FULL PRESCRIBING INFORMATION
WARNING: CARDIOMYOPATHY, INFUSION REACTIONS, EMBRYO-FETAL TOXICITY, and PULMONARY TOXICITY
Cardiomyopathy
Herceptin administration can result in sub-clinical and clinical cardiac failure. The incidence and severity was highest in patients receiving Herceptin with anthracycline-containing chemotherapy regimens.
Evaluate left ventricular function in all patients prior to and during treatment with Herceptin. Discontinue Herceptin treatment in patients receiving adjuvant therapy and withhold Herceptin in patients with metastatic disease for clinically significant decrease in left ventricular function [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
Infusion Reactions; Pulmonary Toxicity
Herceptin administration can result in serious and fatal infusion reactions and pulmonary toxicity. Symptoms usually occur during or within 24 hours of Herceptin administration. Interrupt Herceptin infusion for dyspnea or clinically significant hypotension. Monitor patients until symptoms completely resolve. Discontinue Herceptin for anaphylaxis, angioedema, interstitial pneumonitis, or acute respiratory distress syndrome [see Warnings and Precautions (5.2, 5.4)].
Embryo Fetal Toxicity
Exposure to Herceptin during pregnancy can result in oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Advise patients of these risks and the need for effective contraception [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1, 8.3)].
1 INDICATIONS AND USAGE
1.1 Adjuvant Breast Cancer
Herceptin is indicated for adjuvant treatment of HER2 overexpressing node positive or node negative (ER/PR negative or with one high risk feature [see Clinical Studies (14.1)]) breast cancer
- as part of a treatment regimen consisting of doxorubicin, cyclophosphamide, and either paclitaxel or docetaxel
- as part of a treatment regimen with docetaxel and carboplatin
- as a single agent following multi-modality anthracycline based therapy.
Select patients for therapy based on an FDA-approved companion diagnostic for Herceptin [see Dosage and Administration (2.1)].
1.2 Metastatic Breast Cancer
Herceptin is indicated:
- In combination with paclitaxel for first-line treatment of HER2-overexpressing metastatic breast cancer
- As a single agent for treatment of HER2-overexpressing breast cancer in patients who have received one or more chemotherapy regimens for metastatic disease.
Select patients for therapy based on an FDA-approved companion diagnostic for Herceptin [see Dosage and Administration (2.1)].
1.3 Metastatic Gastric Cancer
Herceptin is indicated, in combination with cisplatin and capecitabine or 5-fluorouracil, for the treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma who have not received prior treatment for metastatic disease.
Select patients for therapy based on an FDA-approved companion diagnostic for Herceptin [see Dosage and Administration (2.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients based on HER2 protein overexpression or HER2 gene amplification in tumor specimens [see Indications and Usage (1) and Clinical Studies (14)]. Assessment of HER2 protein overexpression and HER2 gene amplification should be performed using FDA-approved tests specific for breast or gastric cancers by laboratories with demonstrated proficiency. Information on the FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at: http://www.fda.gov/CompanionDiagnostics.
Assessment of HER2 protein overexpression and HER2 gene amplification in metastatic gastric cancer should be performed using FDA-approved tests specifically for gastric cancers due to differences in gastric vs. breast histopathology, including incomplete membrane staining and more frequent heterogeneous expression of HER2 seen in gastric cancers.
Improper assay performance, including use of suboptimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
2.2 Recommended Doses and Schedules
- Do not administer as an intravenous push or bolus. Do not mix Herceptin with other drugs.
- Do not substitute Herceptin (trastuzumab) for or with ado-trastuzumab emtansine.
Adjuvant Treatment, Breast Cancer
Administer according to one of the following doses and schedules for a total of 52 weeks of Herceptin therapy:
During and following paclitaxel, docetaxel, or docetaxel/carboplatin:
- Initial dose of 4 mg/kg as an intravenous infusion over 90 minutes then at 2 mg/kg as an intravenous infusion over 30 minutes weekly during chemotherapy for the first 12 weeks (paclitaxel or docetaxel) or 18 weeks (docetaxel/carboplatin).
- One week following the last weekly dose of Herceptin, administer Herceptin at 6 mg/kg as an intravenous infusion over 30–90 minutes every three weeks.
As a single agent within three weeks following completion of multi-modality, anthracycline-based chemotherapy regimens:
- Initial dose at 8 mg/kg as an intravenous infusion over 90 minutes
- Subsequent doses at 6 mg/kg as an intravenous infusion over 30–90 minutes every three weeks [see Dosage and Administration (2.3)].
- Extending adjuvant treatment beyond one year is not recommended [see Adverse Reactions (6.1)].
Metastatic Treatment, Breast Cancer
- Administer Herceptin, alone or in combination with paclitaxel, at an initial dose of 4 mg/kg as a 90-minute intravenous infusion followed by subsequent once weekly doses of 2 mg/kg as 30-minute intravenous infusions until disease progression.
Metastatic Gastric Cancer
- Administer Herceptin at an initial dose of 8 mg/kg as a 90-minute intravenous infusion followed by subsequent doses of 6 mg/kg as an intravenous infusion over 30–90 minutes every three weeks until disease progression [see Dosage and Administration (2.3)].
2.3 Important Dosing Considerations
If the patient has missed a dose of Herceptin by one week or less, then the usual maintenance dose (weekly schedule: 2 mg/kg; three-weekly schedule: 6 mg/kg) should be administered as soon as possible. Do not wait until the next planned cycle. Subsequent Herceptin maintenance doses should be administered 7 days or 21 days later according to the weekly or three-weekly schedules, respectively.
If the patient has missed a dose of Herceptin by more than one week, a re-loading dose of Herceptin should be administered over approximately 90 minutes (weekly schedule: 4 mg/kg; three-weekly schedule: 8 mg/kg) as soon as possible. Subsequent Herceptin maintenance doses (weekly schedule: 2 mg/kg; three-weekly schedule 6 mg/kg) should be administered 7 days or 21 days later according to the weekly or three-weekly schedules, respectively.
Infusion Reactions
[See Boxed Warning, Warnings and Precautions (5.2)]
- Decrease the rate of infusion for mild or moderate infusion reactions
- Interrupt the infusion in patients with dyspnea or clinically significant hypotension
- Discontinue Herceptin for severe or life-threatening infusion reactions.
Cardiomyopathy
[See Boxed Warning, Warnings and Precautions (5.1)]
Assess left ventricular ejection fraction (LVEF) prior to initiation of Herceptin and at regular intervals during treatment. Withhold Herceptin dosing for at least 4 weeks for either of the following:
- ≥ 16% absolute decrease in LVEF from pre-treatment values
- LVEF below institutional limits of normal and ≥ 10% absolute decrease in LVEF from pretreatment values.
Herceptin may be resumed if, within 4–8 weeks, the LVEF returns to normal limits and the absolute decrease from baseline is ≤ 15%.
Permanently discontinue Herceptin for a persistent (> 8 weeks) LVEF decline or for suspension of Herceptin dosing on more than 3 occasions for cardiomyopathy.
2.4 Preparation for Administration
To prevent medication errors, it is important to check the vial labels to ensure that the drug being prepared and administered is Herceptin (trastuzumab) and not ado-trastuzumab emtansine.
150 mg Single-dose vial
Reconstitution
Reconstitute each 150 mg vial of Herceptin with 7.4 mL of Sterile Water for Injection (SWFI) (not supplied) to yield a single-dose solution containing 21 mg/mL trastuzumab that delivers 7.15 mL (150 mg trastuzumab).
Use appropriate aseptic technique when performing the following reconstitution steps:
- Using a sterile syringe, slowly inject 7.4 mL of SWFI (not supplied) into the vial containing the lyophilized powder of Herceptin, which has a cake-like appearance. The stream of diluent should be directed into the cake. The reconstituted vial yields a solution for single-dose use, containing 21 mg/mL trastuzumab.
- Swirl the vial gently to aid reconstitution. DO NOT SHAKE.
- Slight foaming of the product may be present upon reconstitution. Allow the vial to stand undisturbed for approximately 5 minutes.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Inspect visually for particulates and discoloration. The solution should be free of visible particulates, clear to slightly opalescent and colorless to pale yellow.
- Use the Herceptin solution immediately following reconstitution with SWFI, as it contains no preservative and is intended for single-dose only. If not used immediately, store the reconstituted Herceptin solution for up to 24 hours at 2°C to 8°C (36°F to 46°F); discard any unused Herceptin after 24 hours. Do not freeze.
Dilution
- Determine the dose (mg) of Herceptin [see Dosage and Administration (2.1)].
- Calculate the volume of the 21 mg/mL reconstituted Herceptin solution needed.
- Withdraw this amount from the vial using a sterile needle and syringe and add it to an infusion bag containing 250 mL of 0.9% Sodium Chloride Injection, USP. DO NOT USE DEXTROSE (5%) SOLUTION.
- Gently invert the bag to mix the solution.
- The solution of Herceptin for infusion diluted in polyvinylchloride or polyethylene bags containing 0.9% Sodium Chloride Injection, USP, should be stored at 2°C to 8°C (36°F to 46°F) for no more than 24 hours prior to use. Discard after 24 hours. This storage time is additional to the time allowed for the reconstituted vials. Do not freeze.
5 WARNINGS AND PRECAUTIONS
5.1 Cardiomyopathy
Herceptin can cause left ventricular cardiac dysfunction, arrhythmias, hypertension, disabling cardiac failure, cardiomyopathy, and cardiac death [see Boxed Warning: Cardiomyopathy]. Herceptin can also cause asymptomatic decline in left ventricular ejection fraction (LVEF).
There is a 4–6 fold increase in the incidence of symptomatic myocardial dysfunction among patients receiving Herceptin as a single agent or in combination therapy compared with those not receiving Herceptin. The highest absolute incidence occurs when Herceptin is administered with an anthracycline.
Withhold Herceptin for ≥ 16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥ 10% absolute decrease in LVEF from pretreatment values [see Dosage and Administration (2.3)]. The safety of continuation or resumption of Herceptin in patients with Herceptin-induced left ventricular cardiac dysfunction has not been studied.
Patients who receive anthracycline after stopping Herceptin may also be at increased risk of cardiac dysfunction [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Cardiac Monitoring
Conduct thorough cardiac assessment, including history, physical examination, and determination of LVEF by echocardiogram or MUGA scan. The following schedule is recommended:
- Baseline LVEF measurement immediately prior to initiation of Herceptin
- LVEF measurements every 3 months during and upon completion of Herceptin
- Repeat LVEF measurement at 4 week intervals if Herceptin is withheld for significant left ventricular cardiac dysfunction [see Dosage and Administration (2.3)]
- LVEF measurements every 6 months for at least 2 years following completion of Herceptin as a component of adjuvant therapy.
In Study 1, 15% (158/1031) of patients discontinued Herceptin due to clinical evidence of myocardial dysfunction or significant decline in LVEF after a median follow-up duration of 8.7 years in the AC-TH arm. In Study 3 (one-year Herceptin treatment), the number of patients who discontinued Herceptin due to cardiac toxicity at 12.6 months median duration of follow-up was 2.6% (44/1678). In Study 4, a total of 2.9% (31/1056) of patients in the TCH arm (1.5% during the chemotherapy phase and 1.4% during the monotherapy phase) and 5.7% (61/1068) of patients in the AC-TH arm (1.5% during the chemotherapy phase and 4.2% during the monotherapy phase) discontinued Herceptin due to cardiac toxicity.
