebo. Patients on GATTEX in both dose groups experienced a 2.5 L/week reduction in PS requirements versus 0.9 L/week for placebo at 24 weeks. Two patients in the GATTEX 0.05 mg/kg/day dose group were weaned off PS by Week 24.
Study 4 (CL0600-005, NCT00172185)
Study 4 was a blinded, uncontrolled extension of Study 3, in which 65 patients from Study 3 received GATTEX for up to an additional 28 weeks of treatment. Of responders in Study 3 who entered Study 4, 75% sustained response on GATTEX after one year of treatment. In the GATTEX 0.05 mg/kg/day dose group, a 20% or greater reduction of PS was achieved in 68% (17/25) of patients. The mean reduction of weekly PN/I.V. volume was 4.9 L/week (52% reduction from baseline) after one year of continuous GATTEX treatment. The patients who had been completely weaned off PN/I.V. support in Study 3 remained off PS through Study 4. During Study 4, an additional patient from Study 3 was weaned off PS.
14.2 Treatment of SBS in Pediatric Patients
Study 5 (TED-C14-006, NCT02682381)
Study 5 was a 24-week, multicenter study conducted in 59 pediatric patients aged 1 year through 17 years with SBS who were dependent on PS. Patients chose whether to receive GATTEX or standard of care (SOC). Patients who chose to receive GATTEX treatment were subsequently randomized in a double-blind manner to 0.025 mg/kg/day (n=24) or 0.05 mg/kg/day (n=26), while 9 patients enrolled in the SOC arm. Randomization to the GATTEX dose groups was stratified by age.
Patients treated with 0.05 mg/kg had a mean age of 6 years at baseline. The most common reasons for intestinal resection leading to SBS were gastroschisis (54%, 14/26), midgut volvulus (23%, 6/26), and necrotizing enterocolitis (12%, 3/26). Stoma was present in 19% (5/26) of patients, and the most common type was jejunostomy (80%, 4/5). The mean length of remaining small intestine was 47 (±28) cm (range: 9 to 120 cm). In the 25 patients who had remaining colon, the colon was in continuity in 22 patients. At baseline, the mean PS volume was 60 (±29) mL/kg/day (range: 24 to 133 mL/kg/day) [8 (±4) L/week (range: 3 to 19 L/week)] and mean PS infusion time was 7 (±1) days/week (range: 5 to 7 days/week) and 11 (±3) hours/day (range: 7 to 20 hours/day).
Results described in Table 3 correspond to the recommended GATTEX dosage of 0.05 mg/kg subcutaneously once daily.
| Efficacy Endpoints | Results |
|---|---|
|
|
| Reduction in PS volume of at least 20%, n/N (%) | 18/26 (69%) |
| Achieved enteral autonomy, n/N (%) | 3/26 (12%) |
| Reduction in PS infusion of ≥1 day/week, n/N (%) | 10/26 (38%) |
| Change in PS volume from baseline (mL/kg/day), mean (SD) and [mean% (SD)] | -23 (18) mL/kg/day [-42% (29%)] |
Study 6 (SHP633-304, NCT02954458)
Study 6 was a prospective, open-label, long-term extension study of pediatric patients who completed Study 5. In the extension study, patients received additional treatment with GATTEX 0.05 mg/kg subcutaneously once daily if they deteriorated or stopped improving after discontinuation of prior GATTEX treatment. Of the 15 patients who initially responded in Study 5 and enrolled in Study 6, 13 patients (87%) required additional treatment with GATTEX. Efficacy results at the end of the first 24-week treatment period in Study 6 (total treatment for a mean of 40 weeks) were similar to those achieved at the end of 24 weeks treatment in Study 5. One additional patient treated with 0.05 mg/kg in Study 5 eventually achieved enteral autonomy during follow-up in Study 6.
16 HOW SUPPLIED/STORAGE AND HANDLING
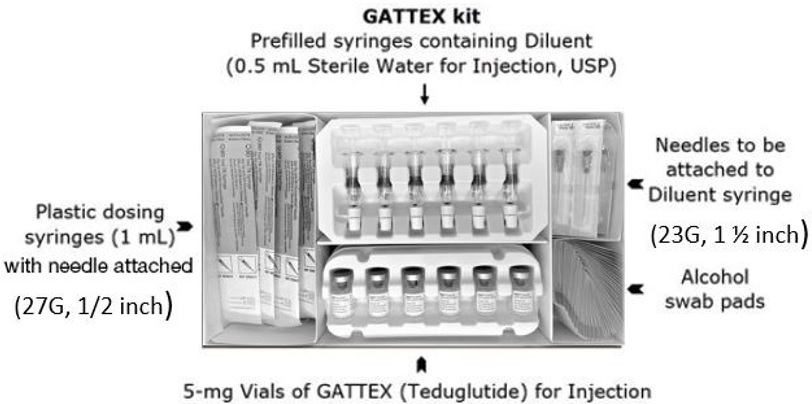
GATTEX (teduglutide) for injection is supplied as 5 mg of teduglutide as a white, lyophilized powder for reconstitution in a sterile, single-dose glass vial with 0.5 mL Sterile Water for Injection in a single-dose prefilled syringe. The product to be dispensed is either a one-vial kit or a 30-vial kit.
One-vial kits are pre-assembled and ready to be used:
GATTEX 5 mg One-vial Kit (NDC 68875-0103-1):
- One single-dose vial of 5 mg teduglutide (NDC 68875-0101-1)
- One disposable prefilled syringe containing 0.5 mL Sterile Water for Injection USP for reconstitution, with a separate needle (23G × 1½ in) to attach to the syringe
- One sterile disposable 1-mL syringe with needle (27G × 1/2 in) for dosing
- Four alcohol swabs
Storage and Handling of One-Vial Kit
Prior to Dispensing:
- Store GATTEX 5 mg Strength one-vial kits refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze. Do not use beyond the expiration date on the label.
After Dispensing by the Pharmacist:
- Store GATTEX 5 mg Strength one-vial kits at room temperature up to 25°C (77°F). Do not freeze. Dispense with a 90-day "use by" dating.
GATTEX is also supplied in 30-vial cartons to be assembled by the dispensing pharmacist into a 30-vial kit by transferring the trays containing 30 vials from a Carton of Drug Vials into a Carton of Ancillary Supplies:
GATTEX 5 mg Carton of Drug Vials (NDC 68875-0101-2):
- Thirty single-dose vials of GATTEX 5 mg (NDC 68875-0101-1)
-
Carton of Ancillary Supplies:
- Thirty disposable prefilled syringes containing diluent (0.5 mL Sterile Water for Injection USP) for reconstitution
- Thirty separate needles (23G × 1½ in) to attach to the syringes for reconstitution
- Thirty sterile disposable 1-mL syringes with needle (27G × 1/2 in)
- Sixty alcohol swabs
The final assembled 30-Vial Kit should contain the items:
GATTEX 5 mg Strength 30-Vial Kit (NDC 68875-0102-1):
- Thirty single-dose vials of 5 mg teduglutide (NDC 68875-0101-1)
- Thirty disposable prefilled syringes containing 0.5 mL Sterile Water for Injection USP for reconstitution, with 30 separate needles (23G × 1½ in) to attach to the syringes
- Thirty sterile disposable 1-mL syringes with needle (27G × 1/2 in) for dosing
- Sixty alcohol swabs
Storage and Handling of 30-Vial Cartons and 30-Vial Kits
Prior to Dispensing:
- Store GATTEX 5 mg vials refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze. Do not use beyond the expiration date on the label.
- Store the Carton of Ancillary Supplies at room temperature up to 25°C (77°F).
After Dispensing by the Pharmacist:
- Store GATTEX 5 mg Strength 30-vial kits at room temperature up to 25°C (77°F). Do not freeze. Dispense with a 90-day "use by" dating.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Acceleration of Neoplastic Growth
Advise patients and their caregivers that they will need to undergo clinical examinations and repeated colonoscopies (or alternate imaging/diagnostics, such as fecal occult blood testing) during treatment with GATTEX to monitor for the development of polyps and/or neoplasia of the GI tract [see Warnings and Precautions (5.1)].
Intestinal Obstruction
Advise patients and their caregivers to immediately contact their healthcare provider if they experience any symptoms suggestive of intestinal or stomal obstruction [see Warnings and Precautions (5.2)].
Biliary and Pancreatic Disease
Advise patients and their caregivers that laboratory assessments will be done periodically while on GATTEX to monitor for the onset or worsening of gallbladder, biliary and pancreatic disease, and to report immediately to their healthcare provider if they develop symptoms suggestive of cholecystitis, cholangitis, cholelithiasis or pancreatic disease [see Warnings and Precautions (5.3)].
Fluid Overload
Advise patients and their caregivers to immediately contact their healthcare provider if they develop fluid overload or symptoms of congestive heart failure while on GATTEX [see Warnings and Precautions (5.4)].
Fluid Imbalance
Advise patients and their caregivers of the risk of fluid and electrolyte imbalance with discontinuation of GATTEX, and to contact their healthcare provider if they develop symptoms suggestive of electrolyte imbalances [see Warnings and Precautions (5.4)].
Increased Absorption of Concomitant Oral Medication
Instruct patients and their caregivers to report to their healthcare provider any concomitant oral medications that they are taking in order to assess any potential for increased absorption during GATTEX treatment of those oral medications requiring titration or with a narrow therapeutic index [see Warnings and Precautions (5.5)].
Lactation
Advise women that breastfeeding is not recommended during treatment with GATTEX [see Use in Specific Populations (8.2)].
GATTEX® and the GATTEX® logo are registered trademarks of Takeda Pharmaceuticals U.S.A., Inc.
©2022 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
Patented: see www.takeda.com/en-us/patents.
Distributed by:
Takeda Pharmaceuticals America, Inc.
Lexington, MA 02421
USA
For information about GATTEX contact:
Takeda Pharmaceuticals America, Inc.
Lexington, MA 02421
USA
1-877-825-3327
www.GATTEX.com
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 10/2022 | ||
| MEDICATION GUIDE GATTEX® (Ga'-tex) (teduglutide) for injection, for subcutaneous use |
|||
| Read this Medication Guide carefully before you start taking GATTEX and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. | |||
| What is the most important information I should know about GATTEX? GATTEX may cause serious side effects, including:
|
|||
|
|
||
If a blockage is found, your healthcare provider may temporarily stop GATTEX.
|
|||
|
|
||
| These are not all the side effects of GATTEX. For more information, see "What are the possible side effects of GATTEX?" | |||
What is GATTEX?
|
|||
| What should I tell my healthcare provider before using GATTEX? Before using GATTEX, tell your healthcare provider about all your medical conditions, including if you or your child:
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||
How should I use GATTEX?
|
|||
| What are the possible side effects of GATTEX? GATTEX may cause serious side effects, including:
|
|||
|
|
||
| The side effects of GATTEX in children and adolescents are similar to those seen in adults. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of GATTEX. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
How should I store GATTEX?
|
|||
| General information about the safe and effective use of GATTEX.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use GATTEX for a condition for which it was not prescribed. Do not give GATTEX to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information about GATTEX talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about GATTEX that is written for health professionals. |
|||
| What are the ingredients in GATTEX? Active ingredient: teduglutide Inactive ingredients: dibasic sodium phosphate heptahydrate, L-histidine, mannitol, and monobasic sodium phosphate monohydrate. Sterile Water for Injection is provided as a diluent. Distributed by: Takeda Pharmaceuticals America, Inc. Lexington, MA 02421 GATTEX® and the GATTEX® logo are registered trademarks of Takeda Pharmaceuticals U.S.A., Inc. ©2022 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved. Patented: see www.takeda.com/en-us/patents. |
|||
Instructions for Use
GATTEX®(Ga'-tex)
(teduglutide)
for injection, for subcutaneous use
5 mg per vial
Read this Instructions for Use before you start using GATTEX and each time you get a refill. There may be new information. Your healthcare provider or nurse should show you how to prepare, measure your dose, and give your injection of GATTEX the right way.
If you cannot give yourself the injection:
- ask your healthcare provider or nurse to help you, or
- ask someone who has been trained by a healthcare provider or nurse to give your injections
Self-administration is not recommended in pediatric patients. In pediatric patients, GATTEX should be injected by:
- a healthcare provider or nurse, or
- a parent or adult caregiver who has been trained by a healthcare provider or nurse to give injections of GATTEX to pediatric patients
Important information:
- Use of the GATTEX 5 mg kit is not recommended in pediatric patients weighing less than 22 pounds (10 kg).
