Label: GENGRAF- cyclosporine capsule





-
NDC Code(s):
0074-0541-30,
0074-3108-32,
0074-3109-32,
0074-6463-32, view more0074-6479-32
- Packager: AbbVie Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 1, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING
Only physicians experienced in management of systemic immunosuppressive therapy for the indicated disease should prescribe Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]). At doses used in solid organ transplantation, only physicians experienced in immunosuppressive therapy and management of organ transplant recipients should prescribe Gengraf®. Patients receiving the drug should be managed in facilities equipped and staffed with adequate laboratory and supportive medical resources. The physician responsible for maintenance therapy should have complete information requisite for the follow-up of the patient.
Gengraf®, a systemic immunosuppressant, may increase the susceptibility to infection and the development of neoplasia. In kidney, liver, and heart transplant patients Gengraf® may be administered with other immunosuppressive agents. Increased susceptibility to infection and the possible development of lymphoma and other neoplasms may result from the increase in the degree of immunosuppression in transplant patients.
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) has increased bioavailability in comparison to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Gengraf® and Sandimmune® are not bioequivalent and cannot be used interchangeably without physician supervision. For a given trough concentration, cyclosporine exposure will be greater with Gengraf® than with Sandimmune®. If a patient who is receiving exceptionally high doses of Sandimmune® is converted to Gengraf®, particular caution should be exercised. Cyclosporine blood concentrations should be monitored in transplant and rheumatoid arthritis patients taking Gengraf® to avoid toxicity due to high concentrations. Dose adjustments should be made in transplant patients to minimize possible organ rejection due to low concentrations. Comparison of blood concentrations in the published literature with blood concentrations obtained using current assays must be done with detailed knowledge of the assay methods employed.
-
SPL UNCLASSIFIED SECTION
For Psoriasis Patients (See also BOXED WARNING above) Psoriasis patients previously treated with PUVA and to a lesser extent, methotrexate or other immunosuppressive agents, UVB, coal tar, or radiation therapy, are at an increased risk of developing skin malignancies when taking Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]).
Cyclosporine, the active ingredient in Gengraf®, in recommended dosages, can cause systemic hypertension and nephrotoxicity. The risk increases with increasing dose and duration of cyclosporine therapy. Renal dysfunction, including structural kidney damage, is a potential consequence of cyclosporine, and therefore, renal function must be monitored during therapy. -
DESCRIPTION
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is a modified oral formulation of cyclosporine that forms an aqueous dispersion in an aqueous environment.
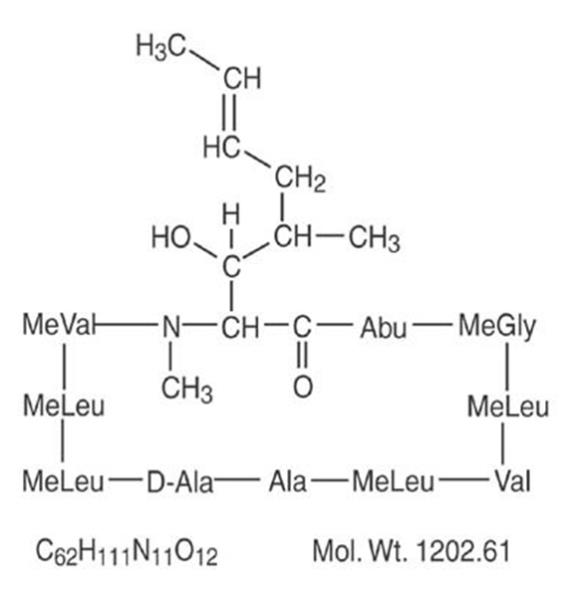
Cyclosporine, the active principle in Gengraf® Capsules, is a cyclic polypeptide immunosuppressant agent consisting of 11 amino acids. It is produced as a metabolite by the fungus species Aphanocladium album.
Chemically, cyclosporine is designated as [R-[R*,R*-(E)]]-cyclic-(L-alanyl-D-alanyl-N-methyl-L-leucyl-N-methyl-L-leucyl-N-methyl-L-valyl-3-hydroxy-N,4-dimethyl-L-2-amino-6-octenoyl-L-α-amino-butyryl-N-methylglycyl-N -methyl-L-leucyl-L-valyl-N-methyl-L-leucyl).
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) are available in 25 mg and 100 mg strengths.
Each 25 mg capsule contains
cyclosporine, 25 mg, alcohol, USP, absolute, 12.8% v/v (10.1% wt/vol.).
Each 100 mg capsule contains
cyclosporine, 100 mg, alcohol, USP, absolute, 12.8% v/v (10.1% wt/vol.).
-
CLINICAL PHARMACOLOGY
Cyclosporine is a potent immunosuppressive agent that in animals prolongs survival of allogeneic transplants involving skin, kidney, liver, heart, pancreas, bone marrow, small intestine, and lung. Cyclosporine has been demonstrated to suppress some humoral immunity and to a greater extent, cell-mediated immune reactions such as allograft rejection, delayed hypersensitivity, experimental allergic encephalomyelitis, Freund's adjuvant arthritis, and graft versus host disease in many animal species for a variety of organs.
The effectiveness of cyclosporine results from specific and reversible inhibition of immunocompetent lymphocytes in the G0- and G1-phase of the cell cycle. T-lymphocytes are preferentially inhibited. The T-helper cell is the main target, although the T-suppressor cell may also be suppressed. Cyclosporine also inhibits lymphokine production and release including interleukin-2.
No effects on phagocytic function (changes in enzyme secretions, chemotactic migration of granulocytes, macrophage migration, carbon clearance in vivo) have been detected in animals. Cyclosporine does not cause bone marrow suppression in animal models or man.
Pharmacokinetics
The immunosuppressive activity of cyclosporine is primarily due to parent drug. Following oral administration, absorption of cyclosporine is incomplete. The extent of absorption of cyclosporine is dependent on the individual patient, the patient population, and the formulation. Elimination of cyclosporine is primarily biliary with only 6% of the dose (parent drug and metabolites) excreted in urine. The disposition of cyclosporine from blood is generally biphasic, with a terminal half-life of approximately 8.4 hours (range 5 to 18 hours). Following intravenous administration, the blood clearance of cyclosporine (assay: HPLC) is approximately 5 to 7 mL/min/kg in adult recipients of renal or liver allografts. Blood cyclosporine clearance appears to be slightly slower in cardiac transplant patients.
The Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) and Gengraf® Oral Solution (cyclosporine oral solution, USP [MODIFIED]) are bioequivalent.
The relationship between administered dose and exposure (area under the concentration versus time curve, AUC) is linear within the therapeutic dose range. The intersubject variability (total, %CV) of cyclosporine exposure (AUC) when cyclosporine (MODIFIED) or Sandimmune® is administered ranges from approximately 20% to 50% in renal transplant patients. This intersubject variability contributes to the need for individualization of the dosing regimen for optimal therapy (See DOSAGE AND ADMINISTRATION). Intrasubject variability of AUC in renal transplant recipients (%CV) was 9% to 21% for cyclosporine (MODIFIED) and 19% to 26% for Sandimmune®. In the same studies, intrasubject variability of trough concentrations (%CV) was 17% to 30% for cyclosporine (MODIFIED) and 16% to 38% for Sandimmune®.
Absorption
Cyclosporine (MODIFIED) has increased bioavailability compared to Sandimmune®. The absolute bioavailability of cyclosporine administered as Sandimmune® is dependent on the patient population, estimated to be less than 10% in liver transplant patients and as great as 89% in some renal transplant patients. The absolute bioavailability of cyclosporine administered as cyclosporine (MODIFIED) has not been determined in adults. In studies of renal transplant, rheumatoid arthritis and psoriasis patients, the mean cyclosporine AUC was approximately 20% to 50% greater and the peak blood cyclosporine concentration (Cmax) was approximately 40% to 106% greater following administration of cyclosporine (MODIFIED) compared to following administration of Sandimmune®. The dose normalized AUC in de novo liver transplant patients administered cyclosporine (MODIFIED) 28 days after transplantation was 50% greater and Cmax was 90% greater than in those patients administered Sandimmune®. AUC and Cmax are also increased (cyclosporine [MODIFIED] relative to Sandimmune®) in heart transplant patients, but data are very limited. Although the AUC and Cmax values are higher on cyclosporine (MODIFIED) relative to Sandimmune®, the predose trough concentrations (dose-normalized) are similar for the two formulations.
Following oral administration of cyclosporine (MODIFIED), the time to peak blood cyclosporine concentrations (Tmax) ranged from 1.5 to 2.0 hours. The administration of food with cyclosporine (MODIFIED) decreases the cyclosporine AUC and Cmax. A high fat meal (669 kcal, 45 grams fat) consumed within one-half hour before cyclosporine (MODIFIED) administration decreased the AUC by 13% and Cmax by 33%. The effects of a low fat meal (667 kcal, 15 grams fat) were similar.
The effect of T-tube diversion of bile on the absorption of cyclosporine from cyclosporine (MODIFIED) was investigated in eleven de novo liver transplant patients. When the patients were administered cyclosporine (MODIFIED) with and without T-tube diversion of bile, very little difference in absorption was observed, as measured by the change in maximal cyclosporine blood concentrations from pre-dose values with the T-tube closed relative to when it was open: 6.9±41% (range -55% to 68%).
Pharmacokinetic Parameters (mean±SD) Patient
PopulationDose/day1
(mg/d)Dose/
weight
(mg/kg/d)AUC2
(ng·hr/mL)Cmax
(ng/mL)Trough3
(ng/mL)CL/F
(mL/min)CL/F
(mL/min/kg)De novo
renal
transplant4
Week 4
(N=37)597±174 7.95±2.81 8772±2089 1802±428 361±129 593±204 7.8±2.9 Stable renal
transplant4
(N=55)344±122 4.10±1.58 6035±2194 1333±469 251±116 492±140 5.9±2.1 De novo liver
transplant5
Week 4
(N=18)458±190 6.89±3.68 7187±2816 1555±740 268±101 577±309 8.6±5.7 De novo
rheumatoid
arthritis6
(N=23)182±55.6 2.37±0.36 2641±877 728±263 96.4±37.7 613±196 8.3±2.8 De novo
psoriasis6
Week 4
(N=18)189±69.8 2.48±0.65 2324±1048 655±186 74.9±46.7 723±186 10.2±3.9 1Total daily dose was divided into two doses administered every 12 hours
2AUC was measured over one dosing interval
3Trough concentration was measured just prior to the morning cyclosporine (MODIFIED) dose, approximately 12 hours after the previous dose
4Assay: TDx specific monoclonal fluorescence polarization immunoassay
5Assay: Cyclo-trac specific monoclonal radioimmunoassay
6Assay: INCSTAR specific monoclonal radioimmunoassayDistribution
Cyclosporine is distributed largely outside the blood volume. The steady state volume of distribution during intravenous dosing has been reported as 3 to 5 L/kg in solid organ transplant recipients. In blood, the distribution is concentration dependent. Approximately 33% to 47% is in plasma, 4% to 9% in lymphocytes, 5% to 12% in granulocytes, and 41% to 58% in erythrocytes. At high concentrations, the binding capacity of leukocytes and erythrocytes becomes saturated. In plasma, approximately 90% is bound to proteins, primarily lipoproteins. Cyclosporine is excreted in human milk. (See PRECAUTIONS, Nursing Mothers)
Metabolism
Cyclosporine is extensively metabolized by the cytochrome P-450 3A enzyme system in the liver, and to a lesser degree in the gastrointestinal tract, and the kidney. The metabolism of cyclosporine can be altered by the coadministration of a variety of agents. (See PRECAUTIONS, Drug Interactions) At least 25 metabolites have been identified from human bile, feces, blood, and urine. The biological activity of the metabolites and their contributions to toxicity are considerably less than those of the parent compound. The major metabolites (M1, M9, and M4N) result from oxidation at the 1-beta, 9-gamma, and 4-N-demethylated positions, respectively. At steady state following the oral administration of Sandimmune®, the mean AUCs for blood concentrations of M1, M9, and M4N are about 70%, 21%, and 7.5% of the AUC for blood cyclosporine concentrations, respectively. Based on blood concentration data from stable renal transplant patients (13 patients administered cyclosporine [MODIFIED] and Sandimmune® in a crossover study), and bile concentration data from de novo liver transplant patients (4 administered cyclosporine [MODIFIED], 3 administered Sandimmune®), the percentage of dose present as M1, M9, and M4N metabolites is similar when either cyclosporine (MODIFIED) or Sandimmune® is administered.
Excretion
Only 0.1% of a cyclosporine dose is excreted unchanged in the urine. Elimination is primarily biliary with only 6% of the dose (parent drug and metabolites) excreted in the urine. Neither dialysis nor renal failure alters cyclosporine clearance significantly.
Drug Interactions
(See PRECAUTIONS, Drug Interactions) When diclofenac or methotrexate was coadministered with cyclosporine in rheumatoid arthritis patients, the AUC of diclofenac and methotrexate, each was significantly increased. (See PRECAUTIONS, Drug Interactions) No clinically significant pharmacokinetic interactions occurred between cyclosporine and aspirin, ketoprofen, piroxicam, or indomethacin.
Specific Populations
Renal Impairment
In a study performed in 4 subjects with end-stage renal disease (creatinine clearance <5 mL/min), an intravenous infusion of 3.5 mg/kg of cyclosporine over 4 hours administered at the end of a hemodialysis session resulted in a mean volume of distribution (Vdss) of 3.49 L/kg and systemic clearance (CL) of 0.369 L/hr/kg. This systemic CL (0.369 L/hr/kg) was approximately two thirds of the mean systemic CL (0.56 L/hr/kg) of cyclosporine in historical control subjects with normal renal function. In 5 liver transplant patients, the mean clearance of cyclosporine on and off hemodialysis was 463 mL/min and 398 mL/min, respectively. Less than 1% of the dose of cyclosporine was recovered in the dialysate.
Hepatic Impairment
Cyclosporine is extensively metabolized by the liver. Since severe hepatic impairment may result in significantly increased cyclosporine exposures, the dosage of cyclosporine may need to be reduced in these patients.
Pediatric Population
Pharmacokinetic data from pediatric patients administered cyclosporine (MODIFIED) or Sandimmune® are very limited. In 15 renal transplant patients aged 3 to 16 years, cyclosporine whole blood clearance after IV administration of Sandimmune® was 10.6±3.7 mL/min/kg (assay: Cyclo-trac specific RIA). In a study of 7 renal transplant patients aged 2 to 16, the cyclosporine clearance ranged from 9.8 to 15.5 mL/min/kg. In 9 liver transplant patients aged 0.6 to 5.6 years, clearance was 9.3±5.4 mL/min/kg (assay: HPLC).
In the pediatric population, cyclosporine (MODIFIED) also demonstrates an increased bioavailability as compared to Sandimmune®. In 7 liver de novo transplant patients aged 1.4 to 10 years, the absolute bioavailability of cyclosporine (MODIFIED) was 43% (range 30% to 68%) and for Sandimmune® in the same individuals absolute bioavailability was 28% (range 17% to 42%).
Pediatric Pharmacokinetic Parameters (mean±SD) Patient Population Dose/day
(mg/d)Dose/weight
(mg/kg/d)AUC1
(ng·hr/mL)Cmax
(ng/mL)CL/F
(mL/min)CL/F
(mL/min/kg)Stable liver transplant2 Age 2 to 8, Dosed TID (N=9) 101±25 5.95±1.32 2163±801 629±219 285±94 16.6±4.3 Age 8 to 15, Dosed BID (N=8) 188±55 4.96±2.09 4272±1462 975±281 378±80 10.2±4.0 Stable liver transplant3 Age 3, Dosed BID (N=1) 120 8.33 5832 1050 171 11.9 Age 8 to 15, Dosed BID (N=5) 158±55 5.51±1.91 4452±2475 1013±635 328±121 11.0±1.9 Stable renal transplant3 Age 7 to 15, Dosed BID (N=5) 328±83 7.37±4.11 6922±1988 1827±487 418±143 8.7±2.9 1AUC was measured over one dosing interval
2Assay: Cyclo-trac specific monoclonal radioimmunoassay
3Assay: TDx specific monoclonal fluorescence polarization immunoassayGeriatric Population
Comparison of single dose data from both normal elderly volunteers (N=18, mean age 69 years) and elderly rheumatoid arthritis patients (N=16, mean age 68 years) to single dose data in young adult volunteers (N=16, mean age 26 years) showed no significant difference in the pharmacokinetic parameters.
-
CLINICAL TRIALS
Rheumatoid Arthritis
The effectiveness of Sandimmune® and cyclosporine (MODIFIED) in the treatment of severe rheumatoid arthritis was evaluated in 5 clinical studies involving a total of 728 cyclosporine treated patients and 273 placebo treated patients.
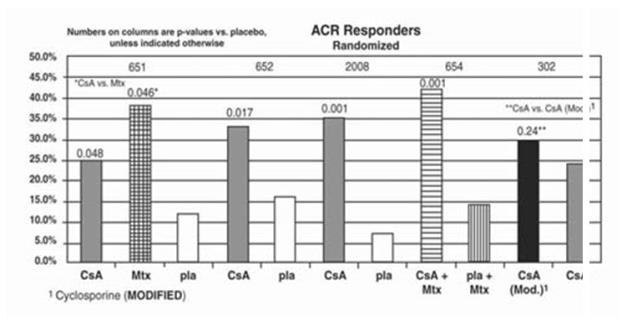
A summary of the results is presented for the "responder" rates per treatment group, with a responder being defined as a patient having completed the trial with a 20% improvement in the tender and the swollen joint count and a 20% improvement in 2 of 4 of investigator global, patient global, disability, and erythrocyte sedimentation rates (ESR) for the Studies 651 and 652 and 3 of 5 of investigator global, patient global, disability, visual analog pain, and ESR for Studies 2008, 654 and 302.
Study 651 enrolled 264 patients with active rheumatoid arthritis with at least 20 involved joints, who had failed at least one major RA drug, using a 3:3:2 randomization to one of the following three groups: (1) cyclosporine dosed at 2.5 to 5 mg/kg/day, (2) methotrexate at 7.5 to 15 mg/week, or (3) placebo. Treatment duration was 24 weeks. The mean cyclosporine dose at the last visit was 3.1 mg/kg/day. See Graph below.
Study 652 enrolled 250 patients with active RA with >6 active painful or tender joints who had failed at least one major RA drug. Patients were randomized using a 3:3:2 randomization to 1 of 3 treatment arms: (1) 1.5 to 5 mg/kg/day of cyclosporine, (2) 2.5 to 5 mg/kg/day of cyclosporine, and (3) placebo. Treatment duration was 16 weeks. The mean cyclosporine dose for group 2 at the last visit was 2.92 mg/kg/day. See Graph below.
Study 2008 enrolled 144 patients with active RA and >6 active joints who had unsuccessful treatment courses of aspirin and gold or Penicillamine. Patients were randomized to 1 of 2 treatment groups (1) cyclosporine 2.5 to 5 mg/kg/day with adjustments after the first month to achieve a target trough level and (2) placebo. Treatment duration was 24 weeks. The mean cyclosporine dose at the last visit was 3.63 mg/kg/day. See Graph below.
Study 654 enrolled 148 patients who remained with active joint counts of 6 or more despite treatment with maximally tolerated methotrexate doses for at least three months. Patients continued to take their current dose of methotrexate and were randomized to receive, in addition, one of the following medications: (1) cyclosporine 2.5 mg/kg/day with dose increases of 0.5 mg/kg/day at weeks 2 and 4 if there was no evidence of toxicity and further increases of 0.5 mg/kg/day at weeks 8 and 16 if a <30% decrease in active joint count occurred without any significant toxicity; dose decreases could be made at any time for toxicity or (2) placebo. Treatment duration was 24 weeks. The mean cyclosporine dose at the last visit was 2.8 mg/kg/day (range: 1.3 to 4.1). See Graph below.
Study 302 enrolled 299 patients with severe active RA, 99% of whom were unresponsive or intolerant to at least one prior major RA drug. Patients were randomized to 1 of 2 treatment groups (1) cyclosporine (MODIFIED) and (2) Sandimmune®, both of which were started at 2.5 mg/kg/day and increased after 4 weeks for inefficacy in increments of 0.5 mg/kg/day to a maximum of 5 mg/kg/day and decreased at any time for toxicity. Treatment duration was 24 weeks. The mean cyclosporine dose at the last visit was 2.91 mg/kg/day (range: 0.72 to 5.17) for cyclosporine (MODIFIED) and 3.27 mg/kg/day (range: 0.73 to 5.68) for Sandimmune®. See Graph below.
-
INDICATIONS AND USAGE
Kidney, Liver, and Heart Transplantation
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is indicated for the prophylaxis of organ rejection in kidney, liver, and heart allogeneic transplants. Cyclosporine (MODIFIED) has been used in combination with azathioprine and corticosteroids.
Rheumatoid Arthritis
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is indicated for the treatment of patients with severe active, rheumatoid arthritis where the disease has not adequately responded to methotrexate. Gengraf® can be used in combination with methotrexate in rheumatoid arthritis patients who do not respond adequately to methotrexate alone.
Psoriasis
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is indicated for the treatment of adult, nonimmunocompromised patients with severe (i.e., extensive and/or disabling), recalcitrant, plaque psoriasis who have failed to respond to at least one systemic therapy (e.g., PUVA, retinoids, or methotrexate) or in patients for whom other systemic therapies are contraindicated, or cannot be tolerated.
While rebound rarely occurs, most patients will experience relapse with Gengraf® as with other therapies upon cessation of treatment.
-
CONTRAINDICATIONS
General
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is contraindicated in patients with a hypersensitivity to cyclosporine or to any of the ingredients of the formulation.
Rheumatoid Arthritis
Rheumatoid arthritis patients with abnormal renal function, uncontrolled hypertension, or malignancies should not receive Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]).
Psoriasis
Psoriasis patients who are treated with Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) should not receive concomitant PUVA or UVB therapy, methotrexate or other immunosuppressive agents, coal tar or radiation therapy. Psoriasis patients with abnormal renal function, uncontrolled hypertension, or malignancies should not receive Gengraf®.
-
WARNINGS
(See also BOXED WARNING)
All Patients
Cyclosporine, the active ingredient of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]), can cause nephrotoxicity and hepatotoxicity. The risk increases with increasing doses of cyclosporine. Renal dysfunction including structural kidney damage is a potential consequence of Gengraf® and therefore renal function must be monitored during therapy. Care should be taken in using cyclosporine with nephrotoxic drugs. (See PRECAUTIONS)
Patients receiving Gengraf® require frequent monitoring of serum creatinine. (See Special Monitoring under DOSAGE AND ADMINISTRATION) Elderly patients should be monitored with particular care, since decreases in renal function also occur with age. If patients are not properly monitored and doses are not properly adjusted, cyclosporine therapy can be associated with the occurrence of structural kidney damage and persistent renal dysfunction.
An increase in serum creatinine and BUN may occur during Gengraf® therapy and reflect a reduction in the glomerular filtration rate. Impaired renal function at any time requires close monitoring, and frequent dosage adjustment may be indicated. The frequency and severity of serum creatinine elevations increase with dose and duration of cyclosporine therapy. These elevations are likely to become more pronounced without dose reduction or discontinuation.
Because Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is not bioequivalent to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP), conversion from Gengraf® to Sandimmune® using a 1:1 ratio (mg/kg/day) may result in lower cyclosporine blood concentrations. Conversion from Gengraf® to Sandimmune® should be made with increased monitoring to avoid the potential of underdosing.
Kidney, Liver, and Heart Transplant
Nephrotoxicity
Cyclosporine, the active ingredient of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]), can cause nephrotoxicity and hepatotoxicity when used in high doses. It is not unusual for serum creatinine and BUN levels to be elevated during cyclosporine therapy. These elevations in renal transplant patients do not necessarily indicate rejection, and each patient must be fully evaluated before dosage adjustment is initiated.
Based on the historical Sandimmune® experience with oral solution, nephrotoxicity associated with cyclosporine had been noted in 25% of cases of renal transplantation, 38% of cases of cardiac transplantation, and 37% of cases of liver transplantation. Mild nephrotoxicity was generally noted 2 to 3 months after renal transplant and consisted of an arrest in the fall of the pre-operative elevations of BUN and creatinine at a range of 35 to 45 mg/dL and 2.0 to 2.5 mg/dL respectively. These elevations were often responsive to cyclosporine dosage reduction.
More overt nephrotoxicity was seen early after transplantation and was characterized by a rapidly rising BUN and creatinine. Since these events are similar to renal rejection episodes, care must be taken to differentiate between them. This form of nephrotoxicity is usually responsive to cyclosporine dosage reduction.
Although specific diagnostic criteria which reliably differentiate renal graft rejection from drug toxicity have not been found, a number of parameters have been significantly associated with one or the other. It should be noted however, that up to 20% of patients may have simultaneous nephrotoxicity and rejection.
