FULL PRESCRIBING INFORMATION
WARNING: ABUSE, MISUSE, AND ADDICTION
DAYTRANA has a high potential for abuse and misuse, which can lead to the development of substance use disorder, including addiction. Misuse and abuse of CNS stimulants, including DAYTRANA, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing DAYTRANA, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug. Throughout DAYTRANA treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction [see Warnings and Precautions (5.1) and Drug Abuse and Dependence (9.2)] .
1 INDICATIONS AND USAGE
DAYTRANA (methylphenidate transdermal system) is indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in pediatric patients 6 to 17 years of age.
2 DOSAGE AND ADMINISTRATION
2.1 Pretreatment Screening
Prior to treating patients with DAYTRANA, assess:
- for the presence of cardiac disease (i.e., perform a careful history, family history of sudden death or ventricular arrhythmia, and physical exam) [see Warnings and Precautions (5.2)].
- the family history and clinically evaluate patients for motor or verbal tics or Tourette’s syndrome before initiating DAYTRANA [see Warnings and Precautions (5.15)].
2.2 Recommended Dosage
It is recommended that DAYTRANA be applied to the hip area 2 hours before an effect is needed and should be removed 9 hours after application. Dosage should be titrated to effect. The recommended dose titration schedule is shown in the table below. Dose titration, final dosage, and wear time should be individualized according to the needs and response of the patient.
|
*Nominal in vivo delivery rate in children and adolescents when applied to the hip, based on a 9-hour wear period. |
||||
| Table 1 DAYTRANA - Recommended Titration Schedule (Patients New to Methylphenidate) | ||||
| Upward Titration, if Response is Not Maximized | ||||
| Week 1 | Week 2 | Week 3 | Week 4 | |
| Transdermal System Size | 12.5 cm 2 | 18.75 cm 2 | 25 cm 2 | 37.5 cm 2 |
| Nominal Delivered Dose* (mg/9 hours) | 10 mg | 15 mg | 20 mg | 30 mg |
| Delivery Rate* | (1.1 mg/hr)* | (1.6 mg/hr)* | (2.2 mg/hr)* | (3.3 mg/hr)* |
Patients converting from another formulation of methylphenidate should follow the above titration schedule due to differences in bioavailability of DAYTRANA compared to other products.
2.3 Application
The parent or caregiver should be encouraged to use the administration chart included with each carton of DAYTRANA to monitor application and removal time, and method of disposal. It is recommended that parents or caregivers apply and remove the transdermal system for children; responsible adolescents may apply or remove the transdermal system themselves if appropriate. If a transdermal system was removed without the parent or caregiver's knowledge, or if a transdermal system is missing from the tray, the parent or caregiver should be encouraged to ask the child when and how the transdermal system was removed. The Medication Guide includes a timetable to calculate when to remove DAYTRANA, based on the 9-hour application time.
The adhesive side of DAYTRANA should be placed on a clean, dry area of the hip. The area selected should not be oily, damaged, or irritated. Apply DAYTRANA to the hip area avoiding the waistline, since clothing may cause the transdermal system to rub off. When applying the transdermal system the next morning, place on the opposite hip at a new site if possible.
If patients or caregivers experience difficulty separating the transdermal system from the release liner or observe transfer of adhesive to the liner, tearing and/or other damage to the transdermal system during removal from the liner, the transdermal system should be discarded and a new transdermal system should be applied. Patients or caregivers should inspect the release liner to ensure that no adhesive containing medication has transferred to the liner. If adhesive transfer has occurred, the transdermal system should be discarded. Refer to the Instructions for Use for recommendations for discarding used DAYTRANA.
DAYTRANA should be applied immediately after opening the individual pouch and removing the protective liner. Do not use if the individual pouch seal is broken or if the transdermal system appears to be damaged. Do not cut transdermal systems. Only intact transdermal systems should be applied. The transdermal system should then be pressed firmly in place with the palm of the hand for approximately 30 seconds, making sure that there is good contact of the transdermal system with the skin, especially around the edges. Exposure to water during bathing, swimming, or showering can affect transdermal system adherence. DAYTRANA should not be applied or re-applied with dressings, tape, or other common adhesives. In the event that a transdermal system does not fully adhere to the skin upon application, or becomes partially or fully detached during wear time, the transdermal system should be discarded and a new transdermal system may be applied at a different site. The total recommended wear time for that day should remain 9 hours regardless of the number of transdermal systems used.
All patients should be advised to avoid exposing the DAYTRANA application site to direct external heat sources, such as hair dryers, heating pads, electric blankets, heated water beds, etc., while wearing the transdermal system [see Warnings and Precautions (5.10)]. When heat is applied to DAYTRANA after transdermal system application, both the rate and the extent of absorption are significantly increased. The temperature-dependent increase in methylphenidate absorption can be greater than 2-fold [see Clinical Pharmacology (12.3)]. This increased absorption can be clinically significant and result in overdose of methylphenidate [see Overdosage (10)].
DAYTRANA should not be stored in refrigerators or freezers.
2.4 Removal of DAYTRANA
DAYTRANA should be peeled off slowly. If necessary, transdermal system removal may be facilitated by gently applying an oil-based product (i.e., petroleum jelly, olive oil, or mineral oil) to the transdermal system edges, gently working the oil underneath the transdermal system edges. If any adhesive remains on the skin following transdermal system removal, an oil-based product may be applied to transdermal system sites in an effort to gently loosen and remove any residual adhesive that remains following transdermal system removal.
In the unlikely event that a transdermal system remains tightly adhered despite these measures, the patient or caregiver should contact the physician or pharmacist. Nonmedical adhesive removers and acetone-based products (i.e., nail polish remover) should not be used to remove DAYTRANA or adhesive.
2.5 Dose/Wear Time Reduction and Discontinuation
DAYTRANA may be removed earlier than 9 hours if a shorter duration of effect is desired or late day side effects appear. Plasma concentrations of d-methylphenidate generally begin declining when the transdermal system is removed, although absorption may continue for several hours. Individualization of wear time may help manage some of the side effects caused by methylphenidate. If aggravation of symptoms or other adverse events occur, the dosage or wear time should be reduced, or, if necessary, the drug should be discontinued. Residual methylphenidate remains in used transdermal systems when worn as recommended.
3 DOSAGE FORM AND STRENGTHS
Four dosage strengths are available:
|
*Nominal in vivo delivery rate in children and adolescents when applied to the hip, based on a 9-hour wear period. |
|||
| Nominal Dose Delivered (mg) Over 9 Hours* | Dosage Rate* (mg/hr) | Transdermal System Size (cm 2) | Methylphenidate Content per Transdermal System (mg) |
| 10 | 1.1 | 12.5 | 27.5 |
| 15 | 1.6 | 18.75 | 41.3 |
| 20 | 2.2 | 25 | 55 |
| 30 | 3.3 | 37.5 | 82.5 |
4 CONTRAINDICATIONS
4.1 Hypersensitivity to Methylphenidate
DAYTRANA is contraindicated in patients known to be hypersensitive to methylphenidate or other components of the product (polyester/ethylene vinyl acetate laminate film backing, acrylic adhesive, silicone adhesive, and fluoropolymer-coated polyester) [see Description (11)] .
5 WARNINGS AND PRECAUTIONS
5.1 Abuse, Misuse, and Addiction
DAYTRANA has a high potential for abuse and misuse. The use of DAYTRANA exposes individuals to the risks of abuse and misuse, which can lead to the development of a substance use disorder, including addiction. DAYTRANA can be diverted for non-medical use into illicit channels or distribution [see Drug Abuse and Dependence (9.2)]. Misuse and abuse of CNS stimulants, including DAYTRANA, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing DAYTRANA, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their caregivers or families about these risks. Advise patients to store DAYTRANA in a safe place, preferably locked, and instruct patients to not give DAYTRANA to anyone else. Throughout DAYTRANA treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
DAYTRANA has special disposal instructions. Instruct patients to find a take back location to dispose of unused or expired DAYTRANA. If a take back program is unavailable, instruct them to:
- Remove DAYTRANA from its pouch, separate it from its liner, fold it in half with the adhesive sides touching each other, and immediately flush the used transdermal system down the toilet, and
- Place the pouch and liner in a container, close the container, and throw out the container in the trash (advise patients not to flush the pouch and liner down the toilet).
5.2 Risks to Patients with Serious Cardiac Disease
Sudden death has been reported in patients with structural cardiac abnormalities or other serious cardiac disease who were treated with CNS stimulants at the recommended ADHD dosage.
Avoid DAYTRANA use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease.
5.3 Increased Blood Pressure and Heart Rate
CNS stimulants may cause an increase in blood pressure (mean increase approximately 2 to 4 mmHg) and heart rate (mean increase approximately 3 to 6 bpm). Some patients may have larger increases.
Monitor all DAYTRANA-treated patients for hypertension and tachycardia.
5.4 Psychiatric Adverse Reactions
Exacerbation of Pre-Existing Psychosis
CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Induction of a Manic Episode in Patients with Bipolar Disease
CNS stimulants may induce a manic or mixed episode in patients. Prior to initiating DAYTRANA treatment, screen patients for risk factors for developing a manic episode (e.g., comorbid or history of depressive symptoms or a family history of suicide, bipolar disorder, or depression).
New Psychotic or Manic Symptoms
CNS stimulants, at the recommended dosages, may cause psychotic or manic symptoms, (e.g., hallucinations, delusional thinking, or mania) in patients without a prior history of psychotic illness or mania. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in approximately 0.1% of CNS stimulant-treated patients compared with 0% of placebo-treated patients. If such symptoms occur, consider discontinuing DAYTRANA.
5.5 Seizures
There is some clinical evidence that stimulants may lower the convulsive threshold in patients with prior history of seizures, in patients with prior EEG abnormalities in absence of seizures, and, very rarely, in patients without a history of seizures and no prior EEG evidence of seizures. In the presence of seizures, the drug should be discontinued.
5.6 Priapism
Prolonged and painful erections, sometimes requiring surgical intervention, have been reported with methylphenidate use in both adult and pediatric male patients. Although priapism was not reported with methylphenidate initiation, it developed after some time on the methylphenidate, often subsequent to an increase in dosage. Priapism also occurred during methylphenidate withdrawal (drug holidays or during discontinuation).
DAYTRANA-treated patients who develop abnormally sustained or frequent and painful erections should seek immediate medical attention.
5.7 Peripheral Vasculopathy, including Raynaud’s phenomenon
Stimulant medications, including DAYTRANA, used to treat ADHD are associated with peripheral vasculopathy, including Raynaud’s phenomenon. Signs and symptoms are usually intermittent and mild; however, sequelae have included digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud’s phenomenon, were observed in post-marketing reports and at therapeutic dosages of CNS stimulants in all age groups throughout the course of treatment. Signs and symptoms generally improved after dosage reduction or discontinuation of the CNS stimulant.
Careful observation for digital changes is necessary during DAYTRANA treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for DAYTRANA-treated patients who develop signs or symptoms of peripheral vasculopathy.
5.8 Long-Term Suppression of Growth in Pediatric Patients
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients.
Careful follow-up of weight and height in children ages 7 to 10 years who were randomized to either methylphenidate or non-medication treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate-treated and non-medication treated children over 36 months (to the ages of 10 to 13 years), suggests that pediatric patients who received methylphenidate for 7 days per week throughout the year had a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this development period.
Closely monitor growth (weight and height) in DAYTRANA-treated pediatric patients. Pediatric patients not growing or gaining height or weight as expected may need to have their treatment interrupted.
5.9 Chemical Leukoderma
DAYTRANA use may result in a persistent loss of skin pigmentation at and around the application site. Loss of pigmentation, in some cases, has been reported at other sites distant from the application site. Chemical leukoderma can mimic the appearance of vitiligo, particularly when the loss of skin pigmentation involves areas distant from the application site. Individuals with a history of vitiligo and/or a family history of vitiligo may be more at risk. Skin depigmentation may persist even after DAYTRANA use is discontinued. Monitor for signs of skin depigmentation, and advise patients to immediately inform their healthcare provider if changes in skin pigmentation occur. Discontinue use of the DAYTRANA in patients with chemical leukoderma.
5.10 Contact Sensitization
In an open-label study of 305 subjects conducted to characterize dermal reactions in children with ADHD treated with DAYTRANA using a 9-hour wear time, one subject (0.3%) was confirmed by patch testing to be sensitized to methylphenidate (allergic contact dermatitis). This subject experienced erythema and edema at DAYTRANA application sites with concurrent urticarial lesions on the abdomen and legs resulting in treatment discontinuation. This subject was not transitioned to oral methylphenidate.
Use of DAYTRANA may lead to contact sensitization. DAYTRANA should be discontinued if contact sensitization is suspected. Erythema is commonly seen with use of DAYTRANA and is not by itself an indication of sensitization. However, contact sensitization should be suspected if erythema is accompanied by evidence of a more intense local reaction (edema, papules, vesicles) that does not significantly improve within 48 hours or spreads beyond the transdermal system site. Confirmation of a diagnosis of contact sensitization (allergic contact dermatitis) may require further diagnostic testing.
Patients sensitized from use of DAYTRANA, as evidenced by development of an allergic contact dermatitis, may develop systemic sensitization or other systemic reactions if methylphenidate-containing products are taken via other routes, e.g., orally. Manifestations of systemic sensitization may include a flare-up of previous dermatitis or of prior positive patch-test sites, or generalized skin eruptions in previously unaffected skin. Other systemic reactions may include headache, fever, malaise, arthralgia, diarrhea, or vomiting. No cases of systemic sensitization have been observed in clinical trials of DAYTRANA.
Patients who develop contact sensitization to DAYTRANA and require oral treatment with methylphenidate should be initiated on oral medication under close medical supervision. It is possible that some patients sensitized to methylphenidate by exposure to DAYTRANA may not be able to take methylphenidate in any form.
5.11 Patients Using External Heat
Patients should be advised to avoid exposing the DAYTRANA application site to direct external heat sources, such as hair dryers, heating pads, electric blankets, heated water beds, etc., while wearing the transdermal system. When heat is applied to DAYTRANA after application, both the rate and extent of absorption are significantly increased. The temperature-dependent increase in methylphenidate absorption can be greater than 2-fold [see Clinical Pharmacology (12.3)]. This increased absorption can be clinically significant and can result in overdose of methylphenidate [see Overdosage (10)].
5.12 Hematologic Monitoring
Periodic CBC, differential, and platelet counts are advised during prolonged therapy.
5.13 Acute Angle Closure Glaucoma
There have been reports of angle closure glaucoma associated with methylphenidate treatment.
Although the mechanism is not clear, DAYTRANA-treated patients considered at risk for acute angle closure glaucoma (e.g., patients with significant hyperopia) should be evaluated by an ophthalmologist.
5.14 Increased Intraocular Pressure and Glaucoma
There have been reports of an elevation of intraocular pressure (IOP) associated with methylphenidate treatment [see Adverse Reactions (6.2)] .
Prescribe DAYTRANA to patients with open-angle glaucoma or abnormally increased IOP only if the benefit of treatment is considered to outweigh the risk. Closely monitor DAYTRANA-treated patients with a history of abnormally increased IOP or open angle glaucoma.
5.15 Motor and Verbal Tics, and Worsening of Tourette’s Syndrome
CNS stimulants, including methylphenidate, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette’s syndrome has also been reported [see Adverse Reactions (6.2)] .
Before initiating DAYTRANA, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor DAYTRANA-treated patients for the emergence or worsening of tics or Tourette’s syndrome, and discontinue treatment if clinically appropriate.
6 ADVERSE REACTIONS
Detailed information on serious and adverse reactions of particular importance is provided in the Boxed Warning and Warnings and Precautions (5) sections:
- Abuse, Misuse, and Addiction [see Boxed Warning]
- Hypersensitivity to Methylphenidate [see Contraindications (4.1)]
- Monoamine Oxidase Inhibitors [see Contraindications (4.2) and Drug Interactions (7.1)]
- Risks to Patients with Serious Cardiac Disease [see Warnings and Precautions (5.2)]
- Increased Blood Pressure and Heart Rate [see Warnings and Precautions (5.3)]
- Psychiatric Adverse Reactions [see Warnings and Precautions (5.4)]
- Seizures [see Warnings and Precautions (5.5)]
- Priapism [see Warnings and Precautions (5.6)]
- Peripheral Vasculopathy [see Warnings and Precautions (5.7)]
- Long-Term Suppression of Growth in Pediatric Patients [see Warnings and Precautions (5.8)]
- Chemical Leukoderma [see Warnings and Precautions (5.9)]
- Contact Sensitization [see Warnings and Precautions (5.10)]
- External Heat [see Warnings and Precautions (5.11)]
- Hematologic Monitoring [see Warnings and Precautions (5.12)]
- Acute Angle Closure Glaucoma [see Warnings and Precautions (5.13)]
- Increased Intraocular Pressure and Glaucoma [see Warnings and Precautions (5.14)]
- Motor and Verbal Tics, and Worsening of Tourette’s Syndrome [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most commonly reported (frequency ≥ 5% and twice the rate of placebo) adverse reactions in a controlled trial in children aged 6-12 included appetite decreased, insomnia, nausea, vomiting, weight decreased, tic, affect lability, and anorexia. The most commonly reported (frequency ≥ 5% and twice the rate of placebo) adverse reactions in a controlled trial in adolescents aged 13-17 were appetite decreased, nausea, insomnia, weight decreased, dizziness, abdominal pain, and anorexia [see Adverse Reactions (6.1)].
The most common (≥ 2% of subjects) adverse reaction associated with discontinuations in double-blind clinical trials in children or adolescents was application site reactions [see Adverse Reactions (6.1)].
The overall DAYTRANA development program included exposure to DAYTRANA in a total of 2,152 participants in clinical trials, including 1,529 children aged 6-12, 223 adolescents aged 13-17, and 400 adults. The 1,752 child and adolescent subjects aged 6-17 years were evaluated in 10 controlled clinical studies, 7 open-label clinical studies, and 5 clinical pharmacology studies. In a combined studies pool of children using DAYTRANA with a wear time of 9 hours, 212 subjects were exposed for ≥ 6 months and 115 were exposed for ≥ 1 year; 85 adolescents have been exposed for ≥ 6 months. Most patients studied were exposed to DAYTRANA transdermal system sizes of 12.5 cm2, 18.75 cm2, 25 cm2 or 37.5 cm2, with a wear time of 9 hours.
In the data presented below, the adverse reactions reported during exposure were obtained primarily by general inquiry at each visit, and were recorded by the clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse reactions without first grouping similar types of events into a smaller number of standardized event categories.
Adverse Reactions in Clinical Studies with Discontinuation of Treatment
In a 7-week double-blind, parallel-group, placebo-controlled study in children with ADHD conducted in the outpatient setting, 7.1% (7/98) of patients treated with DAYTRANA discontinued due to adverse events compared with 1.2% (1/85) receiving placebo. The most commonly reported (≥ 1% and twice the rate of placebo) adverse reactions leading to discontinuation in the DAYTRANA group were application site reaction (2%), tics (1%), headache (1%), and irritability (1%).
In a 7-week double-blind, parallel-group, placebo-controlled study in adolescents with ADHD conducted in the outpatient setting, 5.5% (8/145) of patients treated with DAYTRANA discontinued due to adverse reactions compared with 2.8% (2/72) receiving placebo. The most commonly reported adverse reactions leading to discontinuation in the DAYTRANA group were application site reaction (2%) and decreased appetite/anorexia (1.4%).
Commonly Observed Adverse Reactions in Double-Blind, Placebo-Controlled Trials
Skin Irritation and Application Site Reactions
DAYTRANA is a dermal irritant. In addition to the most commonly reported adverse reactions presented in Table 2, the majority of subjects in those studies had minimal to definite skin erythema at the transdermal system application site. This erythema generally caused no or minimal discomfort and did not usually interfere with therapy or result in discontinuation from treatment. Erythema is not by itself a manifestation of contact sensitization. However, contact sensitization should be suspected if erythema is accompanied by evidence of a more intense local reaction (edema, papules, vesicles) that does not significantly improve within 48 hours or spreads beyond the transdermal system site [see Warnings and Precautions (5.10)].
Most Commonly Reported Adverse Reactions
Table 2 lists treatment-emergent adverse reactions reported in ≥ 1% DAYTRANA-treated children or adolescents with ADHD in two 7 week double-blind, parallel-group, placebo-controlled studies conducted in the outpatient setting. Overall, in these studies, 75.5% of children and 78.6% of adolescents experienced at least 1 adverse event.
|
* Six subjects had affect lability, all judged as mild and described as increased emotionally sensitive, emotionality, emotional instability, emotional lability, and intermittent emotional |
||||
| Table 2 Number (%) of Subjects with Commonly Reported Adverse Reactions (≥ 1% in the DAYTRANA Group) in 7-Week Placebo-controlled Studies in Either Children or Adolescents - Safety Population | ||||
| Adolescents | Children | |||
| System Organ Class Preferred term | Placebo N = 72 | DAYTRANA N = 145 | Placebo N = 85 | DAYTRANA N = 98 |
| Cardiac Disorders | ||||
| Tachycardia | 0 (0) | 1 (0.7) | 0 (0) | 1 (1.0) |
| Gastrointestinal disorders | ||||
| Abdominal pain | 0 (0) | 7 (4.8) | 5 (5.9) | 7 (7.1) |
| Nausea | 2 (2.8) | 14 (9.7) | 2 (2.4) | 12 (12.2) |
| Vomiting | 1 (1.4) | 5 (3.4) | 4 (4.7) | 10 (10.2) |
| Investigations | ||||
| Weight decreased | 1 (1.4) | 8 (5.5) | 0 (0) | 9 (9.2) |
| Metabolism and nutrition disorders | ||||
| Anorexia | 1 (1.4) | 7 (4.8) | 1 (1.2) | 5 (5.1) |
| Decreased appetite | 1 (1.4) | 37 (25.5) | 4 (4.7) | 25 (25.5) |
| Nervous system disorders | ||||
| Dizziness | 1 (1.4) | 8 (5.5) | 1 (1.2) | 0 (0) |
| Headache | 9 (12.5) | 18 (12.4) | 10 (11.8) | 15 (15.3) |
| Psychiatric disorders | ||||
| Affect lability | 1 (1.4) | 0 (0) | 0 (0) | 6 (6.1)* |
| Insomnia | 2 (2.8) | 9 (6.2) | 4 (4.7) | 13 (13.3) |
| Irritability | 5 (6.9) | 16 (11) | 4 (4.7) | 7 (7.1) |
| Tic | 0 (0) | 0 (0) | 0 (0) | 7 (7.1) |
Adverse Reactions in Clinical Studies with the Long-Term Use of DAYTRANA
In a long-term open-label study of up to 12 months duration in 326 children wearing DAYTRANA 9 hours daily, the most common (≥ 10%) adverse reactions were decreased appetite, headache, and weight decreased. A total of 30 subjects (9.2%) were withdrawn from the study due to adverse events and 22 additional subjects (6.7%) discontinued treatment as the result of an application site reaction. Other than application site reactions, affect lability (5 subjects, 1.5%) was the only additional adverse reaction leading to discontinuation reported with a frequency of greater than 1%.
In a long-term open-label study of up to 6 months duration in 162 adolescents wearing DAYTRANA 9 hours daily, the most common (≥ 10%) adverse reactions were decreased appetite and headache. A total of 9 subjects (5.5%) were withdrawn from the study due to adverse events and 3 additional subjects (1.9%) discontinued treatment as the result of an application site reaction. Other adverse reactions leading to discontinuation that occurred with a frequency of greater than 1% included affect lability and irritability (2 subjects each, 1.2%).
6.2 Postmarketing Experience
In addition, the following adverse reactions have been identified during the post-approval use of DAYTRANA. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to DAYTRANA exposure.
Cardiac Disorders: palpitations.
Eye Disorders: visual disturbances, blurred vision, increased intraocular pressure, mydriasis, and accommodation disorder.
General Disorders and Administration Site Disorders: fatigue, application site reactions such as bleeding, bruising, burn, burning, dermatitis, discharge, discoloration, discomfort, dryness, eczema, edema, erosion, erythema, excoriation, exfoliation, fissure, hyperpigmentation, hypopigmentation, induration, infection, inflammation, irritation, pain, papules, paresthesia, pruritus, rash, scab, swelling, ulcer, urticaria, vesicles, and warmth.
Immune System Disorders: hypersensitivity reactions including generalized erythematous and urticarial rashes, allergic contact dermatitis, angioedema, and anaphylaxis.
Investigations: blood pressure increased.
Nervous System Disorders: convulsion, dyskinesia, lethargy, somnolence, serotonin syndrome in combination with serotonergic drugs, and extrapyramidal disorder, motor, and verbal tics.
Psychiatric Disorders: depression, hallucination, nervousness, and libido changes.
Skin and Subcutaneous Tissue Disorders: alopecia.
Adverse Reactions with Oral Methylphenidate Products
Nervousness and insomnia are the most common adverse reactions reported with other methylphenidate products. In children, loss of appetite, abdominal pain, weight loss during prolonged therapy, insomnia, and tachycardia may occur more frequently; however, any of the other adverse reactions listed below may also occur.
Other reactions include:
Cardiac Disorders: angina, arrhythmia, and pulse increased or decreased.
Immune System Disorders: hypersensitivity reactions including skin rash, urticaria, fever, arthralgia, exfoliative dermatitis, erythema multiforme with histopathological findings of necrotizing vasculitis, and thrombocytopenic purpura.
Metabolism and Nutrition Disorders: anorexia and weight loss during prolonged therapy.
Nervous System Disorders: drowsiness, rare reports of Tourette's syndrome and toxic psychosis.
Very rare reports of neuroleptic malignant syndrome (NMS) have been received, and, in most of these, patients were concurrently receiving therapies associated with NMS. In a single report, a ten-year-old boy who had been taking methylphenidate for approximately 18 months experienced an NMS-like event within 45 minutes of ingesting his first dose of venlafaxine. It is uncertain whether this case represented a drug-drug interaction, a response to either drug alone, or some other cause.
Vascular Disorders: blood pressure increased or decreased and cerebral arteritis and/or occlusion.
Although a definite causal relationship has not been established, the following have been reported in patients taking methylphenidate:
Blood and Lymphatic System Disorders: leukopenia and/or anemia.
Hepatobiliary Disorders: abnormal liver function, ranging from transaminase elevation to severe hepatic injury.
Psychiatric Disorders: transient depressed mood.
Skin and Subcutaneous Tissue Disorders: scalp hair loss.
Musculoskeletal and Connective Tissue Disorders: rhabdomyolysis.
7 DRUG INTERACTIONS
7.1 Monoamine Oxidase Inhibitors (MAOI)
Concomitant use of MAOIs and CNS stimulants, including DAYTRANA, can cause hypertensive crisis. Potential outcomes include death, stroke, myocardial infarction, aortic dissection, ophthalmological complications, eclampsia, pulmonary edema, and renal failure [see Contraindications (4.2)]. Concomitant use of DAYTRANA with MAOIs or within 14 days after discontinuing MAOI treatment is contraindicated.
7.2 Antihypertensive Drugs
DAYTRANA may decrease the effectiveness of drugs used to treat hypertension. Monitor blood pressure and adjust the dosage of the antihypertensive drug as needed [see Warnings and Precautions (5.2)].
7.3 Coumarin Anticoagulants, Antidepressants, and Selective Serotonin Reuptake Inhibitors
Human pharmacologic studies have shown that methylphenidate may inhibit the metabolism of coumarin anticoagulants, anticonvulsants (e.g., phenobarbital, phenytoin, primidone), and some tricyclic drugs (e.g., imipramine, clomipramine, desipramine) and selective serotonin reuptake inhibitors. Downward dose adjustments of these drugs may be required when given concomitantly with methylphenidate. It may be necessary to adjust the dosage and monitor plasma drug concentrations (or, in the case of coumarin, coagulation times), when initiating or discontinuing methylphenidate.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ADHD medications, including DAYTRANA, during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for ADHD Medications at 1-866-961-2388 or visit https://womensmentalhealth.org/adhd-medications/.
Risk Summary
Published studies and post-marketing reports on methylphenidate use during pregnancy are insufficient to identify a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the fetus associated with the use of CNS stimulants during pregnancy (see Clinical Considerations).
No effects on morphological development were observed in embryo-fetal development studies with oral administration of methylphenidate to pregnant rats and rabbits during organogenesis. However, spina bifida was observed in rabbits when given oral doses of 200 mg/kg/day. When methylphenidate was administered orally to rats throughout pregnancy and lactation, offspring growth and survival were decreased at maternally toxic doses (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinical recognized pregnancies is 2-4% and 15-20%, respectively.
Fetal/Neonatal adverse reactions
CNS stimulants, such as DAYTRANA, can cause vasoconstriction and thereby decrease placental perfusion. No fetal and/or neonatal adverse reactions have been reported with the use of therapeutic doses of methylphenidate during pregnancy; however, premature delivery and low birth weight infants have been reported in amphetamine-dependent mothers.
Animal Data
Animal reproduction toxicity studies with transdermal methylphenidate have not been performed. In embryo-fetal development studies conducted in rats and rabbits, methylphenidate was administered orally to pregnant animals during the period of organogenesis, at doses up to 100 and 200 mg/kg/day, respectively. No evidence of morphological development effects was found either of the species; however, increased incidences of fetal skeletal variations were observed in rats at 60 mg/kg or greater and an increase in fetal visceral variations was seen in rabbits at the highest dose. In a previous study, methylphenidate was shown to have malformations (increased incidence of fetal spina bifida) in rabbits when given oral doses of 200 mg/kg/day. When methylphenidate was administered orally to rats throughout pregnancy and lactation at doses of up to 60 mg/kg/day, offspring growth and survival were decreased at maternally toxic doses.
In a study in which oral methylphenidate was given to rats throughout pregnancy and lactation at doses up to 60 mg/kg/day, offspring weights and survival were decreased at 40 mg/kg/day and above; these doses caused some maternal toxicity.
8.2 Lactation
Risk Summary
Limited published literature, based on breast milk sampling from five mothers, reports that methylphenidate is present in human milk, which resulted in infant doses of 0.16% to 0.7% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 1.1 and 2.7. There are no reports of adverse effects on the breastfed infant and no effects on milk production. Long-term neurodevelopmental effects on infants from stimulant exposure are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for DAYTRANA and any potential adverse effects on the breastfed infant from DAYTRANA or from the underlying maternal condition.
Clinical Considerations
Monitor breastfeeding infants for adverse reactions, such as agitation, insomnia, anorexia, and reduced weight gain.
8.4 Pediatric Use
The safety and effectiveness of DAYTRANA in pediatric patients less than 6 years have not been established. Long-term effects of methylphenidate in children have not been well established.
The safety and effectiveness of DAYTRANA for the treatment of ADHD have been established in pediatric patients 6 to 17 years.
Long Term Suppression of Growth
Growth should be monitored during treatment with stimulants, including DAYTRANA. Children who are not growing or gaining weight as expected may need to have their treatment interrupted [see Warnings and Precautions (5.8)].
Juvenile Animal Toxicity Data
Rats treated with methylphenidate early in the postnatal period through sexual maturation demonstrated a decrease in spontaneous locomotor activity in adulthood. A deficit in acquisition of a specific learning task was observed in females only.
Studies with transdermal methylphenidate have not been performed in juvenile animals. In a study conducted in young rats, methylphenidate was administered orally at doses of up to 100 mg/kg/day for 9 weeks, starting early in the postnatal period (Postnatal Day 7) and continuing through sexual maturity (Postnatal Week 10). When these animals were tested as adults (Postnatal Weeks 13-14), decreased spontaneous locomotor activity was observed in males and females previously treated with 50 mg/kg/day or greater, and a deficit in the acquisition of a specific learning task was seen in females exposed to the highest dose. The no effect level for juvenile neurobehavioral development in rats was 5 mg/kg/day. The clinical significance of the long-term behavioral effects observed in rats is unknown.
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
DAYTRANA has a high potential for abuse and misuse which can lead to the development of a substance use disorder, including addiction [see Warnings and Precautions (5.1)]. DAYTRANA can be diverted for non-medical use into illicit channels or distribution.
Abuse is the intentional non-therapeutic use of a drug, even once, to achieve a desired psychological or physiological effect. Misuse is the intentional use, for therapeutic purposes, of a drug by an individual in a way other than prescribed by a health care provider or for whom it was not prescribed. Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that may include a strong desire to take the drug, difficulties in controlling drug use (e.g., continuing drug use despite harmful consequences, giving a higher priority to drug use than other activities and obligations), and possible tolerance or physical dependence.
Misuse and abuse of methylphenidate may cause increased heart rate, respiratory rate, or blood pressure; sweating; dilated pupils; hyperactivity; restlessness; insomnia; decreased appetite; loss of coordination; tremors; flushed skin; vomiting; and/or abdominal pain. Anxiety, psychosis, hostility, aggression, and suicidal or homicidal ideation have also been observed with CNS stimulants abuse and/or misuse. Misuse and abuse of CNS stimulants, including DAYTRANA, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
9.3 Dependence
Physical Dependence
DAYTRANA may produce physical dependence. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
Withdrawal signs and symptoms after abrupt discontinuation or dose reduction following prolonged use of CNS stimulants including DAYTRANA include dysphoric mood; depression; fatigue; vivid, unpleasant dreams; insomnia or hypersomnia; increased appetite; and psychomotor retardation or agitation.
Tolerance
DAYTRANA may produce tolerance. Tolerance is a physiological state characterized by a reduced response to a drug after repeated administration (i.e., a higher dose of a drug is required to produce the same effect that was once obtained at a lower dose).
10 OVERDOSAGE
Clinical Effects of Overdose
Overdose of CNS stimulants is characterized by the following sympathomimetic effects:
- Cardiovascular effects including tachyarrhythmias, and hypertension or hypotension. Vasospasm, myocardial infarction, or aortic dissection may precipitate sudden cardiac death. Takotsubo cardiomyopathy may develop.
- CNS effects including psychomotor agitation, confusion, and hallucinations. Serotonin syndrome, seizures, cerebral vascular accidents, and coma may occur.
- Life-threatening hyperthermia (temperatures greater than 104°F) and rhabdomyolysis may develop.
Overdose Management
Consider the possibility of multiple drug ingestion. Because methylphenidate has a large volume of distribution and is rapidly metabolized, dialysis is not useful. Remove all transdermal systems immediately and cleanse the area(s) to remove any remaining adhesive. The continuing absorption of methylphenidate from the skin, even after removal of the transdermal system, should be considered when treating patients with overdose.
Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
11 DESCRIPTION
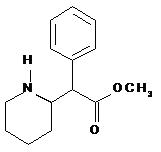
DAYTRANA is an adhesive-based matrix transdermal system containing methylphenidate that is applied to intact skin. The chemical name for methylphenidate is α-phenyl-2-piperidineacetic acid methyl ester. It is a white to off-white powder and is soluble in alcohol, ethyl acetate, and ether. Methylphenidate is practically insoluble in water and petrol ether. Its molecular weight is 233.31. Its empirical formula is C14H19NO2. The structural formula of methylphenidate is:
Transdermal System Components
DAYTRANA contains methylphenidate in a multipolymeric adhesive. The methylphenidate is dispersed in acrylic adhesive that is dispersed in a silicone adhesive. The composition per unit area of all dosage strengths is identical, and the total dose delivered is dependent on the transdermal system size and wear time.
DAYTRANA consists of three layers, as seen in the figure below (cross-section of the transdermal system).
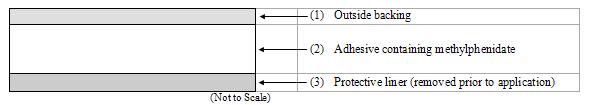
Proceeding from the outer surface toward the surface adhering to the skin, the layers are (1) a polyester/ethylene vinyl acetate laminate film backing, (2) a proprietary adhesive formulation incorporating Noven Pharmaceuticals, Inc.'s DOT Matrix™ transdermal technology consisting of an acrylic adhesive, a silicone adhesive, and methylphenidate, and (3) a fluoropolymer-coated polyester protective liner, which is attached to the adhesive surface and must be removed before the transdermal system can be used.
The active component of the transdermal system is methylphenidate. The remaining components are pharmacologically inactive.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Methylphenidate is a CNS stimulant. Its mode of therapeutic action in ADHD is not known.
12.2 Pharmacodynamics
Methylphenidate is a racemic mixture comprised of the d-and l-enantiomers. The d-enantiomer is more pharmacologically active than the l-enantiomer. Methylphenidate blocks the reuptake of norepinephrine and dopamine into the presynaptic neuron and increases the release of these monoamines into the extraneuronal space.
12.3 Pharmacokinetics
The pharmacokinetics of DAYTRANA when applied to the hip for 9 hours have been studied in ADHD patients 6 to 17 years old.
The amount of methylphenidate absorbed systemically is a function of both wear time and transdermal system size. In patients with ADHD, peak plasma levels of methylphenidate are reached at about 10 hours after single application and 8 hours after repeat transdermal system applications (12.5cm2 to 37.5cm2) when worn up to 9 hours.
On single dosing of children or adolescents with DAYTRANA, there was a delay of, on average, 2 hours before d-methylphenidate was detectable in the circulation. On repeat dosing, low concentrations (1.2-3.0 ng/mL in children and 0.5-1.7ng/mL in adolescents, on average across the dose range) were observed earlier in the profile, due to carry-over effect. Following the application of DAYTRANA once daily with a 9-hour wear time, the mean pharmacokinetic parameters of d-methylphenidate in children and adolescents with ADHD after 4 weeks of therapy are summarized in Table 3.
|
1 Dose maintained fixed for 28 days; 2 Dose escalated at 7 day intervals from 12.5 cm2 through 18.75 cm2 and 25 cm2 to 37.5 cm2; 3 Dose escalated at 7 day intervals from 18 mg through 27 mg and 36 mg to 54 mg; 4 Median (minimum - maximum); t lag = Last Sampling Time Prior to Time of First Quantifiable Plasma Concentration |
||||
| Table 3 Mean Plasma d-Methylphenidate Pharmacokinetic Parameters After Repeated 9-Hour Applications of DAYTRANA or Oral ER-MPH for up to 28 days to Pediatric ADHD Patients (Aged 6 - 17 years) |
||||
| Children | ||||
| Parameter | DAYTRANA 1
12.5cm 2 (N=12) | DAYTRANA 2
37.5cm 2 (N=10) | Oral ER-MPH 3
18mg | Oral ER-MPH 3
54mg |
| C ssmax
(ng/mL) | 15.7 ± 9.39 | 42.9 ± 22.4 | 8.37 ± 4.14 | 26.1 ± 11.2 |
| C ssmin
(ng/mL) | 1.04 ± 1.17 | 1.96 ± 1.73 | 0.708 ± 1.08 | 1.19 ± 1.54 |
| AUC ss
(ng·hr/mL) | 163 ± 101 | 447 ± 230 | 97.7 ± 67.0 | 317 ± 160 |
| t lag
(h) 4 | 0 (0 - 2.0) | 0 (0 - 1.0) | 0 | 0 |
| Adolescents | ||||
| C ssmax
(ng/mL) | 8.32 ± 4.60 | 16.5 ± 6.94 | 5.23 ± 1.72 | 18.0 ± 6.97 |
| C ssmin
(ng/mL) | 0.544 ± 0.383 | 1.02 ± 0.629 | 0.360 ± 0.478 | 1.50 ± 0.937 |
| AUC ss
(ng·hr/mL) | 85.7 ± 50.0 | 167 ± 66.0 | 59.7 ± 19.1 | 216 ± 80.8 |
| t lag
(h) 4 | 0 (0 - 2.0) | 0 (0 - 2.0) | 0 | 0 |
Following administration of DAYTRANA 12.5cm2 to pediatric and adolescent ADHD patients daily for 7 days, there were 13% and 14% increases, respectively, in steady state area under the plasma concentration-time curve (AUC ss) relative to that anticipated on the basis of single dose pharmacokinetics (AUC0 -∞); after 28 days administration, these increments increased to 64% and 76%, respectively. C max increased by nearly 69% and 100% within 4 weeks of daily administration of the starting dose in children and adolescents, respectively.
The observed exposures with DAYTRANA could not be explained by drug accumulation predicted from observed single dose pharmacokinetics and there was no evidence that clearance or rate of elimination changed between single and repeat dosing. Neither were they explainable by differences in dosing patterns between treatments, age, race, or gender. This suggests that transdermal absorption of methylphenidate may increase with repeat dosing with DAYTRANA; on average, steady-state is likely to have been achieved by approximately 14 days of dosing.
In the single- and multiple dose study described above, exposure to l-methylphenidate was 46% of the exposure to d-methylphenidate in children and 40% in adolescents. l-methylphenidate is less pharmacologically active than d-methylphenidate [see Pharmacodynamics (12.2)] .
In a phase 2 PK/PD study in children aged 6-12 years, 2/3 of patients had 2-hour d-MPH concentrations < 5 ng/mL on chronic dosing, and at 3 hours 40% of patients had d-MPH concentrations < 5 ng/mL [see Clinical Studies (14)] .
When DAYTRANA is applied to inflamed skin both the rate and extent of absorption are increased as compared with intact skin. When applied to inflamed skin, lag time is no greater than 1 hour, T max is 4 hours, and both C max and AUC are approximately 3-fold higher.
When heat is applied to DAYTRANA after application, both the rate and the extent of absorption are significantly increased. Median T lag occurs 1 hour earlier, T max occurs 0.5 hours earlier, and median C max and AUC are 2-fold and 2.5-fold higher, respectively.
Application sites other than the hip can have different absorption characteristics and have not been adequately studied in safety or efficacy studies.
Dose Proportionality
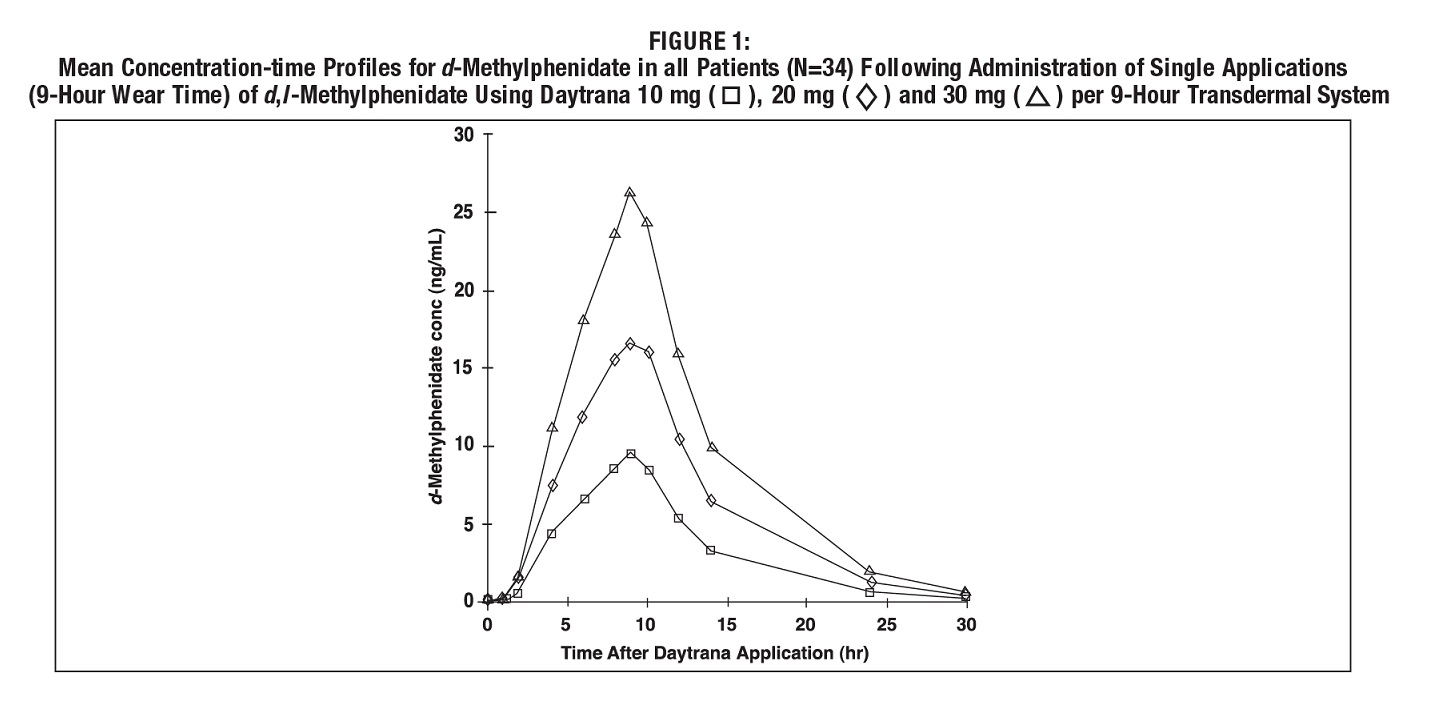
Following a single 9-hour application of DAYTRANA doses of 10 mg / 9 hours to 30 mg / 9 hours transdermal systems to 34 children with ADHD, C max and AUC 0-t of d-methylphenidate were proportional to the transdermal system dose. Mean plasma concentration-time plots are shown in Figure 1. C max of l-methylphenidate was also proportional to the transdermal system dose. AUC 0-t of l-methylphenidate was only slightly greater than proportional to transdermal system dose.
Distribution
Upon removal of DAYTRANA, methylphenidate plasma concentrations in children with ADHD decline in a biexponential manner. This may be due to continued distribution of MPH from the skin after transdermal system removal.
Metabolism and Excretion
Methylphenidate is metabolized primarily by de-esterification to alpha-phenyl-piperidine acetic acid (ritalinic acid), which has little or no pharmacologic activity.
Transdermal administration of methylphenidate exhibits much less first pass effect than oral administration. Consequently, a much lower dose of DAYTRANA on a mg/kg basis compared to oral dosages may still produce higher exposures of d-MPH with transdermal administration compared to oral administration. In addition, very little, if any, l-methylphenidate is systemically available after oral administration due to first pass metabolism, whereas after transdermal administration of racemic methylphenidate exposure to l-methylphenidate is nearly as high as to d-methylphenidate.
The mean elimination t 1/2 from plasma of d-methylphenidate after removal of DAYTRANA in children aged 6 to 12 years and adolescents aged 13-17 years was approximately 4 to 5 hours. The t 1/2 of l-methylphenidate was shorter than for d-methylphenidate and ranged from 1.4 to 2.9 hours, on average.
The C max and AUC of d-methylphenidate were approximately 50% lower in adolescents, compared to children, following either a 1-day or 7-day administration of DAYTRANA (10mg/9hr). Multiple-dose administration of DAYTRANA did not result in significant accumulation of methylphenidate; following 7 days of DAYTRANA administration (10mg/9hr) in children and adolescents, the accumulation index of methylphenidate was 1.1, based on the mean steady state area under the plasma concentration-time curve (AUC ss) relative to that anticipated on the basis of single dose pharmacokinetics (AUC 0-∞).
Food Effects
The pharmacokinetics or the pharmacodynamic food effect performance after application of DAYTRANA has not been studied, but because of the transdermal route of administration, no food effect is expected.
Special Populations
Gender
The pharmacokinetics of methylphenidate after single and repeated doses of DAYTRANA were similar between boys and girls with ADHD, after allowance for differences in body weight.
Race
The influence of race on the pharmacokinetics of methylphenidate after administration of DAYTRANA has not been defined.
Age
The pharmacokinetics of methylphenidate after administration of DAYTRANA have not been studied in children less than 6 years of age.
Renal Impairment
There is no experience with the use of DAYTRANA in patients with renal insufficiency.
Hepatic Impairment
There is no experience with the use of DAYTRANA in patients with hepatic insufficiency.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis/Mutagenesis and Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of transdermal methylphenidate have not been performed. In a lifetime carcinogenicity study of oral methylphenidate carried out in B6C3F1 mice, methylphenidate caused an increase in hepatocellular adenomas and, in males only, an increase in hepatoblastomas, at a daily dose of approximately 60 mg/kg/day. Hepatoblastoma is a relatively rare rodent malignant tumor type. There was no increase in total malignant hepatic tumors. The mouse strain used is sensitive to the development of hepatic tumors and the significance of these results to humans is unknown.
Orally administered methylphenidate did not cause any increases in tumors in a lifetime carcinogenicity study carried out in F344 rats; the highest dose used was approximately 45 mg/kg/day.
In a 24-week oral carcinogenicity study in the transgenic mouse strain p53+/-, which is sensitive to genotoxic carcinogens, there was no evidence of carcinogenicity. In this study, male and female mice were fed diets containing the same concentration of methylphenidate as in the lifetime carcinogenicity study; the high-dose groups were exposed to 60 to 74 mg/kg/day of methylphenidate.
Mutagenesis
Methylphenidate was not mutagenic in the in vitro Ames reverse mutation assay or in the in vitro mouse lymphoma cell forward mutation assay. Sister chromatid exchanges and chromosome aberrations were increased, indicative of a weak clastogenic response, in an in vitro assay in cultured Chinese hamster ovary cells. Methylphenidate was negative in vivo in males and females in the mouse bone marrow micronucleus assay.
Impairment of Fertility
Methylphenidate did not impair fertility in male or female mice that were fed diets containing the drug in an 18-week continuous breeding study. The study was conducted at doses up to 160 mg/kg/day.
14 CLINICAL STUDIES
DAYTRANA was demonstrated to be effective in the treatment of ADHD in two (2) randomized double-blind, placebo-controlled studies in children aged 6 to 12 years and one (1) randomized, double-blind, placebo-controlled study in adolescents aged 13 to 17 years who met Diagnostic and Statistical Manual (DSM-IV-TR®) criteria for ADHD. DAYTRANA wear time was 9 hours in all three (3) studies.
In Study 1, conducted in a classroom setting, symptoms of ADHD were evaluated by school teachers and observers using the Deportment Subscale from the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) rating scale which assesses behavior symptoms in the classroom setting. DAYTRANA was applied for 9 hours before removal. There was a 5-week open-label DAYTRANA dose optimization phase using dosages of 10, 15, 20, and 30 mg / 9 hours, followed by a 2-week randomized, double-blind, placebo-controlled crossover treatment phase using the optimal transdermal system dose for each patient or placebo. The mean differences between DAYTRANA and placebo in change from baseline in SKAMP Deportment Scores were statistically significant in favor of DAYTRANA beginning at 2 hours and remained statistically significant at all subsequent measured time points through 12 hours after application of DAYTRANA.
In Study 2, conducted in the outpatient setting, DAYTRANA or placebo was blindly administered in a flexible-dose design using doses of 10, 15, 20, and 30 mg / 9 hours to achieve an optimal regimen over 5 weeks, followed by a 2-week maintenance period using the optimal transdermal system dose for each patient. Symptoms of ADHD were evaluated by the ADHD-Rating Scale (RS)-IV. DAYTRANA was statistically significantly superior to placebo as measured by the mean change from baseline for the ADHD-RS-IV total score. Although this study was not designed specifically to evaluate dose response, in general there did not appear to be any additional effectiveness accomplished by increasing the transdermal system dose from 20 mg / 9 hours to 30 mg / 9 hours.
In Study 3, conducted in the outpatient setting, DAYTRANA or placebo was blindly administered in a flexible-dose design using doses of 10, 15, 20, and 30 mg / 9 hours during a 5-week dose-optimization phase, followed by a 2-week maintenance period using the optimal transdermal system dose for each patient. Symptoms of ADHD were evaluated using the ADHD-Rating Scale (RS)-IV. DAYTRANA was statistically significantly superior to placebo as measured by the mean change from baseline in the ADHD-RS-IV total score.
16 HOW SUPPLIED/STORAGE AND HANDLING
DAYTRANA is supplied in a sealed tray containing 30 individually pouched transdermal systems. See the chart below for information regarding available strengths.
|
*Nominal in vivo delivery rate per hour in children and adolescents when applied to the hip, based on a 9-hour wear period. **Methylphenidate content in each transdermal system. |
|||||
| Nominal Dose Delivered (mg) Over 9 Hours | Dosage Rate* (mg/hr) | Transdermal System Size (cm 2) | Methylphenidate Content per Transdermal System** (mg) | Transdermal Systems Per Carton | NDC Number |
| 10 | 1.1 | 12.5 | 27.5 | 30 | 68968-5552-3 |
| 15 | 1.6 | 18.75 | 41.3 | 30 | 68968-5553-3 |
| 20 | 2.2 | 25 | 55 | 30 | 68968-5554-3 |
| 30 | 3.3 | 37.5 | 82.5 | 30 | 68968-5555-3 |
Store at 25° C (77° F); excursions permitted to 15-30° C (59-86° F) [see USP Controlled Room Temperature]. Do not store transdermal systems unpouched. Do not store transdermal systems in refrigerators or freezers.
Once the sealed tray is opened, use contents within 2 months. Apply the transdermal system immediately upon removal from the individual protective pouch. For transdermal use only.
See the Patient Counseling Information (17) for specific disposal instructions for unused or expired DAYTRANA.
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Abuse, Misuse, and Addiction
Educate patients and their families about the risks of abuse, misuse, and addiction of DAYTRANA, which can lead to overdose and death, and proper disposal of any unused drug [see Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2), Overdosage (10)]. Advise patients to store DAYTRANA in a safe place, preferably locked, and instruct patients to not give DAYTRANA to anyone else.
Special Disposal Instructions
Advise patients that there are special disposal instructions for unused or expired DAYTRANA [see Warnings and Precautions (5.1)]. Instruct patients to find a take back location to dispose of unused or expired DAYTRANA. If a take back program is unavailable, instruct them to:
- Remove DAYTRANA from its pouch, separate it from its liner, fold it in half with the adhesive sides touching each other, and immediately flush it down the toilet, and
- Place the pouch and liner in a container, close the container, and throw out the container in the trash (advise patients not to flush the pouch and liner down the toilet).
Risks to Patients with Serious Cardiac Disease
Advise patients that there are potential risks to patients with serious cardiac disease, including sudden death, with DAYTRANA use. Instruct patients to contact a healthcare provider immediately if they develop symptoms, such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease [see Warnings and Precautions (5.2)].
Priapism
Advise patients, caregivers, and family members of the possibility of painful or prolonged penile erections (priapism). Instruct the patient to seek immediate medical attention in the event of priapism [see Warnings and Precautions (5.6)].
Circulation problems in fingers and toes [Peripheral vasculopathy, including Raynaud’s phenomenon] [see Warnings and Precautions (5.7)]
- Instruct patients beginning treatment with DAYTRANA about the risk of peripheral vasculopathy, including Raynaud’s Phenomenon, and in associated signs and symptoms: fingers or toes may feel numb, cool, painful, and/or may change color from pale, to blue, to red.
- Instruct patients to report to their physician any new numbness, pain, skin color change, or sensitivity to temperature in fingers or toes.
- Instruct patients to call their physician immediately with any signs of unexplained wounds appearing on fingers or toes while using DAYTRANA.
- Further clinical evaluation (e.g., rheumatology referral) may be appropriate for certain patients.
Long-Term Suppression of Growth in Pediatric Patients
Advise patients that DAYTRANA may cause slowing of growth including weight loss [see Warnings and (5.8)].
Chemical Leukoderma
Advise patients of the possibility of a persistent loss of skin pigmentation at, around and distant from the application site. Advise patients to immediately inform their healthcare provider if changes in skin pigmentation occur [see Warnings and Precautions (5.9)].
Increased Intraocular Pressure (IOP) and Glaucoma
Advise patients that IOP and glaucoma may occur during treatment with DAYTRANA [see Warnings and Precautions (5.14)].
Motor and Verbal Tics, and Worsening of Tourette’s Syndrome
Advise patients that motor and verbal tics and worsening of Tourette’s Syndrome may occur during treatment with DAYTRANA. Instruct patients to notify their healthcare provider if emergence of new tics or worsening of tics or Tourette’s syndrome occurs [see Warnings and Precautions (5.15)].
Important Preparation and Administration Instructions [see Dosage and Administration (2.3)]
- Parents and patients should be informed to apply DAYTRANA to a clean, dry site on the hip, which is not oily, damaged, or irritated. The site of application must be alternated daily. DAYTRANA should not be applied to the waistline, or where tight clothing may rub it.
- If patients or caregivers experience difficulty separating the transdermal system from the release liner or observe tearing and/or other damage to the transdermal system during removal from the liner, the transdermal system should be discarded according to the directions provided in this label, and a new transdermal system should be applied [see Dosage and Administration (2.3)]. Patients or caregivers should inspect the release liner to ensure that no adhesive containing medication has transferred to the liner. If adhesive transfer has occurred, the transdermal system should be discarded.
- DAYTRANA should be applied 2 hours before the desired effect. DAYTRANA should be removed approximately 9 hours after it is applied, although the effects from the transdermal system will last for several more hours. DAYTRANA may be removed earlier than 9 hours if a shorter duration of effect is desired or late day side effects appear.
- The parent or caregiver should be encouraged to use the administration chart included with each carton of DAYTRANA to monitor application and removal time, and method of disposal. The Medication Guide included at the end of this insert also includes a timetable to calculate when to remove DAYTRANA, based on the 9 hour application time.
- Patients or caregivers should avoid touching the adhesive side of the transdermal system during application, in order to avoid absorption of methylphenidate. If they do touch the adhesive side of the transdermal system, they should immediately wash their hands after application.
- In the event that a DAYTRANA does not fully adhere to the skin upon application, or is partially or fully detached during wear time, the transdermal system should be discarded according to the directions provided in this label, and a new transdermal system should be applied [see Dosage and Administration (2.3)]. If a transdermal system is replaced, the total recommended wear time for that day should remain 9 hours, regardless of the number of transdermal systems used.
- DAYTRANA should not be applied or re-applied with dressings, tape, or other common adhesives.
- Exposure to water during bathing, swimming, or showering can affect transdermal system adherence.
- Do not cut transdermal systems. Only intact transdermal systems should be applied.
- If there is an unacceptable duration of appetite loss or insomnia in the evening, taking the transdermal system off earlier may be attempted before decreasing the transdermal system dose.
- Skin redness or itching is common with DAYTRANA and small bumps on the skin may also occur in some patients. If any swelling or blistering occurs the DAYTRANA should not be worn and the patient should be seen by the prescriber. Patients or caregivers should not apply hydrocortisone or other solutions, creams, ointments, or emollients immediately prior to DAYTRANA application, since the effect on transdermal system adhesion and methylphenidate absorption has not been established. The potential adverse effects of topical corticosteroid use during treatment with DAYTRANA are unknown.
Recommended Storage Instructions
Transdermal systems should be stored at 25 degrees Celsius (77 degrees Fahrenheit) with excursions permitted that do not exceed 15 to 30 degrees Celsius (59 to 86 degrees Fahrenheit) [see How Supplied/Storage and Handling (16)]. Patients or caregivers should be advised not to store DAYTRANA in the refrigerator or freezer.
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ADHD medications, including DAYTRANA, during pregnancy [see Use in Specific Populations (8.1)].
Manufactured for: Noven Therapeutics, LLC, Miami, FL 33186.
By: Noven Pharmaceuticals, Inc., Miami, FL 33186.
For more information, call 1-877-567-7857 or visit www.daytrana.com.
DOT Matrix™ is a trademark of Noven Pharmaceuticals, Inc.
DAYTRANA® is a registered trademark of Noven Therapeutics, LLC.
© 2009-2023 Noven Pharmaceuticals, Inc.
102086-22
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. |
Revised 10/2023 |
|
| MEDICATION GUIDE
DAYTRANA® (day-TRON-ah)
|
||
| Important: DAYTRANA is for use on the skin only. | ||
| What is the most important information I should know about DAYTRANA?
DAYTRANA may cause serious side effects, including:
|
||
| What Is DAYTRANA?
DAYTRANA is a central nervous system (CNS) stimulant prescription medication used for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in children 6 to 17 years of age. DAYTRANA may help increase attention and decrease impulsiveness and hyperactivity in children with ADHD. It is not known if DAYTRANA is safe and effective in children younger than 6 years. DAYTRANA is a federally controlled substance (CII) because it contains methylphenidate that can be a target for people who abuse prescription medicines or street drugs. Keep DAYTRANA in a safe place to protect it from theft. Never give your DAYTRANA to anyone else because it may cause death or harm them. Selling or giving away DAYTRANA may harm others and is against the law. |
||
Do not use DAYTRANA if your child:
|
||
Before using DAYTRANA, tell your child's healthcare provider about all of your child's medical conditions, including if your child:
DAYTRANA and some medicines may interact with each other and cause serious side effects. Sometimes the doses of other medicines will need to be changed during treatment with DAYTRANA. Your child's healthcare provider will decide if DAYTRANA can be taken with other medicines. Especially tell your child's healthcare provider if your child takes:
Know the medicines that your child takes. Keep a list of your child's medicines with you to show your child's healthcare provider and pharmacist when your child gets a new medicine. Do not start any new medicine while using DAYTRANA without first talking to your child's healthcare provider. |
||
How should DAYTRANA be used?
|
||
What should your child avoid while using DAYTRANA?
|
||
| What are the possible side effects of DAYTRANA?
DAYTRANA may cause serious side effects, including:
|
||
| The most common side effects of DAYTRANA in children 6 to 12 years old include: | ||
|
|
|
| The most common side effects of DAYTRANA in children 13 to 17 years old include: | ||
|
|
|
|
DAYTRANA may also cause skin problems where it is applied (redness, small bumps, itching) Your child's doctor may do certain blood tests while your child uses DAYTRANA. These are not all the possible side effects of DAYTRANA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store DAYTRANA?
|
||
|
General information about the safe and effective use of DAYTRANA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use DAYTRANA for a condition for which it was not prescribed. Do not give DAYTRANA to other people, even if they have the same symptoms. It may harm them and it is against the law. You can ask your pharmacist or healthcare provider for information about DAYTRANA that is written for healthcare professionals. |
||
| What are the ingredients in DAYTRANA?
Active ingredient: methylphenidate Inactive ingredients: acrylic adhesive, silicone adhesive Manufactured by: Noven Pharmaceuticals, Inc., Miami, FL 33186 For more information, go to www.daytrana.com, or call 1-877-567-7857. |
||
| INSTRUCTIONS FOR USE
DAYTRANA® (day-TRON-ah) (methylphenidate transdermal system) CII |
1. DAYTRANA Dosing Chart
Each carton of DAYTRANA contains a DAYTRANA Dosing Chart to help you keep track of your DAYTRANA transdermal system (patch) including:
- when you apply patch to the skin on your hip each morning
- when you remove the patch
- how and where you threw DAYTRANA away
To use the DAYTRANA Dosing Chart, follow these instructions:
- Each day, when a new patch is applied to your hip, write down the date and time that you applied the patch.
- Use the DAYTRANA schedule below so you can decide when to remove the patch. For example, if the patch is applied to the skin at 6:00 a.m., remove the patch at 3:00 p.m. on the same day. After you remove and throw away the patch, write down the time you removed the patch and how and where you threw it away.
- If the patch you placed on your child is missing, ask your child:
- when the patch came off
- how the patch came off
- where the patch is
DAYTRANA Schedule for 9 Hour Dosing
| If you put the patch on at: | On the same day, remove the patch at: |
| 5:00 a.m. | 2:00 p.m. |
| 6:00 a.m. | 3:00 p.m. |
| 7:00 a.m. | 4:00 p.m. |
| 8:00 a.m. | 5:00 p.m. |
| 9:00 a.m. | 6:00 p.m. |
| 10:00 a.m. | 7:00 p.m. |
| 11:00 a.m. | 8:00 p.m. |
| 12:00 p.m. | 9:00 p.m. |
2. Where to apply DAYTRANA
- Apply patch to your hip area. Do not put the patch near your waist. Clothing and movement may make your patch rub off (See Figure A).
- Use your other hip when you apply a new patch the next morning. Make sure there is no redness, small bumps or itching at the site where the patch is going to be applied.
Figure A
3. Before you apply DAYTRANA
Make sure your skin:
- Is clean (freshly washed), dry, and cool
- Does not have any powder, oil, or lotion
- Does not have any cuts and irritation (rashes, inflammation, redness, or other skin problems).
4. How to apply DAYTRANA
- Open the sealed tray and throw away the small packet (drying agent).
- Each patch is sealed in its own protective pouch.
- Carefully cut the protective pouch open with scissors, being careful not to cut the patch. Do not use patches that have been cut or damaged in any way (See Figure B).
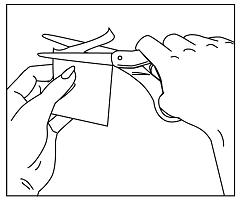
Figure B
- Remove the patch from the protective pouch.
- Look at the patch to make sure it is not damaged. The patch should separate easily from the protective liner. Throw away the patch if the protective liner is hard to remove.
DAYTRANA has 3 layers. The 3 layers are pictured below. The pictures show both sides of the patch:
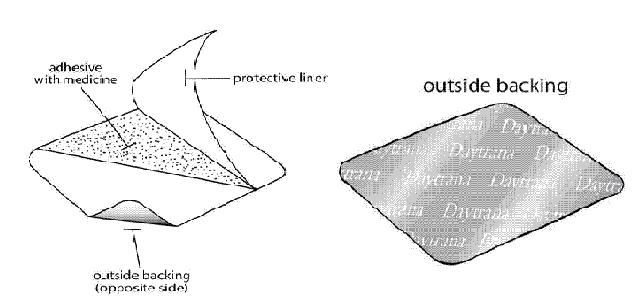
Figure C Figure D
Layers:
- Protective liner: The protective liner is the layer that you remove before you put the patch on (See Figure C).
- Adhesive with medicine: The adhesive with medicine is the layer that sticks to your skin (See Figure C).
- Outside backing: The outside backing is the layer that you see after you put the patch on your skin. The word "Daytrana" is printed on this layer (See Figure D).
- Apply the patch right away after you remove the patch from protective pouch.
- Hold the patch with the hard protective liner facing you. The word DAYTRANA will appear backwards.
- Gently bend the patch along the faint line and slowly peel half the liner, which covers the sticky surface of the patch (See Figure E).
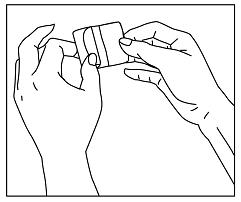
Figure E
- Avoid touching the sticky side of the patch with your fingers.
- If you accidentally touch the sticky side of the patch, apply the patch, then wash your hands right away so that the medicine does not go into the skin on your hands.
- Using the other half of the protective liner as a handle, apply the sticky side of the patch to the selected area of the child's hip (See Figure F).
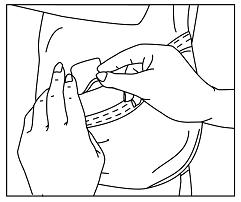
Figure F
- Press the sticky side of the patch firmly into place and smooth it down.
- While you are still holding the sticky side down, gently fold back the other half of the patch.
- Hold an edge of the remaining protective liner and slowly peel it off (See Figure G).
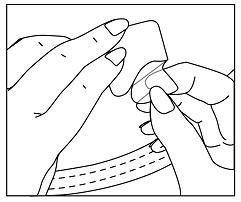
Figure G
- After the protective liner is removed, there should not be any adhesive (glue) sticking to the liner.
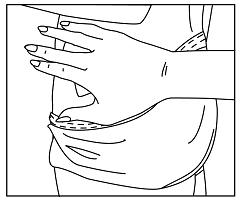
Figure H
- Press the entire patch firmly into place with the palm of your hand over the patch for about 30 seconds (See Figure H).
- Make sure that the patch firmly sticks to your skin.
- Gently rub the edges of the patch with your fingers to make sure the patch sticks to your skin.
- Wash your hands after you apply your patch.
- Write the time you applied your patch on the dosing chart on the carton. Use the dosing schedule so you know what time you should remove your patch.
5. How to remove and throw away DAYTRANA
- When you remove the patch, peel it off slowly. If the patch is too sticky on your skin and you need something to help you remove it:
- Gently apply an oil-based product (petroleum jelly, olive oil, or mineral oil) to the patch edges. Gently spread the oil underneath the patch edges.
- Apply an oil-based product or lotion to your skin if any adhesive (glue) remains after you remove your patch. This will gently loosen and remove any adhesive that is left over.
- If you still cannot easily remove the patch, ask your doctor or pharmacist about what to do for this problem.
- Fold the used patch in half and press it together firmly so that the sticky side sticks to itself. Flush the used patch down the toilet.
- Do not flush the protective pouches or the protective liners down the toilet. These items should be thrown away in a container with a lid.
- Wash your hands after you handle the patch.
- After you remove the patch and throw the patch away, write down the time on the dosing chart.
- Safely throw away any unused patches that are left over from the prescription as soon as they are no longer needed.
To safely throw away the patches:- Remove the leftover patches from their protective pouches and remove the protective liners.
- Either fold the patches in half with the sticky sides together, and flush the patches down the toilet, or
- Dispose of remaining, unused, or expired DAYTRANA by a medicine take-back program at a U.S. Drug Enforcement Administration (DEA) authorized collection site. If no take-back program or DEA authorized collector is available, each unused patch should be removed from its individual pouch, separated from the protective liner, folded in half so that the sticky sides stick together, and flushed down the toilet. Put the pouch and liner in a container with a lid, close the container and throw away the container in the household trash. Do not flush the pouch and liner down the toilet. Visit www.fda.gov/drugdisposal for additional information on disposal of unused medicines.
Manufactured for: Noven Therapeutics, LLC, Miami, FL 33186.
By: Noven Pharmaceuticals, Inc., Miami, FL 33186.
© 2023 Noven Pharmaceuticals, Inc.
This Medication Guide and Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 10/2023
102086-22
PRINCIPAL DISPLAY PANEL - NDC 68968-5552-3 - 10 mg 30 Count Carton

NDC 68968-5552-3
Daytrana®
(methylphenidate transdermal system)
Delivers 10 mg over 9 hours
(1.1 mg/hr)
Patch should be worn for approximately 9 hours
Contains: 30 Patches
CII
Rx only
Noven Therapeutics, LLC
Once the tray is opened, use contents within 2 months.
Manufactured for Noven Therapeutics, LLC, Miami, FL 33186
By Noven Pharmaceuticals, Inc.
©2007, 2010 Noven Pharmaceuticals, Inc.
1-877-567-7857
Pharmacists: Enclosed Medication Guide to be dispensed to each patient.
302188-8 Rev. 9/2016
Each patch contains 27.5 mg of methylphenidate.
Active ingredient release is limited; please see recommended dosing in patient instructions.
Inactive components: acrylic adhesive, coextruded backing film, polyester release liner and silicone adhesive.
For transdermal use only (applied only to skin).
Keep all patches within provided containers and dispense one patch daily. Apply immediately upon removal from pouch.
Do not store unpouched. Do not store patches in refrigerators or freezers.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled Room Temperature]
Important: Keep out of the reach of children.
It is important that this product be disposed of properly. See patient instructions for disposal information.
Dosage and Administration: See package insert.
|
Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method | Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method |
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
PRINCIPAL DISPLAY PANEL - NDC 68968-5553-3 - 15 mg 30 Count Carton

NDC 68968-5553-3
Daytrana®
(methylphenidate transdermal system)
Delivers 15 mg over 9 hours
(1.6 mg/hr)
Patch should be worn for approximately 9 hours
Contains: 30 Patches
CII
Rx only
Noven Therapeutics, LLC
Once the tray is opened, use contents within 2 months.
Manufactured for Noven Therapeutics, LLC, Miami, FL 33186
By Noven Pharmaceuticals, Inc.
©2007, 2010 Noven Pharmaceuticals, Inc.
1-877-567-7857
Pharmacists: Enclosed Medication Guide to be dispensed to each patient.
302189-8 Rev. 9/2016
Each patch contains 41.3 mg of methylphenidate.
Active ingredient release is limited; please see recommended dosing in patient instructions.
Inactive components: acrylic adhesive, coextruded backing film, polyester release liner and silicone adhesive.
For transdermal use only (applied only to skin).
Keep all patches within provided containers and dispense one patch daily. Apply immediately upon removal from pouch.
Do not store unpouched. Do not store patches in refrigerators or freezers.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled Room Temperature]
Important: Keep out of the reach of children.
It is important that this product be disposed of properly. See patient instructions for disposal information.
Dosage and Administration: See package insert.
|
Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method | Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method |
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
PRINCIPAL DISPLAY PANEL - NDC 68968-5554-3 - 20 mg 30 Count Carton

NDC 68968-5554-3
Daytrana®
(methylphenidate transdermal system)
Delivers 20 mg over 9 hours
(2.2 mg/hr)
Patch should be worn for approximately 9 hours
Contains: 30 Patches
CII
Rx only
Noven Therapeutics, LLC
Once the tray is opened, use contents within 2 months.
Manufactured for Noven Therapeutics, LLC, Miami, FL 33186
By Noven Pharmaceuticals, Inc.
©2007, 2010 Noven Pharmaceuticals, Inc.
1-877-567-7857
Pharmacists: Enclosed Medication Guide to be dispensed to each patient.
302190-8 Rev. 9/2016
Each patch contains 55 mg of methylphenidate.
Active ingredient release is limited; please see recommended dosing in patient instructions.
Inactive components: acrylic adhesive, coextruded backing film, polyester release liner and silicone adhesive.
For transdermal use only (applied only to skin).
Keep all patches within provided containers and dispense one patch daily. Apply immediately upon removal from pouch.
Do not store unpouched. Do not store patches in refrigerators or freezers.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled Room Temperature]
Important: Keep out of the reach of children.
It is important that this product be disposed of properly. See patient instructions for disposal information.
Dosage and Administration: See package insert.
|
Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method | Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method |
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
PRINCIPAL DISPLAY PANEL - NDC 68968-5555-3 - 30 mg 30 Count Carton

NDC 68968-5555-3
Daytrana®
(methylphenidate transdermal system)
Delivers 30 mg over 9 hours
(3.3 mg/hr)
Patch should be worn for approximately 9 hours
Contains: 30 Patches
CII
Rx only
Noven Therapeutics, LLC
Once the tray is opened, use contents within 2 months.
Manufactured for Noven Therapeutics, LLC, Miami, FL 33186
By Noven Pharmaceuticals, Inc.
©2007, 2010 Noven Pharmaceuticals, Inc.
1-877-567-7857
Pharmacists: Enclosed Medication Guide to be dispensed to each patient.
302191-8 Rev. 9/2016
Each patch contains 82.5 mg of methylphenidate.
Active ingredient release is limited; please see recommended dosing in patient instructions.
Inactive components: acrylic adhesive, coextruded backing film, polyester release liner and silicone adhesive.
For transdermal use only (applied only to skin).
Keep all patches within provided containers and dispense one patch daily. Apply immediately upon removal from pouch.
Do not store unpouched. Do not store patches in refrigerators or freezers.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled Room Temperature]
Important: Keep out of the reach of children.
It is important that this product be disposed of properly. See patient instructions for disposal information.
Dosage and Administration: See package insert.
|
Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method | Date of Patch Application MM/DD/YYYY | Time Applied | Time Removed | Application Side | Disposal Method |
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |
||
| _AM _PM | _AM _PM | _Right _Left |
_Fold & flush _ Fold & trash | _AM _PM | _AM _PM | _Right _Left | _Fold & flush _ Fold & trash |