FULL PRESCRIBING INFORMATION
WARNING: LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin‑ associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin‑ associated lactic acidosis was characterized by elevated blood lactate levels (>5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally >5 mcg/mL [see Warnings and Precautions (5.1)].
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g. carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided [see Dosage and Administration (2.3), Contraindications (4), Warnings and Precautions (5.1)].
If metformin-associated lactic acidosis is suspected, immediately discontinue RIOMET and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended [see Warnings and Precautions (5.1)].
1 INDICATIONS AND USAGE
RIOMET is indicated as an adjunct to diet and exercise to improve glycemic control in adults and pediatric patients 10 years of age and older with type 2 diabetes mellitus.
2 DOSAGE AND ADMINISTRATION
2.1 Adult Dosage
- •
- Measure the RIOMET dose in the RIOMET specific dosing cup.
- •
- The recommended starting dose of RIOMET is 500 mg (5 mL) orally twice a day or 850 mg (8.5 mL) once a day, given with meals.
- •
- Increase the dose in increments of 500 mg (5 mL) weekly or 850 mg (8.5 mL) every 2 weeks on the basis of glycemic control and tolerability, up to a maximum dose of 2,550 mg (25.5 mL) per day, given in divided doses.
- •
- Doses above 2,000 mg (20 mL) may be better tolerated given in divided doses 3 times a day with meals.
2.2 Pediatric Dosage
- •
- Measure the RIOMET dose in the RIOMET specific dosing cup.
- •
- The recommended starting dose of RIOMET for pediatric patients 10 years of age and older is 500 mg (5 mL) orally twice a day, given with meals.
- •
- Increase dosage in increments of 500 mg (5 mL) weekly on the basis of glycemic control and tolerability, up to a maximum of 2,000 mg (20 mL) per day, given in divided doses twice daily.
2.3 Recommendations for Use in Renal Impairment
- •
- Assess renal function prior to initiation of RIOMET and periodically thereafter.
- •
- RIOMET is contraindicated in patients with an estimated glomerular filtration rate (eGFR) below 30 mL/minute/1.73 m2.
- •
- Initiation of RIOMET in patients with an eGFR between 30 to 45 mL/minute/1.73 m2 is not recommended.
- •
- In patients taking RIOMET whose eGFR later falls below 45 mL/min/1.73 m2, assess the benefit risk of continuing therapy.
- •
- Discontinue RIOMET if the patient’s eGFR later falls below 30 mL/minute/1.73 m2 [see Warnings and Precautions (5.1)].
2.4 Discontinuation for Iodinated Contrast Imaging Procedures
- •
- Discontinue RIOMET at the time of, or prior to, an iodinated contrast imaging procedure in patients with an eGFR between 30 and 60 mL/min/1.73 m2; in patients with a history of liver disease, alcoholism, or heart failure; or in patients who will be administered intra-arterial iodinated contrast. Re-evaluate eGFR 48 hours after the imaging procedure; restart RIOMET if renal function is stable.
3 DOSAGE FORMS AND STRENGTHS
Oral solution: 500 mg per 5 mL (100 mg/mL) clear solution in cherry and strawberry flavor
4 CONTRAINDICATIONS
- 1.
- RIOMET is contraindicated in patients with:
- 2.
- Severe renal impairment (eGFR below 30 mL/min/1.73 m2) [see Warnings and Precautions (5.1)].
- 3.
- Hypersensitivity to metformin.
- 4.
- Acute or chronic metabolic acidosis, including diabetic ketoacidosis, with or without coma.
5 WARNINGS AND PRECAUTIONS
5.1 Lactic Acidosis
- •
- There have been postmarketing cases of metformin-associated lactic acidosis, including fatal cases. These cases had a subtle onset and were accompanied by nonspecific symptoms such as malaise, myalgias, abdominal pain, respiratory distress, or increased somnolence; however, hypotension and resistant bradyarrhythmias have occurred with severe acidosis. Metformin‑associated lactic acidosis was characterized by elevated blood lactate concentrations (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and an increased lactate: pyruvate ratio; metformin plasma levels were generally > 5 mcg/mL. Metformin decreases liver uptake of lactate increasing lactate blood levels which may increase the risk of lactic acidosis, especially in patients at risk.
- •
- If metformin-associated lactic acidosis is suspected, general supportive measures should be instituted promptly in a hospital setting, along with immediate discontinuation of RIOMET. In RIOMET treated patients with a diagnosis or strong suspicion of lactic acidosis, prompt hemodialysis is recommended to correct the acidosis and remove accumulated metformin (metformin hydrochloride is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions). Hemodialysis has often resulted in reversal of symptoms and recovery.
- •
- Educate patients and their families about the symptoms of lactic acidosis and, if these symptoms occur, instruct them to discontinue RIOMET and report these symptoms to their healthcare provider.
- •
- For each of the known and possible risk factors for metformin-associated lactic acidosis, recommendations to reduce the risk of and manage metformin-associated lactic acidosis are provided below:
- •
- Before initiating RIOMET, obtain an estimated glomerular filtration rate (eGFR).
- •
- RIOMET is contraindicated in patients with an eGFR less than 30 mL/min/1.73 m2 [see Contraindications (4)].
- •
- Initiation of RIOMET is not recommended in patients with eGFR between 30 to 45 mL/min/1.73 m2.
- •
- Obtain an eGFR at least annually in all patients taking RIOMET. In patients at risk for the development of renal impairment (e.g., the elderly), renal function should be assessed more frequently.
- •
- In patients taking RIOMET whose eGFR falls below 45 mL/min/1.73 m2, assess the benefit and risk of continuing therapy.
- •
- Renal impairment—The postmarketing metformin-associated lactic acidosis cases primarily occurred in patients with significant renal impairment.
- •
- The risk of metformin accumulation and metformin-associated lactic acidosis increases with the severity of renal impairment because metformin is substantially excreted by the kidney. Clinical recommendations based upon the patient’s renal function include [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]:
- •
- Before initiating RIOMET, obtain an estimated glomerular filtration rate (eGFR).
- •
- RIOMET is contraindicated in patients with an eGFR less than 30 mL/min/1.73 m2 [see Contraindications (4)].
- •
- Initiation of RIOMET is not recommended in patients with eGFR between 30 to 45 mL/min/1.73 m2.
- •
- Obtain an eGFR at least annually in all patients taking RIOMET. In patients at risk for the development of renal impairment (e.g., the elderly), renal function should be assessed more frequently.
- •
- In patients taking RIOMET whose eGFR falls below 45 mL/min/1.73 m2, assess the benefit and risk of continuing therapy.
- •
- Drug interactions — The concomitant use of RIOMET with specific drugs may increase the risk of metformin-associated lactic acidosis: those that impair renal function, result in significant hemodynamic change, interfere with acid-base balance, or increase metformin accumulation. Consider more frequent monitoring of patients [see Drug Interactions (7)].
- •
- Age 65 or greater — The risk of metformin-associated lactic acidosis increases with the patient’s age because elderly patients have a greater likelihood of having hepatic, renal, or cardiac impairment than younger patients. Assess renal function more frequently in elderly patients.
- •
- Radiologic studies with contrast — Administration of intravascular iodinated contrast agents in metformin-treated patients has led to an acute decrease in renal function and the occurrence of lactic acidosis. Stop RIOMET at the time of, or prior to, an iodinated contrast imaging procedure in patients with an eGFR between 30 and 60 mL/min/1.73 m2; in patients with a history of hepatic impairment, alcoholism or heart failure; or in patients who will be administered intra-arterial iodinated contrast. Re-evaluate eGFR 48 hours after the imaging procedure, and restart RIOMET if renal function is stable.
- •
- Surgery and other procedures — Withholding of food and fluids during surgical or other procedures may increase the risk for volume depletion, hypotension, and renal impairment. RIOMET should be temporarily discontinued while patients have restricted food and fluid intake.
- •
- Hypoxic states — Several of the postmarketing cases of metformin-associated lactic acidosis occurred in the setting of acute congestive heart failure (particularly when accompanied by hypoperfusion and hypoxemia). Cardiovascular collapse (shock), acute myocardial infarction, sepsis, and other conditions associated with hypoxemia have been associated with lactic acidosis and may cause prerenal azotemia. When such an event occurs, discontinue RIOMET.
- •
- Excessive alcohol intake — Alcohol potentiates the effect of metformin on lactate metabolism. Patients should be warned against excessive alcohol intake while receiving RIOMET.
- •
- Hepatic impairment — Patients with hepatic impairment have developed cases of metformin-associated lactic acidosis. This may be due to impaired lactate clearance resulting in higher lactate blood levels. Therefore, avoid use of RIOMET in patients with clinical or laboratory evidence of hepatic disease.
5.2 Vitamin B12 Deficiency
- 1.
- In clinical trials of 29-week duration with metformin hydrochloride (HCl) tablets, a decrease to subnormal levels of previously normal serum vitamin B12 levels was observed in approximately 7% of patients. Such decrease, possibly due to interference with B12 absorption from the B12-intrinsic factor complex, may be associated with anemia but appears to be rapidly reversible with discontinuation of metformin or vitamin B12 supplementation. Certain individuals (those with inadequate vitamin B12 or calcium intake or absorption) appear to be predisposed to developing subnormal vitamin B12 levels. Measure hematologic parameters on an annual basis and vitamin B12 at 2 to 3 year intervals in patients on RIOMET and manage any abnormalities [see Adverse Reactions (6.1)].
5.3 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Insulin and insulin secretagogues (e.g., sulfonylurea) are known to cause hypoglycemia. RIOMET may increase the risk of hypoglycemia when combined with insulin and/or an insulin secretagogue. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when used in combination with RIOMET [see Drug Interactions (7)].
6 ADVERSE REACTIONS
- •
- The following adverse reactions are also discussed elsewhere in the labeling:
- •
- Lactic Acidosis [see Boxed Warning and Warnings and Precautions (5.1)]
- •
- Vitamin B12 Deficiency [see Warnings and Precautions (5.2)]
- •
- Hypoglycemia [see Warnings and Precautions (5.3)]
6.1 Clinical Studies Experience
- 1.
- Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
- 2.
- In a U.S. clinical trial of metformin HCl tablets in patients with type 2 diabetes mellitus, a total of 141 patients received metformin HCl tablets up to 2,550 mg per day. Adverse reactions reported in greater than 5% of patients treated with metformin HCl tablets and that were more common than in placebo-treated patients, are listed in Table 1.
- 3.