Nephrotoxicity vs. Rejection Parameter Nephrotoxicity Rejection History Donor >50 years old or hypotensive
Prolonged kidney preservation
Prolonged anastomosis time
Concomitant nephrotoxic drugsAnti-donor immune response
Retransplant patientClinical Often >6 weeks postopb
Prolonged initial nonfunction
(acute tubular necrosis)Often <4 weeks postopb
Fever >37.5°C
Weight gain >0.5 kg
Graft swelling and tenderness
Decrease in daily urine volume
>500 mL (or 50%)Laboratory CyA serum trough level >200 ng/mL
Gradual rise in Cr (<0.15 mg/dL/day)a
Cr plateau <25% above baseline
BUN/Cr ≥20CyA serum trough level <150 ng/mL
Rapid rise in Cr (>0.3 mg/dL/day)a
Cr >25% above baseline
BUN/Cr <20Biopsy Arteriolopathy (medial hypertrophya, hyalinosis, nodular deposits, intimal thickening, endothelial vacuolization, progressive scarring)
Tubular atrophy, isometric vacuolization, isolated calcifications
Minimal edema
Mild focal infiltratesc
Diffuse interstitial fibrosis, often striped formEndovasculitisc (proliferationa, intimal arteritisb, necrosis, sclerosis)
Tubulitis with RBCb and WBCb casts, some irregular vacuolization
Interstitial edemac and hemorrhageb
Diffuse moderate to severe mononuclear infiltratesd
Glomerulitis (mononuclear cells)cAspiration Cytology CyA deposits in tubular and endothelial cells
Fine isometric vacuolization of tubular cellsInflammatory infiltrate with mononuclear phagocytes, macrophages, lymphoblastoid cells, and activated T-cells
These strongly express HLA-DR antigensUrine Cytology Tubular cells with vacuolization and granularization Degenerative tubular cells, plasma cells, and lymphocyturia >20% of sediment Manometry
UltrasonographyIntracapsular pressure <40 mm Hgb
Unchanged graft cross sectional areaIntracapsular pressure >40 mm Hgb
Increase in graft cross sectional area
AP diameter ≥ Transverse diameterMagnetic Resonance Imagery Normal appearance Loss of distinct corticomedullary junction, swelling image intensity of parenchyma approaching that of psoas, loss of hilar fat Radionuclide Scan Normal or generally decreased perfusion
Decrease in tubular function
(131 I-hippuran) > decrease in perfusion
(99m Tc DTPA)Patchy arterial flow
Decrease in perfusion > decrease in tubular function
Increased uptake of Indium 111 labeled platelets or Tc-99m in colloidTherapy Responds to decreased cyclosporine Responds to increased steroids or antilymphocyte globulin ap <0.05, bp <0.01, cp <0.001, dp <0.0001 A form of a cyclosporine-associated nephropathy is characterized by serial deterioration in renal function and morphologic changes in the kidneys. From 5% to 15% of transplant recipients who have received cyclosporine will fail to show a reduction in rising serum creatinine despite a decrease or discontinuation of cyclosporine therapy. Renal biopsies from these patients will demonstrate one or several of the following alterations: tubular vacuolization, tubular microcalcifications, peritubular capillary congestion, arteriolopathy, and a striped form of interstitial fibrosis with tubular atrophy. Though none of these morphologic changes is entirely specific, a diagnosis of cyclosporine-associated structural nephrotoxicity requires evidence of these findings.
When considering the development of cyclosporine-associated nephropathy, it is noteworthy that several authors have reported an association between the appearance of interstitial fibrosis and higher cumulative doses or persistently high circulating trough concentrations of cyclosporine. This is particularly true during the first 6 post-transplant months when the dosage tends to be highest and when, in kidney recipients, the organ appears to be most vulnerable to the toxic effects of cyclosporine. Among other contributing factors to the development of interstitial fibrosis in these patients are prolonged perfusion time, warm ischemia time, as well as episodes of acute toxicity, and acute and chronic rejection. The reversibility of interstitial fibrosis and its correlation to renal function have not yet been determined. Reversibility of arteriolopathy has been reported after stopping cyclosporine or lowering the dosage.
Impaired renal function at any time requires close monitoring, and frequent dosage adjustment may be indicated.
In the event of severe and unremitting rejection, when rescue therapy with pulse steroids and monoclonal antibodies fail to reverse the rejection episode, it may be preferable to switch to alternative immunosuppressive therapy rather than increase the Gengraf® dose to excessive blood concentrations.
Due to the potential for additive or synergistic impairment of renal function, caution should be exercised when coadministering Gengraf® with other drugs that may impair renal function. (See PRECAUTIONS, Drug Interactions)
Thrombotic Microangiopathy
Occasionally patients have developed a syndrome of thrombocytopenia and microangiopathic hemolytic anemia which may result in graft failure. The vasculopathy can occur in the absence of rejection and is accompanied by avid platelet consumption within the graft as demonstrated by Indium 111 labeled platelet studies. Neither the pathogenesis nor the management of this syndrome is clear. Though resolution has occurred after reduction or discontinuation of cyclosporine and 1) administration of streptokinase and heparin or 2) plasmapheresis, this appears to depend upon early detection with Indium 111 labeled platelet scans. (See ADVERSE REACTIONS)
Hyperkalemia
Significant hyperkalemia (sometimes associated with hyperchloremic metabolic acidosis) and hyperuricemia have been seen occasionally in individual patients.
Hepatotoxicity
Cases of hepatotoxicity and liver injury including cholestasis, jaundice, hepatitis, and liver failure have been reported in patients treated with cyclosporine. Most reports included patients with significant co-morbidities, underlying conditions and other confounding factors including infectious complications and comedications with hepatotoxic potential. In some cases, mainly in transplant patients, fatal outcomes have been reported. (See ADVERSE REACTIONS, Postmarketing Experience, Kidney, Liver and Heart Transplantation)
Hepatotoxicity, usually manifested by elevations in hepatic enzymes and bilirubin, was reported in patients treated with cyclosporine in clinical trials: 4% in renal transplantation, 7% in cardiac transplantation, and 4% in liver transplantation. This was usually noted during the first month of therapy when high doses of cyclosporine were used. The chemistry elevations usually decreased with a reduction in dosage.
Malignancies
As in patients receiving other immunosuppressants, those patients receiving cyclosporine are at increased risk for development of lymphomas and other malignancies, particularly those of the skin. Patients taking cyclosporine should be warned to avoid excess ultraviolet light exposure. The increased risk appears related to the intensity and duration of immunosuppression rather than to the use of specific agents. Because of the danger of oversuppression of the immune system resulting in increased risk of infection or malignancy, a treatment regimen containing multiple immunosuppressants should be used with caution. Some malignancies may be fatal. Transplant patients receiving cyclosporine are at increased risk for serious infection with fatal outcome.
Serious Infections
Patients receiving immunosuppressants, including Gengraf®, are at increased risk of developing bacterial, viral, fungal, and protozoal infections, including opportunistic infections. These infections may lead to serious, including fatal, outcomes. (See BOXED WARNING and ADVERSE REACTIONS)
Polyoma Virus Infections
Patients receiving immunosuppressants, including Gengraf®, are at increased risk for opportunistic infections, including polyoma virus infections. Polyoma virus infections in transplant patients may have serious, and sometimes, fatal outcomes. These include cases of JC virus-associated progressive multifocal leukoencephalopathy (PML), and polyoma virus-associated nephropathy (PVAN), especially due to BK virus infection, which have been observed in patients receiving cyclosporine. PVAN is associated with serious outcomes, including deteriorating renal function and renal graft loss, (See ADVERSE REACTIONS, Postmarketing Experience, Kidney, Liver and Heart Transplantation). Patient monitoring may help detect patients at risk for PVAN.
Cases of PML have been reported in patients treated with Gengraf®. PML, which is sometimes fatal, commonly presents with hemiparesis, apathy, confusion, cognitive deficiencies and ataxia. Risk factors for PML include treatment with immunosuppressant therapies and impairment of immune function. In immunosuppressed patients, physicians should consider PML in the differential diagnosis in patients reporting neurological symptoms and consultation with a neurologist should be considered as clinically indicated.
Consideration should be given to reducing the total immunosuppression in transplant patients who develop PML or PVAN. However, reduced immunosuppression may place the graft at risk.
Neurotoxicity
There have been reports of convulsions in adult and pediatric patients receiving cyclosporine, particularly in combination with high dose methylprednisolone.
Encephalopathy, including Posterior Reversible Encephalopathy Syndrome (PRES), has been described both in post-marketing reports and in the literature. Manifestations include impaired consciousness, convulsions, visual disturbances (including blindness), loss of motor function, movement disorders and psychiatric disturbances. In many cases, changes in the white matter have been detected using imaging techniques and pathologic specimens. Predisposing factors such as hypertension, hypomagnesemia, hypocholesterolemia, high-dose corticosteroids, high cyclosporine blood concentrations, and graft-versus-host disease have been noted in many but not all of the reported cases. The changes in most cases have been reversible upon discontinuation of cyclosporine, and in some cases improvement was noted after reduction of dose. It appears that patients receiving liver transplant are more susceptible to encephalopathy than those receiving kidney transplant. Another rare manifestation of cyclosporine-induced neurotoxicity, occurring in transplant patients more frequently than in other indications, is optic disc edema including papilloedema, with possible visual impairment, secondary to benign intracranial hypertension.
Care should be taken in using cyclosporine with nephrotoxic drugs. (See PRECAUTIONS)
Rheumatoid Arthritis
Cyclosporine nephropathy was detected in renal biopsies of 6 out of 60 (10%) rheumatoid arthritis patients after the average treatment duration of 19 months. Only one patient, out of these 6 patients, was treated with a dose ≤4 mg/kg/day. Serum creatinine improved in all but one patient after discontinuation of cyclosporine. The "maximal creatinine increase" appears to be a factor in predicting cyclosporine nephropathy.
There is a potential, as with other immunosuppressive agents, for an increase in the occurrence of malignant lymphomas with cyclosporine. It is not clear whether the risk with cyclosporine is greater than that in rheumatoid arthritis patients or in rheumatoid arthritis patients on cytotoxic treatment for this indication. Five cases of lymphoma were detected: four in a survey of approximately 2,300 patients treated with cyclosporine for rheumatoid arthritis, and another case of lymphoma was reported in a clinical trial. Although other tumors (12 skin cancers, 24 solid tumors of diverse types, and 1 multiple myeloma) were also reported in this survey, epidemiologic analyses did not support a relationship to cyclosporine other than for malignant lymphomas.
Patients should be thoroughly evaluated before and during Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) treatment for the development of malignancies. Moreover, use of Gengraf® therapy with other immunosuppressive agents may induce an excessive immunosuppression which is known to increase the risk of malignancy.
Psoriasis
(See also BOXED WARNING for Psoriasis)
Since cyclosporine is a potent immunosuppressive agent with a number of potentially serious side effects, the risks and benefits of using Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) should be considered before treatment of patients with psoriasis. Cyclosporine, the active ingredient in Gengraf®, can cause nephrotoxicity and hypertension (See PRECAUTIONS) and the risk increases with increasing dose and duration of therapy. Patients who may be at increased risk such as those with abnormal renal function, uncontrolled hypertension or malignancies, should not receive Gengraf®.
Renal dysfunction is a potential consequence of Gengraf®, therefore renal function must be monitored during therapy.
Patients receiving Gengraf® require frequent monitoring of serum creatinine. (See Special Monitoring under DOSAGE AND ADMINISTRATION) Elderly patients should be monitored with particular care, since decreases in renal function also occur with age. If patients are not properly monitored and doses are not properly adjusted, cyclosporine therapy can cause structural kidney damage and persistent renal dysfunction.
An increase in serum creatinine and BUN may occur during Gengraf® therapy and reflects a reduction in the glomerular filtration rate.
Kidney biopsies from 86 psoriasis patients treated for a mean duration of 23 months with 1.2 to 7.6 mg/kg/day of cyclosporine showed evidence of cyclosporine nephropathy in 18/86 (21%) of the patients. The pathology consisted of renal tubular atrophy and interstitial fibrosis. On repeat biopsy of 13 of these patients maintained on various dosages of cyclosporine for a mean of 2 additional years, the number with cyclosporine induced nephropathy rose to 26/86 (30%). The majority of patients (19/26) were on a dose of ≥5.0 mg/kg/day (the highest recommended dose is 4 mg/kg/day). The patients were also on cyclosporine for greater than 15 months (18/26) and/or had a clinically significant increase in serum creatinine for greater than 1 month (21/26). Creatinine levels returned to normal range in 7 of 11 patients in whom cyclosporine therapy was discontinued.
There is an increased risk for the development of skin and lymphoproliferative malignancies in cyclosporine-treated psoriasis patients. The relative risk of malignancies is comparable to that observed in psoriasis patients treated with other immunosuppressive agents.
Tumors were reported in 32 (2.2%) of 1439 psoriasis patients treated with cyclosporine worldwide from clinical trials. Additional tumors have been reported in 7 patients in cyclosporine postmarketing experience. Skin malignancies were reported in 16 (1.1%) of these patients; all but 2 of them had previously received PUVA therapy. Methotrexate was received by 7 patients. UVB and coal tar had been used by 2 and 3 patients, respectively. Seven patients had either a history of previous skin cancer or a potentially predisposing lesion was present prior to cyclosporine exposure. Of the 16 patients with skin cancer, 11 patients had 18 squamous cell carcinomas and 7 patients had 10 basal cell carcinomas.
There were two lymphoproliferative malignancies; one case of non-Hodgkin's lymphoma which required chemotherapy, and one case of mycosis fungoides which regressed spontaneously upon discontinuation of cyclosporine. There were four cases of benign lymphocytic infiltration: 3 regressed spontaneously upon discontinuation of cyclosporine, while the fourth regressed despite continuation of the drug. The remainder of the malignancies, 13 cases (0.9%), involved various organs.
Patients should not be treated concurrently with cyclosporine and PUVA or UVB, other radiation therapy, or other immunosuppressive agents, because of the possibility of excessive immunosuppression and the subsequent risk of malignancies. (See CONTRAINDICATIONS) Patients should also be warned to protect themselves appropriately when in the sun, and to avoid excessive sun exposure. Patients should be thoroughly evaluated before and during treatment for the presence of malignancies remembering that malignant lesions may be hidden by psoriatic plaques. Skin lesions not typical of psoriasis should be biopsied before starting treatment. Patients should be treated with Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) only after complete resolution of suspicious lesions, and only if there are no other treatment options. (See Special Monitoring for Psoriasis Patients)
Special Excipients
Alcohol (ethanol)
The alcohol content (See DESCRIPTION) of Gengraf® should be taken into account when given to patients in whom alcohol intake should be avoided or minimized, e.g., pregnant or breastfeeding women, in patients presenting with liver disease or epilepsy, in alcoholic patients, or pediatric patients. For an adult weighing 70 kg, the maximum daily oral dose would deliver about 1 gram of alcohol which is approximately 6% of the amount of alcohol contained in a standard drink.
-
PRECAUTIONS
General
Hypertension
Cyclosporine is the active ingredient of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]). Hypertension is a common side effect of cyclosporine therapy which may persist. (See ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION for monitoring recommendations) Mild or moderate hypertension is encountered more frequently than severe hypertension and the incidence decreases over time. In recipients of kidney, liver, and heart allografts treated with cyclosporine, antihypertensive therapy may be required. (See Special Monitoring of Rheumatoid Arthritis and Psoriasis Patients) However, since cyclosporine may cause hyperkalemia, potassium-sparing diuretics should not be used. While calcium antagonists can be effective agents in treating cyclosporine-associated hypertension, they can interfere with cyclosporine metabolism. (See Drug Interactions)
Vaccination
During treatment with cyclosporine, vaccination may be less effective; and the use of live attenuated vaccines should be avoided.
Special Monitoring of Rheumatoid Arthritis Patients
Before initiating treatment, a careful physical examination, including blood pressure measurements (on at least two occasions) and two creatinine levels to estimate baseline should be performed. Blood pressure and serum creatinine should be evaluated every 2 weeks during the initial 3 months and then monthly if the patient is stable. It is advisable to monitor serum creatinine and blood pressure always after an increase of the dose of nonsteroidal anti-inflammatory drugs (NSAIDs) and after initiation of new NSAID therapy during Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) treatment. If coadministered with methotrexate, CBC and liver function tests are recommended to be monitored monthly. (See also PRECAUTIONS, General, Hypertension)
In patients who are receiving cyclosporine, the dose of Gengraf® should be decreased by 25% to 50% if hypertension occurs. If hypertension persists, the dose of Gengraf® should be further reduced or blood pressure should be controlled with antihypertensive agents. In most cases, blood pressure has returned to baseline when cyclosporine was discontinued.
In placebo-controlled trials of rheumatoid arthritis patients, systolic hypertension (defined as an occurrence of two systolic blood pressure readings >140 mmHg) and diastolic hypertension (defined as two diastolic blood pressure readings >90 mmHg) occurred in 33% and 19% of patients treated with cyclosporine, respectively. The corresponding placebo rates were 22% and 8%.
Special Monitoring for Psoriasis Patients
Before initiating treatment, a careful dermatological and physical examination, including blood pressure measurements (on at least two occasions) should be performed. Since Gengraf® (cyclosporine capsules, USP [MODIFIED]) is an immunosuppressive agent, patients should be evaluated for the presence of occult infection on their first physical examination and for the presence of tumors initially, and throughout treatment with Gengraf®. Skin lesions not typical for psoriasis should be biopsied before starting Gengraf®. Patients with malignant or premalignant changes of the skin should be treated with Gengraf® only after appropriate treatment of such lesions and if no other treatment option exists.
Baseline laboratories should include serum creatinine (on two occasions), BUN, CBC, serum magnesium, potassium, uric acid, and lipids.
The risk of cyclosporine nephropathy is reduced when the starting dose is low (2.5 mg/kg/day), the maximum dose does not exceed 4.0 mg/kg/day, serum creatinine is monitored regularly while cyclosporine is administered, and the dose of Gengraf® is decreased when the rise in creatinine is greater than or equal to 25% above the patient's pretreatment level. The increase in creatinine is generally reversible upon timely decrease of the dose of Gengraf® or its discontinuation.
Serum creatinine and BUN should be evaluated every 2 weeks during the initial 3 months of therapy and then monthly if the patient is stable. If the serum creatinine is greater than or equal to 25% above the patient's pretreatment level, serum creatinine should be repeated within two weeks. If the change in serum creatinine remains greater than or equal to 25% above baseline, Gengraf® should be reduced by 25% to 50%. If at any time the serum creatinine increases by greater than or equal to 50% above pretreatment level, Gengraf® should be reduced by 25% to 50%. Gengraf® should be discontinued if reversibility (within 25% of baseline) of serum creatinine is not achievable after two dosage modifications. It is advisable to monitor serum creatinine after an increase of the dose of nonsteroidal anti-inflammatory drug and after initiation of new nonsteroidal anti-inflammatory therapy during Gengraf® treatment.
Blood pressure should be evaluated every 2 weeks during the initial 3 months of therapy and then monthly if the patient is stable, or more frequently when dosage adjustments are made. Patients without a history of previous hypertension before initiation of treatment with Gengraf®, should have the drug reduced by 25% to 50% if found to have sustained hypertension. If the patient continues to be hypertensive despite multiple reductions of Gengraf®, then Gengraf® should be discontinued. For patients with treated hypertension, before the initiation of Gengraf® therapy, their medication should be adjusted to control hypertension while on Gengraf®. Gengraf® should be discontinued if a change in hypertension management is not effective or tolerable.
CBC, uric acid, potassium, lipids, and magnesium should also be monitored every 2 weeks for the first 3 months of therapy, and then monthly if the patient is stable or more frequently when dosage adjustments are made. Gengraf® dosage should be reduced by 25% to 50% for any abnormality of clinical concern.
In controlled trials of cyclosporine in psoriasis patients, cyclosporine blood concentrations did not correlate well with either improvement or with side effects such as renal dysfunction.
Information for Patients: Patients should be advised that any change of cyclosporine formulation should be made cautiously and only under physician supervision because it may result in the need for a change in dosage.
Patients should be informed of the necessity of repeated laboratory tests while they are receiving cyclosporine. Patients should be advised of the potential risks during pregnancy and informed of the increased risk of neoplasia. Patients should also be informed of the risk of hypertension and renal dysfunction.
Patients should be advised that during treatment with cyclosporine, vaccination may be less effective and the use of live attenuated vaccines should be avoided.
Patients should be advised to take Gengraf® on a consistent schedule with regard to time of day and relation to meals. Grapefruit and grapefruit juice affect metabolism, increasing blood concentration of cyclosporine, thus should be avoided.
Laboratory Tests
In all patients treated with cyclosporine, renal and liver functions should be assessed repeatedly by measurement of serum creatinine, BUN, serum bilirubin, and liver enzymes. Serum lipids, magnesium, and potassium should also be monitored. Cyclosporine blood concentrations should be routinely monitored in transplant patients (See DOSAGE AND ADMINISTRATION, Blood Concentration Monitoring in Transplant Patients), and periodically monitored in rheumatoid arthritis patients.
Drug Interactions
A. Effect of Drugs and Other Agents on Cyclosporine Pharmacokinetics and/or Safety
All of the individual drugs cited below are well substantiated to interact with cyclosporine. In addition, concomitant use of NSAIDs with cyclosporine, particularly in the setting of dehydration, may potentiate renal dysfunction. Caution should be exercised when using other drugs which are known to impair renal function. (See WARNINGS, Nephrotoxicity)
Drugs That May Potentiate Renal Dysfunction
Antibiotics Antineoplastics Anti-inflammatory Drugs Gastrointestinal Agents ciprofloxacin melphalan azapropazon cimetidine gentamicin colchicine ranitidine tobramycin Antifungals diclofenac vancomycin amphotericin B naproxen Immunosuppressives trimethoprim with sulfamethoxazole ketoconazole sulindac tacrolimus Other Drugs fibric acid derivatives
(e.g.,bezafibrate, fenofibrate)methotrexate During the concomitant use of a drug that may exhibit additive or synergistic renal impairment with cyclosporine, close monitoring of renal function (in particular serum creatinine) should be performed. If a significant impairment of renal function occurs, the dosage of the coadministered drug should be reduced or an alternative treatment considered.
Cyclosporine is extensively metabolized by CYP 3A isoenzymes, in particular CYP3A4, and is a substrate of the multidrug efflux transporter P-glycoprotein. Various agents are known to either increase or decrease plasma or whole blood concentrations of cyclosporine usually by inhibition or induction of CYP3A4 or P-glycoprotein transporter or both. Compounds that decrease cyclosporine absorption such as orlistat should be avoided. Appropriate Gengraf® dosage adjustment to achieve the desired cyclosporine concentrations is essential when drugs that significantly alter cyclosporine concentrations are used concomitantly. (See Blood Concentration Monitoring)
1. Drugs That Increase Cyclosporine Concentrations
Calcium Channel Blockers Antifungals Antibiotics Glucocorticoids Other Drugs Diltiazem fluconazole azithromycin methylprednisolone Allopurinol nicardipine itraconazole clarithromycin Amiodarone verapamil ketoconazole erythromycin Bromocriptine voriconazole quinupristin/ dalfopristin colchicine danazol imatinib metoclopramide nefazodone oral contraceptives HIV Protease inhibitors
The HIV protease inhibitors (e.g., indinavir, nelfinavir, ritonavir, and saquinavir) are known to inhibit cytochrome P-450 3A and thus could potentially increase the concentrations of cyclosporine, however no formal studies of the interaction are available. Care should be exercised when these drugs are administered concomitantly.
Grapefruit juice
Grapefruit and grapefruit juice affect metabolism, increasing blood concentrations of cyclosporine, thus should be avoided.
2. Drugs/Dietary Supplements That Decrease Cyclosporine Concentrations
Antibiotics Anticonvulsants Other Drugs/Dietary Supplements nafcillin carbamazepine bosentan rifampin oxcarbazepine octreotide Phenobarbital orlistat Phenytoin sulfinpyrazone St. John's Wort terbinafine ticlopidine Bosentan
Coadministration of bosentan (250 to 1000 mg every 12 hours based on tolerability) and cyclosporine (300 mg every 12 hours for 2 days then dosing to achieve a Cmin of 200 to 250 ng/mL) for 7 days in healthy subjects resulted in decreases in the cyclosporine mean dose-normalized AUC, Cmax, and trough concentration of approximately 50%, 30%, and 60%, respectively, compared to when cyclosporine was given alone (See also Effect of Cyclosporine on the Pharmacokinetics and/or Safety of Other Drugs or Agents). Coadministration of cyclosporine with bosentan should be avoided.
