ANZEMET- dolasetron mesylate injection
sanofi-aventis U.S. LLC
----------
ANZEMET ® Injection
(dolasetron mesylate)
DESCRIPTION
ANZEMET (dolasetron mesylate) is an antinauseant and antiemetic agent. Chemically, dolasetron mesylate is (2α,6α,8α,9aβ)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl-1H-indole-3-carboxylate monomethanesulfonate, monohydrate. It is a highly specific and selective serotonin subtype 3 (5-HT3) receptor antagonist both in vitro and in vivo. Dolasetron mesylate has the following structural formula:
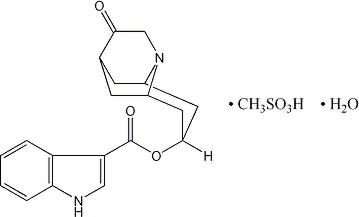
The empirical formula is C19H20N2O3 • CH3SO3H • H2O, with a molecular weight of 438.50. Approximately 74% of dolasetron mesylate monohydrate is dolasetron base.
Dolasetron mesylate monohydrate is a white to off-white powder that is freely soluble in water and propylene glycol, slightly soluble in ethanol, and slightly soluble in normal saline.
ANZEMET Injection is a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each milliliter of ANZEMET Injection contains 20 mg of dolasetron mesylate and 38.2 mg mannitol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.8.
ANZEMET Injection multidose vials contain a clear, colorless, nonpyrogenic, sterile solution for intravenous administration. Each ANZEMET multidose vial contains 25 mL (500 mg) dolasetron mesylate. Each milliliter contains 20 mg dolasetron mesylate, 29 mg mannitol, USP, and 5 mg phenol, USP, with an acetate buffer in water for injection. The pH of the resulting solution is 3.2 to 3.7.
CLINICAL PHARMACOLOGY
Dolasetron mesylate and its active metabolite, hydrodolasetron (MDL 74,156), are selective serotonin 5-HT3 receptor antagonists not shown to have activity at other known serotonin receptors and with low affinity for dopamine receptors. The serotonin 5-HT3 receptors are located on the nerve terminals of the vagus in the periphery and centrally in the chemoreceptor trigger zone of the area postrema.
In healthy volunteers (N=64), dolasetron mesylate in single intravenous doses up to 5 mg/kg produced no effect on pupil size or meaningful changes in EEG tracings. Results from neuropsychiatric tests revealed that dolasetron mesylate did not alter mood or concentration. Multiple daily doses of dolasetron have had no effect on colonic transit in humans. Dolasetron mesylate has no effect on plasma prolactin concentrations.
Effects on Electrocardiogram
QTcF interval was evaluated in a randomized, placebo and active (moxifloxacin 400 mg once-daily) controlled crossover study in 80 healthy adults, with 14 measurements over 24 hours on Day 4. The maximum mean (95% upper confidence bound) differences in QTcF from placebo after baseline-correction were 14.1 (16.1) and 36.6 (38.6) ms for 100 mg and supratherapeutic 300 mg ANZEMET administered intravenously, respectively. ANZEMET 300 mg once daily resulted in approximately 3-fold higher mean Cmax values of dolasetron mesylate and its active metabolite hydrodolasetron on Day 4 compared to those observed with the therapeutic 100 mg ANZEMET dose.
Based on exposure-response analyses in healthy volunteers, QTc interval prolongation appears to be associated with concentrations of hydrodolasetron. Using the established exposure-response relationship, the mean predicted increase (95% upper prediction interval) in QTcF intervals were 22.5 (23.9) and 21.2 (22.6) ms in pediatric and adult cancer patients following a single intravenous dose of 1.8 mg/kg.