- Before you start, check the "Use By" date on your GATTEX kit. Make sure that the "Use By" date has not passed. Do not use anything in the GATTEX kit after the "Use By" date on the kit.
- Give GATTEX within 3 hours after you mix the powder with the Diluent (Sterile Water for Injection).
- Use the syringes and needles provided in the GATTEX kit.
- Do not use a GATTEX vial more than 1 time, even if there is medicine left in the vial.
- Throw away (dispose of) any unused GATTEX after you give your injection.
- Safely throw away GATTEX vials after use.
- Do not re-use syringes or needles. See "Step 7: Dispose of syringes and needles" for information about how to safely throw away needles and syringes.
- To help avoid needle-stick injuries, do not recap needles.
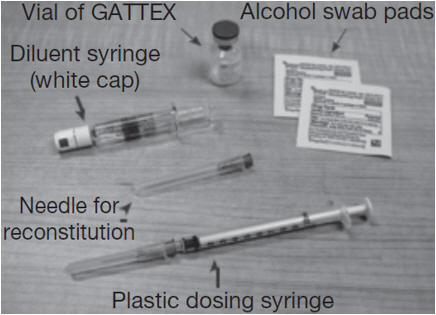
Gather the supplies you will need to prepare GATTEX and to give your injection (See Figure A).
From your GATTEX kit you will need:
|
Step 1: Prepare the injection.
- Choose a well-lit, clean, flat work surface.
- Wash your hands with soap and water.
Step 2: Preparing the Diluent syringe.
- Put the Diluent syringe (See Figure B1) and the 23G, 1½ inch needle in front of you on your work surface.
| Figure B1 |
|
- Hold the Diluent syringe by the barrel. Snap off the white cap (bend the cap sideways until the cap comes off). Only the top portion of the white cap should be snapped off. The lower portion of the cap will remain in place (See Figure B2). Throw the cap away.
| Figure B2 |
|
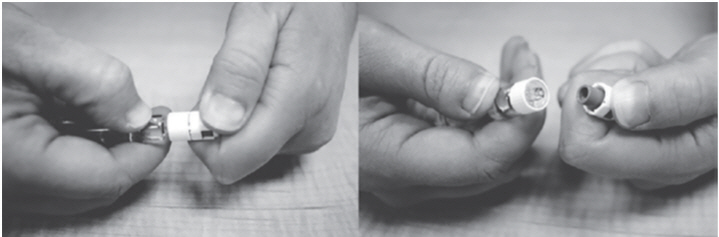
- Remove the 23G, 1½ inch needle from the package. Use the fold in the package to peel back the plastic cover (See Figure C). Leave the plastic cap on the needle.
| Figure C |
|

- Push the open end of the needle onto the end of the Diluent syringe (See Figure D).
Twist the needle clockwise (to the right) until it stops turning.
| Figure D |
|

- When the needle is tightly in place, put the Diluent syringe and needle on your work surface.
Step 3: Mix GATTEX powder with Diluent.
- Remove the green cap from the GATTEX vial. Throw away the green cap.
- Find the gray rubber seal on top of the GATTEX vial (See Figure E).
| Figure E |
|

- Use an alcohol swab pad to clean the gray rubber seal (See Figure F).
- Do not touch the gray rubber seal after you clean it.
| Figure F |
|

- Pick up the Diluent syringe with the needle attached.
- Remove the plastic cap that covers the needle (See Figure G). Throw the cap away.
| Figure G |
|
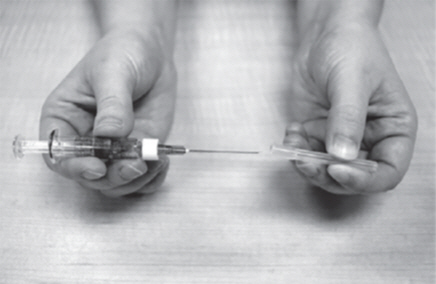
- Hold the GATTEX vial between your thumb and index (pointer) finger (See Figure H). Be careful not to touch the gray rubber seal.
- Push the needle down through the center of the gray rubber seal.
- Slowly push down on the plunger of the Diluent syringe. Empty all the Diluent into the GATTEX vial.
- Leave the needle and Diluent syringe in place.
| Figure H |
|
- Gently tap the barrel of the Diluent syringe with a finger (See Figure I).
- Make sure all the Diluent has gone into the GATTEX vial.
| Figure I |
|
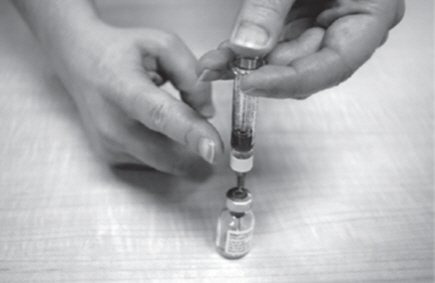
- Remove the Diluent syringe and needle from the GATTEX vial. Let the vial sit for about 30 seconds.
- Do not put the needle cap back on the needle.
- Throw away (dispose of) the Diluent syringe and needle in your sharps disposal container.
- After 30 seconds, place the GATTEX vial between the palms of your hands. Gently roll the vial for about 15 seconds (See Figure J).
- Do not shake the GATTEX vial.
- Do not touch the gray rubber seal. If you do, clean it again with a new alcohol pad.
- Let the GATTEX vial stand on your work surface for about 2 minutes.
| Figure J |
|
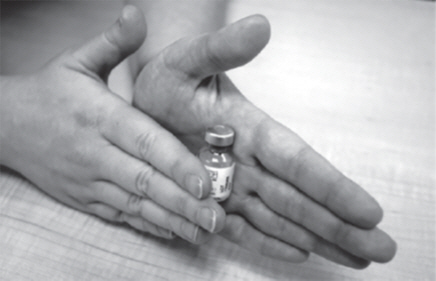
Step 4: Check the mixed GATTEX.
- After 2 minutes, look at the vial of GATTEX. The liquid in the vial should be clear and colorless to pale yellow, and should not have any particles in it.
- If there is any powder in the GATTEX vial that did not dissolve, gently roll the vial between your hands for 15 seconds more.
- Do not shake the GATTEX vial.
- Check the GATTEX vial again for anything that did not dissolve.
- Do not use the GATTEX vial if there is anything in it that did not dissolve. Start from the beginning of this Instructions for Use to prepare a new vial. Use a new GATTEX vial, new Diluent syringe, and a new needle.
Step 5: Draw up your dose of GATTEX.
- Remove the plastic dosing syringe from the package. Use the fold in the package to peel back the plastic cover (See Figure K).
| Figure K |
|
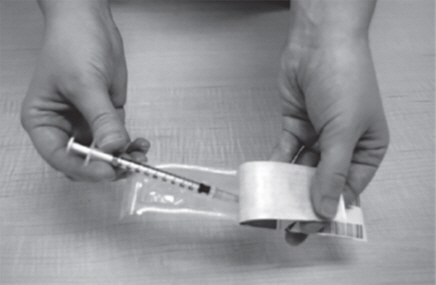
- Remove the needle cap from the plastic dosing syringe (See Figure L).
- Throw the needle cap away. Do not touch the needle or allow it to touch anything.
| Figure L |
|
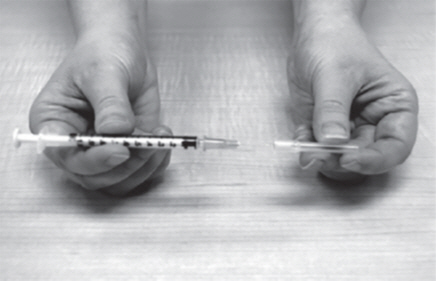
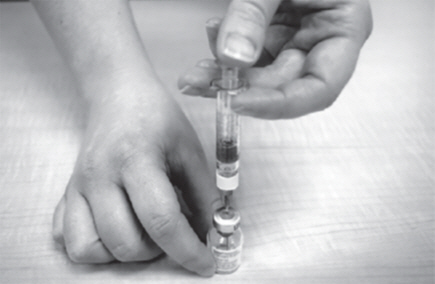
- Carefully pull back on the plunger to the line that matches the dose prescribed by your healthcare provider.
- Use 1 hand to hold the GATTEX vial steady. Use your other hand to insert the needle straight down into the middle of the gray rubber seal on the GATTEX vial (See Figure M). You may feel some resistance as the needle passes through the rubber seal.
- Gently push down the plunger until all of the air has gone from the plastic dosing syringe into the GATTEX vial.
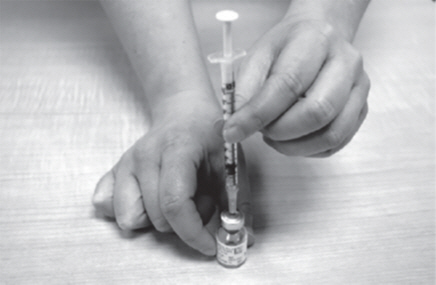
- Turn the GATTEX vial and plastic dosing syringe upside down (See Figure N).
| Figure M |
|
| Figure N |
|
- Hold the GATTEX vial with 1 hand.
- Slowly pull back the plunger of the plastic dosing syringe with your other hand.
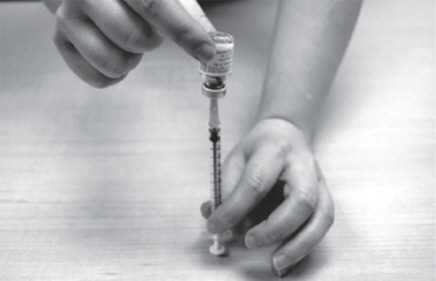
- Fill the plastic dosing syringe until the black tip of the plunger lines up with the mark that matches your prescribed dose (See Figure O).
- Keep the plastic dosing syringe and needle in the GATTEX vial.
| Figure O |
|
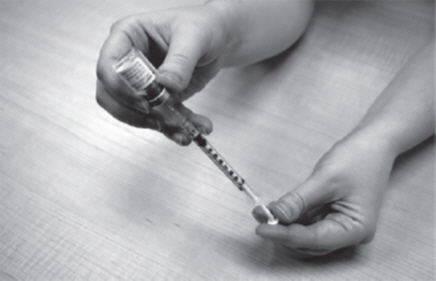
- You may see some bubbles inside the GATTEX vial when the plastic dosing syringe is filled. This is normal. With the needle still in the vial, gently tap the side of the plastic dosing syringe with a finger to make any air bubbles rise to the top (See Figure P).
| Figure P |
|
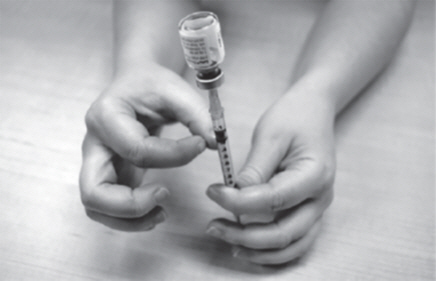
- Slowly push the plunger up until all air bubbles are out of the plastic dosing syringe. Make sure the tip of the needle is in the fluid. Slowly pull back the plunger to draw up the right dose of GATTEX into the plastic dosing syringe.
- Remove the plastic dosing syringe and needle from the GATTEX vial (See Figure Q). Do not touch the needle or allow it to touch anything.
| Figure Q |
|
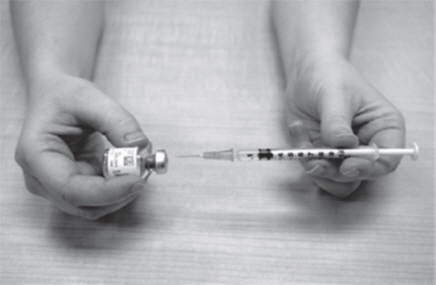
Step 6: Inject GATTEX.
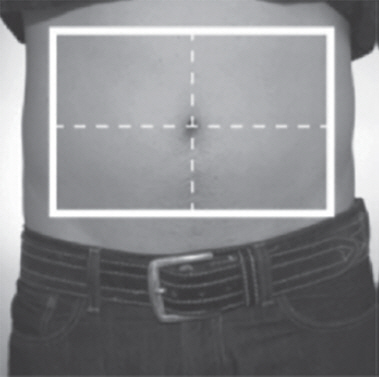
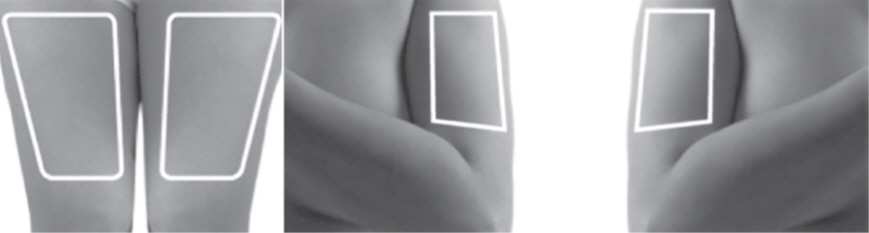
- Choose an injection site on the stomach area (abdomen), thighs, or upper arms.
- Choose a different site to give the injection each day. Do not inject into areas where the skin is tender, bruised, red, or hard. (See Figure R and Figure S)
| Figure R |
|
- Clean the skin where you plan to give the injection with a new alcohol swab pad. Do not touch this area again before giving the injection.
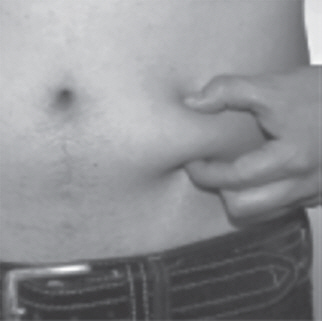
- Use 1 hand to gently pinch up a fold of skin around the injection site (See Figure T).
| Figure T |
|
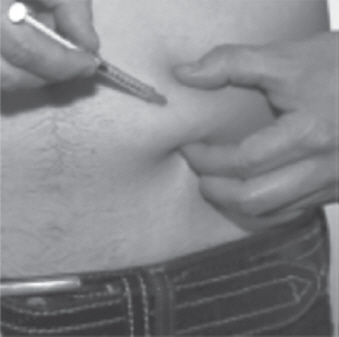
- Use your other hand to hold the plastic dosing syringe. Insert the full length of the needle into the skin at a 45-degree angle with a quick, "dart-like" motion (See Figure U).
| Figure U |
|
- Let go of the skin. Hold the syringe barrel with 1 hand while you slowly push down the plunger until the plastic dosing syringe is empty (See Figure V).
| Figure V |
|
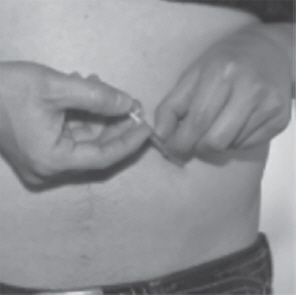
- When the plastic dosing syringe is empty, quickly pull the needle out of your skin. There may be a little bleeding at the injection site. Apply an adhesive bandage to the injection site if needed.
Step 7: Dispose of syringes and needles.
- Do not re-use a syringe or needle.
- To help avoid needle-stick injuries, do not recap a needle.
- Put your needles and syringes in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharp items being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container.
There may be local or state laws about how to throw away syringes and needles. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal. - Do not dispose of your sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your sharps disposal container.
- Throw away the GATTEX vial into the container where you put the syringes and needles.
- If you have any questions, talk to your healthcare provider or pharmacist.
How should I store GATTEX?
- Store GATTEX powder at room temperature up to 77°F (25°C).
- Do not freeze GATTEX.
- Use the GATTEX powder by the expiration date on the "Use By" sticker on the kit.
- Use GATTEX within 3 hours after mixing it.
- Throw away any unused GATTEX that has been mixed, even if there is medicine left in the vial.
- Do not store any GATTEX you have mixed.
Keep GATTEX and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Distributed by:
Takeda Pharmaceuticals America, Inc.
Lexington, MA 02421
USA
1-877-825-3327
GATTEX® and the GATTEX® logo are registered trademarks of Takeda Pharmaceuticals U.S.A., Inc.
©2024 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
Revised: 02/2024
PRINCIPAL DISPLAY PANEL - 5 mg Vial Carton
NDC 68875-0101-2
Gattex®
(teduglutide) for injection
5 mg
5 mg per vial.
For subcutaneous use only.
EACH CARTON CONTAINS:
- Thirty single-dose vials of Gattex®
- Package Insert
- Medication Guide
ATTENTION PHARMACIST:
- For dispensing, transfer the product with vial trays
from this carton into the 30-count patient kit.
Takeda