Boceprevir
Coadministration of boceprevir (800 mg three times daily for 7 days) and cyclosporine (100 mg single dose) in healthy subjects resulted in increases in the mean AUC and Cmax of cyclosporine approximately 2.7-fold and 2-fold, respectively, compared to when cyclosporine was given alone.
Telaprevir
Coadministration of telaprevir (750 mg every 8 hours for 11 days) with cyclosporine (10 mg on day 8) in healthy subjects resulted in increases in the mean dose-normalized AUC and Cmax of cyclosporine approximately 4.5-fold and 1.3-fold, respectively, compared to when cyclosporine (100 mg single dose) was given alone.
St. John's Wort
There have been reports of a serious drug interaction between cyclosporine and the herbal dietary supplement St. John's Wort. This interaction has been reported to produce a marked reduction in the blood concentrations of cyclosporine, resulting in subtherapeutic levels, rejection of transplanted organs, and graft loss.
Rifabutin
Rifabutin is known to increase the metabolism of other drugs metabolized by the cytochrome P-450 system. The interaction between rifabutin and cyclosporine has not been studied. Care should be exercised when these two drugs are administered concomitantly.
B. Effect of Cyclosporine on the Pharmacokinetics and/or Safety of Other Drugs or Agents
Cyclosporine is an inhibitor of CYP3A4 and of multiple drug efflux transporters (e.g., P-glycoprotein) and may increase plasma concentrations of comedications that are substrates of CYP3A4, P-glycoprotein or organic anion transporter proteins.
Cyclosporine may reduce the clearance of digoxin, colchicine, prednisolone, HMG-CoA reductase inhibitors (statins), and, aliskiren, bosentan, dabigatran, repaglinide, NSAIDs, sirolimus, etoposide, and other drugs.
See the full prescribing information of the other drug for further information and specific recommendations. The decision on coadministration of cyclosporine with other drugs or agents should be made by the healthcare provider following the careful assessment of benefits and risks.
Digoxin
Severe digitalis toxicity has been seen within days of starting cyclosporine in several patients taking digoxin. If digoxin is used concurrently with cyclosporine, serum digoxin concentrations should be monitored.
Colchicine
There are reports on the potential of cyclosporine to enhance the toxic effects of colchicine such as myopathy and neuropathy, especially in patients with renal dysfunction. Concomitant administration of cyclosporine and colchicine results in significant increases in colchicine plasma concentrations. If colchicine is used concurrently with cyclosporine, a reduction in the dosage of colchicine is recommended.
HMG-CoA reductase inhibitors (statins)
Literature and postmarketing cases of myotoxicity, including muscle pain and weakness, myositis, and rhabdomyolysis, have been reported with concomitant administration of cyclosporine with lovastatin, simvastatin, atorvastatin, pravastatin, and, rarely fluvastatin. When concurrently administered with cyclosporine, the dosage of these statins should be reduced according to label recommendations. Statin therapy needs to be temporarily withheld or discontinued in patients with signs and symptoms of myopathy or those with risk factors predisposing to severe renal injury, including renal failure, secondary to rhabdomyolysis.
Repaglinide
Cyclosporine may increase the plasma concentrations of repaglinide and thereby increase the risk of hypoglycemia. In 12 healthy male subjects who received two doses of 100 mg cyclosporine capsule orally 12 hours apart with a single dose of 0.25 mg repaglinide tablet (one-half of a 0.5mg tablet) orally 13 hours after the cyclosporine initial dose, the repaglinide mean Cmax and AUC were increased 1.8 fold (range: 0.6 to 3.7 fold) and 2.4 fold (range 1.2 to 5.3 fold), respectively. Close monitoring of blood glucose level is advisable for a patient taking cyclosporine and repaglinide concomitantly.
Ambrisentan
Coadministration of ambrisentan (5 mg daily) and cyclosporine (100 to 150 mg twice daily initially, then dosing to achieve Cmin 150 to 200 ng/mL) for 8 days in healthy subjects resulted in mean increases in ambrisentan AUC and Cmax of approximately 2-fold and 1.5-fold, respectively, compared to ambrisentan alone. When coadministering ambrisentan with cyclosporine, the ambrisentan dose should not be titrated to the recommended maximum daily dose.
Anthracycline antibiotics
High doses of cyclosporine (e.g., at starting intravenous dose of 16 mg/kg/day) may increase the exposure to anthracycline antibiotics (e.g., doxorubicin, mitoxantrone, daunorubicin) in cancer patients.
Aliskiren
Cyclosporine alters the pharmacokinetics of aliskiren, a substrate of P-glycoprotein and CYP3A4. In 14 healthy subjects who received concomitantly single doses of cyclosporine (200 mg) and reduced dose aliskiren (75 mg), the mean Cmax of aliskiren was increased by approximately 2.5-fold (90% CI: 1.96 to 3.17) and the mean AUC by approximately 4.3 fold (90% CI: 3.52 to 5.21), compared to when these subjects received aliskiren alone. The concomitant administration of aliskiren with cyclosporine prolonged the median aliskiren elimination half-life (26 hours versus 43 to 45 hours) and the Tmax (0.5 hours versus 1.5 to 2.0 hours). The mean AUC and Cmax of cyclosporine were comparable to reported literature values. Coadministration of cyclosporine and aliskiren in these subjects also resulted in an increase in the number and/or intensity of adverse events, mainly headache, hot flush, nausea, vomiting, and somnolence. The coadministration of cyclosporine with aliskiren is not recommended.
Bosentan
In healthy subjects, coadministration of bosentan and cyclosporine resulted in time-dependent mean increases in dose-normalized bosentan trough concentrations (i.e., approximately 21-fold on day 1 and 2-fold on day 8 (steady state)) compared to when bosentan was given alone as a single dose on day 1. (See also Effect of Drugs and Other Agents on Cyclosporine Pharmacokinetics and/or Safety) Coadministration of cyclosporine with bosentan should be avoided.
Dabigatran
The effect of cyclosporine on dabigatran concentrations had not been formally studied. Concomitant administration of dabigatran and cyclosporine may result in increased plasma dabigatran concentrations due to the P-gp inhibitory activity of cyclosporine. Coadministration of cyclosporine with dabigatran should be avoided.
Potassium-Sparing Diuretics
Cyclosporine should not be used with potassium-sparing diuretics because hyperkalemia can occur. Caution is also required when cyclosporine is coadministered with potassium sparing drugs (e.g., angiotensin converting enzyme inhibitors, angiotensin II receptor antagonists), potassium-containing drugs as well as in patients on a potassium rich diet. Control of potassium levels in these situations is advisable.
Nonsteroidal Anti-inflammatory Drug (NSAID) Interactions
Clinical status and serum creatinine should be closely monitored when cyclosporine is used with NSAIDs in rheumatoid arthritis patients. (See WARNINGS)
Pharmacodynamic interactions have been reported to occur between cyclosporine and both naproxen and sulindac, in that concomitant use is associated with additive decreases in renal function, as determined by 99mTc-diethylenetriaminepentaacetic acid (DTPA) and (p-aminohippuric acid) PAH clearances. Although concomitant administration of diclofenac does not affect blood concentrations of cyclosporine, it has been associated with approximate doubling of diclofenac blood concentrations and occasional reports of reversible decreases in renal function. Consequently, the dose of diclofenac should be in the lower end of the therapeutic range.
Methotrexate Interaction
Preliminary data indicate that when methotrexate and cyclosporine were coadministered to rheumatoid arthritis patients (N=20), methotrexate concentrations (AUCs) were increased approximately 30% and the concentrations (AUCs) of its metabolite, 7-hydroxy methotrexate, were decreased by approximately 80%. The clinical significance of this interaction is not known. Cyclosporine concentrations do not appear to have been altered (N=6).
Sirolimus
Elevations in serum creatinine were observed in studies using sirolimus in combination with full-dose cyclosporine. This effect is often reversible with cyclosporine dose reduction. Simultaneous coadministration of cyclosporine significantly increases blood levels of sirolimus. To minimize increases in sirolimus concentrations, it is recommended that sirolimus be given 4 hours after cyclosporine administration.
Nifedipine
Frequent gingival hyperplasia when nifedipine is given concurrently with cyclosporine has been reported. The concomitant use of nifedipine should be avoided in patients in whom gingival hyperplasia develops as a side effect of cyclosporine.
Methylprednisolone
Convulsions when high dose methylprednisolone is given concurrently with cyclosporine have been reported.
Other Immunosuppressive Drugs and Agents
Psoriasis patients receiving other immunosuppressive agents or radiation therapy (including PUVA and UVB) should not receive concurrent cyclosporine because of the possibility of excessive immunosuppression.
C. Effect of Cyclosporine on the Efficacy of Live Vaccines
During treatment with cyclosporine, vaccination may be less effective. The use of live vaccines should be avoided.
For additional information on Cyclosporine Drug Interactions please contact AbbVie Inc. Medical Information Department at 1-800-633-9110.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenicity studies were carried out in male and female rats and mice. In the 78-week mouse study, evidence of a statistically significant trend was found for lymphocytic lymphomas in females, and the incidence of hepatocellular carcinomas in mid-dose males significantly exceeded the control value. In the 24-month rat study, pancreatic islet cell adenomas significantly exceeded the control rate in the low dose level. Doses used in the mouse and rat studies were 0.01 to 0.16 times the clinical maintenance dose (6 mg/kg). The hepatocellular carcinomas and pancreatic islet cell adenomas were not dose related. Published reports indicate that co-treatment of hairless mice with UV irradiation and cyclosporine or other immunosuppressive agents shorten the time to skin tumor formation compared to UV irradiation alone.
Cyclosporine was not mutagenic in appropriate test systems. Cyclosporine has not been found to be mutagenic/genotoxic in the Ames Test, the V79-HGPRT Test, the micronucleus test in mice and Chinese hamsters, the chromosome-aberration tests in Chinese hamster bone-marrow, the mouse dominant lethal assay, and the DNA-repair test in sperm from treated mice. A recent study analyzing sister chromatid exchange (SCE) induction by cyclosporine using human lymphocytes in vitro gave indication of a positive effect (i.e., induction of SCE), at high concentrations in this system. In two published research studies, rabbits exposed to cyclosporine in utero (10 mg/kg/day subcutaneously) demonstrated reduced numbers of nephrons, renal hypertrophy, systemic hypertension and progressive renal insufficiency up to 35 weeks of age. Pregnant rats which received 12 mg/kg/day of cyclosporine intravenously (twice the recommended human intravenous dose) had fetuses with an increased incidence of ventricular septal defect. These findings have not been demonstrated in other species and their relevance for humans is unknown.
No impairment in fertility was demonstrated in studies in male and female rats.
Widely distributed papillomatosis of the skin was observed after chronic treatment of dogs with cyclosporine at 9 times the human initial psoriasis treatment dose of 2.5 mg/kg, where doses are expressed on a body surface area basis. This papillomatosis showed a spontaneous regression upon discontinuation of cyclosporine.
An increased incidence of malignancy is a recognized complication of immunosuppression in recipients of organ transplants and patients with rheumatoid arthritis and psoriasis. The most common forms of neoplasms are non-Hodgkin's lymphoma and carcinomas of the skin. The risk of malignancies in cyclosporine recipients is higher than in the normal, healthy population but similar to that in patients receiving other immunosuppressive therapies. Reduction or discontinuance of immunosuppression may cause the lesions to regress.
In psoriasis patients on cyclosporine, development of malignancies, especially those of the skin has been reported. (See WARNINGS) Skin lesions not typical for psoriasis should be biopsied before starting cyclosporine treatment. Patients with malignant or premalignant changes of the skin should be treated with cyclosporine only after appropriate treatment of such lesions and if no other treatment option exists.
Pregnancy
Animal studies have shown reproductive toxicity in rats and rabbits. Cyclosporine gave no evidence of mutagenic or teratogenic effects in the standard test systems with oral application (rats up to 17 mg/kg and rabbits up to 30 mg/kg per day orally). Only at dose levels toxic to dams, were adverse effects seen in reproduction studies in rats. Cyclosporine has been shown to be embryo- and fetotoxic in rats and rabbits following oral administration at maternally toxic doses. Fetal toxicity was noted in rats at 0.8 and rabbits at 5.4 times the transplant doses in humans of 6.0 mg/kg, where dose corrections are based on body surface area. Cyclosporine was embryo- and fetotoxic as indicated by increased pre- and postnatal mortality and reduced fetal weight together with related skeletal retardation.
There are no adequate and well-controlled studies in pregnant women and, therefore, Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) should not be used during pregnancy unless the potential benefit to the mother justifies the potential risk to the fetus.
In pregnant transplant recipients who are being treated with immunosuppressants the risk of premature birth is increased. The following data represent the reported outcomes of 116 pregnancies in women receiving cyclosporine during pregnancy, 90% of whom were transplant patients, and most of whom received cyclosporine throughout the entire gestational period. The only consistent patterns of abnormality were premature birth (gestational period of 28 to 36 weeks) and low birth weight for gestational age. Sixteen fetal losses occurred. Most of the pregnancies (85 of 100) were complicated by disorders; including, preeclampsia, eclampsia, premature labor, abruptio placentae, oligohydramnios, Rh incompatibility, and fetoplacental dysfunction. Pre-term delivery occurred in 47%. Seven malformations were reported in 5 viable infants and in 2 cases of fetal loss. Twenty-eight percent of the infants were small for gestational age. Neonatal complications occurred in 27%. Therefore, the risks and benefits of using Gengraf® during pregnancy should be carefully weighed.
A limited number of observations in children exposed to cyclosporine in utero are available, up to an age of approximately 7 years. Renal function and blood pressure in these children were normal.
Because of the possible disruption of maternal-fetal interaction, the risk/benefit ratio of using Gengraf® in psoriasis patients during pregnancy should carefully be weighed with serious consideration for discontinuation of Gengraf®.
The alcohol content of the Gengraf® formulations should also be taken into account in pregnant women. (See WARNINGS, Special Excipients)
Nursing Mothers
Cyclosporine is present in breast milk. Because of the potential for serious adverse drug reactions in nursing infants from Gengraf®, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Gengraf® contains ethanol. Ethanol will be present in human milk at levels similar to that found in maternal serum and if present in breast milk will be orally absorbed by a nursing infant (See WARNINGS).
Pediatric Use
Although no adequate and well-controlled studies have been completed in children, transplant recipients as young as one year of age have received cyclosporine (MODIFIED) with no unusual adverse effects. The safety and efficacy of cyclosporine (MODIFIED) treatment in children with juvenile rheumatoid arthritis or psoriasis below the age of 18 have not been established.
Geriatric Use
In rheumatoid arthritis clinical trials with cyclosporine, 17.5% of patients were age 65 or older. These patients were more likely to develop systolic hypertension on therapy, and more likely to show serum creatinine rises ≥50% above the baseline after 3 to 4 months of therapy.
Clinical studies of cyclosporine oral solution (MODIFIED) in transplant and psoriasis patients did not include a sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experiences have not identified differences in response between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
Kidney, Liver, and Heart Transplantation
The principal adverse reactions of cyclosporine therapy are renal dysfunction, tremor, hirsutism, hypertension, and gum hyperplasia.
Hypertension
Hypertension, which is usually mild to moderate, may occur in approximately 50% of patients following renal transplantation and in most cardiac transplant patients.
Glomerular Capillary Thrombosis
Glomerular capillary thrombosis has been found in patients treated with cyclosporine and may progress to graft failure. The pathologic changes resembled those seen in the hemolytic-uremic syndrome and included thrombosis of the renal microvasculature, with platelet-fibrin thrombi occluding glomerular capillaries and afferent arterioles, microangiopathic hemolytic anemia, thrombocytopenia, and decreased renal function. Similar findings have been observed when other immunosuppressives have been employed post-transplantation.
Hypomagnesemia
Hypomagnesemia has been reported in some, but not all, patients exhibiting convulsions while on cyclosporine therapy. Although magnesium-depletion studies in normal subjects suggest that hypomagnesemia is associated with neurologic disorders, multiple factors, including hypertension, high dose methylprednisolone, hypocholesterolemia, and nephrotoxicity associated with high plasma concentrations of cyclosporine appear to be related to the neurological manifestations of cyclosporine toxicity.
Clinical Studies
In controlled studies, the nature, severity, and incidence of the adverse events that were observed in 493 transplanted patients treated with cyclosporine (MODIFIED) were comparable with those observed in 208 transplanted patients who received Sandimmune® in these same studies when the dosage of the two drugs was adjusted to achieve the same cyclosporine blood trough concentrations.
Based on the historical experience with Sandimmune®, the following reactions occurred in 3% or greater of 892 patients involved in clinical trials of kidney, heart, and liver transplants.
Randomized Kidney Patients Cyclosporine Patients (Sandimmune®) Body System Adverse Reactions Sandimmune®
(N=227) %Azathioprine
(N=228) %Kidney
(N=705) %Heart
(N=112) %Liver
(N=75) %Genitourinary Renal Dysfunction 32 6 25 38 37 Cardiovascular Hypertension 26 18 13 53 27 Cramps 4 <1 2 <1 0 Skin Hirsutism 21 <1 21 28 45 Acne 6 8 2 2 1 Central Nervous System Tremor 12 0 21 31 55 Convulsions 3 1 1 4 5 Headache 2 <1 2 15 4 Gastrointestinal Gum Hyperplasia 4 0 9 5 16 Diarrhea 3 <1 3 4 8 Nausea/Vomiting 2 <1 4 10 4 Hepatotoxicity <1 <1 4 7 4 Abdominal Discomfort <1 0 <1 7 0 Autonomic Nervous System Paresthesia 3 0 1 2 1 Flushing <1 0 4 0 4 Hematopoietic Leukopenia 2 19 <1 6 0 Lymphoma <1 0 1 6 1 Respiratory Sinusitis <1 0 4 3 7 Miscellaneous Gynecomastia <1 0 <1 4 3 Among 705 kidney transplant patients treated with cyclosporine oral solution (Sandimmune®) in clinical trials, the reason for treatment discontinuation was renal toxicity in 5.4%, infection in 0.9%, lack of efficacy in 1.4%, acute tubular necrosis in 1.0%, lymphoproliferative disorders in 0.3%, hypertension in 0.3%, and other reasons in 0.7% of the patients.
The following reactions occurred in 2% or less of cyclosporine-treated patients: allergic reactions, anemia, anorexia, confusion, conjunctivitis, edema, fever, brittle fingernails, gastritis, hearing loss, hiccups, hyperglycemia, migraine (Gengraf®), muscle pain, peptic ulcer, thrombocytopenia, tinnitus.
The following reactions occurred rarely: anxiety, chest pain, constipation, depression, hair breaking, hematuria, joint pain, lethargy, mouth sores, myocardial infarction, night sweats, pancreatitis, pruritus, swallowing difficulty, tingling, upper GI bleeding, visual disturbance, weakness, weight loss.
Patients receiving immunosuppressive therapies, including cyclosporine and cyclosporine -containing regimens, are at increased risk of infections (viral, bacterial, fungal, parasitic). Both generalized and localized infections can occur. Pre-existing infections may also be aggravated. Fatal outcomes have been reported. (See WARNINGS)
Infectious Complications in Historical Randomized Studies in Renal Transplant Patients Using Sandimmune® Complication Cyclosporine Treatment
(N=227)
% of ComplicationsAzathioprine with Steroids*
(N=228)
% of ComplicationsSepticemia 5.3 4.8 Abscesses 4.4 5.3 Systemic Fungal Infection 2.2 3.9 Local Fungal Infection 7.5 9.6 Cytomegalovirus 4.8 12.3 Other Viral Infections 15.9 18.4 Urinary Tract Infections 21.1 20.2 Wound and Skin Infections 7.0 10.1 Pneumonia 6.2 9.2 *Some patients also received ALG. Postmarketing Experience, Kidney, Liver and Heart Transplantation
Hepatotoxicity
Cases of hepatotoxicity and liver injury including cholestasis, jaundice, hepatitis and liver failure; serious and/or fatal outcomes have been reported. (See WARNINGS, Hepatotoxicity)
Increased Risk of Infections
Cases of JC virus-associated progressive multifocal leukoencephalopathy (PML), sometimes fatal; and polyoma virus-associated nephropathy (PVAN), especially BK virus resulting in graft loss have been reported. (See WARNINGS, Polyoma Virus Infection)
Headache, including Migraine
Cases of migraine have been reported. In some cases, patients have been unable to continue cyclosporine, however, the final decision on treatment discontinuation should be made by the treating physician following the careful assessment of benefits versus risks.
Pain of lower extremities
Isolated cases of pain of lower extremities have been reported in association with cyclosporine. Pain of lower extremities has also been noted as part of Calcineurin-Inhibitor Induced Pain Syndrome (CIPS) as described in the literature.
Rheumatoid Arthritis
The principal adverse reactions associated with the use of cyclosporine in rheumatoid arthritis are renal dysfunction (See WARNINGS), hypertension (See PRECAUTIONS), headache, gastrointestinal disturbances, and hirsutism/hypertrichosis.
In rheumatoid arthritis patients treated in clinical trials within the recommended dose range, cyclosporine therapy was discontinued in 5.3% of the patients because of hypertension and in 7% of the patients because of increased creatinine. These changes are usually reversible with timely dose decrease or drug discontinuation. The frequency and severity of serum creatinine elevations increase with dose and duration of cyclosporine therapy. These elevations are likely to become more pronounced without dose reduction or discontinuation.
The following adverse events occurred in controlled clinical trials:
Cyclosporine (MODIFIED)/Sandimmune® Rheumatoid Arthritis Percentage of Patients with Adverse Events ≥3% in any Cyclosporine Treated Group Studies
651+652+2008Study 302 Study 654 Study 654 Study 302 Studies
651+652
+2008Body
SystemPreferred
TermSandimmune®†
(N=269)Sandimmune®
(N=155)Metho-
trexate &
Sandimmune®
(N=74)Metho-
trexate &
Placebo
(N=73)Cyclosporine
(MODIFIED)
(N=143)Placebo
(N=201)Autonomic Nervous System Disorders Flushing 2% 2% 3% 0% 5% 2% Body As A Whole–General Disorders Accidental Trauma 0% 1% 10% 4% 4% 0% Edema NOS* 5% 14% 12% 4% 10% <1% Fatigue 6% 3% 8% 12% 3% 7% Fever 2% 3% 0% 0% 2% 4% Influenza-like symptoms <1% 6% 1% 0% 3% 2% Pain 6% 9% 10% 15% 13% 4% Rigors 1% 1% 4% 0% 3% 1% Cardiovascular Disorders Arrhythmia 2% 5% 5% 6% 2% 1% Chest Pain 4% 5% 1% 1% 6% 1% Hypertension 8% 26% 16% 12% 25% 2% Central and Peripheral Nervous System Disorders Dizziness 8% 6% 7% 3% 8% 3% Headache 17% 23% 22% 11% 25% 9% Migraine 2% 3% 0% 0% 3% 1% Paresthesia 8% 7% 8% 4% 11% 1% Tremor 8% 7% 7% 3% 13% 4% Gastrointestinal System Disorders Abdominal Pain 15% 15% 15% 7% 15% 10% Anorexia 3% 3% 1% 0% 3% 3% Diarrhea 12% 12% 18% 15% 13% 8% Dyspepsia 12% 12% 10% 8% 8% 4% Flatulence 5% 5% 5% 4% 4% 1% Gastrointestinal Disorder NOS* 0% 2% 1% 4% 4% 0% Gingivitis 4% 3% 0% 0% 0% 1% Gum Hyperplasia 2% 4% 1% 3% 4% 1% Nausea 23% 14% 24% 15% 18% 14% Rectal Hemorrhage 0% 3% 0% 0% 1% 1% Stomatitis 7% 5% 16% 12% 6% 8% Vomiting 9% 8% 14% 7% 6% 5% Hearing and Vestibular Disorders Ear Disorder NOS* 0% 5% 0% 0% 1% 0% Metabolic and Nutritional Disorders Hypomagnesemia 0% 4% 0% 0% 6% 0% Musculoskeletal System Disorders Arthropathy 0% 5% 0% 1% 4% 0% Leg Cramps / Involuntary
Muscle Contractions2% 11% 11% 3% 12% 1% Psychiatric Disorders Depression 3% 6% 3% 1% 1% 2% Insomnia 4% 1% 1% 0% 3% 2% Renal Creatinine elevations ≥30% 43% 39% 55% 19% 48% 13% Creatinine elevations ≥50% 24% 18% 26% 8% 18% 3% Reproductive Disorders, Female Leukorrhea 1% 0% 4% 0% 1% 0% Menstrual Disorder 3% 2% 1% 0% 1% 1% Respiratory System Disorders Bronchitis 1% 3% 1% 0% 1% 3% Coughing 5% 3% 5% 7% 4% 4% Dyspnea 5% 1% 3% 3% 1% 2% Infection NOS* 9% 5% 0% 7% 3% 10% Pharyngitis 3% 5% 5% 6% 4% 4% Pneumonia 1% 0% 4% 0% 1% 1% Rhinitis 0% 3% 11% 10% 1% 0% Sinusitis 4% 4% 8% 4% 3% 3% Upper Respiratory Tract 0% 14% 23% 15% 13% 0% Skin and Appendages Disorders Alopecia 3% 0% 1% 1% 4% 4% Bullous Eruption 1% 0% 4% 1% 1% 1% Hypertrichosis 19% 17% 12% 0% 15% 3% Rash 7% 12% 10% 7% 8% 10% Skin Ulceration 1% 1% 3% 4% 0% 2% Urinary System Disorders Dysuria 0% 0% 11% 3% 1% 2% Micturition Frequency 2% 4% 3% 1% 2% 2% NPN, Increased 0% 19% 12% 0% 18% 0% Urinary Tract Infection 0% 3% 5% 4% 3% 0% Vascular (Extracardiac) Disorders Purpura 3% 4% 1% 1% 2% 0% † Includes patients in 2.5 mg/kg/day dose group only. *NOS=Not Otherwise Specified. In addition, the following adverse events have been reported in 1% to <3% of the rheumatoid arthritis patients in the cyclosporine treatment group in controlled clinical trials.
Autonomic Nervous System: dry mouth, increased sweating
Body as a Whole: allergy, asthenia, hot flushes, malaise, overdose, procedure NOS*, tumor NOS*, weight decrease, weight increase
Cardiovascular: abnormal heart sounds, cardiac failure, myocardial infarction, peripheral ischemia
Central and Peripheral Nervous System: hypoesthesia, neuropathy, vertigo
Endocrine: goiter
Gastrointestinal: constipation, dysphagia, enanthema, eructation, esophagitis, gastric ulcer, gastritis, gastroenteritis, gingival bleeding, glossitis, peptic ulcer, salivary gland enlargement, tongue disorder, tooth disorder
Infection: abscess, bacterial infection, cellulitis, folliculitis, fungal infection, herpes simplex, herpes zoster, renal abscess, moniliasis, tonsillitis, viral infection
Hematologic: anemia, epistaxis, leukopenia, lymphadenopathy
Liver and Biliary System: bilirubinemia
Metabolic and Nutritional: diabetes mellitus, hyperkalemia, hyperuricemia, hypoglycemia
Musculoskeletal System: arthralgia, bone fracture, bursitis, joint dislocation, myalgia, stiffness, synovial cyst, tendon disorder
Neoplasms: breast fibroadenosis, carcinoma
Psychiatric: anxiety, confusion, decreased libido, emotional lability, impaired concentration, increased libido, nervousness, paroniria, somnolence
Reproductive (Female): breast pain, uterine hemorrhage
Respiratory System: abnormal chest sounds, bronchospasm
Skin and Appendages: abnormal pigmentation, angioedema, dermatitis, dry skin, eczema, nail disorder, pruritus, skin disorder, urticaria
Special Senses: abnormal vision, cataract, conjunctivitis, deafness, eye pain, taste perversion, tinnitus, vestibular disorder
Urinary System: abnormal urine, hematuria, increased BUN, micturition urgency, nocturia, polyuria, pyelonephritis, urinary incontinence
*NOS=Not Otherwise Specified
Psoriasis
The principal adverse reactions associated with the use of cyclosporine in patients with psoriasis are renal dysfunction, headache, hypertension, hypertriglyceridemia, hirsutism/hypertrichosis, paresthesia or hyperesthesia, influenza-like symptoms, nausea/vomiting, diarrhea, abdominal discomfort, lethargy, and musculoskeletal or joint pain.
In psoriasis patients treated in US controlled clinical studies within the recommended dose range, cyclosporine therapy was discontinued in 1.0% of the patients because of hypertension and in 5.4% of the patients because of increased creatinine. In the majority of cases, these changes were reversible after dose reduction or discontinuation of cyclosporine.
There has been one reported death associated with the use of cyclosporine in psoriasis. A 27-year-old male developed renal deterioration and was continued on cyclosporine. He had progressive renal failure leading to death.
Frequency and severity of serum creatinine increases with dose and duration of cyclosporine therapy. These elevations are likely to become more pronounced and may result in irreversible renal damage without dose reduction or discontinuation.
Adverse Events Occurring in 3% or More of Psoriasis Patients in Controlled Clinical Trials
Body System* Preferred Term Cyclosporine (MODIFIED)
(N=182)Sandimmune®
(N=185)Infection or Potential Infection 24.7% 24.3% Influenza-Like Symptoms 9.9% 8.1% Upper Respiratory Tract Infections 7.7% 11.3% Cardiovascular System 28.0% 25.4% Hypertension** 27.5% 25.4% Urinary System 24.2% 16.2% Increased Creatinine 19.8% 15.7% Central and Peripheral Nervous System 26.4% 20.5% Headache 15.9% 14.0% Paresthesia 7.1% 4.8% Musculoskeletal System 13.2% 8.7% Arthralgia 6.0% 1.1% Body As a Whole–General 29.1% 22.2% Pain 4.4% 3.2% Metabolic and Nutritional 9.3% 9.7% Reproductive, Female 8.5% (4 of 47 females) 11.5% (6 of 52 females) Resistance Mechanism 18.7% 21.1% Skin and Appendages 17.6% 15.1% Hypertrichosis 6.6% 5.4% Respiratory System 5.0% 6.5% Bronchospasm, Coughing, Dyspnea, Rhinitis 5.0% 4.9% Psychiatric 5.0% 3.8% Gastrointestinal System 19.8% 28.7% Abdominal Pain 2.7% 6.0% Diarrhea 5.0% 5.9% Dyspepsia 2.2% 3.2% Gum Hyperplasia 3.8% 6.0% Nausea 5.5% 5.9% White cell and RES 4.4% 2.7% *Total percentage of events within the system
**Newly occurring hypertension=SBP ≥160 mm Hg and/or DBP ≥90 mm HgThe following events occurred in 1% to less than 3% of psoriasis patients treated with cyclosporine:
Body as a Whole: fever, flushes, hot flushes
Cardiovascular: chest pain
Central and Peripheral Nervous System: appetite increased, insomnia, dizziness, nervousness, vertigo
Gastrointestinal: abdominal distention, constipation, gingival bleeding
Liver and Biliary System: hyperbilirubinemia
Neoplasms: skin malignancies [squamous cell (0.9%) and basal cell (0.4%) carcinomas]
Reticuloendothelial: platelet, bleeding, and clotting disorders, red blood cell disorder
Respiratory: infection, viral and other infection
Skin and Appendages: acne, folliculitis, keratosis, pruritus, rash, dry skin
Urinary System: micturition frequency
Vision: abnormal vision
Mild hypomagnesemia and hyperkalemia may occur but are asymptomatic. Increases in uric acid may occur and attacks of gout have been rarely reported. A minor and dose related hyperbilirubinemia has been observed in the absence of hepatocellular damage. Cyclosporine therapy may be associated with a modest increase of serum triglycerides or cholesterol. Elevations of triglycerides (>750 mg/dL) occur in about 15% of psoriasis patients; elevations of cholesterol (>300 mg/dL) are observed in less than 3% of psoriasis patients. Generally these laboratory abnormalities are reversible upon dose reduction or discontinuation of cyclosporine.
Postmarketing Experience, Psoriasis
Cases of transformation to erythrodermic psoriasis or generalized pustular psoriasis upon either withdrawal or reduction of cyclosporine in patients with chronic plaque psoriasis have been reported.
-
OVERDOSAGE
There is a minimal experience with cyclosporine overdosage. Forced emesis and gastric lavage can be of value up to 2 hours after administration of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]). Transient hepatotoxicity and nephrotoxicity may occur which should resolve following drug withdrawal. Oral doses of cyclosporine up to 10 g (about 150 mg/kg) have been tolerated with relatively minor clinical consequences, such as vomiting, drowsiness, headache, tachycardia and, in a few patients, moderately severe, reversible impairment of renal function. However, serious symptoms of intoxication have been reported following accidental parenteral overdosage with cyclosporine in premature neonates. General supportive measures and symptomatic treatment should be followed in all cases of overdosage. Cyclosporine is not dialyzable to any great extent, nor is it cleared well by charcoal hemoperfusion. The oral dosage at which half of experimental animals are estimated to die is 31 times, 39 times, and >54 times the human maintenance dose for transplant patients (6mg/kg; corrections based on body surface area) in mice, rats, and rabbits.
-
DOSAGE AND ADMINISTRATION
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) has increased bioavailability in comparison to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Gengraf® and Sandimmune® are not bioequivalent and cannot be used interchangeably without physician supervision.
The daily dose of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) should always be given in two divided doses (BID). It is recommended that Gengraf® be administered on a consistent schedule with regard to time of day and relation to meals. Grapefruit and grapefruit juice affect metabolism, increasing blood concentration of cyclosporine, thus should be avoided.
Specific Populations
Renal Impairment in Kidney, Liver, and Heart Transplantation
Cyclosporine undergoes minimal renal elimination and its pharmacokinetics do not appear to be significantly altered in patients with end-stage renal disease who receive routine hemodialysis treatments (See CLINICAL PHARMACOLOGY). However, due to its nephrotoxic potential (See WARNINGS), careful monitoring of renal function is recommended; cyclosporine dosage should be reduced if indicated. (See WARNINGS and PRECAUTIONS)
Renal Impairment in Rheumatoid Arthritis and Psoriasis
Patients with impaired renal function should not receive cyclosporine. (See CONTRAINDICATIONS, WARNINGS and PRECAUTIONS)
Hepatic Impairment
The clearance of cyclosporine may be significantly reduced in severe liver disease patients (See CLINICAL PHARMACOLOGY). Dose reduction may be necessary in patients with severe liver impairment to maintain blood concentrations within the recommended target range (See WARNINGS and PRECAUTIONS).
Newly Transplanted Patients
The initial oral dose of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) can be given 4 to 12 hours prior to transplantation or be given postoperatively. The initial dose of Gengraf® varies depending on the transplanted organ and the other immunosuppressive agents included in the immunosuppressive protocol. In newly transplanted patients, the initial oral dose of Gengraf® is the same as the initial oral dose of Sandimmune®. Suggested initial doses are available from the results of a 1994 survey of the use of Sandimmune® in US transplant centers. The mean ± SD initial doses were 9±3 mg/kg/day for renal transplant patients (75 centers), 8±4 mg/kg/day for liver transplant patients (30 centers), and 7±3 mg/kg/day for heart transplant patients (24 centers). Total daily doses were divided into two equal daily doses. The Gengraf® dose is subsequently adjusted to achieve a pre-defined cyclosporine blood concentration. (See Blood Concentration Monitoring in Transplant Patients, below) If cyclosporine trough blood concentrations are used, the target range is the same for Gengraf® as for Sandimmune®. Using the same trough concentration target range for Gengraf® as for Sandimmune® results in greater cyclosporine exposure when Gengraf® is administered. (See Pharmacokinetics, Absorption) Dosing should be titrated based on clinical assessments of rejection and tolerability. Lower Gengraf® doses may be sufficient as maintenance therapy.
Adjunct therapy with adrenal corticosteroids is recommended initially. Different tapering dosage schedules of prednisone appear to achieve similar results. A representative dosage schedule based on the patient's weight started with 2.0 mg/kg/day for the first 4 days tapered to 1.0 mg/kg/day by 1 week, 0.6 mg/kg/day by 2 weeks, 0.3 mg/kg/day by 1 month, and 0.15 mg/kg/day by 2 months and thereafter as a maintenance dose. Steroid doses may be further tapered on an individualized basis depending on status of patient and function of graft. Adjustments in dosage of prednisone must be made according to the clinical situation.
Conversion from Sandimmune® Soft Gelatin Capsules (Cyclosporine Capsules, USP) to Gengraf® Capsules (Cyclosporine Capsules, USP [MODIFIED]) in Transplant Patients
In transplanted patients who are considered for conversion to Gengraf® from Sandimmune® (cyclosporine), Gengraf® should be started with the same daily dose as was previously used with Sandimmune® (cyclosporine) (1:1 dose conversion). The Gengraf® dose should subsequently be adjusted to attain the pre-conversion cyclosporine blood trough concentration. Using the same trough concentration target range for Gengraf® as for Sandimmune® (cyclosporine) results in greater cyclosporine exposure when Gengraf® is administered. (See Pharmacokinetics, Absorption) Patients with suspected poor absorption of Sandimmune® (cyclosporine) require different dosing strategies. (See Transplant Patients with Poor Absorption of Sandimmune® (Cyclosporine), below) In some patients, the increase in blood trough concentration is more pronounced and may be of clinical significance.