In the thorough QT study, exposure dependent prolongation of the PR and QRS interval was also noted in healthy subjects receiving ANZEMET. The maximum mean (95% upper confidence bound) difference in PR from placebo after baseline-correction was 9.8 (11.6) ms and 33.1 (34.9) ms for 100 mg and supratherapeutic 300 mg ANZEMET, respectively. The maximum mean (95% upper confidence bound) difference in QRS from placebo after baseline-correction was 3.5 (4.5) ms and 13 (14.5) ms for 100 mg and supratherapeutic 300 mg ANZEMET, respectively. Over one-fourth of the subjects treated with the 300 mg dose had an absolute PR over 200 ms and absolute QRS of over 110 ms post-treatment. A change from baseline ≥ 25% was noted in several of these subjects. (see CONTRAINDICATIONS and WARNINGS)
Pharmacokinetics in Humans
Intravenous dolasetron mesylate is rapidly eliminated (t1/2<10 min) and completely metabolized to the most clinically relevant species, hydrodolasetron.
The reduction of dolasetron to hydrodolasetron is mediated by a ubiquitous enzyme, carbonyl reductase. Cytochrome P-450 (CYP)2D6 is primarily responsible for the subsequent hydroxylation of hydrodolasetron and both CYP3A and flavin monooxygenase are responsible for the N-oxidation of hydrodolasetron.
Hydrodolasetron is excreted in the urine unchanged (53.0% of administered intravenous dose). Other urinary metabolites include hydroxylated glucuronides and N-oxide.
Hydrodolasetron appeared rapidly in plasma, with a maximum concentration occurring approximately 0.6 hour after the end of intravenous treatment, and was eliminated with a mean half-life of 7.3 hours (%CV=24) and an apparent clearance of 9.4 mL/min/kg (%CV=28) in 24 adults. Hydrodolasetron is eliminated by multiple routes, including renal excretion and, after metabolism, mainly glucuronidation, and hydroxylation. Hydrodolasetron exhibits linear pharmacokinetics over the intravenous dose range of 50 to 200 mg and they are independent of infusion rate. Doses lower than 50 mg have not been studied. Two thirds of the administered dose is recovered in the urine and one third in the feces. Hydrodolasetron is widely distributed in the body with a mean apparent volume of distribution of 5.8 L/kg (%CV=25, N=24) in adults.
Sixty-nine to 77% of hydrodolasetron is bound to plasma protein. In a study with 14C labeled dolasetron, the distribution of radioactivity to blood cells was not extensive. The binding of hydrodolasetron to α1-acid glycoprotein is approximately 50%. The pharmacokinetics of hydrodolasetron are linear and similar in men and women.
The pharmacokinetics of hydrodolasetron, in special and targeted patient populations following intravenous administration of ANZEMET Injection, are summarized in Table 1. The pharmacokinetics of hydrodolasetron are similar in adult (young and elderly) healthy volunteers. The apparent clearance of hydrodolasetron in pediatric and adolescent patients is 1.4 times to twofold higher than in adults. Following intravenous administration, the apparent clearance of hydrodolasetron remains unchanged with severe hepatic impairment and decreases 47% with severe renal impairment. No dose adjustment is necessary for elderly patients (see PRECAUTIONS, Geriatric Use) or for patients with hepatic or renal impairment.
In a pharmacokinetic study in 18 pediatric patients (2 to 11 years of age) undergoing surgery with general anesthesia and administered a single 1.2 mg/kg intravenous dose of ANZEMET Injection, mean apparent clearance was greater (40%) and terminal half-life shorter (36%) for hydrodolasetron than in healthy adults receiving the same dose.
For 12 pediatric patients, ages 2 to 12 years receiving 1.2 mg/kg ANZEMET Injection diluted in apple or apple-grape juice and administered orally, the mean apparent clearance was 34% greater and half-life was 21% shorter than in healthy adults receiving the same dose.