- Table 1: Adverse Reactions from a Clinical Trial of Metformin HCl Tablets Occurring >5% and More Common than Placebo in Patients with Type 2 Diabetes Mellitus
|
(n = 145) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
- 1.
- Diarrhea led to discontinuation of metformin HCl tablets in 6% of patients. Additionally, the following adverse reactions were reported in ≥ 1% to ≤ 5% of patients treated with metformin HCl tablets and were more commonly reported than placebo: abnormal stools, hypoglycemia, myalgia, lightheaded, dyspnea, nail disorder, rash, sweating increased, taste disorder, chest discomfort, chills, flu syndrome, flushing, palpitation.
- 2.
- Pediatric Patients
- 3.
- In clinical trials with metformin HCl tablets in pediatric patients with type 2 diabetes mellitus, the profile of adverse reactions was similar to that observed in adults.
- 4.
- Laboratory Tests
- 5.
- Vitamin B12 Concentrations
- 6.
- In clinical trials of 29-week duration with metformin HCl tablets, a decrease to subnormal levels of previously normal serum vitamin B12 levels was observed in approximately 7% of patients.
6.2 Postmarketing Experience
- •
- The following adverse reactions have been identified during post approval use of metformin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Cholestatic, hepatocellular, and mixed hepatocellular liver injury have been reported with postmarketing use of metformin.
7 DRUG INTERACTIONS
- 1.
- Table 2 presents clinically significant drug interactions with RIOMET.
- 2.
- Table 2: Clinically Significant Drug Interactions with RIOMET
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
- 1.
- Risk Summary
- 2.
- Limited data with RIOMET in pregnant women are not sufficient to determine a drug-associated risk for major birth defects or miscarriage. Published studies with metformin use during pregnancy have not reported a clear association with metformin and major birth defect or miscarriage risk [see Data]. There are risks to the mother and fetus associated with poorly controlled diabetes mellitus in pregnancy [see Clinical Considerations].
- 3.
- No adverse developmental effects were observed when metformin was administered to pregnant Sprague Dawley rats and rabbits during the period of organogenesis at doses up to 2- and 5‑ times, respectively, a 2,550 mg clinical dose, based on body surface area [see Data].
- 4.
- The estimated background risk of major birth defects is 6 to 10% in women with pre-gestational diabetes mellitus with an HbA1C >7 and has been reported to be as high as 20 to 25% in women with a HbA1C >10. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
- 5.
- Clinical Considerations
- 6.
- Disease-associated maternal and/or embryo/fetal risk
- 7.
- Poorly-controlled diabetes mellitus in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, stillbirth and delivery complications. Poorly controlled diabetes mellitus increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.
- 8.
- Data
- 9.
- Human Data
- 10.
- Published data from post-marketing studies have not reported a clear association with metformin and major birth defects, miscarriage, or adverse maternal or fetal outcomes when metformin was used during pregnancy. However, these studies cannot definitely establish the absence of any metformin-associated risk because of methodological limitations, including small sample size and inconsistent comparator groups.
- 11.
- Animal Data
- 12.
- Metformin hydrochloride did not adversely affect development outcomes when administered to pregnant rats and rabbits at doses up to 600 mg/kg/day. This represents an exposure of about 2 and 5 times a 2,550 mg clinical dose based on body surface area comparisons for rats and rabbits, respectively. Determination of fetal concentrations demonstrated a partial placental barrier to metformin.
8.2 Lactation
- •
- Risk Summary
- •
- Limited published studies report that metformin is present in human milk [see Data]. However, there is insufficient information to determine the effects of metformin on the breastfed infant and no available information on the effects of metformin on milk production. Therefore, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for RIOMET and any potential adverse effects on the breastfed child from RIOMET or from the underlying maternal condition.
- •
- Data
- •
- Published clinical lactation studies report that metformin is present in human milk which resulted in infant doses approximately 0.11% to 1% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 0.13 and 1. However, the studies were not designed to definitely establish the risk of use of metformin during lactation because of small sample size and limited adverse event data collected in infants.
8.3 Females and Males of Reproductive Potential
- •
- Discuss the potential for unintended pregnancy with premenopausal women as therapy with RIOMET may result in ovulation in some anovulatory women.
8.4 Pediatric Use
- 1.
- The safety and effectiveness of RIOMET for the treatment of type 2 diabetes mellitus have been established in pediatric patients 10 to 16 years old. Safety and effectiveness of RIOMET have not been established in pediatric patients less than 10 years old.
- 2.
- Use of RIOMET in pediatric patients 10 to 16 years old for the treatment of type 2 diabetes mellitus is supported by evidence from adequate and well-controlled studies of metformin HCl tablets in adults with additional data from a controlled clinical study of metformin HCl tablets in pediatric patients 10 to 16 years old with type 2 diabetes mellitus, which demonstrated a similar response in glycemic control to that seen in adults [see Clinical Studies (14)]. In this study, adverse reactions were similar to those described in adults. A maximum daily dose of 2,000 mg of RIOMET is recommended. [See Dosage and Administration (2.2).]
8.5 Geriatric Use
- 1.
- Controlled clinical studies of metformin HCl tablets did not include sufficient numbers of elderly patients to determine whether they respond differently from younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy and the higher risk of lactic acidosis. Assess renal function more frequently in elderly patients [see Warnings and Precautions (5.1)].
8.6 Renal Impairment
- •
- Metformin is substantially excreted by the kidney, and the risk of metformin accumulation and lactic acidosis increases with the degree of renal impairment. RIOMET is contraindicated in severe renal impairment, patients with an estimated glomerular filtration rate (eGFR) below 30 mL/min/1.73 m2 [see Dosage and Administration (2.3), Contraindications (4), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
- •
- Overdose of metformin hydrochloride has occurred, including ingestion of amounts greater than 50 grams. Hypoglycemia was reported in approximately 10% of cases, but no causal association with metformin has been established. Lactic acidosis has been reported in approximately 32% of metformin overdose cases [see Warnings and Precautions (5.1)]. Metformin is dialyzable with a clearance of up to 170 mL/min under good hemodynamic conditions. Therefore, hemodialysis may be useful for removal of accumulated drug from patients in whom metformin overdosage is suspected.
11 DESCRIPTION
RIOMET oral solution contains the biguanidine antihyperglycemic agent metformin in the form of monohydrochloride salt. Metformin hydrochloride, is N,N-dimethylimidodicarbonimidic diamide hydrochloride. The structural formula is shown as:
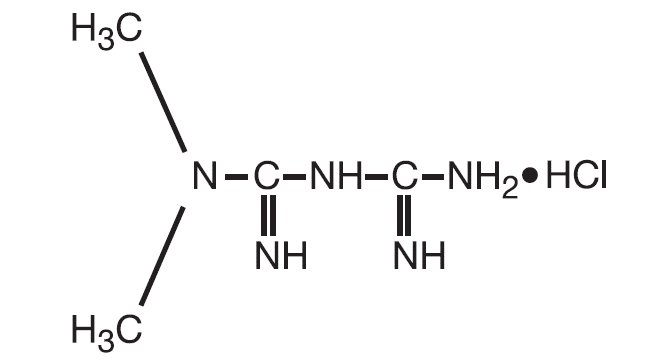
Metformin hydrochloride, USP is a white crystalline powder with a molecular formula of C 4H11N5•HCl and a molecular weight of 165.62. Metformin hydrochloride, USP 2.0 g is soluble in 20 mL of water. The pKa of metformin is 12.4. The pH of a 1% aqueous solution of metformin hydrochloride is 6.68. It is freely soluble in water; slightly soluble in alcohol; practically insoluble in acetone and in methylene chloride.
RIOMET (Cherry Flavor) contains 500 mg of metformin hydrochloride (the equivalent of 389.93 mg metformin) per 5 mL and the following inactive ingredients: Artificial cherry flavor, hydrochloric acid, potassium bicarbonate, purified water, saccharin calcium, and xylitol.
RIOMET (Strawberry Flavor) contains 500 mg of metformin hydrochloride (the equivalent of 389.93 mg metformin) per 5 mL and the following inactive ingredients: Hydrochloric acid, N&A strawberry flavor (propylene glycol and glycerin), potassium bicarbonate, purified water, sucralose, and xylitol.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
- •
- Metformin is an antihyperglycemic agent which improves glucose tolerance in patients with type 2 diabetes mellitus, lowering both basal and postprandial plasma glucose. Metformin decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may decrease.
12.3 Pharmacokinetics
- 1.
- Two pharmacokinetic studies performed in healthy volunteers to evaluate the bioavailability of RIOMET in comparison with metformin HCl tablets under fasting and fed conditions showed that the rate and extent of absorption of RIOMET was found to be comparable to that of metformin HCl tablets under fasting or fed conditions (see Table 3).
|
Table 3: Select Mean (± S.D.) Pharmacokinetic Parameters Following Single Oral Doses of 1000 mg RIOMET and Metformin HCl tablets in healthy, nondiabetic adults (n = 36) under fed and fasting conditions |
|||
|
Formulation |
Cmax (ng/mL) |
AUC0-∞ (ng.h/mL) |
Tmax (h) |
|
Study 1- Fasting state |
|||
|
RIOMET |
1540.1 ± 451.1 |
9069.6 ± 2593.6 |
2.2 ± 0.5 |
|
Metformin HCl Tablets |
1885.1 ± 498.5 |
11100.1 ± 2733.1 |
2.5 ± 0.6 |
|
T/R Ratio X 100 (90% confidence interval) |
81.2 (76.3 to 86.4) |
81.2 (76.9 to 85.6) |
- |
|
Study 2- Fed State |
|||
|
RIOMET |
1235.3 ± 177.7 |
8950.1 ± 1381.2 |
4.1 ± 0.8 |
|
Metformin HCl Tablets |
1361 ± 298.8 |
9307.7 ± 1839.8 |
3.7 ± 0.8 |
|
T/R Ratio X 100 (90% confidence interval) |
91.8 (87.4 to 96.5) |
97.0 (92.9 to 101.2) |
- |
- 1.
- T-test product (RIOMET)
- 2.
- R-reference product (immediate release metformin HCl tablets)
- 3.
- Studies using single oral doses of metformin HCl tablets 500 mg to 1,500 mg, and 850 mg to 2,550 mg, indicate that there is a lack of dose proportionality with increasing doses, which is due to decreased absorption rather than an alteration in elimination. At usual clinical doses and dosing schedules of metformin, steady state plasma concentrations of metformin are reached within 24 to 48 hours and are generally < 1 mcg/mL.
- 4.
- Effect of food: The food-effect study assessed the effects of a high fat/high calorie meal and a low fat/low calorie meal on the bioavailability of RIOMET in comparison with administration in the fasted state, in healthy volunteers. The extent of absorption was increased by approximately 16% and 13% with the low fat/low calorie meal and the high fat/high calorie meal, respectively, compared with the administration in the fasted state. The rate and extent of absorption with high fat/high calorie and low fat/ low calorie meal were similar. The mean tmax was 2.5 hours under fasting conditions as compared to 3.9 hours with both low fat/ low calorie meal and high fat/high calorie meals (see Table 4).
|
Table 4: Select Mean (± S.D.) Metformin Pharmacokinetic Parameters Following Single Oral Doses of 1,000 mg RIOMET in healthy, nondiabetic adults (n = 33) under fed (high fat/high calorie meal and low fat/low calorie meal) and fasting conditions (study 3) |
|||
|
Meal type |
Cmax (ng/mL) |
AUC0-∞ (ng.h/mL) |
tmax (h) |
|
Fasting (F) |
1641.5 ± 551.8 |
9982.9 ± 2544.5 |
2.5 ± 0.9 |
|
Low fat/ low calorie meal (L) |
1525.8 ± 396.7 |
11542.0 ± 2947.5 |
3.9 ± 0.6 |
|
High fat/high calorie meal (H) |
1432.5 ± 346.8 |
11184.5 ± 2446.1 |
3.9 ± 0.8 |
|
L/F Ratio X 100 (90% confidence interval) |
94.6 (84.0 to 106.5) |
115.6 (103.6 to 128.9) |
- |
|
H/F Ratio X 100 (90% confidence interval) |
89.4 (79.4 to 100.6) |
112.6 (100.9 to 125.6) |
- |
|
L/H Ratio X 100 (90% confidence interval) |
105.8 (94.0 to 119.2) |
102.7 (92.0 to 114.6) |
- |
- 1.
- Distribution
- 2.
- The apparent volume of distribution (V/F) of metformin following single oral doses of metformin HCl 850 mg averaged 654 ± 358 L. Metformin is negligibly bound to plasma proteins. Metformin partitions into erythrocytes, most likely as a function of time.
- 3.
- Metabolism
- 4.
- Intravenous single-dose studies in normal subjects demonstrate that metformin is excreted unchanged in the urine and does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion.
- 5.
- Elimination
- 6.
- Renal clearance (see Table 5) is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of metformin elimination. Following oral administration, approximately 90% of the absorbed drug is eliminated via the renal route within the first 24 hours, with a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
- 7.
- Specific Populations
- 8.
- Renal Impairment
- 9.
- In patients with decreased renal function the plasma and blood half-life of metformin is prolonged and the renal clearance is decreased (see Table 5) [See Dosage and Administration (2.3), Contraindications (4), Warnings and Precautions (5.1) and Use in Specific Populations (8.6)].
- 10.
- Hepatic Impairment
- 11.
- No pharmacokinetic studies of metformin have been conducted in patients with hepatic impairment [See Warnings and Precautions (5.1) and Use in Specific Populations (8.7)].
- 12.
- Geriatrics
- 13.
- Limited data from controlled pharmacokinetic studies of metformin HCl tablets in healthy elderly subjects suggest that total plasma clearance of metformin is decreased, the half-life is prolonged, and Cmax is increased, compared to healthy young subjects. It appears that the change in metformin pharmacokinetics with aging is primarily accounted for by a change in renal function (see Table 5). [See Warnings and Precautions (5.1) and Use in Specific Populations (8.5)].
- 14.
- Table 5: Select Mean (±S.D.) Metformin Pharmacokinetic ParametersFollowing Single or Multiple Oral Doses of Metformin HCl Tablets
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
| |||
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
- 1.
- a All doses given fasting except the first 18 doses of the multiple dose studies
- 2.
- bPeak plasma concentration
- 3.
- cTime to peak plasma concentration
- 4.
- d Combined results (average means) of five studies: mean age 32 years (range 23 to 59 years)
- 5.
- e Kinetic study done following dose 19, given fasting
- 6.
- f Elderly subjects, mean age 71 years (range 65 to 81 years)
- 7.
- g CLcr = creatinine clearance normalized to body surface area of 1.73 m2
- 8.
- Pediatrics
- 9.
- After administration of a single oral metformin HCl 500 mg tablet with food, geometric mean metformin Cmax and AUC differed less than 5% between pediatric type 2 diabetic patients (12 to 16 years of age) and gender- and weight-matched healthy adults (20 to 45 years of age), all with normal renal function.
- 10.
- Gender
- 11.
- Metformin pharmacokinetic parameters did not differ significantly between normal subjects and patients with type 2 diabetes mellitus when analyzed according to gender (males = 19, females = 16).
- 12.
- Race
- 13.
- No studies of metformin pharmacokinetic parameters according to race have been performed.
- 14.
- Drug Interactions
- 15.
- In VivoAssessment of Drug Interactions
- 16.
- Table 6: Effect of Coadministered Drug on Plasma Metformin SystemicExposure
|
|
|
|
||
|
|
||||
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
- 1.
- * All metformin HCl and coadministered drugs were given as single doses
- 2.
- † AUC = AUC (INF)
- 3.
- ‡ Ratio of arithmetic means
- 4.
- § At steady state with topiramate 100 mg every 12 hours and metformin 500 mg every 12 hours; AUC = AUC0-12h
- 5.
- Table 7: Effect of Metformin on Coadministered Drug Systemic Exposure
|
|
|
|
||
|
|
||||
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
- 1.
- * All metformin HCl and coadministered drugs were given as single doses
- 2.
- † AUC = AUCinf unless otherwise noted
- 3.
- ‡ Ratio of arithmetic means, p-value of difference < 0.05
- 4.
- § AUC0-24 hr reported
- 5.
- ¶ Ratio of arithmetic means
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
- 1.
- Long-term carcinogenicity studies have been performed in rats (dosing duration of 104 weeks) and mice (dosing duration of 91 weeks) at doses up to and including 900 mg/kg/day and 1,500 mg/kg/day, respectively. These doses are both approximately 3 times the maximum recommended human daily dose of 2,550 mg based on body surface area comparisons. No evidence of carcinogenicity with metformin was found in either male or female mice. Similarly, there was no tumorigenic potential observed with metformin in male rats. There was, however, an increased incidence of benign stromal uterine polyps in female rats treated with 900 mg/kg/day.
- 2.
- There was no evidence of a mutagenic potential of metformin in the following in vitro tests: Ames test (S. typhimurium), gene mutation test (mouse lymphoma cells), or chromosomal aberrations test (human lymphocytes). Results in the in vivo mouse micronucleus test were also negative.
- 3.
- Fertility of male or female rats was unaffected by metformin when administered at doses as high as 600 mg/kg/day, which is approximately 2 times the maximum recommended human daily dose of 2,550 mg based on body surface area comparisons.
14 CLINICAL STUDIES
- 1.
- Adult Clinical Studies
- 2.
- A double-blind, placebo-controlled, multicenter US clinical trial involving obese patients with type
- 3.
- 2 diabetes mellitus whose hyperglycemia was not adequately controlled with dietary management alone (baseline fasting plasma glucose [FPG] of approximately 240 mg/dL) was conducted. Patients were treated with metformin HCl tablets (up to 2,550 mg/day) or placebo for 29 weeks. The results are presented in Table 8.
- 4.
- Table 8: Mean Change in Fasting Plasma Glucose and HbA1c at Week 29Comparing Metformin HCl Tablets vs Placebo in Patients with Type 2Diabetes Mellitus
|
|
|
|
|
|
|
|
|
|
|
|
- 1.
- * Not statistically significant
- 2.
- Mean baseline body weight was 201 lbs and 206 lbs in the metformin HCl tablet and placebo arms, respectively. Mean change in body weight from baseline to week 29 was -1.4 lbs and -2.4 lbs in the metformin HCl tablet and placebo arms, respectively.
- 3.
- A 29-week, double-blind, placebo-controlled study of metformin HCl tablet and glyburide, alone and in combination, was conducted in obese patients with type 2 diabetes mellitus who had failed to achieve adequate glycemic control while on maximum doses of glyburide (baseline FPG of approximately 250 mg/dL). Patients randomized to the combination arm started therapy with metformin HCl tablet 500 mg and glyburide 20 mg. At the end of each week of the first 4 weeks of the trial, these patients had their dosages of metformin HCl increased by 500 mg if they had failed to reach target fasting plasma glucose. After week 4, such dosage adjustments were made monthly, although no patient was allowed to exceed metformin HCl 2,500 mg. Patients in the metformin only arm (metformin HCl plus placebo) discontinued glyburide and followed the same titration schedule. Patients in the glyburide arm continued the same dose of glyburide. At the end of the trial, approximately 70% of the patients in the combination group were taking metformin HCl 2,000 mg/glyburide 20 mg or metformin HCl 2,500 mg/glyburide 20 mg. The results are displayed in Table 9.
- 4.
- Table 9: Mean Change in Fasting Plasma Glucose and HbA1c at Week 29Comparing Metformin HCl Tablets/Glyburide (Comb) vs Glyburide (Glyb) vs Metformin HCl Tablets (GLU): in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Glyburide
|
Comb (n = 213) |
Glyb (n = 209) |
GLU (n = 210) |
p-Values |
|||
|
Glyb vs Comb |
GLU vs Comb |
GLU vs Glyb |
||||
|
|
|
|
NS* 0.001 |
NS* 0.001 |
NS* 0.025 |
|
|
|
|
NS* 0.001 |
NS* 0.001 |
0.007 0.001 |
- 1.
- * Not statistically significant
- 2.
- Mean baseline body weight was 202 lbs, 203 lbs, and 204 lbs in the metformin HCl tablet/glyburide, glyburide, and metformin HCl tablet arms, respectively. Mean change in body weight from baseline to week 29 was 0.9 lbs, -0.7 lbs, and -8.4 lbs in the metformin HCl tablet/glyburide, glyburide, and metformin HCl tablet arms, respectively.
- 3.
- Pediatric Clinical Studies
- 4.
- A double-blind, placebo-controlled study was conducted in pediatric patients aged 10 to 16 years with type 2 diabetes mellitus (mean FPG 182.2 mg/dL), where patients were treated with metformin HCl tablets (up to 2,000 mg/day) for up to 16 weeks (mean duration of treatment 11 weeks). The results are displayed in Table 10.
- 5.
- Table 10: Mean Change in Fasting Plasma Glucose at Week 16 Comparing Metformin HCl Tablets vs Placebo in Pediatric Patientsa with Type 2Diabetes Mellitus
|
|
|
|
|
|
192.3 21.4 |
|
- 1.
- aPediatric patients mean age 13.8 years (range 10 to16 years)
- 2.
- Mean baseline body weight was 205 lbs and 189 lbs in the metformin HCl tablet and placebo arms, respectively. Mean change in body weight from baseline to week 16 was -3.3 lbs and -2.0 lbs in the metformin HCl tablet and placebo arms, respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
- 1.
- RIOMET 500 mg per 5 mL (100 mg/mL) oral solution is supplied in bottles with child-resistant caps and a dosing cup as follows:
|
Flavor |
Appearance |
Size |
NDC |
|
Cherry |
clear, colorless solution |
4 ounce (118 mL) |
10631-206-01 |
|
16 ounce (473 mL) |
10631-206-02 |
||
|
Strawberry |
clear, colorless to light yellow solution |
4 ounce (118 mL) |
10631-238-01 |
|
16 ounce (473 mL) |
10631-238-02 |
17 PATIENT COUNSELING INFORMATION
- 1.
- Advise the patient to read the FDA-approved patient labeling (Patient Information).
- 2.
- Administration:
- 3.
- Instruct patients or caregivers to use the supplied dosing cup to measure the prescribed amount of medication. Inform patients that additional RIOMET dosing cups or oral dosing syringes may be obtained from their pharmacy.
- 4.
- Lactic Acidosis:
- 5.
- Explain the risks of lactic acidosis, its symptoms, and conditions that predispose to its development. Advise patients to discontinue RIOMET immediately and to promptly notify their healthcare provider if unexplained hyperventilation, myalgias, malaise, unusual somnolence or other nonspecific symptoms occur. Counsel patients against excessive alcohol intake and inform patients about importance of regular testing of renal function while receiving RIOMET. Instruct patients to inform their doctor that they are taking RIOMET prior to any surgical or radiological procedure, as temporary discontinuation may be required [see Warnings and Precautions (5.1)].
- 6.
- Hypoglycemia
- 7.
- Inform patients that hypoglycemia may occur when RIOMET is coadministered with oral sulfonylureas and insulin. Explain to patients receiving concomitant therapy the risks of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development [see Warnings and Precautions (5.3)].
- 8.
- Vitamin B12 Deficiency:
- 9.
- Inform patients about importance of regular hematological parameters while receiving RIOMET [see Warnings and Precautions (5.2)].
- 10.
- Females of Reproductive Age:
- 11.
- Inform females that treatment with RIOMET may result in ovulation in some premenopausal anovulatory women which may lead to unintended pregnancy [see Use in Specific Populations (8.3)].
- 12.
- RIOMET is a registered trademark of Sun Pharmaceutical Industries Limited.
- 13.
- Manufactured by:
- 14.
- Mikart, LLC
- 15.
- Atlanta, GA 30318
- 16.
- Distributed by:
- 17.
- Sun Pharmaceutical Industries, Inc.
- 18.
- Cranbury, NJ 08512
- 19.
- November 2018 FDA-11
Patient Medication Information
- •
- PATIENT INFORMATION
- •
- RIOMET (ree oh met)
- •
- (metformin hydrochloride)
- •
- oral solution
- •
- What is the most important information I should know about RIOMET?
- •
- RIOMET can cause serious side effects, including:
- •
- Lactic Acidosis. Metformin hydrochloride, the medicine in RIOMET, can cause a rare, but serious side effect called lactic acidosis (a build-up of lactic acid in the blood) that can cause death. Lactic acidosis is a medical emergency and must be treated in a hospital.
- •
- Stop taking RIOMET and call your healthcare provider right away if you get any of the following symptoms of lactic acidosis:
|
|
|
|
|
|
|
|
- 1.
- You have a higher chance of getting lactic acidosis if you:
- 2.
- have moderate to severe kidney problems. See “Do not take RIOMET if you”
- 3.
- have liver problems.
- 4.
- have congestive heart failure that requires treatment with medicines.
- 5.
- drink a lot of alcohol (very often or short-term “binge” drinking).
- 6.
- get dehydrated (lose a large amount of body fluids). This can happen if you are sick with a fever, vomiting, or diarrhea. Dehydration can also happen when you sweat a lot with activity or exercise and do not drink enough fluids.
- 7.
- have certain x-ray tests with injectable dyes or contrast agents.
- 8.
- have surgery.
- 9.
- have a heart attack, severe infection, or stroke.
- 10.
- are 65 years of age or older.
- 11.
- Tell your healthcare provider if you have any of the problems in the list above.
- 12.
- Tell your healthcare provider that you are taking RIOMET before you have surgery or x-ray tests. Your healthcare provider may need to stop RIOMET for a while if you have surgery or certain x-ray tests.
- 13.
- RIOMET can have other serious side effects. See “What are the possible side effects of RIOMET?”
- 14.
- What is RIOMET?
- 15.
- RIOMET is a prescription medicine that contains metformin hydrochloride. RIOMET is used with diet and exercise to help control high blood sugar (hyperglycemia) in adults and children 10 years of age and older with type 2 diabetes.
- 16.
- It is not known if RIOMET is safe and effective in children under 10 years of age.
- 17.
- Do not take RIOMET if you:
- 18.
- have severe kidney problems.
- 19.
- are allergic to the metformin hydrochloride or any of the ingredients in RIOMET. See the end of this Patient Information leaflet for a complete list of ingredients in RIOMET.
- 20.
- have a condition called metabolic acidosis including diabetic ketoacidosis (high levels of certain acids called “ketones” in your blood or urine).
- 21.
- Before taking RIOMET tell your healthcare provider about all your medical conditions, including if you:
- 22.
- have a history or risk for diabetic ketoacidosis. See “Do not take RIOMET if you?”
- 23.
- have kidney problems.
- 24.
- have liver problems.
- 25.
- have heart problems, including congestive heart failure.
- 26.
- are 65 years or older.
- 27.
- drink alcohol very often or drink a lot of alcohol in short-term “binge” drinking.
- 28.
- are taking insulin or a sulfonylurea medicine.
- 29.
- are pregnant or plan to become pregnant. It is not known if RIOMET will harm your unborn baby. If you are pregnant, talk with your healthcare provider about the best way to control your blood sugar while you are pregnant.
- 30.
- are a premenopausal woman who does not have periods regularly or at all. RIOMET can cause the release of an egg from an ovary in a woman (ovulation). This can increase your chance of getting pregnant.
- 31.
- are breastfeeding or plan to breastfeed. RIOMET can pass into your breast milk. Talk with your healthcare provider about the best way to feed your baby while you take RIOMET.
- 32.
- Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
- 33.
- RIOMET may affect the way other medicines work, and other medicines may affect how RIOMET works.
- 34.
- How should I take RIOMET?
- 35.
- Take RIOMET exactly as your healthcare provider tells you.
- 36.
- Use the Riomet dosing cup to measure your dose. Ask your pharmacist for a dosing cup if you do not have one.
- 37.
- RIOMET should be taken with meals to help decrease an upset stomach.
- 38.
- When your body is under some types of stress, such as fever, trauma (such as a car accident), infection, or surgery, the amount of diabetes medicine that you need may change. Tell your healthcare provider right away if you have any of these problems.
- 39.
- Your healthcare provider should do blood tests to check how well your kidneys are working before and during your treatment with RIOMET.
- 40.
- Your healthcare provider will check your diabetes with regular blood tests, including your blood sugar levels and your hemoglobin A1C.
- 41.
- Low blood sugar (hypoglycemia) can happen more often when RIOMET is taken with certain other diabetes medicines. Talk to your healthcare provider about how to prevent, recognize and manage low blood sugar. See “What are the possible side effects of RIOMET?”
- 42.
- Check your blood sugar as your healthcare provider tells you to.
- 43.
- Stay on your prescribed diet and exercise program while taking RIOMET.
- 44.
- If you take too much RIOMET, call your healthcare provider, or go to the nearest hospital emergency room right away.
- 45.
- What should I avoid while taking RIOMET?
- 46.
- Do not drink a lot of alcoholic drinks while taking RIOMET. This means you should not binge drink for short periods, and you should not drink a lot of alcohol on a regular basis. Alcohol can increase the chance of getting lactic acidosis.
- 47.
- What are the possible side effects of RIOMET?
- 48.
- RIOMET may cause serious side effects, including:
- 49.
- See “What is the most important information I should know about RIOMET?”
- 50.
- Low vitamin B12 (vitamin B12 deficiency). Using RIOMET may cause a decrease in the amount of vitamin B12 in your blood, especially if you have had low vitamin B12 levels before. Your healthcare provider may do blood tests to check your vitamin B12 levels.
- 51.
- Low blood sugar (hypoglycemia). If you take RIOMET with another medicine that can cause lower blood sugar, such as a sulfonylurea or insulin, your risk of getting low blood sugar is higher. The dose of your sulfonylurea medicine or insulin may need to be lowered while you take RIOMET. Signs and symptoms of low blood sugar may include:
|
|
|
|
|
|
|
| |
|
|
- 1.
- Common side effects of RIOMET include:
|
|
|
|
|
|
|
- 1.
- These are not all the possible side effects of RIOMET.
- 2.
- Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
- 3.
- How should I store RIOMET?
- 4.
- Store at room temperature between 59°F to 86°F (15°C to 30°C). See insert.
- 5.
- Keep RIOMET and all medicines out of the reach of children.
- 6.
- General information about the safe and effective use of RIOMET.
- 7.
- Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use RIOMET for a condition for which it was not prescribed. Do not give RIOMET to other people, even if they have the same symptoms that you have. It may harm them.
- 8.
- You can ask your pharmacist or healthcare provider for information about RIOMET that is written for health professionals.
- 9.
- What are the ingredients of RIOMET?
- 10.
- Active ingredients: metformin hydrochloride.
- 11.
- Inactive ingredients (Cherry Flavor): Artificial cherry flavor, hydrochloric acid, potassium bicarbonate, purified water, saccharin calcium, and xylitol.
- 12.
-
Inactive ingredients (Strawberry Flavor): Hydrochloric acid, N&A strawberry flavor (propylene glycol and glycerin), potassium bicarbonate, purified water, sucralose, and xylitol.
Manufactured by:
Mikart, LLC
Atlanta, GA 30318
Distributed by:
Sun Pharmaceutical Industries, Inc.
Cranbury, NJ 08512