Until the blood trough concentration attains the pre-conversion value, it is strongly recommended that the cyclosporine blood trough concentration be monitored every 4 to 7 days after conversion to Gengraf®. In addition, clinical safety parameters such as serum creatinine and blood pressure should be monitored every two weeks during the first two months after conversion. If the blood trough concentrations are outside the desired range and/or if the clinical safety parameters worsen, the dosage of Gengraf® must be adjusted accordingly.
Transplant Patients with Poor Absorption of Sandimmune® (Cyclosporine)
Patients with lower than expected cyclosporine blood trough concentrations in relation to the oral dose of Sandimmune® (cyclosporine) may have poor or inconsistent absorption of cyclosporine from Sandimmune® (cyclosporine). After conversion to Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]), patients tend to have higher cyclosporine concentrations. Due to the increase in bioavailability of cyclosporine following conversion to Gengraf®, the cyclosporine blood trough concentration may exceed the target range. Particular caution should be exercised when converting patients to Gengraf® at doses greater than 10 mg/kg/day. The dose of Gengraf® should be titrated individually based on cyclosporine trough concentrations, tolerability, and clinical response. In this population the cyclosporine blood trough concentration should be measured more frequently, at least twice a week (daily, if initial dose exceeds 10 mg/kg/day) until the concentration stabilizes within the desired range.
Rheumatoid Arthritis
The initial dose of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) is 2.5 mg/kg/day, taken twice daily as a divided (BID) oral dose. Salicylates, NSAIDs, and oral corticosteroids may be continued. (See WARNINGS and PRECAUTIONS, Drug Interactions) Onset of action generally occurs between 4 and 8 weeks. If insufficient clinical benefit is seen and tolerability is good (including serum creatinine less than 30% above baseline), the dose may be increased by 0.5 to 0.75 mg/kg/day after 8 weeks and again after 12 weeks to a maximum of 4 mg/kg/day. If no benefit is seen by 16 weeks of therapy, Gengraf® therapy should be discontinued.
Dose decreases by 25% to 50% should be made at any time to control adverse events, e.g., hypertension elevations in serum creatinine (30% above patient's pretreatment level) or clinically significant laboratory abnormalities. (See WARNINGS and PRECAUTIONS)
If dose reduction is not effective in controlling abnormalities or if the adverse event or abnormality is severe, Gengraf® should be discontinued. The same initial dose and dosage range should be used if Gengraf® is combined with the recommended dose of methotrexate. Most patients can be treated with Gengraf® doses of 3 mg/kg/day or below when combined with methotrexate doses of up to 15 mg/week. (See CLINICAL PHARMACOLOGY, Clinical Trials)
There is limited long-term treatment data. Recurrence of rheumatoid arthritis disease activity is generally apparent within 4 weeks after stopping cyclosporine.
Psoriasis
The initial dose of Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED]) should be 2.5 mg/kg/day. Gengraf® should be taken twice daily, as a divided (1.25 mg/kg BID) oral dose. Patients should be kept at that dose for at least 4 weeks, barring adverse events. If significant clinical improvement has not occurred in patients by that time, the patient's dosage should be increased at 2-week intervals. Based on patient response, dose increases of approximately 0.5 mg/kg/day should be made to a maximum of 4.0 mg/kg/day.
Dose decreases by 25% to 50% should be made at any time to control adverse events, e.g., hypertension, elevations in serum creatinine (≥25% above the patient's pretreatment level), or clinically significant laboratory abnormalities. If dose reduction is not effective in controlling abnormalities, or if the adverse event or abnormality is severe, Gengraf® should be discontinued. (See Special Monitoring of Psoriasis Patients)
Patients generally show some improvement in the clinical manifestations of psoriasis in 2 weeks. Satisfactory control and stabilization of the disease may take 12 to 16 weeks to achieve. Results of a dose-titration clinical trial with Gengraf® indicate that an improvement of psoriasis by 75% or more (based on PASI) was achieved in 51% of the patients after 8 weeks and in 79% of the patients after 16 weeks. Treatment should be discontinued if satisfactory response cannot be achieved after 6 weeks at 4 mg/kg/day or the patient's maximum tolerated dose. Once a patient is adequately controlled and appears stable the dose of Gengraf® should be lowered, and the patient treated with the lowest dose that maintains an adequate response (this should not necessarily be total clearing of the patient). In clinical trials, cyclosporine doses at the lower end of the recommended dosage range were effective in maintaining a satisfactory response in 60% of the patients. Doses below 2.5 mg/kg/day may also be equally effective.
Upon stopping treatment with cyclosporine, relapse will occur in approximately 6 weeks (50% of the patients) to 16 weeks (75% of the patients). In the majority of patients rebound does not occur after cessation of treatment with cyclosporine. Thirteen cases of transformation of chronic plaque psoriasis to more severe forms of psoriasis have been reported. There were 9 cases of pustular and 4 cases of erythrodermic psoriasis. Long term experience with Gengraf® in psoriasis patients is limited and continuous treatment for extended periods greater than one year is not recommended. Alternation with other forms of treatment should be considered in the long term management of patients with this life long disease.
Blood Concentration Monitoring in Transplant Patients
Transplant centers have found blood concentration monitoring of cyclosporine to be an essential component of patient management. Of importance to blood concentration analysis are the type of assay used, the transplanted organ, and other immunosuppressant agents being administered. While no fixed relationship has been established, blood concentration monitoring may assist in the clinical evaluation of rejection and toxicity, dose adjustments, and the assessment of compliance.
Various assays have been used to measure blood concentrations of cyclosporine. Older studies using a nonspecific assay often cited concentrations that were roughly twice those of the specific assays. Therefore, comparison between concentrations in the published literature and an individual patient concentration using current assays must be made with detailed knowledge of the assay methods employed. Current assay results are also not interchangeable and their use should be guided by their approved labeling. A discussion of the different assay methods is contained in Annals of Clinical Biochemistry 1994;31:420-446. While several assays and assay matrices are available, there is a consensus that parent-compound-specific assays correlate best with clinical events. Of these, HPLC is the standard reference, but the monoclonal antibody RIAs and the monoclonal antibody FPIA offer sensitivity, reproducibility, and convenience. Most clinicians base their monitoring on trough cyclosporine concentrations. Applied Pharmacokinetics, Principles of Therapeutic Drug Monitoring (1992) contains a broad discussion of cyclosporine pharmacokinetics and drug monitoring techniques. Blood concentration monitoring is not a replacement for renal function monitoring or tissue biopsies.
-
HOW SUPPLIED
Gengraf® Capsules (cyclosporine capsules, USP [MODIFIED])
25 mg
Oval, white imprinted in blue, 25 mg, and the code OR. Packages of 30 unit-dose blisters. (NDC 0074-3108-32).
100 mg
Oval, white, with two blue stripes, imprinted in blue, 100 mg, and the code OT. Packages of 30 unit-dose blisters. (NDC 0074-3109-32).
Store and Dispense
In the original unit-dose container at controlled room temperature 68°-77°F (20°-25°C). (See USP Controlled Room Temperature).
Sandimmune® is a registered trademark of Novartis Pharmaceuticals Corporation.
© AbbVie Inc. 2000-2021
AbbVie Inc., North Chicago, IL 60064, U.S.A.
20067177 February, 2021
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
PRINCIPAL DISPLAY PANEL
NDC 0074–3108–32
30 Capsules
GENGRAF®
Capsules
(cyclosporine capsules, USP
[MODIFIED])
25 mg
WARNING: Gengraf®Capsules (cyclosporine capsules, USP [MODIFIED]) is NOT BIOEQUIVALENT to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Do NOT use interchangeably without a physician’s supervision.
abbvie Rx only
![NDC 0074–3108–32
30 Capsules
GENGRAF®
Capsules
(cyclosporine capsules, USP
[MODIFIED])
25 mg
WARNING: Gengraf®Capsules (cyclosporine capsules, USP [MODIFIED]) is NOT BIOEQUIVALENT to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Do NOT use interchangeably without a physician’s supervision.
abbvie Rx only](/dailymed/image.cfm?name=gengraf-capsules-spl-05.jpg&setid=e896cbfe-4745-4088-82a3-8dc10b75c41f)
- PRINCIPAL DISPLAY PANEL
-
PRINCIPAL DISPLAY PANEL
NDC 0074–3109–32
30 Capsules
GENGRAF®
Capsules
(cyclosporine capsules, USP
[MODIFIED])
100 mg
WARNING: Gengraf®Capsules (cyclosporine capsules, USP [MODIFIED]) is NOT BIOEQUIVALENT to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Do NOT use interchangeably without a physician’s supervision.
abbvie Rx only
![NDC 0074–3109–32
30 Capsules
GENGRAF®
Capsules
(cyclosporine capsules, USP
[MODIFIED])
100 mg
WARNING: Gengraf®Capsules (cyclosporine capsules, USP [MODIFIED]) is NOT BIOEQUIVALENT to Sandimmune® Soft Gelatin Capsules (cyclosporine capsules, USP). Do NOT use interchangeably without a physician’s supervision.
abbvie Rx only](/dailymed/image.cfm?name=gengraf-capsules-spl-07.jpg&setid=e896cbfe-4745-4088-82a3-8dc10b75c41f)
-
INGREDIENTS AND APPEARANCE
GENGRAF
cyclosporine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-0541 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYCLOSPORINE (UNII: 83HN0GTJ6D) (CYCLOSPORINE - UNII:83HN0GTJ6D) CYCLOSPORINE 50 mg Inactive Ingredients Ingredient Name Strength POLYOXYL 35 CASTOR OIL (UNII: 6D4M1DAL6O) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SORBITAN MONOOLEATE (UNII: 06XEA2VD56) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color white Score no score Shape OVAL Size 19mm Flavor Imprint Code 50;mg;OS Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-0541-30 5 in 1 CARTON 06/20/2016 04/27/2018 1 6 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065003 06/20/2016 04/27/2018 GENGRAF
cyclosporine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-6479 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYCLOSPORINE (UNII: 83HN0GTJ6D) (CYCLOSPORINE - UNII:83HN0GTJ6D) CYCLOSPORINE 100 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) POLYOXYL 35 CASTOR OIL (UNII: 6D4M1DAL6O) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SORBITAN MONOOLEATE (UNII: 06XEA2VD56) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color white Score no score Shape OVAL (White with two blue stripes) Size 23mm Flavor Imprint Code 100;mg;OT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-6479-32 5 in 1 CARTON 05/24/2010 09/16/2017 1 6 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065003 05/24/2010 09/16/2017 GENGRAF
cyclosporine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-6463 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYCLOSPORINE (UNII: 83HN0GTJ6D) (CYCLOSPORINE - UNII:83HN0GTJ6D) CYCLOSPORINE 25 mg Inactive Ingredients Ingredient Name Strength POLYOXYL 35 CASTOR OIL (UNII: 6D4M1DAL6O) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SORBITAN MONOOLEATE (UNII: 06XEA2VD56) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color white Score no score Shape OVAL Size 16mm Flavor Imprint Code 25;mg;OR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-6463-32 3 in 1 CARTON 05/24/2010 08/31/2017 1 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065003 05/24/2010 08/31/2017 GENGRAF
cyclosporine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-3108 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYCLOSPORINE (UNII: 83HN0GTJ6D) (CYCLOSPORINE - UNII:83HN0GTJ6D) CYCLOSPORINE 25 mg Inactive Ingredients Ingredient Name Strength POLYOXYL 35 CASTOR OIL (UNII: 6D4M1DAL6O) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SORBITAN MONOOLEATE (UNII: 06XEA2VD56) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color white Score no score Shape OVAL Size 16mm Flavor Imprint Code 25;mg;OR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-3108-32 3 in 1 CARTON 11/01/2015 1 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065003 11/01/2015 GENGRAF
cyclosporine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-3109 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYCLOSPORINE (UNII: 83HN0GTJ6D) (CYCLOSPORINE - UNII:83HN0GTJ6D) CYCLOSPORINE 100 mg Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) POLYOXYL 35 CASTOR OIL (UNII: 6D4M1DAL6O) POLYSORBATE 80 (UNII: 6OZP39ZG8H) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SORBITAN MONOOLEATE (UNII: 06XEA2VD56) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color white Score no score Shape OVAL (White with two blue stripes) Size 23mm Flavor Imprint Code 100;mg;OT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-3109-32 5 in 1 CARTON 11/01/2015 1 6 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065003 11/01/2015 Labeler - AbbVie Inc. (078458370)