| Age (years) | Dose | CLapp
(mL/min/kg) | t1/2
(h) | Cmax
(ng/mL) |
|
|---|---|---|---|---|---|
| CLapp: apparent clearance t1/2: terminal elimination half-life ( ): coefficient of variation in % | |||||
|
|||||
| Young Healthy Volunteers (N=24) | 19–40 | 100 mg | 9.4 (28%) | 7.3 (24%) | 320 (25%) |
| Elderly Healthy Volunteers (N=15) | 65–75 | 2.4 mg/kg | 8.3 (30%) | 6.9 (22%) | 620 (31%) |
| Pediatric Surgery Patients (N=18) | 2–11 | 1.2 mg/kg | 13.1 (47%) | 4.8 (23%) | 255 (22%) |
| Patients with Severe Renal Impairment (N=12) (Creatinine clearance ≤10 mL/min) | 28–74 | 200 mg | 5.0 (33%) | 10.9 (30%) | 867 (31%) |
| Patients with Severe Hepatic Impairment (N=3) | 42–52 | 150 mg | 9.6 (19%) | 11.7 (22%) | 396 (45%) |
Clinical Studies
Prevention of Postoperative Nausea and Vomiting
ANZEMET Injection administered intravenously at a dose of 12.5 mg approximately 15 minutes before the cessation of general balanced anesthesia (short-acting barbiturate, nitrous oxide, narcotic and analgesic, and skeletal muscle relaxant) was significantly more effective than placebo in preventing postoperative nausea and vomiting. No increased efficacy was seen with higher doses.
One trial compared single intravenous ANZEMET Injection doses of 12.5, 25, 50, and 100 mg with placebo in 635 women surgical patients undergoing laparoscopic procedures. ANZEMET Injection at a dose of 12.5 mg was statistically superior to placebo for complete response (no vomiting, no rescue medication) (p=.0003). Complete response rates were 50% and 31%, respectively.
Another trial compared single intravenous ANZEMET Injection doses of 12.5, 25, 50, and 100 mg with placebo in 1030 (722 women and 308 men) surgical patients. In women, the 12.5 mg dose was statistically superior to placebo for complete response. The complete response rates were 50% and 40%, respectively. However, in men, there was no statistically significant difference in complete response between any ANZEMET dose and placebo.
Treatment of Postoperative Nausea and/or Vomiting
Two randomized, double-blinded trials compared single intravenous ANZEMET Injection doses of 12.5, 25, 50, and 100 mg with placebo in 124 male and 833 female patients who had undergone surgery with general balanced anesthesia and presented with early postoperative nausea or vomiting requiring antiemetic treatment.
In both studies, the 12.5 mg intravenous dose of ANZEMET was statistically superior to placebo for complete response (no vomiting, no escape medication). No significant increased efficacy was seen with higher doses.
INDICATIONS AND USAGE
ANZEMET Injection is indicated for the following:
- (1)
-
the prevention of postoperative nausea and vomiting (PONV) in adults and children 2 years and older. As with other antiemetics, routine prophylaxis is not recommended for patients in whom there is little expectation that nausea and/or vomiting will occur postoperatively. In patients where nausea and/or vomiting must be avoided postoperatively, ANZEMET Injection is recommended even where the incidence of postoperative nausea and/or vomiting is low.
When prophylaxis has failed, a repeat dose should not be initiated as rescue therapy.
- (2)
- the treatment of postoperative nausea and/or vomiting in adults and children 2 years and older.
CONTRAINDICATIONS
ANZEMET Injection is contraindicated in patients known to have hypersensitivity to the drug.
ANZEMET Injection solution administered intravenously is contraindicated in adult and pediatric patients for the prevention of nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy due to dose dependent QT prolongation. Mean QTc effects over 20 ms are expected in this patient population (see CLINICAL PHARMACOLOGY and WARNINGS).
WARNINGS
QTc Interval Prolongation
ANZEMET prolongs the QT interval in a dose dependent fashion. Torsade de Pointes has been reported during post-marketing experience. Avoid ANZEMET in patients with congenital long QT syndrome, hypokalemia or hypomagnesemia. Hypokalemia and hypomagnesemia must be corrected prior to ANZEMET administration. Monitor these electrolytes after administration as clinically indicated. Use ECG monitoring in patients with congestive heart failure and bradycardia (see CLINICAL PHARMACOLOGY).
PR and QRS Interval Prolongation
ANZEMET has been shown to cause dose dependent prolongation of the PR and QRS interval and reports of second or third degree atrioventricular block, cardiac arrest and serious ventricular arrhythmias including fatalities in both adult and pediatric patients. At particular risk are patients with underlying structural heart disease and preexisting conduction system abnormalities, elderly, patients with sick sinus syndrome, patients with atrial fibrillation with slow ventricular response, patients with myocardial ischemia or patients receiving drugs known to prolong the PR interval (such as verapamil) and QRS interval (e.g., flecainide or quinidine). ANZEMET should be used with caution and with ECG monitoring in these patients. ANZEMET should be avoided in patients with complete heart block or at risk for complete heart block, unless they have an implanted pacemaker (see CLINICAL PHARMACOLOGY).
