MIACALCIN- calcitonin salmon injection, solution
Novartis Pharmaceuticals Corporation
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MIACALCIN injection safely and effectively. See full prescribing information for MIACALCIN injection.
MIACALCIN® (calcitonin-salmon) injection, synthetic, for subcutaneous or intramuscular use Initial U.S. Approval: 1975 INDICATIONS AND USAGEMiacalcin synthetic injection is a calcitonin, indicated for the following conditions:
Limitations of Use: DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSHypersensitivity to calcitonin-salmon or any of the excipients (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions are nausea with or without vomiting (10%), injection site inflammation (10%), and flushing of the face or hands (2-5%) (6)
DRUG INTERACTIONSConcomitant use of calcitonin-salmon and lithium may lead to a reduction in plasma lithium concentrations due to increased urinary clearance of lithium. The dose of lithium may require adjustment (7) USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION. Revised: 2/2015 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Treatment of Paget’s Disease of Bone
Miacalcin injection is indicated for the treatment of symptomatic Paget’s disease of bone in patients with moderate to severe disease characterized by polyostotic involvement with elevated serum alkaline phosphatase and urinary hydroxyproline excretion. There is no evidence that the prophylactic use of calcitonin-salmon is beneficial in asymptomatic patients. Miacalcin injection should be used only in patients who do not respond to alternative treatments or for whom such treatments are not suitable (e.g., patients for whom other therapies are contraindicated or for patients who are intolerant or unwilling to use other therapies).
1.2 Treatment of Hypercalcemia
Miacalcin injection is indicated for the early treatment of hypercalcemic emergencies, along with other appropriate agents, when a rapid decrease in serum calcium is required, until more specific treatment of the underlying disease can be accomplished. It may also be added to existing therapeutic regimens for hypercalcemia such as intravenous fluids and furosemide, oral phosphate or corticosteroids, or other agents.
1.3 Treatment of Postmenopausal Osteoporosis
Miacalcin injection is indicated for the treatment of postmenopausal osteoporosis in women greater than 5 years postmenopause. The evidence of efficacy for calcitonin-salmon injection is based on increases in total body calcium observed in clinical trials. Fracture reduction efficacy has not been demonstrated. Miacalcin injection should be reserved for patients for whom alternative treatments are not suitable (e.g., patients for whom other therapies are contraindicated or for patients who are intolerant or unwilling to use other therapies).
2 DOSAGE AND ADMINISTRATION
2.1 Paget’s Disease of Bone
The recommended dose of Miacalcin injection for treatment of symptomatic Paget's disease of bone is 100 International Units (0.5 mL) per day administered subcutaneously or intramuscularly.
2.2 Hypercalcemia
The recommended starting dose of Miacalcin injection for early treatment of hypercalcemia is 4 International Units/kg body weight every 12 hours by subcutaneous or intramuscular injection. If the response to this dose is not satisfactory after one or two days, the dose may be increased to 8 International Units/kg every 12 hours. If the response remains unsatisfactory after two more days, the dose may be further increased to a maximum of 8 International Units/kg every 6 hours.
2.3 Postmenopausal Osteoporosis
The recommended dose of Miacalcin injection for treatment of postmenopausal osteoporosis in women greater than 5 years postmenopause is 100 International Units (0.5 mL) per day administered subcutaneously or intramuscularly. The minimum effective dose of Miacalcin injection for the prevention of vertebral bone mineral density loss has not been established.
2.4 Preparation and Administration
Visually inspect Miacalcin vials. Miacalcin injection is a clear, colorless, solution. If the solution is not clear and colorless, or contains any particles, or if the vial is damaged, do not administer the solution.
If the volume of Miacalcin injection to be injected exceeds 2 mL, intramuscular injection is preferable and the total dose should be distributed across multiple sites of injection.
Instruct patients to use sterile injection technique when administering Miacalcin injection, and to dispose of needles properly.
3 DOSAGE FORMS AND STRENGTHS
Miacalcin injection is available as a clear, colorless, sterile solution of synthetic calcitonin-salmon in individual 2 mL multi-dose vials containing 200 International Units per mL.
4 CONTRAINDICATIONS
Hypersensitivity to calcitonin-salmon or any of the excipients. Reactions have included anaphylaxis with death, bronchospasm, and swelling of the tongue or throat [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions have been reported in patients receiving Miacalcin injection, e.g., bronchospasm, swelling of the tongue or throat, anaphylactic shock, and death due to anaphylaxis. Appropriate medical support and monitoring measures should be readily available when Miacalcin injection is administered. If anaphylaxis or other severe hypersensitivity/allergic reactions occur, initiate appropriate treatment [see Contraindications (4)].
For patients with suspected hypersensitivity to calcitonin-salmon, skin testing should be considered prior to treatment utilizing a dilute, sterile solution of Miacalcin injection. Healthcare providers may wish to refer patients who require skin testing to an allergist. A detailed skin testing protocol is available from the Medical Services Department of Novartis Pharmaceuticals Corporation.
5.2 Hypocalcemia
Hypocalcemia associated with tetany (i.e., muscle cramps, twitching) and seizure activity has been reported with Miacalcin injection therapy. Hypocalcemia must be corrected before initiating therapy. Other disorders affecting mineral metabolism (such as vitamin D deficiency) should also be effectively treated. In patients at risk for hypocalcemia, provisions for parenteral calcium administration should be available during the first several administrations of calcitonin-salmon and serum calcium and symptoms of hypocalcemia should be monitored. Use of Miacalcin injection for the treatment of Paget’s disease or postmenopausal osteoporosis is recommended in conjunction with an adequate intake of calcium and vitamin D [see Dosage and Administration (2.5)].
5.3 Malignancy
In a meta-analysis of 21 randomized, controlled clinical trials with calcitonin-salmon (nasal spray or investigational oral formulations), the overall incidence of malignancies reported was higher among calcitonin-salmon-treated patients (4.1%) compared with placebo-treated patients (2.9%). This suggests an increased risk of malignancies in calcitonin-salmon-treated patients compared to placebo-treated patients. It is not possible to exclude an increased risk when calcitonin-salmon is administered long-term subcutaneously, intramuscularly, or intravenously. The benefits for the individual patient should be carefully considered against possible risks [see Adverse Reactions (6.1)].
5.4 Antibody Formation
Circulating antibodies to calcitonin-salmon have been reported with Miacalcin injection. The possibility of antibody formation should be considered in any patient with an initial response to Miacalcin injection who later stops responding to treatment [see Adverse Reactions (6.3)].
5.5 Urine Sediment Abnormalities
Coarse granular casts and casts containing renal tubular epithelial cells were reported in young adult volunteers at bed rest who were given injectable calcitonin-salmon to study the effect of immobilization on osteoporosis. There was no other evidence of renal abnormality and the urine sediment normalized after calcitonin-salmon was stopped. Periodic examinations of urine sediment should be considered.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Hypersensitivity Reactions, including anaphylaxis [see Warnings and Precautions (5.1)]
- Hypocalcemia [see Warnings and Precautions (5.2)]
- Malignancy [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of calcitonin-salmon injection was assessed in open-label trials several months to two years in duration. The most common adverse reactions are discussed below.
Nausea: Nausea with or without vomiting has been noted in about 10% of patients treated with calcitonin-salmon. It is most evident when treatment is first initiated and tends to decrease or disappear with continued administration.
Dermatologic Reactions: Local inflammatory reactions at the site of subcutaneous or intramuscular injection have been reported in about 10% of patients. Flushing of face or hands occurred in about 2%-5% of patients. Skin rashes and pruritus of the ear lobes have also been reported.
Other Adverse Reactions: Nocturia, feverish sensation, pain in the eyes, poor appetite, abdominal pain, pedal edema, and salty taste have been reported in patients treated with calcitonin-salmon injection.
Malignancy
A meta-analysis of 21 randomized, controlled clinical trials with calcitonin-salmon (nasal spray or investigational oral formulations) was conducted to assess the risk of malignancies in calcitonin-salmon-treated patients compared to placebo-treated patients. The trials in the meta-analysis ranged in duration from 6 months to 5 years and included a total of 10883 patients (6151 treated with calcitonin-salmon and 4732 treated with placebo). The overall incidence of malignancies reported in these 21 trials was higher among calcitonin-salmon-treated patients (254/6151 or 4.1%) compared with placebo-treated patients (137/4732 or 2.9%). Findings were similar when analyses were restricted to the 18 nasal spray only trials [calcitonin-salmon 122/2712 (4.5%); placebo 30/1309 (2.3%)].
The meta-analysis results suggest an increased risk of overall malignancies in calcitonin-salmon-treated patients compared to placebo-treated patients when all 21 trials are included and when the analysis is restricted to the 18 nasal spray only trials (see Table 1). It is not possible to exclude an increased risk when calcitonin-salmon is administered by the subcutaneous, intramuscular, or intravenous route because these routes of administration were not investigated in the meta-analysis. The increased malignancy risk seen with the meta-analysis was heavily influenced by a single large 5-year trial, which had an observed risk difference of 3.4% [95% CI (0.4%, 6.5%)]. Imbalances in risks were still observed when analyses excluded basal cell carcinoma (see Table 1); the data were not sufficient for further analyses by type of malignancy. A mechanism for these observations has not been identified. Although a definitive causal relationship between calcitonin-salmon use and malignancies cannot be established from this meta-analysis, the benefits for the individual patient should be carefully evaluated against all possible risks [see Warnings and Precautions (5.3)].
| 1 The overall adjusted risk difference is the difference between the percentage of patients who had any malignancy (or malignancy excluding basal cell carcinoma) in calcitonin-salmon and placebo treatment groups, using the Mantel-Haenszel (MH) fixed-effect method. A risk difference of 0 is suggestive of no difference in malignancy risks between the treatment groups. 2 The corresponding 95% confidence interval for the overall adjusted risk difference also based on MH fixed-effect method. |
|||
| Patients | Malignancies | Risk Difference1
(%) | 95% Confidence Interval2 (%) |
| All (nasal spray + oral) | All | 1.0 | (0.3, 1.6) |
| All (nasal spray + oral) | Excluding basal cell carcinoma | 0.5 | (-0.1, 1.2) |
| All (nasal spray only) | All | 1.4 | (0.3, 2.6) |
| All (nasal spray only) | Excluding basal cell carcinoma | 0.8 | (-0.2, 1.8) |
6.2 Postmarketing Experience
Because postmarketing adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been reported during post-approval use of Miacalcin injection.
Allergic / Hypersensitivity Reactions: Serious hypersensitivity reactions have been reported in patients receiving calcitonin-salmon injection, e.g., bronchospasm, swelling of the tongue or throat, anaphylactic shock, and death due to anaphylaxis.
Skin and subcutaneous tissue disorders: Urticaria
Hypocalcemia: Hypocalcemia with tetany (i.e. muscle cramps, twitching) and seizure activity have been reported.
Body as a Whole: influenza-like symptoms, fatigue, edema (facial, peripheral, and generalized)
Musculoskeletal: arthralgia, musculoskeletal pain
Cardiovascular: hypertension
Gastrointestinal: abdominal pain, diarrhea
Urinary System: polyuria
Nervous System: dizziness, headache, paresthesia, tremor
Vision: visual disturbance
6.3 Immunogenicity
Consistent with the potentially immunogenic properties of medicinal products containing peptides, administration of Miacalcin may trigger the development of anti-calcitonin antibodies. Circulating antibodies to calcitonin-salmon after 2-18 months of treatment have been reported in about one-half of the patients with Paget’s disease in whom antibody studies were done. In some cases, high antibody titers are found; these patients usually will have a loss of response to treatment [see Warnings and Precautions (5.4)].
The incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of a positive antibody test result may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of antibodies among different calcitonin-salmon products may be misleading.
7 DRUG INTERACTIONS
No formal drug interaction studies have been performed with Miacalcin injection.