Among 64 patients receiving adjuvant chemotherapy (Studies 1 and 2) who developed congestive heart failure, one patient died of cardiomyopathy, one patient died suddenly without documented etiology, and 33 patients were receiving cardiac medication at last follow-up. Approximately 24% of the surviving patients had recovery to a normal LVEF (defined as ≥50%) and no symptoms on continuing medical management at the time of last follow-up. Incidence of congestive heart failure (CHF) is presented in Table 1. The safety of continuation or resumption of Herceptin in patients with Herceptin-induced left ventricular cardiac dysfunction has not been studied.
| Incidence of CHF | |||
| Study | Regimen | Herceptin | Control |
|
|||
| 1 & 2* | AC† → Paclitaxel+Herceptin | 3.2% (64/2000)‡ | 1.3% (21/1655) |
| 3§ | Chemo → Herceptin | 2% (30/1678) | 0.3% (5/1708) |
| 4 | AC† → Docetaxel+Herceptin | 2% (20/1068) | 0.3% (3/1050) |
| 4 | Docetaxel+Carbo+Herceptin | 0.4% (4/1056) | 0.3% (3/1050) |
In Study 3 (one-year Herceptin treatment), at a median follow-up duration of 8 years, the incidence of severe CHF (NYHA III & IV) was 0.8%, and the rate of mild symptomatic and asymptomatic left ventricular dysfunction was 4.6%.
| Incidence | |||||
| NYHA I-IV | NYHA III-IV | ||||
| Study | Event | Herceptin | Control | Herceptin | Control |
| 5 (AC)† | Cardiac Dysfunction | 28% | 7% | 19% | 3% |
| 5 (paclitaxel) | Cardiac Dysfunction | 11% | 1% | 4% | 1% |
| 6 | Cardiac Dysfunction‡ | 7% | N/A | 5% | N/A |
In Study 4, the incidence of NCI-CTC Grade 3/4 cardiac ischemia/infarction was higher in the Herceptin containing regimens (AC-TH: 0.3% (3/1068) and TCH: 0.2% (2/1056)) as compared to none in AC-T.
5.2 Infusion Reactions
Infusion reactions consist of a symptom complex characterized by fever and chills, and on occasion included nausea, vomiting, pain (in some cases at tumor sites), headache, dizziness, dyspnea, hypotension, rash, and asthenia [see Adverse Reactions (6.1)].
In post-marketing reports, serious and fatal infusion reactions have been reported. Severe reactions, which include bronchospasm, anaphylaxis, angioedema, hypoxia, and severe hypotension, were usually reported during or immediately following the initial infusion. However, the onset and clinical course were variable, including progressive worsening, initial improvement followed by clinical deterioration, or delayed post-infusion events with rapid clinical deterioration. For fatal events, death occurred within hours to days following a serious infusion reaction.
Interrupt Herceptin infusion in all patients experiencing dyspnea, clinically significant hypotension, and intervention of medical therapy administered (which may include epinephrine, corticosteroids, diphenhydramine, bronchodilators, and oxygen). Patients should be evaluated and carefully monitored until complete resolution of signs and symptoms. Permanent discontinuation should be strongly considered in all patients with severe infusion reactions.
There are no data regarding the most appropriate method of identification of patients who may safely be retreated with Herceptin after experiencing a severe infusion reaction. Prior to resumption of Herceptin infusion, the majority of patients who experienced a severe infusion reaction were pre-medicated with antihistamines and/or corticosteroids. While some patients tolerated Herceptin infusions, others had recurrent severe infusion reactions despite pre-medications.
5.3 Embryo-Fetal Toxicity
Herceptin can cause fetal harm when administered to a pregnant woman. In post marketing reports, use of Herceptin during pregnancy resulted in cases of oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death.
Verify the pregnancy status of females of reproductive potential prior to the initiation of Herceptin. Advise pregnant women and females of reproductive potential that exposure to Herceptin during pregnancy or within 7 months prior to conception can result in fetal harm. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of Herceptin [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.3)].
5.4 Pulmonary Toxicity
Herceptin use can result in serious and fatal pulmonary toxicity. Pulmonary toxicity includes dyspnea, interstitial pneumonitis, pulmonary infiltrates, pleural effusions, non-cardiogenic pulmonary edema, pulmonary insufficiency and hypoxia, acute respiratory distress syndrome, and pulmonary fibrosis. Such events can occur as sequelae of infusion reactions [see Warnings and Precautions (5.2)]. Patients with symptomatic intrinsic lung disease or with extensive tumor involvement of the lungs, resulting in dyspnea at rest, appear to have more severe toxicity.
5.5 Exacerbation of Chemotherapy-Induced Neutropenia
In randomized, controlled clinical trials, the per-patient incidences of NCI-CTC Grade 3–4 neutropenia and of febrile neutropenia were higher in patients receiving Herceptin in combination with myelosuppressive chemotherapy as compared to those who received chemotherapy alone. The incidence of septic death was similar among patients who received Herceptin and those who did not [see Adverse Reactions (6.1)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Cardiomyopathy [see Warnings and Precautions (5.1)]
- Infusion Reactions [see Warnings and Precautions (5.2)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.3)]
- Pulmonary Toxicity [see Warnings and Precautions (5.4)]
- Exacerbation of Chemotherapy-Induced Neutropenia [see Warnings and Precautions (5.5)]
The most common adverse reactions in patients receiving Herceptin in the adjuvant and metastatic breast cancer setting are fever, nausea, vomiting, infusion reactions, diarrhea, infections, increased cough, headache, fatigue, dyspnea, rash, neutropenia, anemia, and myalgia. Adverse reactions requiring interruption or discontinuation of Herceptin treatment include CHF, significant decline in left ventricular cardiac function, severe infusion reactions, and pulmonary toxicity [see Dosage and Administration (2.3)].
In the metastatic gastric cancer setting, the most common adverse reactions (≥ 10%) that were increased (≥ 5% difference) in the Herceptin arm as compared to the chemotherapy alone arm were neutropenia, diarrhea, fatigue, anemia, stomatitis, weight loss, upper respiratory tract infections, fever, thrombocytopenia, mucosal inflammation, nasopharyngitis, and dysgeusia. The most common adverse reactions which resulted in discontinuation of treatment on the Herceptin-containing arm in the absence of disease progression were infection, diarrhea, and febrile neutropenia.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adjuvant Breast Cancer Studies
The data below reflect exposure to one-year Herceptin therapy across three randomized, open-label studies, Studies 1, 2, and 3, with (n = 3678) or without (n = 3363) trastuzumab in the adjuvant treatment of breast cancer.
The data summarized in Table 3 below, from Study 3, reflect exposure to Herceptin in 1678 patients; the median treatment duration was 51 weeks and median number of infusions was 18. Among the 3386 patients enrolled in the observation and one-year Herceptin arms of Study 3 at a median duration of follow-up of 12.6 months in the Herceptin arm, the median age was 49 years (range: 21 to 80 years), 83% of patients were Caucasian, and 13% were Asian.
| One Year Herceptin | Observation | |
| Adverse Reaction | (n = 1678) | (n = 1708) |
|
Cardiac | ||
| Hypertension | 64 (4%) | 35 (2%) |
| Dizziness | 60 (4%) | 29 (2%) |
| Ejection Fraction Decreased | 58 (3.5%) | 11 (0.6%) |
| Palpitations | 48 (3%) | 12 (0.7%) |
| Cardiac Arrhythmias‡ | 40 (3%) | 17 (1%) |
| Cardiac Failure Congestive | 30 (2%) | 5 (0.3%) |
| Cardiac Failure | 9 (0.5%) | 4 (0.2%) |
| Cardiac Disorder | 5 (0.3%) | 0 (0%) |
| Ventricular Dysfunction | 4 (0.2%) | 0 (0%) |
|
Respiratory Thoracic Mediastinal Disorders |
||
| Cough | 81 (5%) | 34 (2%) |
| Influenza | 70 (4%) | 9 (0.5%) |
| Dyspnea | 57 (3%) | 26 (2%) |
| URI | 46 (3%) | 20 (1%) |
| Rhinitis | 36 (2%) | 6 (0.4%) |
| Pharyngolaryngeal Pain | 32 (2%) | 8 (0.5%) |
| Sinusitis | 26 (2%) | 5 (0.3%) |
| Epistaxis | 25 (2%) | 1 (0.06%) |
| Pulmonary Hypertension | 4 (0.2%) | 0 (0%) |
| Interstitial Pneumonitis | 4 (0.2%) | 0 (0%) |
|
Gastrointestinal Disorders |
||
| Diarrhea | 123 (7%) | 16 (1%) |
| Nausea | 108 (6%) | 19 (1%) |
| Vomiting | 58 (3.5%) | 10 (0.6%) |
| Constipation | 33 (2%) | 17 (1%) |
| Dyspepsia | 30 (2%) | 9 (0.5%) |
| Upper Abdominal Pain | 29 (2%) | 15 (1%) |
|
Musculoskeletal & Connective Tissue Disorders |
||
| Arthralgia | 137 (8%) | 98 (6%) |
| Back Pain | 91 (5%) | 58 (3%) |
| Myalgia | 63 (4%) | 17 (1%) |
| Bone Pain | 49 (3%) | 26 (2%) |
| Muscle Spasm | 46 (3%) | 3 (0.2%) |
|
Nervous System Disorders |
||
| Headache | 162 (10%) | 49 (3%) |
| Paraesthesia | 29 (2%) | 11 (0.6%) |
|
Skin & Subcutaneous Tissue Disorders |
||
| Rash | 70 (4%) | 10 (0.6%) |
| Nail Disorders | 43 (2%) | 0 (0%) |
| Pruritus | 40 (2%) | 10 (0.6%) |
|
General disorders |
||
| Pyrexia | 100 (6%) | 6 (0.4%) |
| Edema Peripheral | 79 (5%) | 37 (2%) |
| Chills | 85 (5%) | 0 (0%) |
| Asthenia | 75 (4.5%) | 30 (2%) |
| Influenza-like Illness | 40 (2%) | 3 (0.2%) |
| Sudden Death | 1 (0.06%) | 0 (0%) |
|
Infections |
||
| Nasopharyngitis | 135 (8%) | 43 (3%) |
| UTI | 39 (3%) | 13 (0.8%) |
|
Immune System Disorders |
||
| Hypersensitivity | 10 (0.6%) | 1 (0.06%) |
| Autoimmune Thyroiditis | 4 (0.3%) | 0 (0%) |
In Study 3, a comparison of 3-weekly Herceptin treatment for two years versus one year was also performed. The rate of asymptomatic cardiac dysfunction was increased in the 2-year Herceptin treatment arm (8.1% versus 4.6% in the one-year Herceptin treatment arm). More patients experienced at least one adverse reaction of Grade 3 or higher in the 2-year Herceptin treatment arm (20.4%) compared with the one-year Herceptin treatment arm (16.3%).
The safety data from Studies 1 and 2 were obtained from 3655 patients, of whom 2000 received Herceptin; the median treatment duration was 51 weeks. The median age was 49 years (range: 24–80); 84% of patients were White, 7% Black, 4% Hispanic, and 3% Asian.
In Study 1, only Grade 3–5 adverse events, treatment-related Grade 2 events, and Grade 2–5 dyspnea were collected during and for up to 3 months following protocol-specified treatment. The following non-cardiac adverse reactions of Grade 2–5 occurred at an incidence of at least 2% greater among patients receiving Herceptin plus chemotherapy as compared to chemotherapy alone: fatigue (29.5% vs. 22.4%), infection (24.0% vs. 12.8%), hot flashes (17.1% vs. 15.0%), anemia (12.3% vs. 6.7%), dyspnea (11.8% vs. 4.6%), rash/desquamation (10.9% vs. 7.6%), leukopenia (10.5% vs. 8.4%), neutropenia (6.4% vs. 4.3%), headache (6.2% vs. 3.8%), pain (5.5% vs. 3.0%), edema (4.7% vs. 2.7%), and insomnia (4.3% vs. 1.5%). The majority of these events were Grade 2 in severity.
In Study 2, data collection was limited to the following investigator-attributed treatment-related adverse reactions: NCI-CTC Grade 4 and 5 hematologic toxicities, Grade 3–5 non-hematologic toxicities, selected Grade 2–5 toxicities associated with taxanes (myalgia, arthralgias, nail changes, motor neuropathy, and sensory neuropathy) and Grade 1–5 cardiac toxicities occurring during chemotherapy and/or Herceptin treatment. The following non-cardiac adverse reactions of Grade 2–5 occurred at an incidence of at least 2% greater among patients receiving Herceptin plus chemotherapy as compared to chemotherapy alone: arthralgia (12.2% vs. 9.1%), nail changes (11.5% vs. 6.8%), dyspnea (2.4% vs. 0.2%), and diarrhea (2.2% vs. 0%). The majority of these events were Grade 2 in severity.
Safety data from Study 4 reflect exposure to Herceptin as part of an adjuvant treatment regimen from 2124 patients receiving at least one dose of study treatment [AC-TH: n = 1068; TCH: n = 1056]. The overall median treatment duration was 54 weeks in both the AC-TH and TCH arms. The median number of infusions was 26 in the AC-TH arm and 30 in the TCH arm, including weekly infusions during the chemotherapy phase and every three week dosing in the monotherapy period. Among these patients, the median age was 49 years (range 22 to 74 years). In Study 4, the toxicity profile was similar to that reported in Studies 1, 2, and 3 with the exception of a low incidence of CHF in the TCH arm.
Metastatic Breast Cancer Studies
The data below reflect exposure to Herceptin in one randomized, open-label study, Study 5, of chemotherapy with (n = 235) or without (n = 234) trastuzumab in patients with metastatic breast cancer, and one single-arm study (Study 6; n = 222) in patients with metastatic breast cancer. Data in Table 4 are based on Studies 5 and 6.
Among the 464 patients treated in Study 5, the median age was 52 years (range: 25–77 years). Eighty-nine percent were White, 5% Black, 1% Asian, and 5% other racial/ethnic groups. All patients received 4 mg/kg initial dose of Herceptin followed by 2 mg/kg weekly. The percentages of patients who received Herceptin treatment for ≥ 6 months and ≥ 12 months were 58% and 9%, respectively.
Among the 352 patients treated in single agent studies (213 patients from Study 6), the median age was 50 years (range 28–86 years), 86% were White, 3% were Black, 3% were Asian, and 8% in other racial/ethnic groups. Most of the patients received 4 mg/kg initial dose of Herceptin followed by 2 mg/kg weekly. The percentages of patients who received Herceptin treatment for ≥ 6 months and ≥ 12 months were 31% and 16%, respectively.
| Single Agent* n = 352 | Herceptin + Paclitaxel n = 91 | Paclitaxel Alone n = 95 | Herceptin + AC†
n = 143 | AC† Alone n = 135 |
||
| Body as a Whole | ||||||
| Pain | 47% | 61% | 62% | 57% | 42% | |
| Asthenia | 42% | 62% | 57% | 54% | 55% | |
| Fever | 36% | 49% | 23% | 56% | 34% | |
| Chills | 32% | 41% | 4% | 35% | 11% | |
| Headache | 26% | 36% | 28% | 44% | 31% | |
| Abdominal pain | 22% | 34% | 22% | 23% | 18% | |
| Back pain | 22% | 34% | 30% | 27% | 15% | |
| Infection | 20% | 47% | 27% | 47% | 31% | |
| Flu syndrome | 10% | 12% | 5% | 12% | 6% | |
| Accidental injury | 6% | 13% | 3% | 9% | 4% | |
| Allergic reaction | 3% | 8% | 2% | 4% | 2% | |
| Cardiovascular | ||||||
| Tachycardia | 5% | 12% | 4% | 10% | 5% | |
| Congestive heart failure | 7% | 11% | 1% | 28% | 7% | |
| Digestive | ||||||
| Nausea | 33% | 51% | 9% | 76% | 77% | |
| Diarrhea | 25% | 45% | 29% | 45% | 26% | |
| Vomiting | 23% | 37% | 28% | 53% | 49% | |
| Nausea and vomiting | 8% | 14% | 11% | 18% | 9% | |
| Anorexia | 14% | 24% | 16% | 31% | 26% | |
| Heme & Lymphatic | ||||||
| Anemia | 4% | 14% | 9% | 36% | 26% | |
| Leukopenia | 3% | 24% | 17% | 52% | 34% | |
| Metabolic | ||||||
| Peripheral edema | 10% | 22% | 20% | 20% | 17% | |
| Edema | 8% | 10% | 8% | 11% | 5% | |
| Musculoskeletal | ||||||
| Bone pain | 7% | 24% | 18% | 7% | 7% | |
| Arthralgia | 6% | 37% | 21% | 8% | 9% | |
| Nervous | ||||||
| Insomnia | 14% | 25% | 13% | 29% | 15% | |
| Dizziness | 13% | 22% | 24% | 24% | 18% | |
| Paresthesia | 9% | 48% | 39% | 17% | 11% | |
| Depression | 6% | 12% | 13% | 20% | 12% | |
| Peripheral neuritis | 2% | 23% | 16% | 2% | 2% | |
| Neuropathy | 1% | 13% | 5% | 4% | 4% | |
| Respiratory | ||||||
| Cough increased | 26% | 41% | 22% | 43% | 29% | |
| Dyspnea | 22% | 27% | 26% | 42% | 25% | |
| Rhinitis | 14% | 22% | 5% | 22% | 16% | |
| Pharyngitis | 12% | 22% | 14% | 30% | 18% | |
| Sinusitis | 9% | 21% | 7% | 13% | 6% | |
| Skin | ||||||
| Rash | 18% | 38% | 18% | 27% | 17% | |
| Herpes simplex | 2% | 12% | 3% | 7% | 9% | |
| Acne | 2% | 11% | 3% | 3% | < 1% | |
| Urogenital | ||||||
| Urinary tract infection | 5% | 18% | 14% | 13% | 7% | |
Metastatic Gastric Cancer
The data below are based on the exposure of 294 patients to Herceptin in combination with a fluoropyrimidine (capecitabine or 5-FU) and cisplatin (Study 7). In the Herceptin plus chemotherapy arm, the initial dose of Herceptin 8 mg/kg was administered on Day 1 (prior to chemotherapy) followed by 6 mg/kg every 21 days until disease progression. Cisplatin was administered at 80 mg/m2 on Day 1 and the fluoropyrimidine was administered as either capecitabine 1000 mg/m2 orally twice a day on Days 1-14 or 5-fluorouracil 800 mg/m2/day as a continuous intravenous infusion Days 1 through 5. Chemotherapy was administered for six 21-day cycles. Median duration of Herceptin treatment was 21 weeks; median number of Herceptin infusions administered was eight.
| Herceptin + FC (N = 294) N (%) | FC (N = 290) N (%) |
|||
| Body System/ Adverse Event | All Grades | Grades 3/4 | All Grades | Grades 3/4 |
| Investigations | ||||
| Neutropenia | 230 (78) | 101 (34) | 212 (73) | 83 (29) |
| Hypokalemia | 83 (28) | 28 (10) | 69 (24) | 16 (6) |
| Anemia | 81 (28) | 36 (12) | 61 (21) | 30 (10) |
| Thrombocytopenia | 47 (16) | 14 (5) | 33 (11) | 8 (3) |
| Blood and Lymphatic System Disorders | ||||
| Febrile Neutropenia | — | 15 ( 5) | — | 8 (3) |
| Gastrointestinal Disorders | ||||
| Diarrhea | 109 (37) | 27 (9) | 80 (28) | 11 (4) |
| Stomatitis | 72 (24) | 2 (1) | 43 (15) | 6 (2) |
| Dysphagia | 19 (6) | 7 (2) | 10 (3) | 1 (≤ 1) |
| Body as a Whole | ||||
| Fatigue | 102 (35) | 12 (4) | 82 (28) | 7 (2) |
| Fever | 54 (18) | 3 (1) | 36 (12) | 0 (0) |
| Mucosal Inflammation | 37 (13) | 6 (2) | 18 (6) | 2 (1) |
| Chills | 23 (8) | 1 (≤ 1) | 0 (0) | 0 (0) |
| Metabolism and Nutrition Disorders | ||||
| Weight Decrease | 69 (23) | 6 (2) | 40 (14) | 7 (2) |
| Infections and Infestations | ||||
| Upper Respiratory Tract Infections | 56 (19) | 0 (0) | 29 (10) | 0 (0) |
| Nasopharyngitis | 37 (13) | 0 (0) | 17 (6) | 0 (0) |
| Renal and Urinary Disorders | ||||
| Renal Failure and Impairment | 53 (18) | 8 (3) | 42 (15) | 5 (2) |
| Nervous System Disorders | ||||
| Dysgeusia | 28 (10) | 0 (0) | 14 (5) | 0 (0) |
The following subsections provide additional detail regarding adverse reactions observed in clinical trials of adjuvant breast cancer, metastatic breast cancer, metastatic gastric cancer, or post-marketing experience.
Cardiomyopathy
Serial measurement of cardiac function (LVEF) was obtained in clinical trials in the adjuvant treatment of breast cancer. In Study 3, the median duration of follow-up was 12.6 months (12.4 months in the observation arm; 12.6 months in the 1-year Herceptin arm); and in Studies 1 and 2, 7.9 years in the AC-T arm, 8.3 years in the AC-TH arm. In Studies 1 and 2, 6% of all randomized patients with post-AC LVEF evaluation were not permitted to initiate Herceptin following completion of AC chemotherapy due to cardiac dysfunction (LVEF < LLN or ≥ 16 point decline in LVEF from baseline to end of AC). Following initiation of Herceptin therapy, the incidence of new-onset dose-limiting myocardial dysfunction was higher among patients receiving Herceptin and paclitaxel as compared to those receiving paclitaxel alone in Studies 1 and 2, and in patients receiving one-year Herceptin monotherapy compared to observation in Study 3 (see Table 6, Figures 1 and 2). The per-patient incidence of new-onset cardiac dysfunction, as measured by LVEF, remained similar when compared to the analysis performed at a median follow-up of 2.0 years in the AC-TH arm. This analysis also showed evidence of reversibility of left ventricular dysfunction, with 64.5% of patients who experienced symptomatic CHF in the AC-TH group being asymptomatic at latest follow-up, and 90.3% having full or partial LVEF recovery.
| LVEF < 50% and Absolute Decrease from Baseline | Absolute LVEF Decrease | ||||
| LVEF < 50% | ≥ 10% decrease | ≥ 16% decrease | < 20% and ≥ 10% | ≥ 20% | |
|
|||||
| Studies 1 & 2†,‡ | |||||
| AC→TH (n = 1856) | 23.1% (428) | 18.5% (344) | 11.2% (208) | 37.9% (703) | 8.9% (166) |
| AC→T (n = 1170) | 11.7% (137) | 7.0% (82) | 3.0% (35) | 22.1% (259) | 3.4% (40) |
| Study 3§ | |||||
| Herceptin (n = 1678) | 8.6% (144) | 7.0% (118) | 3.8% (64) | 22.4% (376) | 3.5% (59) |
| Observation (n = 1708) | 2.7% (46) | 2.0% (35) | 1.2% (20) | 11.9% (204) | 1.2% (21) |
| Study 4¶ | |||||
| TCH (n = 1056) | 8.5% (90) | 5.9% (62) | 3.3% (35) | 34.5% (364) | 6.3% (67) |
| AC→TH (n = 1068) | 17% (182) | 13.3% (142) | 9.8% (105) | 44.3% (473) | 13.2% (141) |
| AC→T (n = 1050) | 9.5% (100) | 6.6% (69) | 3.3% (35) | 34% (357) | 5.5% (58) |
Figure 1
Studies 1 and 2: Cumulative Incidence of Time to First LVEF Decline of ≥ 10 Percentage Points from Baseline and to Below 50% with Death as a Competing Risk Event
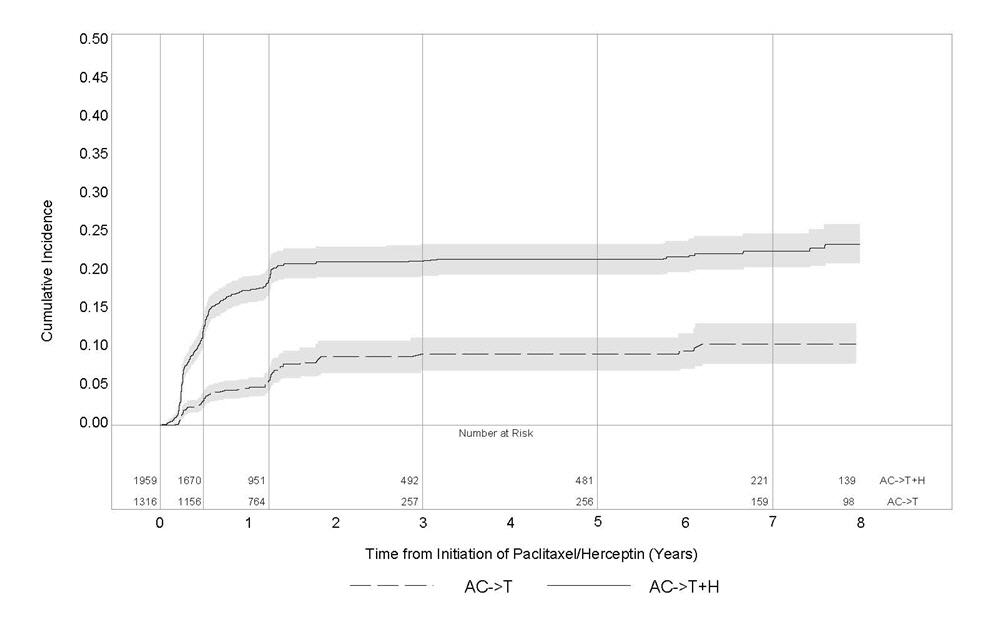
Time 0 is initiation of paclitaxel or Herceptin + paclitaxel therapy.
Figure 2
Study 3: Cumulative Incidence of Time to First LVEF Decline of ≥ 10 Percentage Points from Baseline and to Below 50% with Death as a Competing Risk Event
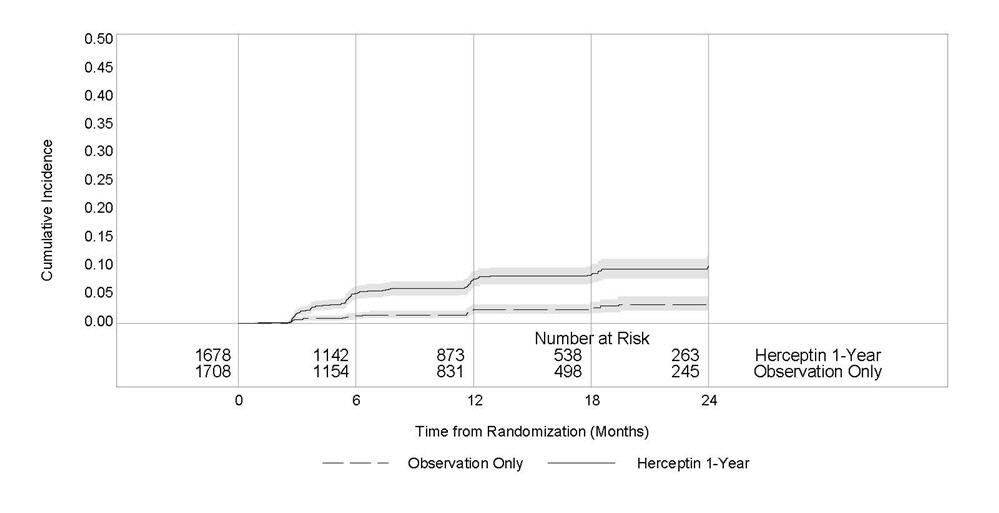
Time 0 is the date of randomization.
Figure 3
Study 4: Cumulative Incidence of Time to First LVEF Decline of ≥10 Percentage Points from Baseline and to Below 50% with Death as a Competing Risk Event
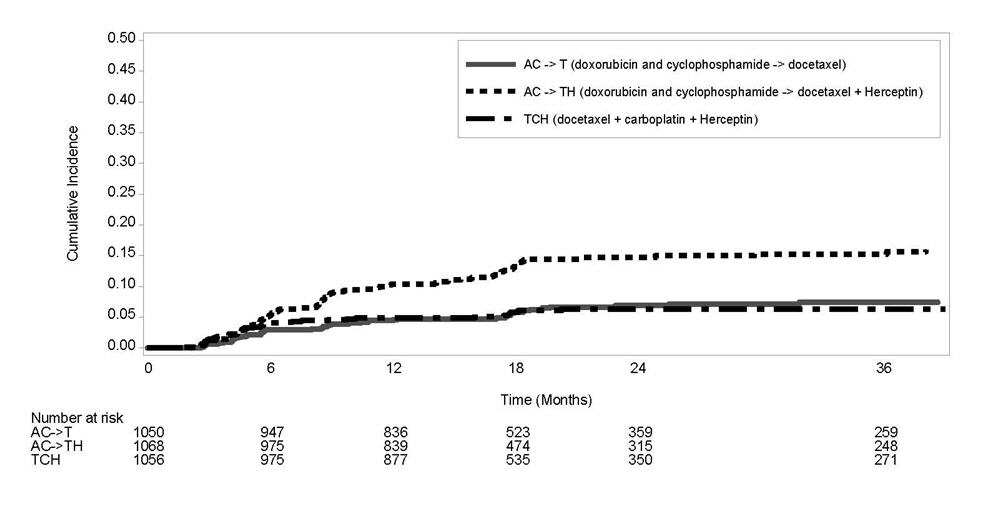
Time 0 is the date of randomization.
The incidence of treatment emergent congestive heart failure among patients in the metastatic breast cancer trials was classified for severity using the New York Heart Association classification system (I–IV, where IV is the most severe level of cardiac failure) (see Table 2). In the metastatic breast cancer trials, the probability of cardiac dysfunction was highest in patients who received Herceptin concurrently with anthracyclines.
In Study 7, 5.0% of patients in the Herceptin plus chemotherapy arm compared to 1.1% of patients in the chemotherapy alone arm had LVEF value below 50% with a ≥ 10% absolute decrease in LVEF from pretreatment values.
Infusion Reactions
During the first infusion with Herceptin, the symptoms most commonly reported were chills and fever, occurring in approximately 40% of patients in clinical trials. Symptoms were treated with acetaminophen, diphenhydramine, and meperidine (with or without reduction in the rate of Herceptin infusion); permanent discontinuation of Herceptin for infusion reactions was required in < 1% of patients. Other signs and/or symptoms may include nausea, vomiting, pain (in some cases at tumor sites), rigors, headache, dizziness, dyspnea, hypotension, elevated blood pressure, rash, and asthenia. Infusion reactions occurred in 21% and 35% of patients, and were severe in 1.4% and 9% of patients, on second or subsequent Herceptin infusions administered as monotherapy or in combination with chemotherapy, respectively. In the post-marketing setting, severe infusion reactions, including hypersensitivity, anaphylaxis, and angioedema have been reported.
Anemia
In randomized controlled clinical trials, the overall incidence of anemia (30% vs. 21% [Study 5]), of selected NCI-CTC Grade 2–5 anemia (12.3% vs. 6.7% [Study 1]), and of anemia requiring transfusions (0.1% vs. 0 patients [Study 2]) were increased in patients receiving Herceptin and chemotherapy compared with those receiving chemotherapy alone. Following the administration of Herceptin as a single agent (Study 6), the incidence of NCI-CTC Grade 3 anemia was < 1%. In Study 7 (metastatic gastric cancer), on the Herceptin containing arm as compared to the chemotherapy alone arm, the overall incidence of anemia was 28% compared to 21% and of NCI-CTC Grade 3/4 anemia was 12.2% compared to 10.3%.
Neutropenia
In randomized controlled clinical trials in the adjuvant setting, the incidence of selected NCI-CTC Grade 4–5 neutropenia (1.7% vs. 0.8% [Study 2]) and of selected Grade 2–5 neutropenia (6.4% vs. 4.3% [Study 1]) were increased in patients receiving Herceptin and chemotherapy compared with those receiving chemotherapy alone. In a randomized, controlled trial in patients with metastatic breast cancer, the incidences of NCI-CTC Grade 3/4 neutropenia (32% vs. 22%) and of febrile neutropenia (23% vs. 17%) were also increased in patients randomized to Herceptin in combination with myelosuppressive chemotherapy as compared to chemotherapy alone. In Study 7 (metastatic gastric cancer) on the Herceptin containing arm as compared to the chemotherapy alone arm, the incidence of NCI-CTC Grade 3/4 neutropenia was 36.8% compared to 28.9%; febrile neutropenia 5.1% compared to 2.8%.
Infection
The overall incidences of infection (46% vs. 30% [Study 5]), of selected NCI-CTC Grade 2–5 infection/febrile neutropenia (24.3% vs. 13.4% [Study 1]) and of selected Grade 3–5 infection/febrile neutropenia (2.9% vs. 1.4% [Study 2]) were higher in patients receiving Herceptin and chemotherapy compared with those receiving chemotherapy alone. The most common site of infections in the adjuvant setting involved the upper respiratory tract, skin, and urinary tract.
In Study 4, the overall incidence of infection was higher with the addition of Herceptin to AC-T but not to TCH [44% (AC-TH), 37% (TCH), 38% (AC-T)]. The incidences of NCI-CTC Grade 3-4 infection were similar [25% (AC-TH), 21% (TCH), 23% (AC-T)] across the three arms.
In a randomized, controlled trial in treatment of metastatic breast cancer, the reported incidence of febrile neutropenia was higher (23% vs. 17%) in patients receiving Herceptin in combination with myelosuppressive chemotherapy as compared to chemotherapy alone.
Pulmonary Toxicity
Adjuvant Breast Cancer
Among women receiving adjuvant therapy for breast cancer, the incidence of selected NCI-CTC Grade 2–5 pulmonary toxicity (14.3% vs. 5.4% [Study 1]) and of selected NCI-CTC Grade 3–5 pulmonary toxicity and spontaneous reported Grade 2 dyspnea (3.4% vs. 0.9% [Study 2]) was higher in patients receiving Herceptin and chemotherapy compared with chemotherapy alone. The most common pulmonary toxicity was dyspnea (NCI-CTC Grade 2–5: 11.8% vs. 4.6% [Study 1]; NCI-CTC Grade 2–5: 2.4% vs. 0.2% [Study 2]).
Pneumonitis/pulmonary infiltrates occurred in 0.7% of patients receiving Herceptin compared with 0.3% of those receiving chemotherapy alone. Fatal respiratory failure occurred in 3 patients receiving Herceptin, one as a component of multi-organ system failure, as compared to 1 patient receiving chemotherapy alone.
In Study 3, there were 4 cases of interstitial pneumonitis in the one-year Herceptin treatment arm compared to none in the observation arm at a median follow-up duration of 12.6 months.
Metastatic Breast Cancer
Among women receiving Herceptin for treatment of metastatic breast cancer, the incidence of pulmonary toxicity was also increased. Pulmonary adverse events have been reported in the post-marketing experience as part of the symptom complex of infusion reactions. Pulmonary events include bronchospasm, hypoxia, dyspnea, pulmonary infiltrates, pleural effusions, non-cardiogenic pulmonary edema, and acute respiratory distress syndrome. For a detailed description, see Warnings and Precautions (5.4).
Thrombosis/Embolism
In 4 randomized, controlled clinical trials, the incidence of thrombotic adverse events was higher in patients receiving Herceptin and chemotherapy compared to chemotherapy alone in three studies (2.6% vs. 1.5% [Study 1], 2.5% and 3.7% vs. 2.2% [Study 4] and 2.1% vs. 0% [Study 5]).
Diarrhea
Among women receiving adjuvant therapy for breast cancer, the incidence of NCI-CTC Grade 2–5 diarrhea (6.7% vs. 5.4% [Study 1]) and of NCI-CTC Grade 3–5 diarrhea (2.2% vs. 0% [Study 2]), and of Grade 1–4 diarrhea (7% vs. 1% [Study 3; one-year Herceptin treatment at 12.6 months median duration of follow-up]) were higher in patients receiving Herceptin as compared to controls. In Study 4, the incidence of Grade 3–4 diarrhea was higher [5.7% AC-TH, 5.5% TCH vs. 3.0% AC-T] and of Grade 1–4 was higher [51% AC-TH, 63% TCH vs. 43% AC-T] among women receiving Herceptin. Of patients receiving Herceptin as a single agent for the treatment of metastatic breast cancer, 25% experienced diarrhea. An increased incidence of diarrhea was observed in patients receiving Herceptin in combination with chemotherapy for treatment of metastatic breast cancer.
Renal Toxicity
In Study 7 (metastatic gastric cancer) on the Herceptin-containing arm as compared to the chemotherapy alone arm the incidence of renal impairment was 18% compared to 14.5%. Severe (Grade 3/4) renal failure was 2.7% on the Herceptin-containing arm compared to 1.7% on the chemotherapy only arm. Treatment discontinuation for renal insufficiency/failure was 2% on the Herceptin-containing arm and 0.3% on the chemotherapy only arm.
In the post-marketing setting, rare cases of nephrotic syndrome with pathologic evidence of glomerulopathy have been reported. The time to onset ranged from 4 months to approximately 18 months from initiation of Herceptin therapy. Pathologic findings included membranous glomerulonephritis, focal glomerulosclerosis, and fibrillary glomerulonephritis. Complications included volume overload and congestive heart failure.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. Among 903 women with metastatic breast cancer, human anti-human antibody (HAHA) to Herceptin was detected in one patient using an enzyme-linked immunosorbent assay (ELISA). This patient did not experience an allergic reaction. Samples for assessment of HAHA were not collected in studies of adjuvant breast cancer.
The incidence of antibody formation is highly dependent on the sensitivity and the specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Herceptin with the incidence of antibodies to other products may be misleading.
6.3 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of Herceptin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Infusion reaction [see Warnings and Precautions (5.2)]
- Oligohydramnios or oligohydramnios sequence, including pulmonary hypoplasia, skeletal abnormalities, and neonatal death [see Warnings and Precautions (5.3)]
- Glomerulopathy [see Adverse Reactions (6.1)]
- Immune thrombocytopenia
- Tumor lysis syndrome (TLS): Cases of possible TLS have been reported in patients treated with Herceptin. Patients with significant tumor burden (e.g. bulky metastases) may be at a higher risk. Patients could present with hyperuricemia, hyperphosphatemia, and acute renal failure which may represent possible TLS. Providers should consider additional monitoring and/or treatment as clinically indicated.
7 DRUG INTERACTIONS
Patients who receive anthracycline after stopping Herceptin may be at increased risk of cardiac dysfunction because of trastuzumab's long washout period based on population PK analysis [see Clinical Pharmacology (12.3)]. If possible, physicians should avoid anthracycline-based therapy for up to 7 months after stopping Herceptin. If anthracyclines are used, the patient's cardiac function should be monitored carefully.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Pharmacovigilance Program
There is a pregnancy pharmacovigilance program for Herceptin. If Herceptin is administered during pregnancy, or if a patient becomes pregnant while receiving Herceptin or within 7 months following the last dose of Herceptin, health care providers and patients should immediately report Herceptin exposure to Genentech at 1-888-835-2555.
Risk Summary
Herceptin can cause fetal harm when administered to a pregnant woman. In post-marketing reports, use of Herceptin during pregnancy resulted in cases of oligohydramnios and of oligohydramnios sequence, manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death [see Data]. Apprise the patient of the potential risks to a fetus. There are clinical considerations if Herceptin is used in a pregnant woman or if a patient becomes pregnant within 7 months following the last dose of Herceptin [see Clinical Considerations].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
In post-marketing reports, use of Herceptin during pregnancy resulted in cases of oligohydramnios and of oligohydramnios sequence, manifesting in the fetus as pulmonary hypoplasia, skeletal abnormalities, and neonatal death. These case reports described oligohydramnios in pregnant women who received Herceptin either alone or in combination with chemotherapy. In some case reports, amniotic fluid index increased after Herceptin was stopped. In one case, Herceptin therapy resumed after amniotic index improved and oligohydramnios recurred.
Animal Data
In studies where trastuzumab was administered to pregnant Cynomolgus monkeys during the period of organogenesis at doses up to 25 mg/kg given twice weekly (up to 25 times the recommended weekly human dose of 2 mg/kg), trastuzumab crossed the placental barrier during the early (Gestation Days 20 to 50) and late (Gestation Days 120 to 150) phases of gestation. The resulting concentrations of trastuzumab in fetal serum and amniotic fluid were approximately 33% and 25%, respectively, of those present in the maternal serum but were not associated with adverse developmental effects.
8.2 Lactation
Risk Summary
There is no information regarding the presence of trastuzumab in human milk, the effects on the breastfed infant, or the effects on milk production. Published data suggest human IgG is present in human milk but does not enter the neonatal and infant circulation in substantial amounts.
Trastuzumab was present in the milk of lactating Cynomolgus monkeys but not associated with neonatal toxicity [see Data]. Consider the developmental and health benefits of breastfeeding along with the mother's clinical need for Herceptin treatment and any potential adverse effects on the breastfed child from Herceptin or from the underlying maternal condition. This consideration should also take into account the trastuzumab wash out period of 7 months [see Clinical Pharmacology (12.3)].
Data
In lactating Cynomolgus monkeys, trastuzumab was present in breast milk at about 0.3% of maternal serum concentrations after pre- (beginning Gestation Day 120) and post-partum (through Post-partum Day 28) doses of 25 mg/kg administered twice weekly (25 times the recommended weekly human dose of 2 mg/kg of Herceptin). Infant monkeys with detectable serum levels of trastuzumab did not exhibit any adverse effects on growth or development from birth to 1 month of age.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of Herceptin.
Contraception
Females
Herceptin can cause embryo-fetal harm when administered during pregnancy.
Advise females of reproductive potential to use effective contraception during treatment with Herceptin and for 7 months following the last dose of Herceptin [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.3)].
8.4 Pediatric Use
The safety and effectiveness of Herceptin in pediatric patients have not been established.
8.5 Geriatric Use
Herceptin has been administered to 386 patients who were 65 years of age or over (253 in the adjuvant treatment and 133 in metastatic breast cancer treatment settings). The risk of cardiac dysfunction was increased in geriatric patients as compared to younger patients in both those receiving treatment for metastatic disease in Studies 5 and 6, or adjuvant therapy in Studies 1 and 2. Limitations in data collection and differences in study design of the 4 studies of Herceptin in adjuvant treatment of breast cancer preclude a determination of whether the toxicity profile of Herceptin in older patients is different from younger patients. The reported clinical experience is not adequate to determine whether the efficacy improvements (ORR, TTP, OS, DFS) of Herceptin treatment in older patients is different from that observed in patients < 65 years of age for metastatic disease and adjuvant treatment.
In Study 7 (metastatic gastric cancer), of the 294 patients treated with Herceptin, 108 (37%) were 65 years of age or older, while 13 (4.4%) were 75 and over. No overall differences in safety or effectiveness were observed.
10 OVERDOSAGE
There is no experience with overdosage in human clinical trials. Single doses higher than 8 mg/kg have not been tested.
11 DESCRIPTION
Trastuzumab is a humanized IgG1 kappa monoclonal antibody that selectively binds with high affinity to the extracellular domain of the human epidermal growth factor receptor 2 protein, HER2. Trastuzumab is produced by recombinant DNA technology in a mammalian cell (Chinese Hamster Ovary) culture.
Herceptin (trastuzumab) for injection is a sterile, white to pale yellow, preservative-free lyophilized powder with a cake-like appearance, for intravenous administration.
Each single-dose vial of Herceptin delivers 150 mg trastuzumab, 136.2 mg α,α-trehalose dihydrate, 3.4 mg L-histidine HCl monohydrate, 2.2 mg L-histidine, and 0.6 mg polysorbate 20. Reconstitution with 7.4 mL of sterile water for injection (SWFI) yields a solution containing 21 mg/mL trastuzumab that delivers 7.15 mL (150 mg trastuzumab), at a pH of approximately 6.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The HER2 (or c-erbB2) proto-oncogene encodes a transmembrane receptor protein of 185 kDa, which is structurally related to the epidermal growth factor receptor. Herceptin has been shown, in both in vitro assays and in animals, to inhibit the proliferation of human tumor cells that overexpress HER2.
Herceptin is a mediator of antibody-dependent cellular cytotoxicity (ADCC). In vitro, Herceptin-mediated ADCC has been shown to be preferentially exerted on HER2 overexpressing cancer cells compared with cancer cells that do not overexpress HER2.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effects of trastuzumab on electrocardiographic (ECG) endpoints, including QTc interval duration, were evaluated in patients with HER2 positive solid tumors. Trastuzumab had no clinically relevant effect on the QTc interval duration and there was no apparent relationship between serum trastuzumab concentrations and change in QTcF interval duration in patients with HER2 positive solid tumors.
12.3 Pharmacokinetics
The pharmacokinetics of trastuzumab was evaluated in a pooled population pharmacokinetic (PK) model analysis of 1,582 subjects with primarily breast cancer and metastatic gastric cancer (MGC) receiving intravenous Herceptin. Total trastuzumab clearance increases with decreasing concentrations due to parallel linear and non-linear elimination pathways.
Although the average trastuzumab exposure was higher following the first cycle in breast cancer patients receiving the three-weekly schedule compared to the weekly schedule of Herceptin, the average steady-state exposure was essentially the same at both dosages. The average trastuzumab exposure following the first cycle and at steady state as well as the time to steady state was higher in breast cancer patients compared to MGC patients at the same dosage; however, the reason for this exposure difference is unknown. Additional predicted trastuzumab exposure and PK parameters following the first Herceptin cycle and at steady state exposure are described in Tables 7 and 8, respectively.
Population PK based simulations indicate that following discontinuation of Herceptin, concentrations in at least 95% of breast cancer and MGC patients will decrease to approximately 3% of the population predicted steady-state trough serum concentration (approximately 97% washout) by 7 months [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1, 8.3)].
| Schedule | Primary tumor type | N | Cmin
(µg/mL) | Cmax
(µg/mL) | AUC0-21days
(µg.day/mL) |
|---|---|---|---|---|---|
| 8 mg/kg + 6 mg/kg q3w | Breast cancer | 1195 | 29.4 (5.8 - 59.5) | 178 (117 - 291) | 1373 (736 - 2245) |
| MGC | 274 | 23.1 (6.1 - 50.3) | 132 (84.2 - 225) | 1109 (588 - 1938) |
|
| 4 mg/kg + 2 mg/kg qw | Breast cancer | 1195 | 37.7 (12.3 - 70.9) | 88.3 (58 - 144) | 1066 (586 - 1754) |
| Schedule | Primary tumor type | N | Cmin,ss*
(µg/mL) | Cmax,ss†
(µg/mL) | AUCss, 0-21 days
(µg.day/mL) | Time to steady-state (week) | Total CL range at steady-state (L/day) |
|---|---|---|---|---|---|---|---|
| 8 mg/kg + 6 mg/kg q3w | Breast cancer | 1195 | 47.4 (5 - 115) | 179 (107 - 309) | 1794 (673 - 3618) | 12 | 0.173 - 0.283 |
| MGC | 274 | 32.9 (6.1 - 88.9) | 131 (72.5 - 251) | 1338 (557 - 2875) | 9 | 0.189 - 0.337 | |
| 4 mg/kg + 2 mg/kg qw | Breast cancer | 1195 | 66.1 (14.9 - 142) | 109 (51.0 - 209) | 1765 (647 - 3578) | 12 | 0.201 - 0.244 |
Specific Populations
Based on a population pharmacokinetic analysis, no clinically significant differences were observed in the pharmacokinetics of trastuzumab based on age (< 65 (n = 1294); ≥ 65 (n = 288)), race (Asian (n = 264); non-Asian (n = 1324)) and renal impairment (mild (creatinine clearance [CLcr] 60 to 90 mL/min) (n = 636) or moderate (CLcr 30 to 60 mL/min) (n = 133)). The pharmacokinetics of trastuzumab in patients with severe renal impairment, end-stage renal disease with or without hemodialysis, or hepatic impairment is unknown.
Drug Interaction Studies
There have been no formal drug interaction studies performed with Herceptin in humans. Clinically significant interactions between Herceptin and concomitant medications used in clinical trials have not been observed.
Paclitaxel and doxorubicin: Concentrations of paclitaxel and doxorubicin and their major metabolites (i.e., 6-α hydroxyl-paclitaxel [POH], and doxorubicinol [DOL], respectively) were not altered in the presence of trastuzumab when used as combination therapy in clinical trials. Trastuzumab concentrations were not altered as part of this combination therapy.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Herceptin has not been tested for carcinogenic potential.
No evidence of mutagenic activity was observed when trastuzumab was tested in the standard Ames bacterial and human peripheral blood lymphocyte mutagenicity assays at concentrations of up to 5000 mcg/mL. In an in vivo micronucleus assay, no evidence of chromosomal damage to mouse bone marrow cells was observed following bolus intravenous doses of up to 118 mg/kg of trastuzumab.
A fertility study was conducted in female Cynomolgus monkeys at doses up to 25 times the weekly recommended human dose of 2 mg/kg of trastuzumab and has revealed no evidence of impaired fertility, as measured by menstrual cycle duration and female sex hormone levels.
14 CLINICAL STUDIES
14.1 Adjuvant Breast Cancer
The safety and efficacy of Herceptin in women receiving adjuvant chemotherapy for HER2 overexpressing breast cancer were evaluated in an integrated analysis of two randomized, open-label, clinical trials (Studies 1 and 2) with a total of 4063 women at the protocol-specified final overall survival analysis, a third randomized, open-label, clinical trial (Study 3) with a total of 3386 women at definitive Disease-Free Survival analysis for one-year Herceptin treatment versus observation, and a fourth randomized, open-label clinical trial with a total of 3222 patients (Study 4).
Studies 1 and 2
In Studies 1 and 2, breast tumor specimens were required to show HER2 overexpression (3+ by IHC) or gene amplification (by FISH). HER2 testing was verified by a central laboratory prior to randomization (Study 2) or was required to be performed at a reference laboratory (Study 1). Patients with a history of active cardiac disease based on symptoms, abnormal electrocardiographic, radiologic, or left ventricular ejection fraction findings or uncontrolled hypertension (diastolic > 100 mm Hg or systolic > 200 mm Hg) were not eligible.
Patients were randomized (1:1) to receive doxorubicin and cyclophosphamide followed by paclitaxel (AC→paclitaxel) alone or paclitaxel plus Herceptin (AC→paclitaxel + Herceptin). In both trials, patients received four 21-day cycles of doxorubicin 60 mg/m2 and cyclophosphamide 600 mg/m2. Paclitaxel was administered either weekly (80 mg/m2) or every 3 weeks (175 mg/m2) for a total of 12 weeks in Study 1; paclitaxel was administered only by the weekly schedule in Study 2. Herceptin was administered at 4 mg/kg on the day of initiation of paclitaxel and then at a dose of 2 mg/kg weekly for a total of 52 weeks. Herceptin treatment was permanently discontinued in patients who developed congestive heart failure, or persistent/recurrent LVEF decline [see Dosage and Administration (2.3)]. Radiation therapy, if administered, was initiated after the completion of chemotherapy. Patients with ER+ and/or PR+ tumors received hormonal therapy. The primary endpoint of the combined efficacy analysis was Disease-Free Survival (DFS), defined as the time from randomization to recurrence, occurrence of contralateral breast cancer, other second primary cancer, or death. The secondary endpoint was overall survival (OS).
A total of 3752 patients were included in the joint efficacy analysis of the primary endpoint of DFS following a median follow-up of 2.0 years in the AC→paclitaxel + Herceptin arm. The pre-planned final OS analysis from the joint analysis included 4063 patients and was performed when 707 deaths had occurred after a median follow-up of 8.3 years in the AC→paclitaxel + Herceptin arm. The data from both arms in Study 1 and two of the three study arms in Study 2 were pooled for efficacy analyses. The patients included in the primary DFS analysis had a median age of 49 years (range, 22–80 years; 6% > 65 years), 84% were white, 7% black, 4% Hispanic, and 4% Asian/Pacific Islander. Disease characteristics included 90% infiltrating ductal histology, 38% T1, 91% nodal involvement, 27% intermediate and 66% high grade pathology, and 53% ER+ and/or PR+ tumors. Similar demographic and baseline characteristics were reported for the efficacy evaluable population, after 8.3 years of median follow-up in the AC→paclitaxel + Herceptin arm.
Study 3
In Study 3, breast tumor specimens were required to show HER2 overexpression (3+ by IHC) or gene amplification (by FISH) as determined at a central laboratory. Patients with node-negative disease were required to have ≥ T1c primary tumor. Patients with a history of congestive heart failure or LVEF < 55%, uncontrolled arrhythmias, angina requiring medication, clinically significant valvular heart disease, evidence of transmural infarction on ECG, poorly controlled hypertension (systolic > 180 mm Hg or diastolic > 100 mm Hg) were not eligible.
Study 3 was designed to compare one and two years of three-weekly Herceptin treatment versus observation in patients with HER2 positive EBC following surgery, established chemotherapy and radiotherapy (if applicable). Patients were randomized (1:1:1) upon completion of definitive surgery, and at least four cycles of chemotherapy to receive no additional treatment, or one year of Herceptin treatment or two years of Herceptin treatment. Patients undergoing a lumpectomy had also completed standard radiotherapy. Patients with ER+ and/or PgR+ disease received systemic adjuvant hormonal therapy at investigator discretion. Herceptin was administered with an initial dose of 8 mg/kg followed by subsequent doses of 6 mg/kg once every three weeks. The main outcome measure was Disease-Free Survival (DFS), defined as in Studies 1 and 2.
A protocol specified interim efficacy analysis comparing one-year Herceptin treatment to observation was performed at a median follow-up duration of 12.6 months in the Herceptin arm and formed the basis for the definitive DFS results from this study. Among the 3386 patients randomized to the observation (n = 1693) and Herceptin one-year (n = 1693) treatment arms, the median age was 49 years (range 21–80), 83% were Caucasian, and 13% were Asian. Disease characteristics: 94% infiltrating ductal carcinoma, 50% ER+ and/or PgR+, 57% node positive, 32% node negative, and in 11% of patients, nodal status was not assessable due to prior neo-adjuvant chemotherapy. Ninety-six percent (1055/1098) of patients with node-negative disease had high risk features: among the 1098 patients with node-negative disease, 49% (543) were ER− and PgR−, and 47% (512) were ER and/or PgR+ and had at least one of the following high risk features: pathological tumor size greater than 2 cm, Grade 2–3, or age < 35 years. Prior to randomization, 94% of patients had received anthracycline-based chemotherapy regimens.
After the definitive DFS results comparing observation to one-year Herceptin treatment were disclosed, a prospectively planned analysis that included comparison of one year versus two years of Herceptin treatment at a median follow-up duration of 8 years was performed. Based on this analysis, extending Herceptin treatment for a duration of two years did not show additional benefit over treatment for one year [Hazard Ratios of two-years Herceptin versus one-year Herceptin treatment in the intent to treat (ITT) population for Disease-Free Survival (DFS) = 0.99 (95% CI: 0.87, 1.13), p-value = 0.90 and Overall Survival (OS) = 0.98 (0.83, 1.15); p-value = 0.78].
Study 4
In Study 4, breast tumor specimens were required to show HER2 gene amplification (FISH+ only) as determined at a central laboratory. Patients were required to have either node-positive disease, or node-negative disease with at least one of the following high-risk features: ER/PR-negative, tumor size > 2 cm, age < 35 years, or histologic and/or nuclear Grade 2 or 3. Patients with a history of CHF, myocardial infarction, Grade 3 or 4 cardiac arrhythmia, angina requiring medication, clinically significant valvular heart disease, poorly controlled hypertension (diastolic > 100 mm Hg), any T4 or N2, or known N3 or M1 breast cancer were not eligible.
Patients were randomized (1:1:1) to receive doxorubicin and cyclophosphamide followed by docetaxel (AC-T), doxorubicin and cyclophosphamide followed by docetaxel plus Herceptin (AC-TH), or docetaxel and carboplatin plus Herceptin (TCH). In both the AC-T and AC-TH arms, doxorubicin 60 mg/m2 and cyclophosphamide 600 mg/m2 were administered every 3 weeks for four cycles; docetaxel 100 mg/m 2 was administered every 3 weeks for four cycles. In the TCH arm, docetaxel 75 mg/m2 and carboplatin (at a target AUC of 6 mg/mL/min as a 30- to 60-minute infusion) were administered every 3 weeks for six cycles. Herceptin was administered weekly (initial dose of 4 mg/kg followed by weekly dose of 2 mg/kg) concurrently with either T or TC, and then every 3 weeks (6 mg/kg) as monotherapy for a total of 52 weeks. Radiation therapy, if administered, was initiated after completion of chemotherapy. Patients with ER+ and/or PR+ tumors received hormonal therapy. Disease-Free Survival (DFS) was the main outcome measure.
Among the 3222 patients randomized, the median age was 49 (range 22 to 74 years; 6% ≥ 65 years). Disease characteristics included 54% ER+ and/or PR+ and 71% node positive. Prior to randomization, all patients underwent primary surgery for breast cancer.
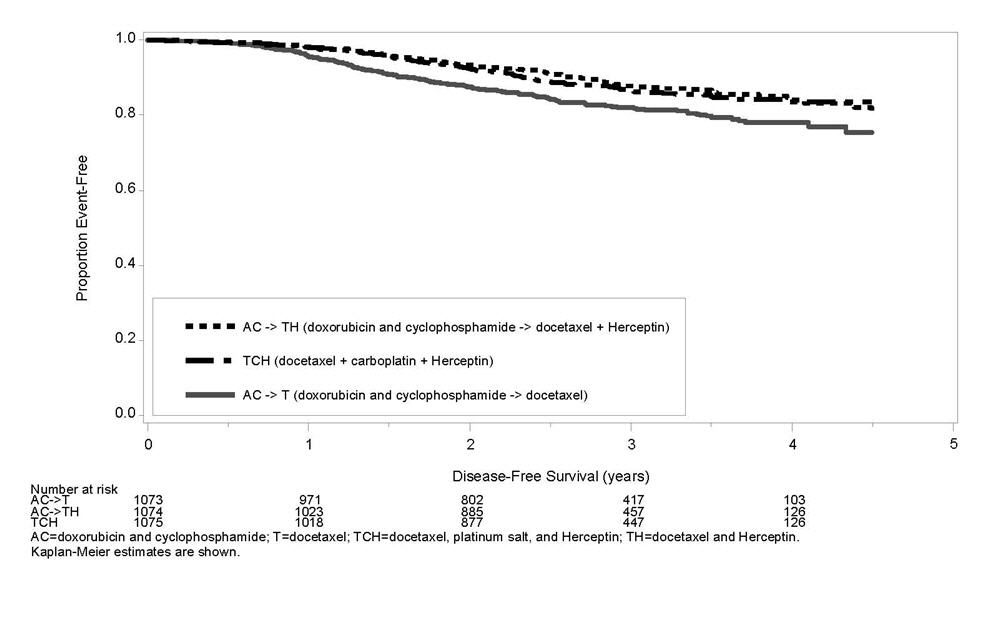
The results for DFS for the integrated analysis of Studies 1 and 2, Study 3, and Study 4 and OS results for the integrated analysis of Studies 1 and 2, and Study 3 are presented in Table 9. For Studies 1 and 2, the duration of DFS following a median follow-up of 2.0 years in the AC→TH arm is presented in Figure 4, and the duration of OS after a median follow-up of 8.3 years in the AC→TH arm is presented in Figure 5. The duration of DFS for Study 4 is presented in Figure 6. Across all four studies, at the time of definitive DFS analysis, there were insufficient numbers of patients within each of the following subgroups to determine if the treatment effect was different from that of the overall patient population: patients with low tumor grade, patients within specific ethnic/racial subgroups (Black, Hispanic, Asian/Pacific Islander patients), and patients > 65 years of age. For Studies 1 and 2, the OS hazard ratio was 0.64 (95% CI: 0.55, 0.74). At 8.3 years of median follow up [AC→TH], the survival rate was estimated to be 86.9% in the AC→TH arm and 79.4% in the AC→T arm. The final OS analysis results from Studies 1 and 2 indicate that OS benefit by age, hormone receptor status, number of positive lymph nodes, tumor size and grade, and surgery/radiation therapy was consistent with the treatment effect in the overall population. In patients ≤ 50 years of age (n = 2197), the OS hazard ratio was 0.65 (95% CI: 0.52, 0.81) and in patients > 50 years of age (n = 1866), the OS hazard ratio was 0.63 (95% CI: 0.51, 0.78). In the subgroup of patients with hormone receptor-positive disease (ER-positive and/or PR-positive) (n = 2223), the hazard ratio for OS was 0.63 (95% CI: 0.51, 0.78). In the subgroup of patients with hormone receptor-negative disease (ER-negative and PR-negative) (n = 1830), the hazard ratio for OS was 0.64 (95% CI: 0.52, 0.80). In the subgroup of patients with tumor size ≤ 2 cm (n = 1604), the hazard ratio for OS was 0.52 (95% CI: 0.39, 0.71). In the subgroup of patients with tumor size > 2 cm (n = 2448), the hazard ratio for OS was 0.67 (95% CI: 0.56, 0.80).
| DFS events | DFS Hazard ratio (95% CI) p-value | Deaths (OS events) | OS Hazard ratio p-value |
|
|---|---|---|---|---|
| CI = confidence interval. | ||||
|
||||
| Studies 1 + 2* | ||||
| AC→TH (n = 1872)† (n = 2031)‡ | 133† | 0.48†,§
(0.39, 0.59) p< 0.0001¶ | 289‡ | 0.64‡,§
(0.55, 0.74) p< 0.0001¶ |
| AC→T (n = 1880)† (n = 2032)‡ | 261† | 418‡ | ||
| Study 3# | ||||
| Chemo→ Herceptin (n = 1693) | 127 | 0.54 (0.44, 0.67) p< 0.0001Þ | 31 | 0.75 p = NSß |
| Chemo→ Observation (n = 1693) | 219 | 40 | ||
| Study 4à | ||||
| TCH (n = 1075) | 134 | 0.67 (0.54 – 0.84) p=0.0006¶,è | 56 | |
| AC→TH (n = 1074) | 121 | 0.60 (0.48 – 0.76) p< 0.0001¶,à | 49 | |
| AC→T (n = 1073) | 180 | 80 | ||
Figure 4
Duration of Disease-Free Survival in Patients with Adjuvant Treatment of Breast Cancer (Studies 1 and 2)
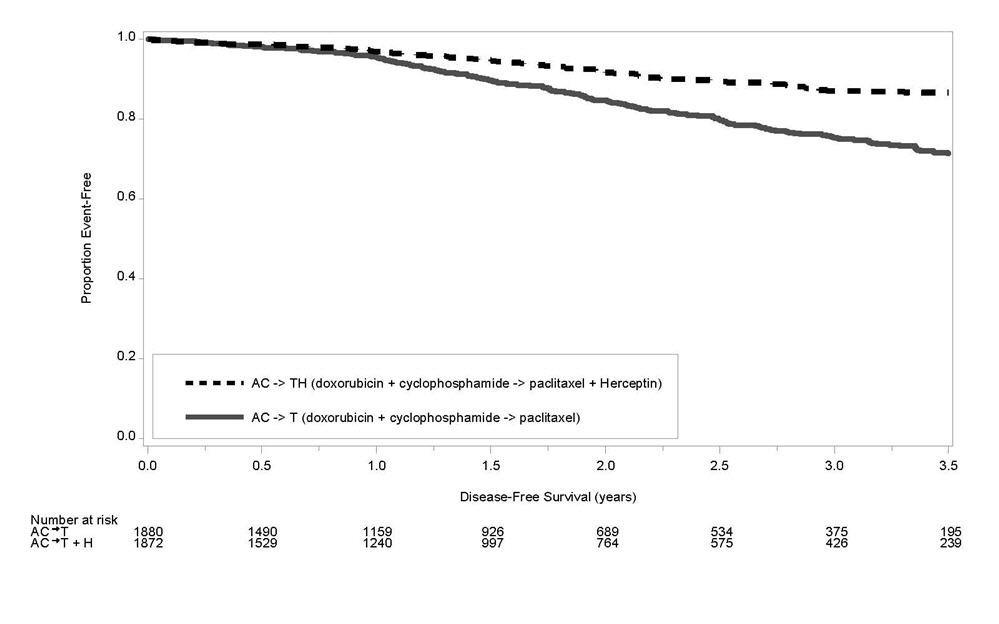
| Figure 5
Duration of Overall Survival in Patients with Adjuvant Treatment of Breast Cancer (Studies 1 and 2) |
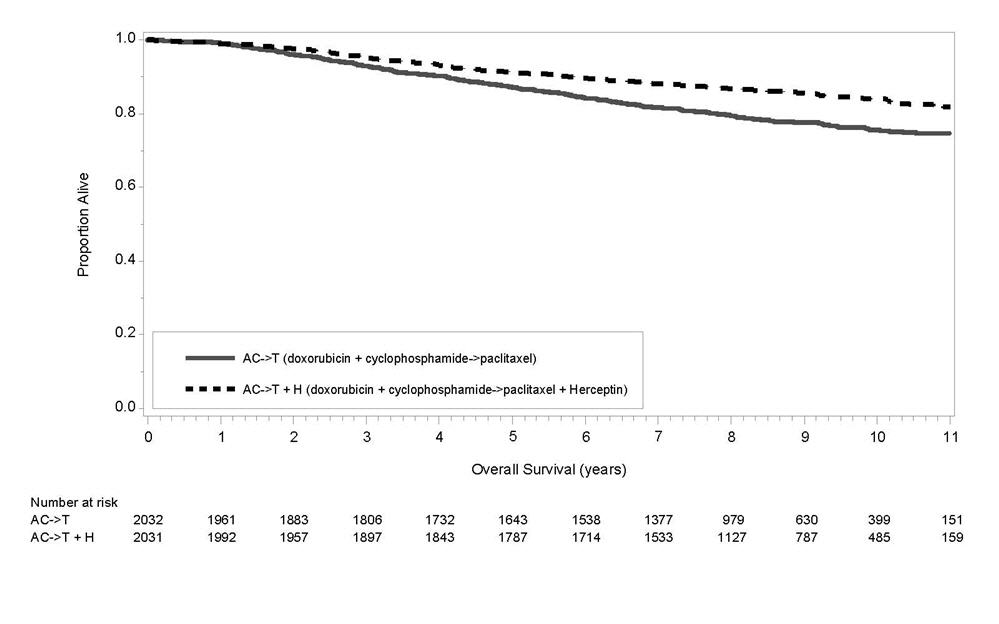 |
Figure 6
Duration of Disease-Free Survival in Patients with Adjuvant Treatment of Breast Cancer (Study 4)
Exploratory analyses of DFS as a function of HER2 overexpression or gene amplification were conducted for patients in Studies 2 and 3, where central laboratory testing data were available. The results are shown in Table 10. The number of events in Study 2 was small with the exception of the IHC 3+/FISH+ subgroup, which constituted 81% of those with data. Definitive conclusions cannot be drawn regarding efficacy within other subgroups due to the small number of events. The number of events in Study 3 was adequate to demonstrate significant effects on DFS in the IHC 3+/FISH unknown and the FISH+/IHC unknown subgroups.
| Study 2 | Study 3* | |||
| HER2 Assay Result† | Number of Patients | Hazard Ratio DFS (95% CI) | Number of Patients | Hazard Ratio DFS (95% CI) |
| IHC 3+ | ||||
| FISH (+) | 1170 | 0.42 (0.27, 0.64) | 91 | 0.56 (0.13, 2.50) |
| FISH (−) | 51 | 0.71 (0.04, 11.79) | 8 | — |
| FISH Unknown | 51 | 0.69 (0.09, 5.14) | 2258 | 0.53 (0.41, 0.69) |
| IHC < 3+ / FISH (+) | 174 | 1.01 (0.18, 5.65) | 299‡ | 0.53 (0.20, 1.42) |
| IHC unknown / FISH (+) | — | — | 724 | 0.59 (0.38, 0.93) |
14.2 Metastatic Breast Cancer
The safety and efficacy of Herceptin in treatment of women with metastatic breast cancer were studied in a randomized, controlled clinical trial in combination with chemotherapy (Study 5, n = 469 patients) and an open-label, single agent clinical trial (Study 6, n = 222 patients). Both trials studied patients with metastatic breast cancer whose tumors overexpress the HER2 protein. Patients were eligible if they had 2 or 3 levels of overexpression (based on a 0 to 3 scale) by immunohistochemical assessment of tumor tissue performed by a central testing lab.
Previously Untreated Metastatic Breast Cancer (Study 5)
Study 5 was a multicenter, randomized, open-label clinical trial conducted in 469 women with metastatic breast cancer who had not been previously treated with chemotherapy for metastatic disease. Tumor specimens were tested by IHC (Clinical Trial Assay, CTA) and scored as 0, 1+, 2+, or 3+, with 3+ indicating the strongest positivity. Only patients with 2+ or 3+ positive tumors were eligible (about 33% of those screened). Patients were randomized to receive chemotherapy alone or in combination with Herceptin given intravenously as a 4 mg/kg loading dose followed by weekly doses of Herceptin at 2 mg/kg. For those who had received prior anthracycline therapy in the adjuvant setting, chemotherapy consisted of paclitaxel (175 mg/m2 over 3 hours every 21 days for at least six cycles); for all other patients, chemotherapy consisted of anthracycline plus cyclophosphamide (AC: doxorubicin 60 mg/m2 or epirubicin 75 mg/m2 plus 600 mg/m2 cyclophosphamide every 21 days for six cycles). Sixty-five percent of patients randomized to receive chemotherapy alone in this study received Herceptin at the time of disease progression as part of a separate extension study.
Based upon the determination by an independent response evaluation committee, the patients randomized to Herceptin and chemotherapy experienced a significantly longer median time to disease progression, a higher overall response rate (ORR), and a longer median duration of response as compared with patients randomized to chemotherapy alone. Patients randomized to Herceptin and chemotherapy also had a longer median survival (see Table 11). These treatment effects were observed both in patients who received Herceptin plus paclitaxel and in those who received Herceptin plus AC; however the magnitude of the effects was greater in the paclitaxel subgroup.
| Combined Results | Paclitaxel Subgroup | AC Subgroup | ||||
| Herceptin + All Chemotherapy (n = 235) | All Chemotherapy (n = 234) | Herceptin + Paclitaxel (n = 92) | Paclitaxel (n = 96) | Herceptin + AC*
(n = 143) | AC (n = 138) |
|
| Primary Endpoint | ||||||
| Median TTP (mos)†, ‡ | 7.2 | 4.5 | 6.7 | 2.5 | 7.6 | 5.7 |
| 95% CI | 7, 8 | 4, 5 | 5, 10 | 2, 4 | 7, 9 | 5, 7 |
| p-value§ | < 0.0001 | < 0.0001 | 0.002 | |||
| Secondary Endpoints | ||||||
| Overall Response Rate† | 45 | 29 | 38 | 15 | 50 | 38 |
| 95% CI | 39, 51 | 23, 35 | 28, 48 | 8, 22 | 42, 58 | 30, 46 |
| p-value¶ | < 0.001 | < 0.001 | 0.10 | |||
| Median Resp Duration (mos)†,‡ | 8.3 | 5.8 | 8.3 | 4.3 | 8.4 | 6.4 |
| 25%, 75% Quartile | 6, 15 | 4, 8 | 5 ,11 | 4, 7 | 6, 15 | 4, 8 |
| Med Survival (mos)‡ | 25.1 | 20.3 | 22.1 | 18.4 | 26.8 | 21.4 |
| 95% CI | 22, 30 | 17, 24 | 17, 29 | 13, 24 | 23, 33 | 18, 27 |
| p-value§ | 0.05 | 0.17 | 0.16 | |||
Data from Study 5 suggest that the beneficial treatment effects were largely limited to patients with the highest level of HER2 protein overexpression (3+) (see Table 12).
| HER2 Assay Result | Number of Patients (N) | Relative Risk* for Time to Disease Progression (95% CI) | Relative Risk * for Mortality (95% CI) |
| CTA 2+ or 3+ | 469 | 0.49 (0.40, 0.61) | 0.80 (0.64, 1.00) |
| FISH (+) † | 325 | 0.44 (0.34, 0.57) | 0.70 (0.53, 0.91) |
| FISH (−)† | 126 | 0.62 (0.42, 0.94) | 1.06 (0.70, 1.63) |
| CTA 2+ | 120 | 0.76 (0.50, 1.15) | 1.26 (0.82, 1.94) |
| FISH (+) | 32 | 0.54 (0.21, 1.35) | 1.31 (0.53, 3.27) |
| FISH (–) | 83 | 0.77 (0.48, 1.25) | 1.11 (0.68, 1.82) |
| CTA 3+ | 349 | 0.42 (0.33, 0.54) | 0.70 (0.51, 0.90) |
| FISH (+) | 293 | 0.42 (0.32, 0.55) | 0.67 (0.51, 0.89) |
| FISH (–) | 43 | 0.43 (0.20, 0.94) | 0.88 (0.39, 1.98) |
Previously Treated Metastatic Breast Cancer (Study 6)
Herceptin was studied as a single agent in a multicenter, open-label, single-arm clinical trial (Study 6) in patients with HER2 overexpressing metastatic breast cancer who had relapsed following one or two prior chemotherapy regimens for metastatic disease. Of 222 patients enrolled, 66% had received prior adjuvant chemotherapy, 68% had received two prior chemotherapy regimens for metastatic disease, and 25% had received prior myeloablative treatment with hematopoietic rescue. Patients were treated with a loading dose of 4 mg/kg IV followed by weekly doses of Herceptin at 2 mg/kg IV.
The ORR (complete response + partial response), as determined by an independent Response Evaluation Committee, was 14%, with a 2% complete response rate and a 12% partial response rate. Complete responses were observed only in patients with disease limited to skin and lymph nodes. The overall response rate in patients whose tumors tested as CTA 3+ was 18% while in those that tested as CTA 2+, it was 6%.
14.3 Metastatic Gastric Cancer
The safety and efficacy of Herceptin in combination with cisplatin and a fluoropyrimidine (capecitabine or 5-fluorouracil) were studied in patients previously untreated for metastatic gastric or gastroesophageal junction adenocarcinoma (Study 7). In this open-label, multi-center trial, 594 patients were randomized 1:1 to Herceptin in combination with cisplatin and a fluoropyrimidine (FC+H) or chemotherapy alone (FC). Randomization was stratified by extent of disease (metastatic vs. locally advanced), primary site (gastric vs. gastroesophageal junction), tumor measurability (yes vs. no), ECOG performance status (0,1 vs. 2), and fluoropyrimidine (capecitabine vs. 5-fluorouracil). All patients were either HER2 gene amplified (FISH+) or HER2 overexpressing (IHC 3+). Patients were also required to have adequate cardiac function (e.g., LVEF > 50%).
On the Herceptin-containing arm, Herceptin was administered as an IV infusion at an initial dose of 8 mg/kg followed by 6 mg/kg every 3 weeks until disease progression. On both study arms cisplatin was administered at a dose of 80 mg/m2 Day 1 every 3 weeks for 6 cycles as a 2 hour IV infusion. On both study arms, capecitabine was administered at 1000 mg/m2 dose orally twice daily (total daily dose 2000 mg/m2) for 14 days of each 21 day cycle for 6 cycles. Alternatively, continuous intravenous infusion (CIV) 5-fluorouracil was administered at a dose of 800 mg/m2/day from Day 1 through Day 5 every three weeks for 6 cycles.
The median age of the study population was 60 years (range: 21-83); 76% were male; 53% were Asian, 38% Caucasian, 5% Hispanic, 5% other racial/ethnic groups; 91% had ECOG PS of 0 or 1; 82% had primary gastric cancer and 18% had primary gastroesophageal adenocarcinoma. Of these patients, 23% had undergone prior gastrectomy, 7% had received prior neoadjuvant and/or adjuvant therapy, and 2% had received prior radiotherapy.
The main outcome measure of Study 7 was overall survival (OS), analyzed by the unstratified log-rank test. The final OS analysis based on 351 deaths was statistically significant (nominal significance level of 0.0193). An updated OS analysis was conducted at one year after the final analysis. The efficacy results of both the final and the updated analyses are summarized in Table 13 and Figure 7.
| FC Arm N = 296 | FC + H Arm N = 298 |
|
|
||
| Definitive (Second Interim) Overall Survival | ||
| No. Deaths (%) | 184 (62.2%) | 167 (56.0%) |
| Median | 11.0 | 13.5 |
| 95% CI (mos.) | (9.4, 12.5) | (11.7, 15.7) |
| Hazard Ratio | 0.73 | |
| 95% CI | (0.60, 0.91) | |
| p value*, two sided | 0.0038 | |
| Updated Overall Survival | ||
| No. Deaths (%) | 227 (76.7%) | 221 (74.2%) |
| Median | 11.7 | 13.1 |
| 95% CI (mos.) | (10.3, 13.0) | (11.9, 15.1) |
| Hazard Ratio | 0.80 | |
| 95% CI | (0.67, 0.97) | |
Figure 7
Updated Overall Survival in Patients with Metastatic Gastric Cancer (Study 7)
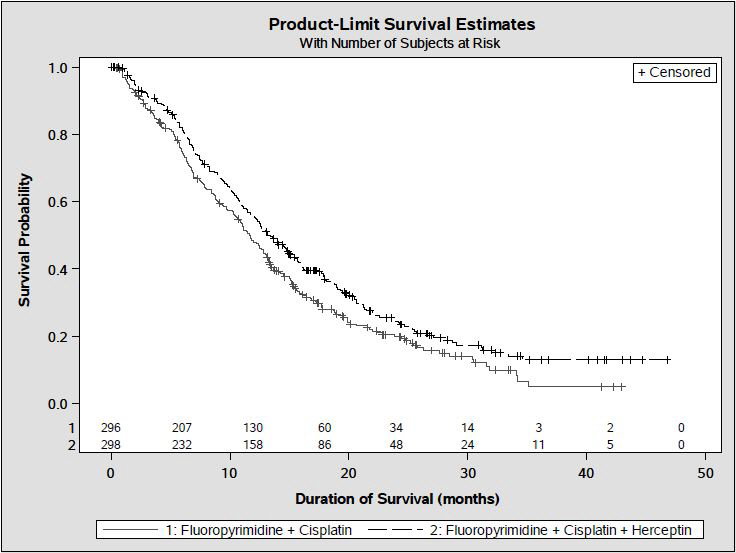
An exploratory analysis of OS in patients based on HER2 gene amplification (FISH) and protein overexpression (IHC) testing is summarized in Table 14.
| FC (N = 296)* | FC + H (N = 298)† |
|
|
||
| FISH+ / IHC 0, 1+ subgroup (N=133) | ||
| No. Deaths / n (%) | 57/71 (80%) | 56/62 (90%) |
| Median OS Duration (mos.) | 8.8 | 8.3 |
| 95% CI (mos.) | (6.4, 11.7) | (6.2, 10.7) |
| Hazard ratio (95% CI) | 1.33 (0.92, 1.92) | |
| FISH+ / IHC2+ subgroup (N=160) | ||
| No. Deaths / n (%) | 65/80 (81%) | 64/80 (80%) |
| Median OS Duration (mos.) | 10.8 | 12.3 |
| 95% CI (mos.) | (6.8, 12.8) | (9.5, 15.7) |
| Hazard ratio (95% CI) | 0.78 (0.55, 1.10) | |
| FISH+ or FISH- / IHC3+‡ subgroup (N=294) | ||
| No. Deaths / n (%) | 104/143 (73%) | 96/151 (64%) |
| Median OS Duration (mos.) | 13.2 | 18.0 |
| 95% CI (mos.) | (11.5, 15.2) | (15.5, 21.2) |
| Hazard ratio (95% CI) | 0.66 (0.50, 0.87) | |
17 PATIENT COUNSELING INFORMATION
Cardiomyopathy
- Advise patients to contact a health care professional immediately for any of the following: new onset or worsening shortness of breath, cough, swelling of the ankles/legs, swelling of the face, palpitations, weight gain of more than 5 pounds in 24 hours, dizziness or loss of consciousness [see Boxed Warning: Cardiomyopathy].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential that Herceptin exposure during pregnancy or within 7 months prior to conception can result in fetal harm. Advise female patients to contact their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations (8.1)].
- Advise women who are exposed to Herceptin during pregnancy or who become pregnant within 7 months following the last dose of Herceptin that there is a pregnancy pharmacovigilance program that monitor pregnancy outcomes. Encourage these patients to report their pregnancy to Genentech [see Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of Herceptin [see Use in Specific Populations (8.3)].