Serotonin Syndrome
The development of serotonin syndrome has been reported with 5-HT3 receptor antagonists. Most reports have been associated with concomitant use of serotonergic drugs (e.g., selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors, mirtazapine, fentanyl, lithium, tramadol, and intravenous methylene blue). Some of the reported cases were fatal. Serotonin syndrome occurring with overdose of another 5-HT3 receptor antagonist alone has also been reported. The majority of reports of serotonin syndrome related to 5-HT3 receptor antagonist use occurred in a post-anesthesia care unit or an infusion center.
Symptoms associated with serotonin syndrome may include the following combination of signs and symptoms: mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, with or without gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome, especially with concomitant use of Anzemet and other serotonergic drugs. If symptoms of serotonin syndrome occur, discontinue Anzemet and initiate supportive treatment. Patients should be informed of the increased risk of serotonin syndrome, especially if Anzemet is used concomitantly with other serotonergic drugs (see DRUG INTERACTIONS, Patient Counseling Information).
PRECAUTIONS
General
Dolasetron should be administered with caution in patients who have or may develop prolongation of cardiac conduction intervals, particularly QTc. These include patients with hypokalemia or hypomagnesemia, patients taking diuretics with potential for inducing electrolyte abnormalities, patients with congenital QT syndrome, patients taking anti-arrhythmic drugs or other drugs which lead to QT prolongation, and cumulative high dose anthracycline therapy.
Cross hypersensitivity reactions have been reported in patients who received other selective 5-HT3 receptor antagonists. These reactions have not been seen with dolasetron mesylate.
Drug Interactions
The potential for clinically significant drug-drug interactions posed by dolasetron and hydrodolasetron appears to be low for drugs commonly used in surgery, because hydrodolasetron is eliminated by multiple routes. See PRECAUTIONS, General for information about potential interaction with other drugs that prolong the QTc interval.
When oral dolasetron (200 mg once daily) was coadministered with cimetidine (300 mg four times daily) for 7 days, the systemic exposure (i.e., AUC) of hydrodolasetron increased by 24% and the maximum plasma concentration of hydrodolasetron increased by 15%. When oral dolasetron (200 mg once daily) was coadministered with rifampin (600 mg once daily) for 7 days, the systemic exposure of hydrodolasetron decreased by 28% and the maximum plasma concentration of hydrodolasetron decreased by 17%.
Caution should be exercised when ANZEMET Injection is coadministered with drugs that prolong ECG intervals and/or cause hypokalemia or hypomagnesemia (see WARNINGS).
In patients taking furosemide, nifedipine, diltiazem, ACE inhibitors, verapamil, glyburide, and propranolol, no effect was shown on the clearance of hydrodolasetron. Clearance of hydrodolasetron decreased by about 27% when dolasetron mesylate was administered intravenously concomitantly with atenolol. ANZEMET did not influence anesthesia recovery time in patients.
Serotonin syndrome (including altered mental status, autonomic instability, and neuromuscular symptoms) has been described following the concomitant use of 5-HT3 receptor antagonists and other serotonergic drugs, including selective serotonin reuptake inhibitors (SSRIs) and serotonin and noradrenaline reuptake inhibitors (SNRIs).
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 24-month carcinogenicity study, there was a statistically significant (P<0.001) increase in the incidence of combined hepatocellular adenomas and carcinomas in male mice treated with 150 mg/kg/day and above. In this study, mice (CD-1) were treated orally with dolasetron mesylate 75, 150 or 300 mg/kg/day (225, 450 or 900 mg/m2/day). For a 50 kg person of average height (1.46 m2 body surface area), these doses represent 3.4, 6.8 and 13.5 times the recommended clinical dose (66.6 mg/m2, intravenous) on a body surface area basis. No increase in liver tumors was observed at a dose of 75 mg/kg/day in male mice and at doses up to 300 mg/kg/day in female mice.
In a 24-month rat (Sprague-Dawley) carcinogenicity study, oral dolasetron mesylate was not tumorigenic at doses up to 150 mg/kg/day (900 mg/m2/day, 13.5 times the recommended human dose based on body surface area) in male rats and 300 mg/kg/day (1800 mg/m2/day, 27 times the recommended human dose based on body surface area) in female rats.
Dolasetron mesylate was not genotoxic in the Ames test, the rat lymphocyte chromosomal aberration test, the Chinese hamster ovary (CHO) cell (HGPRT) forward mutation test, the rat hepatocyte unscheduled DNA synthesis (UDS) test or the mouse micronucleus test.
Dolasetron mesylate was found to have no effect on fertility and reproductive performance at oral doses up to 100 mg/kg/day (600 mg/m2/day, 9 times the recommended human dose based on body surface area) in female rats and up to 400 mg/kg/day (2400 mg/m2/day, 36 times the recommended human dose based on body surface area) in male rats.
Teratogenic Effects
Pregnancy Category B
Teratology studies have not revealed evidence of impaired fertility or harm to the fetus due to dolasetron mesylate. These studies have been performed in pregnant rats at intravenous doses up to 60 mg/kg/day (5.4 times the recommended human dose based on body surface area) and pregnant rabbits at intravenous doses up to 20 mg/kg/day (3.2 times the recommended human dose based on body surface area). There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether dolasetron mesylate is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ANZEMET Injection is administered to a nursing woman.
Pediatric Use
Prevention of chemotherapy-induced nausea and vomiting (CINV)
Dolasetron is contraindicated in pediatric patients for the prevention of nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy. (see CONTRAINDICATIONS).
Prevention and treatment of post-operative nausea and vomiting (PONV)
Safety and effectiveness in pediatric patients (2 years and older) for prevention and treatment of postoperative nausea and vomiting is based on pharmacokinetic studies and efficacy data in adults. Safety and effectiveness in pediatric patients under 2 years of age have not been established.
Two open-label, noncomparative pharmacokinetic studies have been performed in a total of 30 pediatric patients undergoing surgery with general anesthesia. These patients received ANZEMET Injection either intravenously or orally in juice. Pediatric patients from 2 to 12 years of age participated in these trials, which included an intravenous ANZEMET Injection dose of 1.2 mg/kg, and an oral dose of 1.2 mg/kg. There is no experience in pediatric patients under 2 years of age. Overall, ANZEMET Injection was well tolerated in these pediatric patients. No efficacy information was collected in the pediatric postoperative nausea and vomiting studies.
Geriatric Use
Prevention of chemotherapy-induced nausea and vomiting (CINV)
Dolasetron is contraindicated in geriatric patients for prevention of nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy (see CONTRAINDICATIONS).
Prevention and treatment of post-operative nausea and vomiting (PONV)
Controlled clinical studies in the prevention and treatment of post-operative nausea and vomiting did not include sufficient numbers of patients aged 65 years or older – only 57 (2%) geriatric patients (43 received intravenous ANZEMET Injection) out of 3289 total patients participated in the controlled PONV trials – to determine whether they respond differently from younger patients. Other reported clinical experiences have not identified differences in responses between geriatric and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Elderly patients are at particular risk for prolongation of the PR, QRS, and QT interval; therefore, caution should be exercised and ECG monitoring should be performed when using ANZEMET for prevention of postoperative nausea and vomiting in this population (see WARNINGS).
The pharmacokinetics, including clearance of intravenous ANZEMET Injection, in elderly and younger patients are similar (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Humans). Dosage adjustment is not needed in patients over the age of 65.
ADVERSE REACTIONS
Postoperative Patients
In controlled clinical trials with 2550 adult patients, headache and dizziness were reported more frequently with 12.5 mg ANZEMET Injection than with placebo. Rates of other adverse events were similar. Following is a listing of all adverse events reported in ≥2% of patients receiving either placebo or 12.5 mg ANZEMET Injection for the prevention or treatment of postoperative nausea and vomiting in controlled clinical trials (Table 2).
| Event | ANZEMET Injection 12.5 mg (n=615) | Placebo (n=739) |
|---|---|---|
| Headache | 58 (9.4%) | 51 (6.9%) |
| Dizziness | 34 (5.5%) | 23 (3.1%) |
| Drowsiness | 15 (2.4%) | 18 (2.4%) |
| Pain | 15 (2.4%) | 21 (2.8%) |
| Urinary Retention | 12 (2.0%) | 16 (2.2%) |
In clinical trials, the following reported adverse events, assessed by investigators as treatment-related or causality unknown, occurred following oral or intravenous administration of ANZEMET in < 2% of adult patients undergoing surgery:
Body as a Whole: Chills/shivering.
Cardiovascular: Sinus arrhythmia, hypotension, orthostatic hypotension. The following events also occurred and with a similar frequency as placebo and/or active comparator: Mobitz I AV block, chest pain, syncope, severe bradycardia, and palpitations. See PRECAUTIONS section for information on potential effects on ECG.
In addition, the following asymptomatic treatment-emergent ECG changes were seen at rates less than or equal to those for active or placebo controls: bradycardia, tachycardia, T wave change, ST-T wave change, extrasystole (APCs or VPCs), bundle branch block (left and right).
Furthermore, severe hypotension, bradycardia and syncope have been reported immediately or closely following IV administration.
Dermatologic: Rash.
Gastrointestinal System: Constipation, dyspepsia, abdominal pain.
Hearing, Taste and Vision: Taste perversion, abnormal vision.
Hypersensitivity: Anaphylactic reaction, urticaria.
Liver and Biliary System: Transient increases in AST (SGOT) and/or ALT (SGPT). The increases did not appear to be related to dose or duration of therapy and were not associated with symptomatic hepatic disease. Similar increases were seen with patients receiving active comparator.
Musculoskeletal: Myalgia, arthralgia.
Nervous System: Vertigo; flushing, paraesthesia.
Psychiatric: Agitation, anxiety, abnormal dreaming.
Respiratory System: Bronchospasm.
Vascular (Extracardiac): Local pain or burning on IV administration.
OVERDOSAGE
There is no known specific antidote for dolasetron mesylate, and patients with suspected overdose should be managed with supportive therapy. Individual doses as large as 5 mg/kg intravenously or 400 mg orally have been safely given to healthy volunteers or cancer patients.
Following a suspected overdose of ANZEMET Injection, a patient found to have second-degree or higher AV conduction block with ECG should undergo cardiac telemetry monitoring.
It is not known if dolasetron mesylate is removed by hemodialysis or peritoneal dialysis.
Single intravenous doses of dolasetron mesylate at 160 mg/kg in male mice and 140 mg/kg in female mice and rats of both sexes (6.3 to 12.6 times the recommended human dose based on body surface area) were lethal. Symptoms of acute toxicity were tremors, depression and convulsions.
A 59-year-old man with metastatic melanoma and no known pre-existing cardiac conditions developed severe hypotension and dizziness 40 minutes after receiving a 15 minute intravenous infusion of 1000 mg (13 mg/kg) of dolasetron mesylate. Treatment for the overdose consisted of infusion of 500 mL of a plasma expander, dopamine, and atropine. The patient had normal sinus rhythm and prolongation of PR, QRS and QTc intervals on an ECG recorded 2 hours after the infusion. The patient's blood pressure was normal 3 hours after the event and the ECG intervals returned to baseline on follow-up. The patient was released from the hospital 6 hours after the event.
A 7-year-old boy received 6 mg/kg dolasetron mesylate orally before surgery. No symptoms occurred and no treatment was required.
DOSAGE AND ADMINISTRATION
The recommended dose of ANZEMET Injection should not be exceeded.
Prevention or Treatment of Postoperative Nausea and/or Vomiting
Adults
The recommended intravenous dosage of ANZEMET Injection is 12.5 mg given as a single dose approximately 15 minutes before the cessation of anesthesia (prevention) or as soon as nausea or vomiting presents (treatment).
Pediatric Patients
Intravenous Administration
The recommended intravenous dosage in pediatric patients 2 to 16 years of age is 0.35 mg/kg, with a maximum dose of 12.5 mg, given as a single dose approximately 15 minutes before the cessation of anesthesia or as soon as nausea or vomiting presents. Safety and effectiveness in pediatric patients under 2 years of age have not been established.
Oral Administration of the Intravenous Product
ANZEMET Injection solution may be mixed into apple or apple-grape juice for oral dosing in pediatric patients. When ANZEMET Injection solution is administered orally, the recommended oral dosage in pediatric patients 2 to 16 years of age is 1.2 mg/kg up to a maximum 100-mg dose given within 2 hours before surgery. The diluted product may be kept up to 2 hours at room temperature before use.
Use in the Elderly, in Renal Failure Patients, or in Hepatically Impaired Patients
No dosage adjustment is recommended; however, ECG monitoring is recommended for elderly and renally impaired patients (see WARNINGS and CLINICAL PHARMACOLOGY, Pharmacokinetics in Humans).
ADMINISTRATION
ANZEMET Injection can be safely infused intravenously as rapidly as 30 seconds or diluted in a compatible intravenous solution (see below) to 50 mL and infused over a period of up to 15 minutes. ANZEMET Injection should not be mixed with other drugs. Flush the infusion line before and after administration of ANZEMET Injection.
STABILITY
After dilution, ANZEMET Injection is stable under normal lighting conditions at room temperature for 24 hours or under refrigeration for 48 hours with the following compatible intravenous fluids: 0.9% sodium chloride injection, 5% dextrose injection, 5% dextrose and 0.45% sodium chloride injection, 5% dextrose and Lactated Ringer's injection, Lactated Ringer's injection, and 10% mannitol injection. Although ANZEMET Injection is chemically and physically stable when diluted as recommended, sterile precautions should be observed because diluents generally do not contain preservative. After dilution, do not use beyond 24 hours, or 48 hours if refrigerated.
Parenteral drug products should be inspected visually for particulate matter and discoloration before administration whenever solution and container permit.
HOW SUPPLIED
ANZEMET Injection (dolasetron mesylate) is supplied as a clear, colorless solution in single and multidose vials.
| ANZEMET® Injection (dolasetron mesylate) 20 mg/mL |
||
|---|---|---|
| Strength | Description | NDC Number |
| 12.5 mg | 0.625mL single use vial (Box of 6) | 0088-1208-06 |
| 100 mg/5 mL | 5mL single-use vial | 0088-1206-32 |
| 500 mg/25 mL | 25 mL multidose vial | 0088-1209-26 |
PATIENT COUNSELING INFORMATION
Patients should be informed that ANZEMET may cause serious cardiac arrhythmias such as QT prolongation or heart block. Patients should be instructed to tell their health care provider right away if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode.
Patients should be informed that the chances of developing serious cardiac arrhythmias such as QT prolongation and Torsade de Pointes or heart block are higher in the following people:
- Patients with a personal or family history of abnormal heart rhythms, such as congenital long QT syndrome
- Patients with a personal history of sick sinus syndrome, atrial fibrillation with slow ventricular response or myocardial ischemia
- Patients who take medications that may prolong the PR interval, such as certain antihypertensives or medications that may prolong the QRS interval, such as antiarrythmic medications
- Patients who take medications, such as diuretics, which may cause electrolyte abnormalities
- Patients with hypokalemia or hypomagnesemia
- Elderly patients
ANZEMET should be avoided in these patients, since they may be more at risk for cardiac arrhythmias such as QT prolongation and Torsade de Pointes.
Advise patients of the possibility of serotonin syndrome with concomitant use of Anzemet and another serotonergic agent such as medications to treat depression and migraines. Advise patients to seek immediate medical attention if the following symptoms occur: changes in mental status, autonomic instability, neuromuscular symptoms with or without gastrointestinal symptoms.
Prescribing information as of October 2014
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
A SANOFI COMPANY
©2014 sanofi-aventis U.S. LLC
PRINCIPAL DISPLAY PANEL - 12.5mg Vial Label
NDC 0088-1208-06
Anzemet® Injection
dolasetron mesylate
injection
12.5mg/0.625mL



| ANZEMET
dolasetron mesylate injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ANZEMET
dolasetron mesylate injection |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| ANZEMET
dolasetron mesylate injection |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (783243835) |