Concomitant use of calcitonin-salmon and lithium may lead to a reduction in plasma lithium concentrations due to increased urinary clearance of lithium. The dose of lithium may require adjustment.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C:
Risk Summary
There are no adequate and well-controlled studies in pregnant women. Miacalcin injection should be used during pregnancy only if the potential benefit justifies the use as compared with potential risks to the patient and fetus. Based on animal data, Miacalcin is predicted to have low probability of increasing the risk of adverse developmental outcomes above background risk.
Animal Data
Calcitonin-salmon has been shown to cause a decrease in fetal birth weights in rabbits when given by subcutaneous injection in doses 4-18 times the parenteral dose recommended for human use (of 54 International Units/m2).
No embryo/fetal toxicities related to Miacalcin were reported from maternal subcutaneous daily doses in rats up to 80 International Units /kg/day from gestation day 6 to 15.
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. No studies have been conducted to assess the impact of Miacalcin on milk production in humans, its presence in human breast milk, or its effects on the breastfed child. Because many drugs are excreted in human milk, caution should be exercised when Miacalcin is administered to a nursing woman. Calcitonin has been shown to inhibit lactation in rats.
8.5 Geriatric Use
Clinical studies of Miacalcin injection did not include sufficient numbers of subjects aged 65 years and older to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10 OVERDOSAGE
The pharmacologic actions of Miacalcin injection suggest that hypocalcemic tetany could occur in overdose. Therefore, provisions for parenteral administration of calcium should be available for the treatment of overdose.
A dose of calcitonin-salmon l000 International Units subcutaneously may produce nausea and vomiting. Doses of 32 International Units per kg per day for 1-2 days demonstrate no other adverse effects. Data on chronic high-dose administration are insufficient to assess toxicity.
11 DESCRIPTION
Calcitonin is a polypeptide hormone secreted by the parafollicular cells of the thyroid gland in mammals and by the ultimobranchial gland of birds and fish.
Miacalcin (calcitonin-salmon) injection, synthetic is a synthetic polypeptide of 32 amino acids in the same linear sequence that is found in calcitonin of salmon origin. This is shown by the following graphic formula:
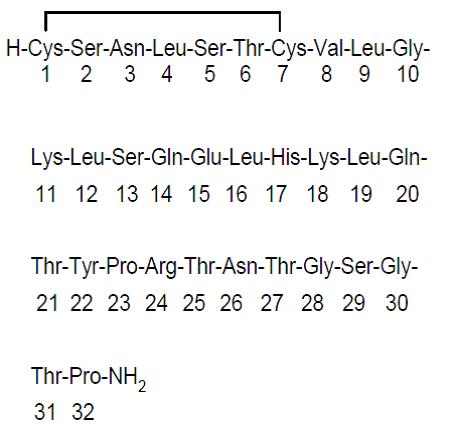
It is provided in sterile solution for subcutaneous or intramuscular injection. Each milliliter contains: calcitonin-salmon 200 International Units.
Inactive Ingredients (per mL): acetic acid, USP, 2.25 mg; phenol, USP, 5.0 mg; sodium acetate trihydrate, USP, 2.0 mg; sodium chloride, USP, 7.5 mg; water for injection, USP.
The activity of Miacalcin injection is stated in International Units based on bioassay in comparison with the International Reference Preparation of calcitonin-salmon for Bioassay, distributed by the National Institute for Biological Standards and Control, Holly Hill, London.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Calcitonin-salmon is a calcitonin receptor agonist. Calcitonin-salmon acts primarily on bone, but direct renal effects and actions on the gastrointestinal tract are also recognized. Calcitonin-salmon appears to have actions essentially identical to calcitonins of mammalian origin, but its potency per mg is greater and it has a longer duration of action.
The actions of calcitonin on bone and its role in normal human bone physiology are still not completely elucidated, although calcitonin receptors have been discovered in osteoclasts and osteoblasts.
12.2 Pharmacodynamics
Bone
Single injections of calcitonin-salmon caused a marked transient inhibition of the ongoing bone resorptive process. With prolonged use, there is a persistent, smaller decrease in the rate of bone resorption. Histologically, this is associated with a decreased number of osteoclasts and an apparent decrease in their resorptive activity.
In healthy adults, who have a relatively low rate of bone resorption, the administration of exogenous calcitonin-salmon results in decreases in serum calcium within the limits of the normal range. In healthy children and in patients whose bone resorption is more rapid, decreases in serum calcium are more pronounced in response to calcitonin-salmon.
Kidney
Studies with injectable calcitonin-salmon show increases in the excretion of filtered phosphate, calcium, and sodium by decreasing their tubular reabsorption.
Gastrointestinal Tract
Some evidence from studies with injectable preparations suggests that calcitonin-salmon may have effects on the gastrointestinal tract. Short-term administration of injectable calcitonin-salmon results in marked transient decreases in the volume and acidity of gastric juice and in the volume and the trypsin and amylase content of pancreatic juice. Whether these effects continue to be elicited after each injection of calcitonin-salmon during chronic therapy has not been investigated.
12.3 Pharmacokinetics
The absolute bioavailability of calcitonin-salmon is approximately 66% and 71% after intramuscular or subcutaneous injection, respectively. After subcutaneous administration, peak plasma levels are reached in approximately 23 minutes. The terminal half-life is approximately 58 minutes for intramuscular administration and 59 to 64 minutes for subcutaneous administration. The apparent volume of distribution is 0.15-0.3 L/kg.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
The incidence of pituitary adenomas was increased in rats after one and two years of subcutaneous exposure to synthetic calcitonin-salmon. The significance of this finding to humans is unknown because pituitary adenomas are very common in rats as they age, the pituitary adenomas did not transform into metastatic tumors, there were no other clear treatment- related neoplasms, and synthetic calcitonin-salmon related neoplasms were not observed in mice after two years of dosing.
Rat findings:
The only clear neoplastic finding in rats dosed subcutaneously with calcitonin-salmon was an increase in the incidence of pituitary adenomas in male Fisher 344 rats and female Sprague Dawley rats after one year of dosing and male Sprague Dawley rats dosed for one and two years. In female Sprague Dawley rats, the incidence of pituitary adenomas after two years was high in all treatment groups (between 80% and 92% including the control groups) such that a treatment-related effect could not be distinguished from natural background incidence. The lowest dose in male Sprague Dawley rats that developed an increased incidence of pituitary adenomas after two years of dosing (1.7 International Units/kg/day) is approximately 1/6th of the maximum recommended subcutaneous dose in humans (100 International Units/day) based on body surface area conversion between rats and humans. The findings suggest that calcitonin-salmon reduced the latency period for development of non-functioning pituitary adenomas.
Mouse findings:
No carcinogenicity potential was evident in male or female mice dosed subcutaneously for two years with synthetic calcitonin-salmon at doses up to 800 International Units/kg/day. The 800 International Units/kg/day dose is approximately 39 times the maximum recommended subcutaneous dose in humans (100 International Units/day) based on body surface area conversion between mice and humans.
Mutagenesis
Synthetic calcitonin-salmon tested negative for mutagenicity using Salmonella typhimurium (5 strains) and Escherichia coli (2 strains), with and without rat liver metabolic activation, and was not clastogenic in a chromosome aberration test in Chinese Hamster V79 cells. There was no evidence that calcitonin-salmon was clastogenic in the in vivo mouse micronucleus test.
Fertility
Effects of calcitonin-salmon on fertility have not been assessed in animals.
14 CLINICAL STUDIES
14.1 Paget’s Disease of Bone
The trials used for the basis of approval for calcitonin-salmon injection for treatment of Paget’s disease of bone were conducted in patients with moderate to severe disease characterized by polyostotic involvement with elevated serum alkaline phosphatase and urinary hydroxyproline excretion. In open-label clinical trials of several months to two years duration with historical controls, biochemical abnormalities were substantially improved (more than 30% reduction) in about 2/3 of patients studied and bone pain was improved in a similar fraction. A small number of documented instances of reversal of neurologic deficits have occurred, including improvement in the basilar compression syndrome, and improvement of spinal cord and spinal nerve lesions. There is too little experience to predict the likelihood of improvement of any given neurologic lesion. Hearing loss is improved infrequently (4 of 29 patients studied by audiometry). Patients with increased cardiac output due to extensive Paget’s disease of bone have had measured decreases in cardiac output while receiving calcitonin-salmon. The number of treated patients in this category is too small to predict how likely such a result will be.
There is no evidence that the prophylactic use of calcitonin-salmon is beneficial in asymptomatic patients.
14.2 Hypercalcemia
In four open-label clinical trials enrolling 53 patients, calcitonin-salmon has been shown to lower elevated serum calcium levels of patients with carcinoma (with or without metastases), multiple myeloma, and primary hyperparathyroidism (lesser response). These patients were treated with calcitonin-salmon only when other methods of lowering serum calcium (hydration, oral phosphate, corticosteroids) were unsuccessful or unsuitable. With patients’ pre-therapy serum calcium levels as controls, reduction in serum calcium was evident within 1-2 hours of administration. The peak effect occurred within 24-48 hours of injection and administration of calcitonin-salmon every 12 hours maintained a hypocalcemic effect for approximately 5-8 days, the time period evaluated for most patients in the clinical trials. The average reduction of 8-hour post-injection serum calcium was approximately 9% (2-3 mg/dL). Patients with higher values of serum calcium tended to show greater reductions during calcitonin-salmon treatment.
14.3 Postmenopausal Osteoporosis
The trials used for the basis of approval for calcitonin-salmon injection for treatment of postmenopausal osteoporosis were two randomized, open-label, 2-year studies in postmenopausal women 50–74 years of age with total body calcium <85% of expected normal, and vertebral osteopenia (by x-ray criteria) and/or at least one atraumatic compression fracture. The primary efficacy endpoint was total body calcium measured by neutron activation analysis. Patients were randomized to calcitonin-salmon injection 100 International Units daily (subcutaneously or intramuscularly) at bedtime, or control. All subjects received daily supplements of 1200 mg calcium carbonate and 400 International Units of vitamin D.
In both studies, total body calcium increased from baseline with calcitonin-salmon therapy at 1 year, followed by a trend to decreasing total body calcium (still above baseline) at 2 years.
Thoracic and lumbar spine X-rays (AP/lateral) were obtained yearly. For the two studies combined (34 calcitonin-salmon and 35 control subjects), in the first year there was a total of 6 new vertebral compression fractures in the calcitonin-salmon group and 5 in the control group. In the second year there were 7 new fractures in each group.
No evidence currently exists to indicate whether Miacalcin injection decreases the risk of osteoporotic fracture. A controlled study, which was prematurely discontinued, failed to demonstrate any benefit of calcitonin-salmon on fracture rate.
No adequate controlled trials have examined the effect of calcitonin-salmon injection on vertebral bone mineral density beyond 1 year of treatment. Therefore, the minimum effective dose of Miacalcin injection for prevention of vertebral bone mineral density loss has not been established.
In clinical studies of postmenopausal osteoporosis, bone biopsy and radial bone mass assessments at baseline and after 26 months of daily injectable calcitonin-salmon indicate that calcitonin therapy results in the formation of normal bone.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Miacalcin (calcitonin-salmon) injection, synthetic is available as a sterile solution in individual 2 mL multi-dose vials containing 200 International Units per mL .......................NDC 0078-0149-23
Storage and Handling
Store in refrigerator between 2°C-8°C (36°F-46°F).
17 PATIENT COUNSELING INFORMATION
- Instruct patients and other persons who may administer Miacalcin injection in sterile injection technique. Also instruct patients to dispose of needles properly [see Dosage and Administration (2.4)].
- Inform patients of the potential increase in risk of malignancy [see Warnings and Precautions (5.3)].
- Advise patients with postmenopausal osteoporosis or Paget’s disease of bone to maintain an adequate calcium (at least 1000 mg elemental calcium per day) and vitamin D (at least 400 International Units per day) intake [see Dosage and Administration (2.5)].
- Instruct patients to seek emergency medical help or go to the nearest hospital emergency room right away if they develop any signs or symptoms of a serious allergic reaction [see Warnings and Precautions (5.1)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2015-50
February 2015
| MIACALCIN
calcitonin salmon injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |