FAMVIR- famciclovir tablet, film coated
Novartis Pharmaceuticals Corporation
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FAMVIR® safely and effectively. See full prescribing information for FAMVIR.
FAMVIR (famciclovir) tablets, for oral use Initial U.S. Approval: 1994 INDICATIONS AND USAGEFAMVIR, a prodrug of penciclovir, is a deoxynucleoside analog DNA polymerase inhibitor indicated for: Immunocompetent Adult Patients (1.1)
Human Immunodeficiency Virus (HIV)-Infected Adult Patients (1.2)
Limitation of Use The efficacy and safety of FAMVIR have not been established for:
DOSAGE AND ADMINISTRATION
Patients with renal impairment: Adjust dose based on creatinine clearance. (2.3) DOSAGE FORMS AND STRENGTHSTablets: 125 mg, 250 mg, 500 mg (3) CONTRAINDICATIONSKnown hypersensitivity to the product, its components, or Denavir® (penciclovir cream). (4) WARNINGS AND PRECAUTIONSADVERSE REACTIONSThe most common adverse events reported in at least 1 indication by greater than 10% of adult patients are headache and nausea. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSProbenecid: May increase penciclovir levels. Monitor for evidence of penciclovir toxicity. (7.2) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 12/2018 |
|||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Immunocompetent Adult Patients
Herpes labialis (cold sores): FAMVIR is indicated for the treatment of recurrent herpes labialis in adult patients.
Genital herpes:
Recurrent episodes: FAMVIR is indicated for the treatment of recurrent episodes of genital herpes. The efficacy of FAMVIR when initiated more than 6 hours after onset of symptoms or lesions has not been established.
Suppressive therapy: FAMVIR is indicated for chronic suppressive therapy of recurrent episodes of genital herpes in adult patients. The efficacy and safety of FAMVIR for the suppression of recurrent genital herpes beyond 1 year have not been established.
Herpes zoster (shingles): FAMVIR is indicated for the treatment of herpes zoster in adult patients. The efficacy of FAMVIR when initiated more than 72 hours after onset of rash has not been established.
1.2 HIV-Infected Adult Patients
Recurrent orolabial or genital herpes: FAMVIR is indicated for the treatment of recurrent episodes of orolabial or genital herpes in HIV-infected adults. The efficacy of FAMVIR when initiated more than 48 hours after onset of symptoms or lesions has not been established.
Limitation of Use
The efficacy and safety of FAMVIR have not been established for:
- Patients with first episode of genital herpes
- Patients with ophthalmic zoster
- Immunocompromised patients other than for the treatment of recurrent orolabial or genital herpes in HIV-infected patients
- Black and African American patients with recurrent genital herpes
2 DOSAGE AND ADMINISTRATION
FAMVIR may be taken with or without food.
2.1 Dosing Recommendation in Immunocompetent Adult Patients
Herpes labialis (cold sores): The recommended dosage of FAMVIR for the treatment of recurrent herpes labialis is 1500 mg as a single dose. Therapy should be initiated at the first sign or symptom of herpes labialis (e.g., tingling, itching, burning, pain, or lesion).
Genital herpes:
Recurrent episodes: The recommended dosage of FAMVIR for the treatment of recurrent episodes of genital herpes is 1000 mg twice daily for 1 day. Therapy should be initiated at the first sign or symptom of a recurrent episode (e.g., tingling, itching, burning, pain, or lesion).
Suppressive therapy: The recommended dosage of FAMVIR for chronic suppressive therapy of recurrent episodes of genital herpes is 250 mg twice daily.
Herpes zoster (shingles): The recommended dosage of FAMVIR for the treatment of herpes zoster is 500 mg every 8 hours for 7 days. Therapy should be initiated as soon as herpes zoster is diagnosed.
2.2 Dosing Recommendation in HIV-Infected Adult Patients
Recurrent orolabial or genital herpes: The recommended dosage of FAMVIR for the treatment of recurrent orolabial or genital herpes in HIV-infected patients is 500 mg twice daily for 7 days. Therapy should be initiated at the first sign or symptom of a recurrent episode (e.g., tingling, itching, burning, pain, or lesion).
2.3 Dosing Recommendation in Patients with Renal Impairment
Dosage recommendations for adult patients with renal impairment are provided in Table 1 [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
| Indication and Normal Dosage
Regimen | Creatinine Clearance
(mL/min) | Adjusted Dosage
Regimen Dose (mg) | Dosing Interval |
| Single-Day Dosing Regimens | |||
| Recurrent Genital Herpes 1000 mg every 12 hours for 1 day | ≥ 60 | 1000 | every 12 hours for 1 day |
| 40-59 | 500 | every 12 hours for 1 day | |
| 20-39 | 500 | single dose | |
| < 20 | 250 | single dose | |
| HD* | 250 | single dose following dialysis |
|
| Recurrent Herpes Labialis 1500 mg single dose | ≥ 60 | 1500 | single dose |
| 40-59 | 750 | single dose | |
| 20-39 | 500 | single dose | |
| < 20 | 250 | single dose | |
| HD* | 250 | single dose following dialysis |
|
| Multiple-Day Dosing Regimens | |||
| Herpes Zoster 500 mg every 8 hours | ≥ 60 | 500 | every 8 hours |
| 40-59 | 500 | every 12 hours | |
| 20-39 | 500 | every 24 hours | |
| < 20 | 250 | every 24 hours | |
| HD* | 250 | following each dialysis | |
| Suppression of Recurrent Genital Herpes 250 mg every 12 hours | ≥ 40 | 250 | every 12 hours |
| 20-39 | 125 | every 12 hours | |
| < 20 | 125 | every 24 hours | |
| HD* | 125 | following each dialysis | |
| Recurrent Orolabial or Genital Herpes in HIV-Infected Patients 500 mg every 12 hours | ≥ 40 | 500 | every 12 hours |
| 20-39 | 500 | every 24 hours | |
| < 20 | 250 | every 24 hours | |
| HD* | 250 | following each dialysis | |
*Hemodialysis
3 DOSAGE FORMS AND STRENGTHS
FAMVIR tablets are available in 3 strengths:
- 125 mg: White, round film-coated, biconvex, beveled edges, debossed with “FAMVIR” on one side and “125” on the other side
- 250 mg: White, round film-coated, biconvex, beveled edges, debossed with “FAMVIR” on one side and “250” on the other side
- 500 mg: White, oval film-coated, biconvex, debossed with “FAMVIR” on one side and “500” on the other side
4 CONTRAINDICATIONS
FAMVIR is contraindicated in patients with known hypersensitivity to the product, its components, or Denavir® (penciclovir cream).
5 WARNINGS AND PRECAUTIONS
5.1 Acute Renal Failure
Cases of acute renal failure have been reported in patients with underlying renal disease who have received inappropriately high doses of FAMVIR for their level of renal function. Dosage reduction is recommended when administering FAMVIR to patients with renal impairment [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
6 ADVERSE REACTIONS
Acute renal failure is discussed in greater detail in other sections of the label [see Warnings and Precautions (5)].
The most common adverse events reported in at least 1 indication by greater than 10% of adult patients treated with FAMVIR are headache and nausea.
6.1 Clinical Trials Experience in Adult Patients
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Immunocompetent patients: The safety of FAMVIR has been evaluated in active- and placebo-controlled clinical studies involving 816 FAMVIR-treated patients with herpes zoster (FAMVIR, 250 mg three times daily to 750 mg three times daily); 163 FAMVIR-treated patients with recurrent genital herpes (FAMVIR, 1000 mg twice daily); 1,197 patients with recurrent genital herpes treated with FAMVIR as suppressive therapy (125 mg once daily to 250 mg three times daily) of which 570 patients received FAMVIR (open-labeled and/or double-blind) for at least 10 months; and 447 FAMVIR-treated patients with herpes labialis (FAMVIR, 1500 mg once daily or 750 mg twice daily). Table 2 lists selected adverse events.
| Incidence | |||||||||
| Herpes Zoster† | Recurrent
Genital Herpes‡ | Genital Herpes-
Suppression§ | Herpes Labialis‡ | ||||||
| FAMVIR | Placebo | FAMVIR | Placebo | FAMVIR | Placebo | FAMVIR | Placebo | ||
| (n=273) | (n=146) | (n=163) | (n=166) | (n=458) | (n=63) | (n=447) | (n=254) | ||
| Events | % | % | % | % | % | % | % | % | |
| Nervous System | |||||||||
| Headache | 22.7 | 17.8 | 13.5 | 5.4 | 39.3 | 42.9 | 8.5 | 6.7 | |
| Paresthesia | 2.6 | 0.0 | 0.0 | 0.0 | 0.9 | 0.0 | 0.0 | 0.0 | |
| Migraine | 0.7 | 0.7 | 0.6 | 0.6 | 3.1 | 0.0 | 0.2 | 0.0 | |
| Gastrointestinal | |||||||||
| Nausea | 12.5 | 11.6 | 2.5 | 3.6 | 7.2 | 9.5 | 2.2 | 3.9 | |
| Diarrhea | 7.7 | 4.8 | 4.9 | 1.2 | 9.0 | 9.5 | 1.6 | 0.8 | |
| Vomiting | 4.8 | 3.4 | 1.2 | 0.6 | 3.1 | 1.6 | 0.7 | 0.0 | |
| Flatulence | 1.5 | 0.7 | 0.6 | 0.0 | 4.8 | 1.6 | 0.2 | 0.0 | |
| Abdominal Pain | 1.1 | 3.4 | 0.0 | 1.2 | 7.9 | 7.9 | 0.2 | 0.4 | |
| Body as a Whole | |||||||||
| Fatigue | 4.4 | 3.4 | 0.6 | 0.0 | 4.8 | 3.2 | 1.6 | 0.4 | |
| Skin and Appendages | |||||||||
| Pruritus | 3.7 | 2.7 | 0.0 | 0.6 | 2.2 | 0.0 | 0.0 | 0.0 | |
| Rash | 0.4 | 0.7 | 0.0 | 0.0 | 3.3 | 1.6 | 0.0 | 0.0 | |
| Reproductive (Female) | |||||||||
| Dysmenorrhea | 0.0 | 0.7 | 1.8 | 0.6 | 7.6 | 6.3 | 0.4 | 0.0 | |
*Patients may have entered into more than one clinical trial.
†7 days of treatment
‡1 day of treatment
§daily treatment
Table 3 lists selected laboratory abnormalities in genital herpes suppression trials.
| Parameter | FAMVIR
(n=660)† % | Placebo
(n=210)† % |
| Anemia (<0.8 x NRL) | 0.1 | 0.0 |
| Leukopenia (<0.75 x NRL) | 1.3 | 0.9 |
| Neutropenia (<0.8 x NRL) | 3.2 | 1.5 |
| AST (SGOT) (>2 x NRH) | 2.3 | 1.2 |
| ALT (SGPT) (>2 x NRH) | 3.2 | 1.5 |
| Total Bilirubin (>1.5 x NRH) | 1.9 | 1.2 |
| Serum Creatinine (>1.5 x NRH) | 0.2 | 0.3 |
| Amylase (>1.5 x NRH) | 1.5 | 1.9 |
| Lipase (>1.5 x NRH) | 4.9 | 4.7 |
*Percentage of patients with laboratory abnormalities that were increased or decreased from baseline and were outside of specified ranges.
†n values represent the minimum number of patients assessed for each laboratory parameter.
NRH=Normal Range High.
NRL=Normal Range Low.
HIV-infected patients: In HIV-infected patients, the most frequently reported adverse events for FAMVIR (500 mg twice daily; n=150) and acyclovir (400 mg, 5x/day; n=143), respectively, were headache (17% vs. 15%), nausea (11% vs. 13%), diarrhea (7% vs. 11%), vomiting (5% vs. 4%), fatigue (4% vs. 2%), and abdominal pain (3% vs. 6%).
6.2 Postmarketing Experience
The adverse events listed below have been reported during post-approval use of FAMVIR. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Blood and lymphatic system disorders: Thrombocytopenia
Hepatobiliary disorders: Abnormal liver function tests, cholestatic jaundice
Immune system disorders: Anaphylactic shock, anaphylactic reaction
Nervous system disorders: Dizziness, somnolence, seizure
Psychiatric disorders: Confusion (including delirium, disorientation, and confusional state occurring predominantly in the elderly), hallucinations
Skin and subcutaneous tissue disorders: Urticaria, erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, angioedema (e.g., face, eyelid, periorbital, and pharyngeal edema), hypersensitivity vasculitis
Cardiac disorders: Palpitations
7 DRUG INTERACTIONS
7.1 Potential for FAMVIR to Affect Other Drugs
The steady-state pharmacokinetics of digoxin were not altered by concomitant administration of multiple doses of famciclovir (500 mg three times daily). No clinically significant effect on the pharmacokinetics of zidovudine, its metabolite zidovudine glucuronide, or emtricitabine was observed following a single oral dose of 500 mg famciclovir coadministered with zidovudine or emtricitabine.
An in vitro study using human liver microsomes suggests that famciclovir is not an inhibitor of CYP3A4 enzymes.
7.2 Potential for Other Drugs to Affect Penciclovir
No clinically significant alterations in penciclovir pharmacokinetics were observed following single-dose administration of 500 mg famciclovir after pretreatment with multiple doses of allopurinol, cimetidine, theophylline, zidovudine, promethazine, when given shortly after an antacid (magnesium and aluminum hydroxide), or concomitantly with emtricitabine. No clinically significant effect on penciclovir pharmacokinetics was observed following multiple-dose (three times daily) administration of famciclovir (500 mg) with multiple doses of digoxin.
Concurrent use with probenecid or other drugs significantly eliminated by active renal tubular secretion may result in increased plasma concentrations of penciclovir.
The conversion of 6-deoxy penciclovir to penciclovir is catalyzed by aldehyde oxidase. Interactions with other drugs metabolized by this enzyme and/or inhibiting this enzyme could potentially occur. Clinical interaction studies of famciclovir with cimetidine and promethazine, in vitro inhibitors of aldehyde oxidase, did not show relevant effects on the formation of penciclovir. Raloxifene, a potent aldehyde oxidase inhibitor in vitro, could decrease the formation of penciclovir. However, a clinical drug-drug interaction study to determine the magnitude of interaction between penciclovir and raloxifene has not been conducted.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from pharmacovigilance reports with FAMVIR use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the fetus associated with untreated herpes simplex virus during pregnancy (see Clinical Considerations). After oral administration, famciclovir (prodrug) is converted to penciclovir (active drug). In animal reproduction studies with famciclovir, no evidence of adverse developmental outcomes was observed at systemic exposures of penciclovir (AUC) slightly higher than those at the maximum recommended human dose (MRHD) of FAMVIR (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Novartis Pharmaceuticals Corporation maintains a pregnancy reporting system to monitor maternal-fetal outcomes of pregnant women exposed to FAMVIR. Healthcare providers are encouraged to report pregnancies and pregnancy outcomes to the Novartis Adverse Event reporting line at 1-888-NOW-NOVA (669-6682).
Clinical Considerations
Disease-associated maternal and/or embryo-fetal risk
The risk of neonatal herpes infection varies from 30% to 50% for genital herpes simplex virus (HSV) infections that occur in late pregnancy (third trimester), whereas in early pregnancy, infection carries a risk of about 1%. A primary herpes outbreak during the first trimester of pregnancy has been associated with neonatal chorioretinitis, microcephaly and, in rare cases, skin lesions. In very rare cases, transplacental transmission can occur resulting in congenital infection, including microcephaly, hepatosplenomegaly, intrauterine growth restriction and stillbirth. Co-infection with HSV increases the risk of perinatal HIV transmission in women who had a clinical diagnosis of genital herpes during pregnancy.
Data
Animal Data
Famciclovir was administered orally to pregnant rats and rabbits (up to 1000 mg/kg/day) on gestation Day(s) 6 to 15, and to rats on gestation Day 15 to lactation/post-partum Day 25. No adverse effects on embryo-fetal (rats and rabbits) or pre/post-natal (rats) development were observed up to the highest dose tested. During organogenesis, systemic exposures of penciclovir (active metabolite) were 3.4 times (rats) and 1.6 times (rabbits) the human systemic exposure of penciclovir based on AUC at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of famciclovir (prodrug) or penciclovir (active drug) in human milk, the effects on the breastfed infant, or the effects on milk production. Animal data indicate that penciclovir is present in the milk of lactating rats (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for FAMVIR and any potential adverse effects on the breastfed infant from FAMVIR or from the underlying maternal condition.
Data
Penciclovir was the primary drug-related component excreted into the milk of lactating rats following a single oral dose of 40 mg per kg on lactation Day 12, with milk concentrations of up to approximately 8 times that of maternal plasma concentrations observed 0.5 hours postdose.
8.3 Females and Males of Reproductive Potential
Infertility
Decreased fertility, due to testicular toxicity, was observed in male animals following repeated administration of famciclovir or penciclovir [see Nonclinical Toxicology (13.1)].
In two placebo-controlled studies, 130 men with a history of recurrent genital herpes received either oral FAMVIR (250 mg twice daily; n=66) or placebo (n=64) therapy for 18 weeks. The men were otherwise healthy and had a normal sperm profile prior to treatment. There was no evidence of significant effects on sperm count, motility or morphology during FAMVIR treatment or during an 8-week follow-up.
8.4 Pediatric Use
The efficacy of FAMVIR has not been established in pediatric patients. The pharmacokinetic profile and safety of famciclovir (experimental granules mixed with OraSweet® or tablets) were studied in 3 open-label studies.
Study 1 was a single-dose pharmacokinetic and safety study in infants 1 month to less than 1 year of age who had an active herpes simplex virus (HSV) infection or who were at risk for HSV infection. Eighteen subjects were enrolled and received a single dose of famciclovir experimental granules mixed with OraSweet based on the patient’s body weight (doses ranged from 25 mg to 175 mg). These doses were selected to provide penciclovir systemic exposures similar to the penciclovir systemic exposures observed in adults after administration of 500 mg famciclovir. The efficacy and safety of famciclovir have not been established as suppressive therapy in infants following neonatal HSV infections. In addition, the efficacy cannot be extrapolated from adults to infants because there is no similar disease in adults. Therefore, famciclovir is not recommended in infants.
Study 2 was an open-label, single-dose pharmacokinetic, multiple-dose safety study of famciclovir experimental granules mixed with OraSweet in children 1 to less than 12 years of age with clinically suspected HSV or varicella zoster virus (VZV) infection. Fifty-one subjects were enrolled in the pharmacokinetic part of the study and received a single body weight adjusted dose of famciclovir (doses ranged from 125 mg to 500 mg). These doses were selected to provide penciclovir systemic exposures similar to the penciclovir systemic exposures observed in adults after administration of 500 mg famciclovir. Based on the pharmacokinetic data observed with these doses in children, a new weight-based dosing algorithm was designed and used in the multiple-dose safety part of the study. Pharmacokinetic data were not obtained with the revised weight-based dosing algorithm.
A total of 100 patients were enrolled in the multiple-dose safety part of the study; 47 subjects with active or latent HSV infection and 53 subjects with chickenpox. Patients with active or latent HSV infection received famciclovir twice a day for 7 days. The daily dose of famciclovir ranged from 150 mg to 500 mg twice daily depending on the patient’s body weight. Patients with chickenpox received famciclovir three times daily for 7 days. The daily dose of famciclovir ranged from 150 mg to 500 mg three times daily depending on the patient’s body weight. The clinical adverse events and laboratory test abnormalities observed in this study were similar to these seen in adults. The available data are insufficient to support the use of famciclovir for the treatment of children 1 to less than 12 years of age with chickenpox or infections due to HSV for the following reasons:
Chickenpox: The efficacy of famciclovir for the treatment of chickenpox has not been established in either pediatric or adult patients. Famciclovir is approved for the treatment of herpes zoster in adult patients. However, extrapolation of efficacy data from adults with herpes zoster to children with chickenpox would not be appropriate. Although chickenpox and herpes zoster are caused by the same virus, the diseases are different.
Genital herpes: Clinical information on genital herpes in children is limited. Therefore, efficacy data from adults cannot be extrapolated to this population. Further, famciclovir has not been studied in children 1 to less than 12 years of age with recurrent genital herpes. None of the children in Study 2 had genital herpes.
Herpes labialis: There are no pharmacokinetic and safety data in children 1 to less than 12 years of age to support a famciclovir dose that provides penciclovir systemic exposures comparable to the penciclovir systemic exposures in adults after a single dose administration of 1500 mg. Moreover, no efficacy data have been obtained in children 1 to less than 12 years of age with recurrent herpes labialis.
Study 3 was an open-label, single-arm study to evaluate the pharmacokinetics, safety, and antiviral activity of a single 1500 mg dose (three 500 mg tablets) of famciclovir in children 12 to less than 18 years of age with recurrent herpes labialis. A total of 53 subjects were enrolled in the study; 10 subjects in the pharmacokinetic part of the study and 43 subjects in the non-pharmacokinetic part of the study. All enrolled subjects weighed greater than or equal to 40 kg. The 43 subjects enrolled in the non-pharmacokinetic part of the study had active recurrent herpes labialis and received a single 1500 mg dose of famciclovir within 24 hours after the onset of symptoms (median time to treatment initiation was 21 hours). The safety profile of famciclovir observed in this study was similar to that seen in adults. The median time to healing of patients with non-aborted lesions was 5.9 days.
In a phase 3 trial in adults in which patients received a single 1500 mg dose of famciclovir or placebo, the median time to healing among patients with non-aborted lesions was 4.4 days in the famciclovir 1500 mg single-dose group and 6.2 days in the placebo group. Of note, in the adult study treatment was initiated by patients within 1 hour after the onset of symptoms [see Clinical Studies (14.1)]. Based on the efficacy results in Study 3, famciclovir is not recommended in children 12 to less than 18 years of age with recurrent herpes labialis.
8.5 Geriatric Use
Of 816 patients with herpes zoster in clinical studies who were treated with FAMVIR, 248 (30.4%) were greater than or equal to 65 years of age, and 103 (13%) were greater than or equal to 75 years of age. No overall differences were observed in the incidence or types of adverse events between younger and older patients. Of 610 patients with recurrent herpes simplex (type 1 or type 2) in clinical studies who were treated with FAMVIR, 26 (4.3%) were greater than 65 years of age, and 7 (1.1%) were greater than 75 years of age. Clinical studies of FAMVIR in patients with recurrent genital herpes did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently compared to younger subjects.
No famciclovir dosage adjustment based on age is recommended unless renal function is impaired [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]. In general, appropriate caution should be exercised in the administration and monitoring of FAMVIR in elderly patients reflecting the greater frequency of decreased renal function and concomitant use of other drugs.
8.6 Patients with Renal Impairment
Apparent plasma clearance, renal clearance, and the plasma-elimination rate constant of penciclovir decreased linearly with reductions in renal function. After the administration of a single 500 mg famciclovir oral dose (n=27) to healthy volunteers and to volunteers with varying degrees of renal impairment (CLCR ranged from 6.4 to 138.8 mL/min), the following results were obtained (Table 4):
| Parameter
(mean ± S.D.) | CLCR† ≥60
(mL/min) (n=15) | CLCR 40-59
(mL/min) (n=5) | CLCR 20-39
(mL/min) (n=4) | CLCR <20
(mL/min) (n=3) |
| CLCR (mL/min) | 88.1 ± 20.6 | 49.3 ± 5.9 | 26.5 ± 5.3 | 12.7 ± 5.9 |
| CLR (L/hr) | 30.1 ± 10.6 | 13.0 ± 1.3‡ | 4.2 ± 0.9 | 1.6 ± 1.0 |
| CL/F§ (L/hr) | 66.9 ± 27.5 | 27.3 ± 2.8 | 12.8 ± 1.3 | 5.8 ± 2.8 |
| Half-life (hr) | 2.3 ± 0.5 | 3.4 ± 0.7 | 6.2 ± 1.6 | 13.4 ± 10.2 |
† CLCR is measured creatinine clearance.
‡ n=4.
§ CL/F consists of bioavailability factor and famciclovir to penciclovir conversion factor.
In a multiple-dose study of famciclovir conducted in subjects with varying degrees of renal impairment (n=18), the pharmacokinetics of penciclovir were comparable to those after single doses.
A dosage adjustment is recommended for patients with renal impairment [see Dosage and Administration (2.3)].
8.7 Patients with Hepatic Impairment
Mild or moderate hepatic impairment (chronic hepatitis [n=6], chronic ethanol abuse [n=8], or primary biliary cirrhosis [n=1]) had no effect on the extent of availability (AUC) of penciclovir following a single dose of 500 mg famciclovir. However, there was a 44% decrease in penciclovir mean maximum plasma concentration (Cmax) and the time to maximum plasma concentration (tmax) was increased by 0.75 hours in patients with hepatic impairment compared to normal volunteers. No dosage adjustment is recommended for patients with mild or moderate hepatic impairment. The pharmacokinetics of penciclovir has not been evaluated in patients with severe hepatic impairment. Conversion of famciclovir to the active metabolite penciclovir may be impaired in these patients resulting in a lower penciclovir plasma concentrations, and thus possibly a decrease of efficacy of famciclovir [see Clinical Pharmacology (12)].
8.8 Black and African American Patients
In a randomized, double-blind, placebo-controlled trial conducted in 304 immunocompetent black and African American adults with recurrent genital herpes there was no difference in median time to healing between patients receiving FAMVIR or placebo. In general, the adverse reaction profile was similar to that observed in other FAMVIR clinical trials for adult patients [see Adverse Reactions (6.1)]. The relevance of these study results to other indications in black and African American patients is unknown [see Clinical Studies (14.2)].
10 OVERDOSAGE
Appropriate symptomatic and supportive therapy should be given. Penciclovir is removed by hemodialysis.
11 DESCRIPTION
The active ingredient in FAMVIR tablets is famciclovir, an orally administered prodrug of the antiviral agent penciclovir. Chemically, famciclovir is known as 2-[2-(2-amino-9H-purin-9-yl)ethyl]-1,3-propanediol diacetate. Its molecular formula is C14H19N5O4; its molecular weight is 321.3. It is a synthetic acyclic guanine derivative and has the following structure
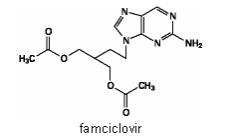
Famciclovir is a white to pale yellow solid. It is freely soluble in acetone and methanol, and sparingly soluble in ethanol and isopropanol. At 25°C famciclovir is freely soluble (greater than 25% w/v) in water initially, but rapidly precipitates as the sparingly soluble (2%-3% w/v) monohydrate. Famciclovir is not hygroscopic below 85% relative humidity. Partition coefficients are: octanol/water (pH 4.8) P=1.09 and octanol/phosphate buffer (pH 7.4) P=2.08.
FAMVIR tablets contain 125 mg, 250 mg, or 500 mg of famciclovir, together with the following inactive ingredients: hydroxypropyl cellulose, hydroxypropyl methylcellulose, lactose, magnesium stearate, polyethylene glycols, sodium starch glycolate and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Famciclovir is an orally administered prodrug of the anti-alpha herpes viral agent penciclovir [see Microbiology (12.4)].
12.3 Pharmacokinetics
Famciclovir is the diacetyl 6-deoxy analog of the active antiviral compound penciclovir. Following oral administration famciclovir undergoes rapid and extensive metabolism to penciclovir and little or no famciclovir is detected in plasma or urine. Penciclovir is predominantly eliminated unchanged by the kidney. Therefore, the dose of FAMVIR needs to be adjusted in patients with different degrees of renal impairment [see Dosage and Administration (2.3)].
Pharmacokinetics in adults:
Absorption and Bioavailability: The absolute bioavailability of penciclovir is 77 ± 8% as determined following the administration of a 500 mg famciclovir oral dose and a 400 mg penciclovir intravenous dose to 12 healthy male subjects.
Penciclovir concentrations increased in proportion to dose over a famciclovir dose range of 125 mg to 1000 mg administered as a single dose. Table 5 shows the mean pharmacokinetic parameters of penciclovir after single administration of FAMVIR to healthy male volunteers.
| Dose | AUC (0-inf)† (mcg hr/mL) | Cmax‡ (mcg/mL) | tmax§ (h) |
| 125 mg | 2.24 | 0.8 | 0.9 |
| 250 mg | 4.48 | 1.6 | 0.9 |
| 500 mg | 8.95 | 3.3 | 0.9 |
| 1000 mg | 17.9 | 6.6 | 0.9 |
* Based on pharmacokinetic data from 17 studies
† AUC (0-inf) (mcg hr/mL) = area under the plasma concentration-time profile extrapolated to infinity.
‡ Cmax (mcg/mL) = maximum observed plasma concentration.
§ tmax (h) = time to Cmax.
Following oral single-dose administration of 500 mg famciclovir to 7 patients with herpes zoster, the AUC (mean ± SD), Cmax, and tmax were 12.1±1.7 mcg hr/mL, 4.0±0.7 mcg/mL, and 0.7±0.2 hours, respectively. The AUC of penciclovir was approximately 35% greater in patients with herpes zoster as compared to healthy volunteers. Some of this difference may be due to differences in renal function between the 2 groups.
There is no accumulation of penciclovir after the administration of 500 mg famciclovir three times daily for 7 days.
Penciclovir Cmax decreased approximately 50% and tmax was delayed by 1.5 hours when a capsule formulation of famciclovir was administered with food (nutritional content was approximately 910 Kcal and 26% fat). There was no effect on the extent of availability (AUC) of penciclovir. There was an 18% decrease in Cmax and a delay in tmax of about 1 hour when famciclovir was given 2 hours after a meal as compared to its administration 2 hours before a meal. Because there was no effect on the extent of systemic availability of penciclovir, FAMVIR can be taken without regard to meals.
Distribution: The volume of distribution (Vdβ) was 1.08±0.17 L/kg in 12 healthy male subjects following a single intravenous dose of penciclovir at 400 mg administered as a 1-hour intravenous infusion. Penciclovir is less than 20% bound to plasma proteins over the concentration range of 0.1 to 20 mcg/mL. The blood/plasma ratio of penciclovir is approximately 1.
Metabolism: Following oral administration, famciclovir is deacetylated and oxidized to form penciclovir. Metabolites that are inactive include 6-deoxy penciclovir, monoacetylated penciclovir, and 6-deoxy monoacetylated penciclovir (5%, less than 0.5% and less than 0.5% of the dose in the urine, respectively). Little or no famciclovir is detected in plasma or urine. An in vitro study using human liver microsomes demonstrated that cytochrome P450 does not play an important role in famciclovir metabolism. The conversion of 6-deoxy penciclovir to penciclovir is catalyzed by aldehyde oxidase. Cimetidine and promethazine, in vitro inhibitors of aldehyde oxidase, did not show relevant effects on the formation of penciclovir in vivo [see Drug Interactions (7.2)].
Elimination: Approximately 94% of administered radioactivity was recovered in urine over 24 hours (83% of the dose was excreted in the first 6 hours) after the administration of 5 mg/kg radiolabeled penciclovir as a 1-hour infusion to 3 healthy male volunteers. Penciclovir accounted for 91% of the radioactivity excreted in the urine.
Following the oral administration of a single 500 mg dose of radiolabeled famciclovir to 3 healthy male volunteers, 73% and 27% of administered radioactivity were recovered in urine and feces over 72 hours, respectively. Penciclovir accounted for 82% and 6-deoxy penciclovir accounted for 7% of the radioactivity excreted in the urine. Approximately 60% of the administered radiolabeled dose was collected in urine in the first 6 hours.
After intravenous administration of penciclovir in 48 healthy male volunteers, mean ± SD total plasma clearance of penciclovir was 36.6±6.3 L/hr (0.48±0.09 L/hr/kg). Penciclovir renal clearance accounted for 74.5±8.8% of total plasma clearance.
Renal clearance of penciclovir following the oral administration of a single 500 mg dose of famciclovir to 109 healthy male volunteers was 27.7±7.6 L/hr. Active tubular secretion contributes to the renal elimination of penciclovir.
The plasma elimination half-life of penciclovir was 2.0±0.3 hours after intravenous administration of penciclovir to 48 healthy male volunteers and 2.3±0.4 hours after oral administration of 500 mg famciclovir to 124 healthy male volunteers. The half-life in 17 patients with herpes zoster was 2.8±1.0 hours and 2.7±1.0 hours after single and repeated doses, respectively.
Special populations:
Geriatric patients: Based on cross study comparison, penciclovir AUC was 40% higher and penciclovir renal clearance was 22% lower in elderly subjects (n=18, age 65 to 79 years) as compared with younger subjects. Some of this difference may be due to differences in renal function between the 2 groups. No famciclovir dosage adjustment based on age is recommended unless renal function is impaired [see Dosage and Administration (2.3), Use in Specific Populations (8.5)].
Patients with renal impairment: In subjects with varying degrees of renal impairment, apparent plasma clearance, renal clearance, and the plasma-elimination rate constant of penciclovir decreased linearly with reductions in renal function, after both single and repeated dosing [see Use in Specific Populations (8.6)]. A dosage adjustment is recommended for patients with renal impairment [see Dosage and Administration (2.3)].
Patients with hepatic impairment: Mild or moderate hepatic impairment had no effect on the extent of availability (AUC) of penciclovir [see Use in Specific Populations (8.7)]. No dosage adjustment is recommended for patients with mild or moderate hepatic impairment. The effect of severe hepatic impairment on the pharmacokinetics of penciclovir has not been evaluated.
HIV-infected patients: Following oral administration of a single dose of 500 mg famciclovir to HIV-positive patients, the pharmacokinetic parameters of penciclovir were comparable to those observed in healthy subjects.
Gender: The pharmacokinetics of penciclovir were evaluated in 18 healthy male and 18 healthy female volunteers after single-dose oral administration of 500 mg famciclovir. AUC of penciclovir was 9.3±1.9 mcg hr/mL and 11.1±2.1 mcg hr/mL in males and females, respectively. Penciclovir renal clearance was 28.5±8.9 L/hr and 21.8±4.3 L/hr, respectively. These differences were attributed to differences in renal function between the 2 groups. No famciclovir dosage adjustment based on gender is recommended.
Race: A retrospective evaluation was performed to compare the pharmacokinetic parameters obtained in black and Caucasian subjects after single and repeat once-daily, twice-daily, or three times-daily administration of famciclovir 500 mg. Data from a study in healthy volunteers (single dose), a study in subjects with varying degrees of renal impairment (single and repeat dose) and a study in subjects with hepatic impairment (single dose) did not indicate any significant differences in the pharmacokinetics of penciclovir between black and Caucasian subjects.
12.4 Microbiology
Mechanism of action: Famciclovir is a prodrug of penciclovir, which has demonstrated inhibitory activity against herpes simplex virus types 1 (HSV-1) and 2 (HSV-2) and varicella zoster virus (VZV). In cells infected with HSV-1, HSV-2 or VZV, the viral thymidine kinase phosphorylates penciclovir to a monophosphate form that, in turn, is converted by cellular kinases to the active form penciclovir triphosphate. Biochemical studies demonstrate that penciclovir triphosphate inhibits HSV-2 DNA polymerase competitively with deoxyguanosine triphosphate. Consequently, α-herpes viral DNA synthesis and, therefore, replication are selectively inhibited. Penciclovir triphosphate has an intracellular half-life of 10 hours in HSV-1-, 20 hours in HSV-2- and 7 hours in VZV-infected cells grown in culture. However, the clinical significance of the intracellular half-life is unknown.
Antiviral activity: In cell culture studies, penciclovir has antiviral activity against the following herpes viruses: HSV-1, HSV-2 and VZV. The antiviral activity of penciclovir against wild type strains grown on human foreskin fibroblasts was assessed with a plaque reduction assay and staining with crystal violet 3 days post-infection for HSV and 10 days post-infection for VZV. The median EC50 values of penciclovir against laboratory and clinical isolates of HSV-1, HSV-2, and VZV were 2 µM (range 1.2 to 2.4 µM, n=7), 2.6 µM (range 1.6 to 11 µM, n=6), and 34 µM (range 6.7 to 71 µM, n=6), respectively.
Resistance:
In Cell Culture
Penciclovir-resistant HSV-1 and HSV-2 strains were isolated in cell culture. Penciclovir-resistant mutants of HSV and VZV resulted from mutations in the viral thymidine kinase (TK) and DNA polymerase (POL) genes. Frameshifts were commonly isolated and result in premature truncation of the HSV TK product with decreased enzymatic activity and consequent decreased susceptibility to penciclovir. Mutations in the viral TK gene may lead to complete loss of TK activity (TK negative), reduced levels of TK activity (TK partial), or alteration in the ability of viral TK to phosphorylate the drug without an equivalent loss in the ability to phosphorylate thymidine (TK altered). In cell culture, the following resistance-associated substitutions in TK of HSV-1 and HSV-2 were observed: HSV-1 TK G6C, F13L, H142Y, G200D, L205S, S254Stop, V267G, and T287M; HSV-2 TK G39E, F191L, E226K, and T288M. The median EC50 values observed in a plaque reduction assay with penciclovir resistant HSV-1, HSV-2, and VZV were 69 µM (range 14 to 115 µM, n=6), 46 µM (range 4 to > 395 µM, n=9), and 92 µM (range 51 to 148 µM, n=4), respectively.
Resistance and Cross-resistance in Clinical Isolates from HSV-Infected Patients
Clinical HSV-1 and HSV-2 isolates obtained from patients who failed treatment with acyclovir for their α-herpesvirus infections were evaluated for genotypic changes in the TK and POL genes. These HSV isolates had frameshift mutations leading to loss of thymidine kinase or had substitutions in the viral thymidine kinase and viral DNA polymerase. Phenotypic analysis of these clinical isolates confirmed resistance to penciclovir and acyclovir. These and other resistance-associated substitutions reported in the literature, or observed in clinical trials, are listed in Table 6. The list is not all inclusive and additional changes will likely be identified in HSV variants isolated from patients who fail penciclovir containing regimens. The possibility of viral resistance to penciclovir should be considered in patients who fail to respond or experience recurrent viral shedding during therapy.
| HSV-1 | TK | G6C, R32H, R51W, Y53C/H, H58N, G59W, G61A, S74Stop, E83K, P84L, T103P, Q104Stop, D116N, M121R, I143V, R163H, L170P, Y172C, A174P, R176Q/W, Q185R, A189V, G200D, G206R, L208H, R216C, R220H, R222C/H, FS 224, Y239S, T245M, Q250Stop, S254Stop, R256W, Q261Stop, R281Stop, T287M, L315S, M322K, C336Y |
| HSV-2 | TK | G39E, R51W, Y53N, R177W*, R221H, T288M* |
| HSV-1 | POL | A657T, D672N, V715G, A719V, S724N, E798K, V813M, N815S, Y818C, G841S, R842S, F891C, V958L |
| HSV-2 | POL | - |
*These substitutions were also observed in penciclovir-treated patients.
Note: Many additional pathways to penciclovir resistance likely exist.
Cross-resistance has been observed among HSV isolates carrying foscarnet resistance-associated substitutions (Table 7).
| HSV-1 | POL | D672N, S724N, E798K, V813M, Y818C, F891C, V958L |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Two-year dietary carcinogenicity studies with famciclovir were conducted in rats and mice. An increase in the incidence of mammary adenocarcinoma (a common tumor in animals of this strain) was seen in female rats receiving the high dose of 600 mg/kg/day (1.1 to 4.5x the human systemic exposure at the recommended total daily oral dose ranging between 500 mg and 2000 mg, based on area under the plasma concentration curve comparisons [24 hr AUC] for penciclovir). No increases in tumor incidence were reported in male rats treated at doses up to 240 mg/kg/day (0.7 to 2.7x the human AUC), or in male and female mice at doses up to 600 mg/kg/day (0.3 to 1.2x the human AUC).
Mutagenesis: Famciclovir and penciclovir (the active metabolite of famciclovir) were tested for genotoxic potential in a battery of in vitro and in vivo assays. Famciclovir and penciclovir were negative in in vitro tests for gene mutations in bacteria (S. typhimurium and E. coli) and unscheduled DNA synthesis in mammalian HeLa 83 cells (at doses up to 10,000 and 5,000 mcg/plate, respectively). Famciclovir was also negative in the L5178Y mouse lymphoma assay (5000 mcg/mL), the in vivo mouse micronucleus test (4800 mg/kg), and rat dominant lethal study (5000 mg/kg). Famciclovir induced increases in polyploidy in human lymphocytes in vitro in the absence of chromosomal damage (1200 mcg/mL). Penciclovir was positive in the L5178Y mouse lymphoma assay for gene mutation/chromosomal aberrations, with and without metabolic activation (1000 mcg/mL). In human lymphocytes, penciclovir caused chromosomal aberrations in the absence of metabolic activation (250 mcg/mL). Penciclovir caused an increased incidence of micronuclei in mouse bone marrow in vivo when administered intravenously at doses highly toxic to bone marrow (500 mg/kg), but not when administered orally.
Impairment of fertility: Testicular toxicity was observed in rats, mice, and dogs following repeated administration of famciclovir or penciclovir. Testicular changes included atrophy of the seminiferous tubules, reduction in sperm count, and/or increased incidence of sperm with abnormal morphology or reduced motility. The degree of toxicity to male reproduction was related to dose and duration of exposure. In male rats, decreased fertility was observed after 10 weeks of dosing at 500 mg/kg/day (1.4 to 5.7x the human AUC). The no observable effect level for sperm and testicular toxicity in rats following chronic administration (26 weeks) was 50 mg/kg/day (0.15 to 0.6x the human systemic exposure based on AUC comparisons). Testicular toxicity was observed following chronic administration to mice (104 weeks) and dogs (26 weeks) at doses of 600 mg/kg/day (0.3 to 1.2x the human AUC) and 150 mg/kg/day (1.3 to 5.1x the human AUC), respectively.
Famciclovir had no effect on general reproductive performance or fertility in female rats at doses up to 1000 mg/kg/day (2.7 to 10.8x the human AUC).
14 CLINICAL STUDIES
14.1 Herpes Labialis (Cold Sores)
A randomized, double-blind, placebo-controlled trial was conducted in 701 immunocompetent adults with recurrent herpes labialis. Patients self-initiated therapy within 1 hour of first onset of signs or symptoms of a recurrent herpes labialis episode with FAMVIR 1500 mg as a single dose (n=227), FAMVIR 750 mg twice daily (n=220) or placebo (n=254) for 1 day. The median time to healing among patients with non-aborted lesions (progressing beyond the papule stage) was 4.4 days in the FAMVIR 1500 mg single-dose group (n=152) as compared to 6.2 days in the placebo group (n=168). The median difference in time to healing between the placebo and FAMVIR 1500 mg treated groups was 1.3 days (95% CI: 0.6–2.0). No differences in proportion of patients with aborted lesions (not progressing beyond the papule stage) were observed between patients receiving FAMVIR or placebo: 33% for FAMVIR 1500 mg single dose and 34% for placebo. The median time to loss of pain and tenderness was 1.7 days in FAMVIR 1500 mg single dose-treated patients vs. 2.9 days in placebo-treated patients.
14.2 Genital Herpes
Recurrent episodes: A randomized, double-blind, placebo-controlled trial was conducted in 329 immunocompetent adults with recurrent genital herpes. Patients self-initiated therapy within 6 hours of the first sign or symptom of a recurrent genital herpes episode with either FAMVIR 1000 mg twice daily (n=163) or placebo (n=166) for 1 day. The median time to healing among patients with non-aborted lesions (progressing beyond the papule stage) was 4.3 days in FAMVIR-treated patients (n=125) as compared to 6.1 days in placebo-treated patients (n=145). The median difference in time to healing between the placebo and FAMVIR-treated groups was 1.2 days (95% CI: 0.5 to 2.0). Twenty-three percent of FAMVIR-treated patients had aborted lesions (no lesion development beyond erythema) vs. 13% in placebo-treated patients. The median time to loss of all symptoms (e.g., tingling, itching, burning, pain, or tenderness) was 3.3 days in FAMVIR-treated patients vs. 5.4 days in placebo-treated patients.
A randomized (2:1), double-blind, placebo-controlled trial was conducted in 304 immunocompetent black and African American adults with recurrent genital herpes. Patients self-initiated therapy within 6 hours of the first sign or symptom of a recurrent genital herpes episode with either FAMVIR 1000 mg twice daily (n=206) or placebo (n=98) for 1 day. The median time to healing among patients with non-aborted lesions was 5.4 days in FAMVIR-treated patients (n=152) as compared to 4.8 days in placebo-treated patients (n=78). The median difference in time to healing between the placebo and FAMVIR-treated groups was -0.26 days (95% CI: -0.98 to 0.40).
Suppressive therapy: Two randomized, double-blind, placebo-controlled, 12-month trials were conducted in 934 immunocompetent adults with a history of 6 or more recurrences of genital herpes episodes per year. Comparisons included FAMVIR 125 mg three times daily, 250 mg twice daily, 250 mg three times daily, and placebo. At 12 months, 60% to 65% of patients were still receiving FAMVIR and 25% were receiving placebo treatment. Recurrence rates at 6 and 12 months in patients treated with the 250 mg twice daily dose are shown in Table 8.
| Recurrence Rates
at 6 Months | Recurrence Rates
at 12 Months |
|||
| FAMVIR
250 mg twice daily (n=236) | Placebo
(n=233) | FAMVIR
250 mg twice daily (n=236) | Placebo
(n=233) |
|
| Recurrence-free | 39% | 10% | 29% | 6% |
| Recurrences† | 47% | 74% | 53% | 78% |
| Lost to follow-up‡ | 14% | 16% | 17% | 16% |
†Based on patient reported data; not necessarily confirmed by a physician.
‡Patients recurrence-free at time of last contact prior to withdrawal.
FAMVIR-treated patients had approximately one-fifth the median number of recurrences as compared to placebo-treated patients. Higher doses of FAMVIR were not associated with an increase in efficacy.
14.3 Recurrent Orolabial or Genital Herpes in HIV-Infected Patients
A randomized, double-blind trial compared famciclovir 500 mg twice daily for 7 days (n=150) with oral acyclovir 400 mg 5 times daily for 7 days (n=143) in HIV-infected patients with recurrent orolabial or genital herpes treated within 48 hours of lesion onset. Approximately 40% of patients had a CD4+ count below 200 cells/mm3, 54% of patients had anogenital lesions and 35% had orolabial lesions. Famciclovir therapy was comparable to oral acyclovir in reducing new lesion formation and in time to complete healing.
14.4 Herpes Zoster (Shingles)
Two randomized, double-blind trials, 1 placebo-controlled and 1 active-controlled, were conducted in 964 immunocompetent adults with uncomplicated herpes zoster. Treatment was initiated within 72 hours of first lesion appearance and was continued for 7 days.
In the placebo-controlled trial, 419 patients were treated with either FAMVIR 500 mg three times daily (n=138), FAMVIR 750 mg three times daily (n=135) or placebo (n=146). The median time to full crusting was 5 days among FAMVIR 500 mg-treated patients as compared to 7 days in placebo-treated patients. The times to full crusting, loss of vesicles, loss of ulcers, and loss of crusts were shorter for FAMVIR 500 mg-treated patients than for placebo-treated patients in the overall study population. The effects of FAMVIR were greater when therapy was initiated within 48 hours of rash onset; it was also more profound in patients 50 years of age or older. Among the 65.2% of patients with at least 1 positive viral culture, FAMVIR treated patients had a shorter median duration of viral shedding than placebo-treated patients (1 day and 2 days, respectively).
There were no overall differences in the duration of pain before rash healing between FAMVIR- and placebo-treated groups. In addition, there was no difference in the incidence of pain after rash healing (postherpetic neuralgia) between the treatment groups. In the 186 patients (44.4% of total study population) who developed postherpetic neuralgia, the median duration of postherpetic neuralgia was shorter in patients treated with FAMVIR 500 mg than in those treated with placebo (63 days and 119 days, respectively). No additional efficacy was demonstrated with higher dose of FAMVIR.
In the active-controlled trial, 545 patients were treated with 1 of 3 doses of FAMVIR three times daily or with acyclovir 800 mg five times daily. Times to full lesion crusting and times to loss of acute pain were comparable for all groups and there were no statistically significant differences in the time to loss of postherpetic neuralgia between FAMVIR and acyclovir-treated groups.
16 HOW SUPPLIED/STORAGE AND HANDLING
FAMVIR tablets are supplied as film-coated tablets as follows: 125 mg in bottles of 30; 250 mg in bottles of 30; 500 mg in bottles of 30, and Single Unit Packages of 50 (intended for institutional use only).
- FAMVIR 125 mg tablet:
White, round film-coated, biconvex, beveled edges, debossed with “FAMVIR” on one side and “125” on the other.
125 mg 30’s………………………………………………………………………………………… NDC 0078-0366-15
- FAMVIR 250 mg tablet:
White, round film-coated, biconvex, beveled edges, debossed with “FAMVIR” on one side and “250” on the other.
250 mg 30’s………………………………………………………………………………………… NDC 0078-0367-15
- FAMVIR 500 mg tablet:
White, oval film-coated, biconvex, debossed with “FAMVIR” on one side and “500” on the other.
500 mg 30’s………………………………………………………………………………………… NDC 0078-0368-15
500 mg SUP 50’s……………………………………………………………………………...…… NDC 0078-0368-64
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
There is no evidence that FAMVIR will affect the ability of a patient to drive or to use machines. However, patients who experience dizziness, somnolence, confusion or other central nervous system disturbances while taking FAMVIR should refrain from driving or operating machinery.
Because FAMVIR contains lactose (FAMVIR 125 mg, 250 mg, and 500 mg tablets contain lactose 26.9 mg, 53.7 mg and 107.4 mg, respectively), patients with rare hereditary problems of galactose intolerance, a severe lactase deficiency or glucose-galactose malabsorption should be advised to discuss with their healthcare provider before taking FAMVIR.
Herpes Labialis (Cold Sores)
Patients should be advised to initiate treatment at the earliest sign or symptom of a recurrence of cold sores (e.g., tingling, itching, burning, pain, or lesion). Patients should be instructed that treatment for cold sores should not exceed 1 dose. Patients should be informed that FAMVIR is not a cure for cold sores.
Genital Herpes
Patients should be informed that FAMVIR is not a cure for genital herpes. There are no data evaluating whether FAMVIR will prevent transmission of infection to others. Because genital herpes is a sexually transmitted disease, patients should avoid contact with lesions or intercourse when lesions and/or symptoms are present to avoid infecting partners. Genital herpes is frequently transmitted in the absence of symptoms through asymptomatic viral shedding. Therefore, patients should be counseled to use safer sex practices.
If episodic therapy for recurrent genital herpes is indicated, patients should be advised to initiate therapy at the first sign or symptom of an episode.
There are no data on safety or effectiveness of chronic suppressive therapy of longer than 1-year duration.
Herpes Zoster (Shingles)
There are no data on treatment initiated more than 72 hours after onset of zoster rash. Patients should be advised to initiate treatment as soon as possible after a diagnosis of herpes zoster.
Pregnancy
Report pregnancies and pregnancy outcomes to the Novartis Adverse Event reporting line at 1-888-NOW-NOVA (669-6682).
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
T2018-143
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: December 2018 | |
|
PATIENT INFORMATION
FAMVIR® (Fam’-veer) (famciclovir) tablets |
||
| What is FAMVIR?
FAMVIR is a prescription antiviral medicine used: |
||
It is not known if FAMVIR is safe and effective for:
It is not known if FAMVIR is effective in children. | ||
| Do not take FAMVIR if you are allergic to any of its ingredients or to Denavir® (penciclovir cream). See the end of this Patient Information leaflet for a complete list of ingredients in FAMVIR. | ||
Before you take FAMVIR, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
||
How should I take FAMVIR?
|
||
| What should I avoid while taking FAMVIR?
You should avoid driving or operating machinery if you get dizziness, sleepiness or confusion during treatment with FAMVIR. |
||
| What are the possible side effects of FAMVIR?
The most common side effects of FAMVIR are headache and nausea. These are not all the possible side effects of FAMVIR. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store FAMVIR?
|
||
| General information about the safe and effective use of FAMVIR
FAMVIR is not a cure for cold sores or genital herpes. It is not known if FAMVIR can stop the spread of herpes to others. If you are sexually active, you can pass herpes to your partner even if you are taking FAMVIR. Herpes can be passed to your partner even if you do not have active symptoms. You should continue to practice safer sex to lower the chances of spreading genital herpes to others. Do not have sexual contact with your partner during an outbreak of genital herpes or if you have any symptoms of genital herpes. Ask your healthcare provider for more information about safer sex practices. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FAMVIR for a condition for which it was not prescribed. Do not give FAMVIR to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about FAMVIR that is written for health professionals. |
||
| What are the ingredients in FAMVIR?
Active ingredient: famciclovir Inactive ingredients: hydroxypropyl cellulose, hydroxypropyl methylcellulose, lactose, magnesium stearate, polyethylene glycols, sodium starch glycolate, and titanium dioxide Distributed by: © Novartis T2018-144 For more information about FAMVIR, call 1-888-669-6682. |
||
PRINCIPAL DISPLAY PANEL
Package Label – 125 mg
Rx Only NDC 0078-0366-15
FAMVIR® (famciclovir) Tablets
125 mg per tablet
30 Tablets



| FAMVIR
famciclovir tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| FAMVIR
famciclovir tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| FAMVIR
famciclovir tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |