Label: OFEV- nintedanib capsule


- NDC Code(s): 0597-0143-60, 0597-0145-60
- Packager: Boehringer Ingelheim Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 1, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OFEV safely and effectively. See full prescribing information for OFEV.
OFEV® (nintedanib capsules), for oral use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
Warnings and Precautions, Nephrotic Range Proteinuria (5.8) 01/2022 INDICATIONS AND USAGE
OFEV is a kinase inhibitor indicated in adults for:
DOSAGE AND ADMINISTRATION
- Recommended dosage: 150 mg taken orally twice daily approximately 12 hours apart taken with food. (2.2)
- Recommended dosage in patients with mild hepatic impairment (Child Pugh A): 100 mg taken orally twice daily approximately 12 hours apart taken with food. (2.3, 8.6)
- Consider temporary dose reduction to 100 mg, treatment interruption, or discontinuation for management of adverse reactions. (2.4, 5.2, 5.3, 6)
- Prior to treatment initiation, conduct liver function tests in all patients and a pregnancy test in females of reproductive potential. (2.1, 5.2, 5.4)
DOSAGE FORMS AND STRENGTHS
Capsules: 150 mg and 100 mg (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hepatic impairment: OFEV is not recommended for use in patients with moderate or severe hepatic impairment. In patients with mild hepatic impairment (Child Pugh A), the recommended dosage is 100 mg twice daily approximately 12 hours apart taken with food. Consider treatment interruption, or discontinuation for management of adverse reactions in these patients. (2.3, 2.4, 5.1, 8.6, 12.3)
- Elevated liver enzymes and drug-induced liver injury: ALT, AST, and bilirubin elevations have occurred with OFEV, including cases of drug-induced liver injury. In the postmarketing period, non-serious and serious cases of drug-induced liver injury, including severe liver injury with fatal outcome, have been reported. The majority of hepatic events occur within the first three months of treatment. Liver enzyme and bilirubin increases were reversible with dose modification or interruption in the majority of cases. Monitor ALT, AST, and bilirubin prior to initiation of treatment, at regular intervals during the first three months of treatment, and periodically thereafter or as clinically indicated. Temporary dosage reductions or discontinuations may be required. (2.1, 2.4, 5.2)
- Gastrointestinal disorders: Diarrhea, nausea, and vomiting have occurred with OFEV. Treat patients at first signs with adequate hydration and antidiarrheal medicine (e.g., loperamide) or anti-emetics. Discontinue OFEV if severe diarrhea, nausea, or vomiting persists despite symptomatic treatment. (5.3)
- Embryo-Fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use highly effective contraception. Advise women taking oral hormonal contraceptives experiencing vomiting, diarrhea, or other conditions where the drug absorption may be reduced to use alternative highly effective contraception. (5.4, 8.1, 8.3)
- Arterial thromboembolic events have been reported. Use caution when treating patients at higher cardiovascular risk including known coronary artery disease. (5.5)
- Bleeding events have been reported. Use OFEV in patients with known bleeding risk only if anticipated benefit outweighs the potential risk. (5.6)
- Gastrointestinal perforation has been reported. Use OFEV with caution when treating patients with recent abdominal surgery, previous history of diverticular disease or receiving concomitant corticosteroids or NSAIDs. Discontinue OFEV in patients who develop gastrointestinal perforation. Only use OFEV in patients with known risk of gastrointestinal perforation if the anticipated benefit outweighs the potential risk. (5.7)
- Nephrotic range proteinuria has been reported. Consider treatment interruption in patients who develop new or worsening proteinuria. (5.8)
ADVERSE REACTIONS
Most common adverse reactions (≥5%) are: diarrhea, nausea, abdominal pain, vomiting, liver enzyme elevation, decreased appetite, headache, weight decreased, and hypertension. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Boehringer Ingelheim Pharmaceuticals, Inc. at (800) 542-6257 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Coadministration of P-gp and CYP3A4 inhibitors may increase nintedanib exposure. Monitor patients closely for tolerability of OFEV. (7.1)
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding is not recommended. (8.2)
- Renal impairment: The safety and efficacy of OFEV have not been studied in patients with severe renal impairment and end-stage renal disease. (8.7, 12.3)
- Smokers: Decreased exposure has been noted in smokers which may alter the efficacy profile of OFEV. (8.8)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Idiopathic Pulmonary Fibrosis
1.2 Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype
1.3 Systemic Sclerosis-Associated Interstitial Lung Disease
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to OFEV Administration
2.2 Recommended Dosage
2.3 Recommended Dosage for Patients with Hepatic Impairment
2.4 Dosage Modification due to Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Impairment
5.2 Elevated Liver Enzymes and Drug-Induced Liver Injury
5.3 Gastrointestinal Disorders
5.4 Embryo-Fetal Toxicity
5.5 Arterial Thromboembolic Events
5.6 Risk of Bleeding
5.7 Gastrointestinal Perforation
5.8 Nephrotic Range Proteinuria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 P-glycoprotein (P-gp) and CYP3A4 Inhibitors and Inducers
7.2 Anticoagulants
7.3 Pirfenidone
7.4 Bosentan
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
8.8 Smokers
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Idiopathic Pulmonary Fibrosis
14.2 Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype
14.3 Systemic Sclerosis-Associated Interstitial Lung Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Idiopathic Pulmonary Fibrosis
OFEV is indicated for the treatment of adults with idiopathic pulmonary fibrosis (IPF).
1.2 Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype
OFEV is indicated for the treatment of adults with chronic fibrosing interstitial lung diseases (ILDs) with a progressive phenotype [see Clinical Studies (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to OFEV Administration
Conduct liver function tests in all patients and a pregnancy test in females of reproductive potential prior to initiating treatment with OFEV [see Warnings and Precautions (5.2, 5.4)].
2.2 Recommended Dosage
The recommended dosage of OFEV is 150 mg taken orally twice daily administered approximately 12 hours apart.
Administration Information
OFEV capsules should be taken with food [see Clinical Pharmacology (12.3)] and swallowed whole with liquid. OFEV capsules should not be chewed because of a bitter taste.
OFEV capsules should not be opened or crushed. If contact with the content of the capsule occurs, wash hands immediately and thoroughly. The effect of chewing or crushing of the capsule on the pharmacokinetics of nintedanib is not known.
2.3 Recommended Dosage for Patients with Hepatic Impairment
Mild Hepatic Impairment
In patients with mild hepatic impairment (Child Pugh A), the recommended dosage of OFEV is 100 mg orally twice daily approximately 12 hours apart taken with food [see Use in Specific Populations (8.6)].
Moderate or Severe Hepatic Impairment
Treatment with OFEV is not recommended [see Warnings and Precautions (5.1) and Use in Specific Populations (8.6)].
2.4 Dosage Modification due to Adverse Reactions
In addition to symptomatic treatment, if applicable, the management of adverse reactions of OFEV may require dose reduction or temporary interruption until the specific adverse reaction resolves to levels that allow continuation of therapy. OFEV treatment may be resumed at the full dosage (150 mg twice daily), or at the reduced dosage (100 mg twice daily), which subsequently may be increased to the full dosage. If a patient does not tolerate 100 mg twice daily, discontinue treatment with OFEV [see Warnings and Precautions (5.2, 5.3, 5.5, 5.7) and Adverse Reactions (6.1)].
Elevated Liver Enzymes
Dose modifications or interruptions may be necessary for liver enzyme elevations. Conduct liver function tests (aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin) prior to initiation of treatment with OFEV, at regular intervals during the first three months of treatment, and periodically thereafter or as clinically indicated. Measure liver tests promptly in patients who report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. Discontinue OFEV in patients with AST or ALT greater than 3 times the upper limit of normal (ULN) with signs or symptoms of liver injury and for AST or ALT elevations greater than 5 times the upper limit of normal. For AST or ALT greater than 3 times to less than 5 times the ULN without signs of liver damage, interrupt treatment or reduce OFEV to 100 mg twice daily. Once liver enzymes have returned to baseline values, treatment with OFEV may be reintroduced at a reduced dosage (100 mg twice daily), which subsequently may be increased to the full dosage (150 mg twice daily) [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
In patients with mild hepatic impairment (Child Pugh A), consider treatment interruption, or discontinuation for management of adverse reactions.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Impairment
Treatment with OFEV is not recommended in patients with moderate (Child Pugh B) or severe (Child Pugh C) hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. Patients with mild hepatic impairment (Child Pugh A) can be treated with a reduced dose of OFEV [see Dosage and Administration (2.3)].
5.2 Elevated Liver Enzymes and Drug-Induced Liver Injury
Cases of drug-induced liver injury (DILI) have been observed with OFEV treatment. In the clinical trials and postmarketing period, non-serious and serious cases of DILI were reported. Cases of severe liver injury with fatal outcome have been reported in the postmarketing period. The majority of hepatic events occur within the first three months of treatment. In clinical trials, administration of OFEV was associated with elevations of liver enzymes (ALT, AST, ALKP, GGT) and bilirubin. Liver enzyme and bilirubin increases were reversible with dose modification or interruption in the majority of cases. In IPF studies (Study 1, Study 2, and Study 3), the majority (94%) of patients with ALT and/or AST elevations had elevations less than 5 times ULN and the majority (95%) of patients with bilirubin elevations had elevations less than 2 times ULN. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), the majority (95%) of patients with ALT and/or AST elevations had elevations less than 5 times ULN and the majority (94%) of patients with bilirubin elevations had elevations less than 2 times ULN. In the SSc-ILD study (Study 4), a maximum ALT and/or AST greater than or equal to 3 times ULN was observed for 4.9% of patients in the OFEV group and for 0.7% of patients in the placebo group [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. Patients with a low body weight (less than 65 kg), Asian, and female patients may have a higher risk of elevations in liver enzymes. Nintedanib exposure increased with patient age, which may also result in a higher risk of increased liver enzymes [see Clinical Pharmacology (12.3)].
Conduct liver function tests (ALT, AST, and bilirubin) prior to initiation of treatment with OFEV, at regular intervals during the first three months of treatment, and periodically thereafter or as clinically indicated. Measure liver tests promptly in patients who report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. Dosage modifications or interruption may be necessary for liver enzyme elevations [see Dosage and Administration (2.1, 2.4)].
5.3 Gastrointestinal Disorders
Diarrhea
In clinical trials, diarrhea was the most frequent gastrointestinal event reported. In most patients, the event was of mild to moderate intensity and occurred within the first 3 months of treatment. In IPF studies (Study 1, Study 2, and Study 3), diarrhea was reported in 62% versus 18% of patients treated with OFEV and placebo, respectively [see Adverse Reactions (6.1)]. Diarrhea led to permanent dose reduction in 11% of patients treated with OFEV compared to 0 placebo-treated patients. Diarrhea led to discontinuation of OFEV in 5% of the patients compared to less than 1% of placebo-treated patients. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), diarrhea was reported in 67% versus 24% of patients treated with OFEV and placebo, respectively [see Adverse Reactions (6.1)]. Diarrhea led to permanent dose reduction in 16% of patients treated with OFEV compared to less than 1% of placebo-treated patients. Diarrhea led to discontinuation of OFEV in 6% of the patients compared to less than 1% of placebo-treated patients. In the SSc-ILD study (Study 4), diarrhea was reported in 76% versus 32% of patients treated with OFEV and placebo, respectively [see Adverse Reactions (6.1)]. Diarrhea led to permanent dose reduction in 22% of patients treated with OFEV compared to 1% of placebo-treated patients. Diarrhea led to discontinuation of OFEV in 7% of the patients compared to 0.3% of placebo-treated patients.
Dosage modifications or treatment interruptions may be necessary in patients with adverse reactions of diarrhea. Treat diarrhea at first signs with adequate hydration and antidiarrheal medication (e.g., loperamide), and consider dose reduction or treatment interruption if diarrhea continues [see Dosage and Administration (2.4)]. OFEV treatment may be resumed at the full dosage (150 mg twice daily), or at the reduced dosage (100 mg twice daily), which subsequently may be increased to the full dosage. If severe diarrhea persists despite symptomatic treatment, discontinue treatment with OFEV.
Nausea and Vomiting
In IPF studies (Study 1, Study 2, and Study 3), nausea was reported in 24% versus 7% and vomiting was reported in 12% versus 3% of patients treated with OFEV and placebo, respectively. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), nausea was reported in 29% versus 9% and vomiting was reported in 18% versus 5% of patients treated with OFEV and placebo, respectively. In the SSc-ILD study (Study 4), nausea was reported in 32% versus 14% and vomiting was reported in 25% versus 10% of patients treated with OFEV and placebo, respectively [see Adverse Reactions (6.1)]. In most patients, these events were of mild to moderate intensity. In IPF studies (Study 1, Study 2, and Study 3), nausea led to discontinuation of OFEV in 2% of patients and vomiting led to discontinuation of OFEV in 1% of the patients. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), nausea led to discontinuation of OFEV in less than 1% of patients and vomiting led to discontinuation of OFEV in 1% of the patients. In the SSc-ILD study (Study 4), nausea led to discontinuation of OFEV in 2% of patients and vomiting led to discontinuation of OFEV in 1% of the patients.
For nausea or vomiting that persists despite appropriate supportive care including anti-emetic therapy, dose reduction or treatment interruption may be required [see Dosage and Administration (2.4)]. OFEV treatment may be resumed at the full dosage (150 mg twice daily), or at the reduced dosage (100 mg twice daily), which subsequently may be increased to the full dosage. If severe nausea or vomiting does not resolve, discontinue treatment with OFEV.
5.4 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, OFEV can cause fetal harm when administered to a pregnant woman. Nintedanib caused embryo-fetal deaths and structural abnormalities in rats and rabbits when administered during organogenesis at less than (rats) and approximately 5 times (rabbits) the maximum recommended human dose (MRHD) in adults. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to avoid becoming pregnant while receiving treatment with OFEV and to use highly effective contraception at initiation of, during treatment, and at least 3 months after the last dose of OFEV. Nintedanib does not change the exposure to oral contraceptive containing ethinylestradiol and levonorgestrel in patients with SSc-ILD. However, the efficacy of oral hormonal contraceptives may be compromised by vomiting and/or diarrhea or other conditions where the drug absorption may be reduced. Advise women taking oral hormonal contraceptives experiencing these conditions to use alternative highly effective contraception. Verify pregnancy status prior to treatment with OFEV and during treatment as appropriate [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1, 12.3)].
5.5 Arterial Thromboembolic Events
Arterial thromboembolic events have been reported in patients taking OFEV. In IPF studies (Study 1, Study 2, and Study 3), arterial thromboembolic events were reported in 2.5% of patients treated with OFEV and less than 1% of placebo-treated patients. Myocardial infarction was the most common adverse reaction under arterial thromboembolic events, occurring in 1.5% of OFEV-treated patients compared to less than 1% of placebo-treated patients. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), arterial thromboembolic events were reported in less than 1% of patients in both treatment arms. Myocardial infarction was observed in less than 1% of patients in both treatment arms. In the SSc-ILD study (Study 4), arterial thromboembolic events were reported in 0.7% of patients in both treatment arms. There were 0 cases of myocardial infarction in OFEV-treated patients compared to 0.7% of placebo-treated patients.
Use caution when treating patients at higher cardiovascular risk including known coronary artery disease. Consider treatment interruption in patients who develop signs or symptoms of acute myocardial ischemia.
5.6 Risk of Bleeding
Based on the mechanism of action (VEGFR inhibition), OFEV may increase the risk of bleeding. In IPF studies (Study 1, Study 2, and Study 3), bleeding events were reported in 10% of patients treated with OFEV and in 7% of patients treated with placebo. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), bleeding events were reported in 11% of patients treated with OFEV and in 13% of patients treated with placebo. In the SSc-ILD study (Study 4), bleeding events were reported in 11% of patients treated with OFEV and in 8% of patients treated with placebo. In clinical trials, epistaxis was the most frequent bleeding event reported.
In the postmarketing period non-serious and serious bleeding events, some of which were fatal, have been observed.
Use OFEV in patients with known risk of bleeding only if the anticipated benefit outweighs the potential risk.
5.7 Gastrointestinal Perforation
Based on the mechanism of action, OFEV may increase the risk of gastrointestinal perforation. In IPF studies (Study 1, Study 2, and Study 3), gastrointestinal perforation was reported in less than 1% of patients treated with OFEV, compared to 0 cases in the placebo-treated patients. In the chronic fibrosing ILDs with a progressive phenotype study (Study 5), gastrointestinal perforation was not reported in any patients in any treatment arm. In the SSc-ILD study (Study 4), no cases of gastrointestinal perforation were reported in patients treated with OFEV or in placebo-treated patients.
In the postmarketing period, cases of gastrointestinal perforations have been reported, some of which were fatal.
Use caution when treating patients who have had recent abdominal surgery, previous history of diverticular disease or receiving concomitant corticosteroids or NSAIDs. Discontinue therapy with OFEV in patients who develop gastrointestinal perforation. Only use OFEV in patients with known risk of gastrointestinal perforation if the anticipated benefit outweighs the potential risk.
5.8 Nephrotic Range Proteinuria
Cases of proteinuria within the nephrotic range have been reported in the postmarketing period. Histological findings, when available, were consistent with glomerular microangiopathy with or without renal thrombi. Improvement in proteinuria has been observed after OFEV was discontinued; however, in some cases, residual proteinuria persisted. Consider treatment interruption in patients who develop new or worsening proteinuria.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Elevated Liver Enzymes and Drug-Induced Liver Injury [see Warnings and Precautions (5.2)]
- Gastrointestinal Disorders [see Warnings and Precautions (5.3)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.4)]
- Arterial Thromboembolic Events [see Warnings and Precautions (5.5)]
- Risk of Bleeding [see Warnings and Precautions (5.6)]
- Gastrointestinal Perforation [see Warnings and Precautions (5.7)]
- Nephrotic Range Proteinuria [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of OFEV was evaluated in over 1000 IPF patients, 332 patients with chronic fibrosing ILDs with a progressive phenotype, and over 280 patients with SSc-ILD. Over 200 IPF patients were exposed to OFEV for more than 2 years in clinical trials.
Idiopathic Pulmonary Fibrosis
OFEV was studied in three randomized, double-blind, placebo-controlled, 52-week trials. In the phase 2 (Study 1) and phase 3 (Study 2 and Study 3) trials, 723 patients with IPF received OFEV 150 mg twice daily and 508 patients received placebo. The median duration of exposure was 10 months for patients treated with OFEV and 11 months for patients treated with placebo. Subjects ranged in age from 42 to 89 years (median age of 67 years). Most patients were male (79%) and Caucasian (60%).
The most frequent serious adverse reactions reported in patients treated with OFEV, more than placebo, were bronchitis (1.2% vs. 0.8%) and myocardial infarction (1.5% vs. 0.4%). The most common adverse events leading to death in patients treated with OFEV, more than placebo, were pneumonia (0.7% vs. 0.6%), lung neoplasm malignant (0.3% vs. 0%), and myocardial infarction (0.3% vs. 0.2%). In the predefined category of major adverse cardiovascular events (MACE) including MI, fatal events were reported in 0.6% of OFEV-treated patients and 1.8% of placebo-treated patients.
Adverse reactions leading to permanent dose reductions were reported in 16% of OFEV-treated patients and 1% of placebo-treated patients. The most frequent adverse reaction that led to permanent dose reduction in the patients treated with OFEV was diarrhea (11%).
Adverse reactions leading to discontinuation were reported in 21% of OFEV-treated patients and 15% of placebo-treated patients. The most frequent adverse reactions that led to discontinuation in OFEV-treated patients were diarrhea (5%), nausea (2%), and decreased appetite (2%).
The most common adverse reactions with an incidence of greater than or equal to 5% and more frequent in the OFEV than placebo treatment group are listed in Table 1.
Table 1 Adverse Reactions Occurring in ≥5% of OFEV-treated Patients with Idiopathic Pulmonary Fibrosis and More Commonly Than Placebo in Study 1, Study 2, and Study 3 Adverse Reaction OFEV, 150 mg
n=723Placebo
n=508a Includes abdominal pain, abdominal pain upper, abdominal pain lower, gastrointestinal pain and abdominal tenderness.
b Includes gamma-glutamyltransferase increased, hepatic enzyme increased, alanine aminotransferase increased, aspartate aminotransferase increased, hepatic function abnormal, liver function test abnormal, transaminase increased, blood alkaline phosphatase-increased, alanine aminotransferase abnormal, aspartate aminotransferase abnormal, and gamma-glutamyltransferase abnormal.
c Includes hypertension, blood pressure increased, hypertensive crisis, and hypertensive cardiomyopathy.Gastrointestinal disorders Diarrhea 62% 18% Nausea 24% 7% Abdominal paina 15% 6% Vomiting 12% 3% Hepatobiliary disorders Liver enzyme elevationb 14% 3% Metabolism and nutrition disorders Decreased appetite 11% 5% Nervous system disorders Headache 8% 5% Investigations Weight decreased 10% 3% Vascular disorders Hypertensionc 5% 4% In addition, hypothyroidism was reported in patients treated with OFEV, more than placebo (1.1% vs. 0.6%). Alopecia was also reported in more patients treated with OFEV than placebo (0.8% vs. 0.4%).
Combination with Pirfenidone
Concomitant treatment with nintedanib and pirfenidone was investigated in an exploratory open-label, randomized (1:1) trial of nintedanib 150 mg twice daily with add-on pirfenidone (titrated to 801 mg three times a day) compared to nintedanib 150 mg twice daily alone in 105 randomized patients for 12 weeks. The primary endpoint was the percentage of patients with gastrointestinal adverse events from baseline to Week 12. Gastrointestinal adverse events were in line with the established safety profile of each component and were experienced in 37 (70%) patients treated with pirfenidone added to nintedanib versus 27 (53%) patients treated with nintedanib alone.
Diarrhea, nausea, vomiting, and abdominal pain (includes upper abdominal pain, abdominal discomfort, and abdominal pain) were the most frequent adverse events reported in 20 (38%) versus 16 (31%), in 22 (42%) versus 6 (12%), in 15 (28%) versus 6 (12%), and in 15 (28%) versus 7 (14%) patients treated with pirfenidone added to nintedanib versus nintedanib alone, respectively. More subjects reported AST or ALT elevations (greater than or equal to 3× the upper limit of normal) when using pirfenidone in combination with nintedanib (n=3 (6%)) compared to nintedanib alone (n=0) [see Warnings and Precautions (5.2, 5.3)].
Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype
OFEV was studied in a phase 3, double-blind, placebo-controlled trial (Study 5) in which 663 patients with chronic fibrosing ILDs with a progressive phenotype were randomized to receive OFEV 150 mg twice daily (n=332) or placebo (n=331) for at least 52 weeks. At 52 weeks, the median duration of exposure was 12 months for patients in both treatment arms. Subjects ranged in age from 27 to 87 years (median age of 67 years). The majority of patients were Caucasian (74%) or Asian (25%). Most patients were male (54%).
The most frequent serious adverse event reported in patients treated with OFEV, more than placebo, was pneumonia (4% vs. 3%). Adverse events leading to death were reported in 3% of patients treated with OFEV and in 5% of patients treated with placebo. No pattern was identified in the adverse events leading to death.
Adverse reactions leading to permanent dose reductions were reported in 33% of OFEV-treated patients and 4% of placebo-treated patients. The most frequent adverse reaction that led to permanent dose reduction in the patients treated with OFEV was diarrhea (16%).
Adverse reactions leading to discontinuation were reported in 20% of OFEV-treated patients and 10% of placebo-treated patients. The most frequent adverse reaction that led to discontinuation in OFEV-treated patients was diarrhea (6%).
The safety profile in patients with chronic fibrosing ILDs with a progressive phenotype treated with OFEV was consistent with that observed in IPF patients. In addition, the following adverse events were reported in OFEV more than placebo in chronic progressive fibrosing ILD: nasopharyngitis (13% vs. 12%), upper respiratory tract infection (7% vs 6%), urinary tract infection (6% vs. 4%), fatigue (10% vs. 6%), and back pain (6% vs. 5%).
Systemic Sclerosis-Associated Interstitial Lung Disease
OFEV was studied in a phase 3, randomized, double-blind, placebo-controlled trial (Study 4) in which 576 patients with SSc-ILD received OFEV 150 mg twice daily (n=288) or placebo (n=288). Patients were to receive treatment for at least 52 weeks; individual patients were treated for up to 100 weeks. The median duration of exposure was 15 months for patients treated with OFEV and 16 months for patients treated with placebo. Subjects ranged in age from 20 to 79 years (median age of 55 years). Most patients were female (75%). Patients were mostly Caucasian (67%), Asian (25%), or Black (6%). At baseline, 49% of patients were on stable therapy with mycophenolate.
The most frequent serious adverse events reported in patients treated with OFEV, more than placebo, were interstitial lung disease (2.4% nintedanib vs. 1.7% placebo) and pneumonia (2.8% nintedanib vs. 0.3% placebo). Within 52 weeks, 5 patients treated with OFEV (1.7%) and 4 patients treated with placebo (1.4%) died. There was no pattern among adverse events leading to death in either treatment arm.
Adverse reactions leading to permanent dose reductions were reported in 34% of OFEV-treated patients and 4% of placebo-treated patients. The most frequent adverse reaction that led to permanent dose reduction in the patients treated with OFEV was diarrhea (22%).
Adverse reactions leading to discontinuation were reported in 16% of OFEV-treated patients and 9% of placebo-treated patients. The most frequent adverse reactions that led to discontinuation in OFEV-treated patients were diarrhea (7%), nausea (2%), vomiting (1%), abdominal pain (1%), and interstitial lung disease (1%).
The safety profile in patients treated with OFEV with or without mycophenolate at baseline was comparable.
The most common adverse reactions with an incidence of greater than or equal to 5% in OFEV-treated patients and more commonly than in placebo are listed in Table 2.
Table 2 Adverse Reactions Occurring in ≥5% of OFEV-treated Patients with Systemic Sclerosis-Associated Interstitial Lung Disease and More Commonly Than Placebo in Study 4 Adverse Reaction OFEV, 150 mg
n=288Placebo
n=288a Includes abdominal pain, abdominal pain upper, abdominal pain lower, and esophageal pain. b Includes alanine aminotransferase increased, gamma-glutamyltransferase increased, aspartate aminotransferase increased, hepatic enzyme increased, blood alkaline phosphatase increased, transaminase increased, and hepatic function abnormal. c Includes hypertension, blood pressure increased, and hypertensive crisis. Diarrhea 76% 32% Nausea 32% 14% Vomiting 25% 10% Skin ulcer 18% 17% Abdominal paina 18% 11% Liver enzyme elevationb 13% 3% Weight decreased 12% 4% Fatigue 11% 7% Decreased appetite 9% 4% Headache 9% 8% Pyrexia 6% 5% Back pain 6% 4% Dizziness 6% 4% Hypertensionc 5% 2% In addition, alopecia was reported in patients treated with OFEV, more than placebo (1.4% vs. 1.0%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of OFEV. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse reactions have been identified during postapproval use of OFEV: drug-induced liver injury [see Warnings and Precautions (5.2)], non-serious and serious bleeding events, some of which were fatal [see Warnings and Precautions (5.6)], proteinuria [see Warnings and Precautions (5.8)], pancreatitis, thrombocytopenia, rash, pruritus.
-
7 DRUG INTERACTIONS
7.1 P-glycoprotein (P-gp) and CYP3A4 Inhibitors and Inducers
Nintedanib is a substrate of P-gp and, to a minor extent, CYP3A4 [see Clinical Pharmacology (12.3)]. Coadministration with oral doses of a P-gp and CYP3A4 inhibitor, ketoconazole, increased exposure to nintedanib by 60%. Concomitant use of P-gp and CYP3A4 inhibitors (e.g., erythromycin) with OFEV may increase exposure to nintedanib [see Clinical Pharmacology (12.3)]. In such cases, patients should be monitored closely for tolerability of OFEV. Management of adverse reactions may require interruption, dose reduction, or discontinuation of therapy with OFEV [see Dosage and Administration (2.4)].
Coadministration with oral doses of a P-gp and CYP3A4 inducer, rifampicin, decreased exposure to nintedanib by 50%. Concomitant use of P-gp and CYP3A4 inducers (e.g., carbamazepine, phenytoin, and St. John's wort) with OFEV should be avoided as these drugs may decrease exposure to nintedanib [see Clinical Pharmacology (12.3)].
7.2 Anticoagulants
Nintedanib is a VEGFR inhibitor and may increase the risk of bleeding. Monitor patients on full anticoagulation therapy closely for bleeding and adjust anticoagulation treatment as necessary [see Warnings and Precautions (5.6)].
7.3 Pirfenidone
In a multiple-dose study conducted to assess the pharmacokinetic effects of concomitant treatment with nintedanib and pirfenidone, the coadministration of nintedanib with pirfenidone did not alter the exposure of either agent [see Clinical Pharmacology (12.3)]. Therefore, no dose adjustment is necessary during concomitant administration of nintedanib with pirfenidone.
7.4 Bosentan
Coadministration of nintedanib with bosentan did not alter the pharmacokinetics of nintedanib [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], OFEV can cause fetal harm when administered to a pregnant woman. There are no data on the use of OFEV during pregnancy. In animal studies of pregnant rats and rabbits treated during organogenesis, nintedanib caused embryo-fetal deaths and structural abnormalities at less than (rats) and approximately 5 times (rabbits) the maximum recommended human dose [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and miscarriage in clinically recognized pregnancies is 15% to 20%.
Data
Animal Data
In animal reproduction toxicity studies, nintedanib caused embryo-fetal deaths and structural abnormalities in rats and rabbits at less than and approximately 5 times the maximum recommended human dose (MRHD) in adults (on a plasma AUC basis at maternal oral doses of 2.5 and 15 mg/kg/day in rats and rabbits, respectively). Malformations included abnormalities in the vasculature, urogenital, and skeletal systems. Vasculature anomalies included missing or additional major blood vessels. Skeletal anomalies included abnormalities in the thoracic, lumbar, and caudal vertebrae (e.g., hemivertebra, missing, or asymmetrically ossified), ribs (bifid or fused), and sternebrae (fused, split, or unilaterally ossified). In some fetuses, organs in the urogenital system were missing. In rabbits, a significant change in sex ratio was observed in fetuses (female:male ratio of approximately 71%:29%) at approximately 15 times the MRHD in adults (on an AUC basis at a maternal oral dose of 60 mg/kg/day). Nintedanib decreased post-natal viability of rat pups during the first 4 post-natal days when dams were exposed to less than the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
8.2 Lactation
Risk Summary
There is no information on the presence of nintedanib in human milk, the effects on the breast-fed infant or the effects on milk production. Nintedanib and/or its metabolites are present in the milk of lactating rats [see Data]. Because of the potential for serious adverse reactions in nursing infants from OFEV, advise women that breastfeeding is not recommended during treatment with OFEV.
8.3 Females and Males of Reproductive Potential
Based on findings from animal studies and its mechanism of action, OFEV can cause fetal harm when administered to a pregnant woman and may reduce fertility in females of reproductive potential [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1, 12.3), and Nonclinical Toxicology (13.1)]. Counsel patients on pregnancy prevention and planning.
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to treatment with OFEV and during treatment as appropriate [see Dosage and Administration (2.1), Warnings and Precautions (5.4), and Use in Specific Populations (8.1)].
Contraception
OFEV can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to avoid becoming pregnant while receiving treatment with OFEV. Advise females of reproductive potential to use highly effective contraception at initiation of, during treatment, and for at least 3 months after taking the last dose of OFEV. Nintedanib does not change the exposure to oral contraceptive containing ethinylestradiol and levonorgestrel in patients with SSc-ILD. However, the efficacy of oral hormonal contraceptives may be compromised by vomiting and/or diarrhea or other conditions where the drug absorption may be reduced. Advise women taking oral hormonal contraceptives experiencing these conditions to use alternative highly effective contraception.
Infertility
Based on animal data, OFEV may reduce fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
Of the total number of subjects in phase 2 and 3 clinical studies of OFEV in IPF (Study 1, Study 2, and Study 3), 61% were 65 and over, while 16% were 75 and over. In the chronic fibrosing ILDs with a progressive phenotype clinical study (Study 5), 61% were 65 and over, while 19% were 75 and older. In SSc-ILD (Study 4), 21.4% were 65 and over, while 1.9% were 75 and older. In phase 3 studies, no overall differences in effectiveness were observed between subjects who were 65 and over and younger subjects; no overall differences in safety were observed between subjects who were 65 and over or 75 and over and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
Nintedanib is predominantly eliminated via biliary/fecal excretion (greater than 90%). In a PK study performed in patients with hepatic impairment (Child Pugh A, Child Pugh B), exposure to nintedanib was increased [see Clinical Pharmacology (12.3)]. In patients with mild hepatic impairment (Child Pugh A), the recommended dosage of OFEV is 100 mg twice daily [see Dosage and Administration (2.3)]. Monitor for adverse reactions and consider treatment interruption, or discontinuation for management of adverse reactions in these patients [see Dosage and Administration (2.4)]. Treatment of patients with moderate (Child Pugh B) and severe (Child Pugh C) hepatic impairment with OFEV is not recommended [see Warnings and Precautions (5.1)].
8.7 Renal Impairment
Based on a single-dose study, less than 1% of the total dose of nintedanib is excreted via the kidney [see Clinical Pharmacology (12.3)]. Adjustment of the starting dose in patients with mild to moderate renal impairment is not required. The safety, efficacy, and pharmacokinetics of nintedanib have not been studied in patients with severe renal impairment (less than 30 mL/min CrCl) and end-stage renal disease.
8.8 Smokers
Smoking was associated with decreased exposure to OFEV [see Clinical Pharmacology (12.3)], which may alter the efficacy profile of OFEV. Encourage patients to stop smoking prior to treatment with OFEV and to avoid smoking when using OFEV.
-
10 OVERDOSAGE
In IPF trials, one patient was inadvertently exposed to a dose of 600 mg daily for a total of 21 days. A non-serious adverse event (nasopharyngitis) occurred and resolved during the period of incorrect dosing, with no onset of other reported events. Overdosage was also reported in two patients in oncology studies who were exposed to a maximum of 600 mg twice daily for up to 8 days. Adverse events reported were consistent with the existing safety profile of OFEV. Both patients recovered. In case of overdosage, interrupt treatment and initiate general supportive measures as appropriate.
-
11 DESCRIPTION
OFEV capsules contain nintedanib, a kinase inhibitor [see Mechanism of Action (12.1)]. Nintedanib is presented as the ethanesulfonate salt (esylate), with the chemical name 1H-Indole-6-carboxylic acid, 2,3-dihydro-3-[[[4-[methyl[(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-,methyl ester, (3Z)-, ethanesulfonate (1:1).
Its structural formula is:

Nintedanib esylate is a bright yellow powder with an empirical formula of C31H33N5O4∙C2H6O3S and a molecular weight of 649.76 g/mol.
OFEV capsules for oral administration are available in 2 dose strengths containing 100 mg or 150 mg of nintedanib (equivalent to 120.40 mg or 180.60 mg nintedanib ethanesulfonate, respectively). The inactive ingredients of OFEV are the following: Fill Material: triglycerides, hard fat, lecithin. Capsule Shell: gelatin, glycerol, titanium dioxide, red ferric oxide, yellow ferric oxide, black ink.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nintedanib is a small molecule that inhibits multiple receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs). Nintedanib inhibits the following RTKs: platelet-derived growth factor receptor (PDGFR) α and β, fibroblast growth factor receptor (FGFR) 1-3, vascular endothelial growth factor receptor (VEGFR) 1-3, colony stimulating factor 1 receptor (CSF1R), and Fms-like tyrosine kinase-3 (FLT-3). These kinases except for FLT-3 have been implicated in pathogenesis of interstitial lung diseases (ILD). Nintedanib binds competitively to the adenosine triphosphate (ATP) binding pocket of these kinases and blocks the intracellular signaling cascades, which have been demonstrated to be involved in the pathogenesis of fibrotic tissue remodeling in ILD. Nintedanib also inhibits the following nRTKs: Lck, Lyn and Src kinases. The contribution of FLT-3 and nRTK inhibition to nintedanib efficacy in ILD is unknown.
12.3 Pharmacokinetics
The PK properties of nintedanib were similar in healthy volunteers, patients with IPF, patients with chronic fibrosing ILDs with a progressive phenotype, patients with SSc-ILD, and cancer patients. The PK of nintedanib is linear. Dose proportionality was shown by an increase of nintedanib exposure with increasing doses (dose range 50 to 450 mg once daily and 150 to 300 mg twice daily). Accumulation upon multiple administrations in patients with IPF was 1.76-fold for AUC. Steady-state plasma concentrations were achieved within one week of dosing. Nintedanib trough concentrations remained stable for more than one year. The inter-individual variability in the PK of nintedanib was moderate to high (coefficient of variation of standard PK parameters in the range of 30% to 70%), intra-individual variability low to moderate (coefficients of variation below 40%).
Absorption
Nintedanib reached maximum plasma concentrations approximately 2 to 4 hours after oral administration as a soft gelatin capsule under fed conditions. The absolute bioavailability of a 100 mg dose was 4.7% (90% CI: 3.62 to 6.08) in healthy volunteers. Absorption and bioavailability are decreased by transporter effects and substantial first-pass metabolism.
After food intake, nintedanib exposure increased by approximately 20% compared to administration under fasted conditions (90% CI: 95.3% to 152.5%) and absorption was delayed (median tmax fasted: 2.00 hours; fed: 3.98 hours), irrespective of the food type.
Distribution
Nintedanib follows bi-phasic disposition kinetics. After intravenous infusion, a high volume of distribution which was larger than total body volume (Vss: 1050 L) was observed.
The in vitro protein binding of nintedanib in human plasma was high, with a bound fraction of 97.8%. Serum albumin is considered to be the major binding protein. Nintedanib is preferentially distributed in plasma with a blood to plasma ratio of 0.87.
Elimination
The effective half-life of nintedanib in patients with IPF was 9.5 hours (gCV 31.9%). Total plasma clearance after intravenous infusion was high (CL: 1390 mL/min; gCV 28.8%). Urinary excretion of unchanged drug within 48 hours was about 0.05% of the dose after oral and about 1.4% of the dose after intravenous administration; the renal clearance was 20 mL/min.
Metabolism
The prevalent metabolic reaction for nintedanib is hydrolytic cleavage by esterases resulting in the free acid moiety BIBF 1202. BIBF 1202 is subsequently glucuronidated by UGT enzymes, namely UGT 1A1, UGT 1A7, UGT 1A8, and UGT 1A10 to BIBF 1202 glucuronide. Only a minor extent of the biotransformation of nintedanib consisted of CYP pathways, with CYP3A4 being the predominant enzyme involved. The major CYP-dependent metabolite could not be detected in plasma in the human absorption, distribution, metabolism, and elimination study. In vitro, CYP-dependent metabolism accounted for about 5% compared to about 25% ester cleavage.
Excretion
The major route of elimination of drug-related radioactivity after oral administration of [14C] nintedanib was via fecal/biliary excretion (93.4% of dose), and the majority of OFEV was excreted as BIBF 1202. The contribution of renal excretion to the total clearance was low (0.65% of dose). The overall recovery was considered complete (above 90%) within 4 days after dosing.
Specific Populations
Age, Body Weight, and Sex
Based on population PK analysis, age and body weight were correlated with nintedanib exposure. However, the effects on exposure are not sufficient to warrant a dose adjustment. There was no influence of sex on the exposure of nintedanib.
Patients with Renal Impairment
Based on a population PK analysis of data from 933 patients with IPF, exposure to nintedanib was not influenced by mild (CrCl: 60 to 90 mL/min; n=399) or moderate (CrCl: 30 to 60 mL/min; n=116) renal impairment. Data in severe renal impairment (CrCl below 30 mL/min) was limited.
Patients with Hepatic Impairment
A dedicated single-dose phase I pharmacokinetics study of OFEV compared 8 subjects with mild hepatic impairment (Child Pugh A) and 8 subjects with moderate hepatic impairment (Child Pugh B) to 17 subjects with normal hepatic function. In subjects with mild hepatic impairment, the mean exposure to nintedanib was 2.4-fold higher based on Cmax (90% CI: 1.6 to 3.6) and 2.2-fold higher based on AUC0-inf (90% CI: 1.4 to 3.5). In subjects with moderate hepatic impairment, exposure was 6.9-fold higher based on Cmax (90% CI: 4.4 to 11.0) and 7.6-fold higher based on AUC0-inf (90% CI: 5.1 to 11.3). Subjects with severe hepatic impairment (Child Pugh C) have not been studied.
Drug Interaction Studies
Potential for Nintedanib to Affect Other Drugs
Effect of nintedanib coadministration on pirfenidone AUC and Cmax was evaluated in a multiple-dose study. Nintedanib did not have an effect on the exposure of pirfenidone.
Fifteen female patients with SSc-ILD received a single dose of a combination of 30 mcg ethinylestradiol and 150 mcg levonorgestrel before and after twice daily dosing of 150 mg nintedanib for at least 10 days. Co-administration of nintedanib did not change the exposure of ethinylestradiol and levonorgestrel [see Warnings and Precautions (5.4) and Use in Specific Populations (8.3)].
In in vitro studies, nintedanib was shown not to be an inhibitor of OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, or MRP-2. In vitro studies also showed that nintedanib has weak inhibitory potential on OCT-1, BCRP, and P-gp; these findings are considered to be of low clinical relevance. Nintedanib and its metabolites, BIBF 1202 and BIBF 1202 glucuronide, did not inhibit or induce CYP enzymes in vitro.
Potential for Other Drugs to Affect Nintedanib
Nintedanib is a substrate of P-gp and, to a minor extent, CYP3A4. Coadministration with the P-gp and CYP3A4 inhibitor, ketoconazole, increased exposure to nintedanib 1.61-fold based on AUC and 1.83-fold based on Cmax in a dedicated drug-drug interaction study. In a drug-drug interaction study with the P-gp and CYP3A4 inducer, rifampicin, exposure to nintedanib decreased to 50.3% based on AUC and to 60.3% based on Cmax upon coadministration with rifampicin compared to administration of nintedanib alone.
Effect of pirfenidone coadministration on nintedanib AUC and Cmax was evaluated in a multiple-dose drug-drug interaction study. Pirfenidone did not have an effect on the exposure of nintedanib. Concomitant treatment with nintedanib and pirfenidone was also investigated in a separate trial, which was an exploratory open-label, randomized (1:1) trial of nintedanib 150 mg twice daily with add-on pirfenidone (titrated to 801 mg three times a day) compared to nintedanib 150 mg twice daily alone in 105 randomized patients for 12 weeks. Similar nintedanib trough plasma concentrations were observed when comparing patients receiving nintedanib alone with patients receiving nintedanib with add-on pirfenidone.
Healthy volunteers received a single dose of 150 mg nintedanib before and after multiple dosing of 125 mg bosentan twice daily at steady state. Coadministration of nintedanib with bosentan did not alter the pharmacokinetics of nintedanib.
Nintedanib displays a pH-dependent solubility profile with increased solubility at acidic pH less than 3. However, in the clinical trials, coadministration with proton pump inhibitors or histamine H2 antagonists did not influence the exposure (trough concentrations) of nintedanib.
In in vitro studies, nintedanib was shown not to be a substrate of OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, MRP-2, or BCRP. In vitro studies also showed that nintedanib was a substrate of OCT-1; these findings are considered to be of low clinical relevance.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year oral carcinogenicity studies of nintedanib in rats and mice have not revealed any evidence of carcinogenic potential. Nintedanib was dosed up to 10 and 30 mg/kg/day in rats and mice, respectively. These doses were less than and approximately 4 times the MRHD on a plasma drug AUC basis.
Nintedanib was negative for genotoxicity in the in vitro bacterial reverse mutation assay, the mouse lymphoma cell forward mutation assay, and the in vivo rat micronucleus assay.
In rats, nintedanib reduced female fertility at exposure levels approximately 3 times the MRHD (on an AUC basis at an oral dose of 100 mg/kg/day). Effects included increases in resorption and post-implantation loss, and a decrease in gestation index. Changes in the number and size of corpora lutea in the ovaries were observed in chronic toxicity studies in rats and mice. An increase in the number of females with resorptions only was observed at exposures approximately equal to the MRHD (on an AUC basis at an oral dose of 20 mg/kg/day). Nintedanib had no effects on male fertility in rats at exposure levels approximately 3 times the MRHD (on an AUC basis at an oral dose of 100 mg/kg/day).
-
14 CLINICAL STUDIES
14.1 Idiopathic Pulmonary Fibrosis
The clinical efficacy of OFEV has been studied in 1231 patients with IPF in one phase 2 (Study 1 [NCT00514683]) and two phase 3 studies (Study 2 [NCT01335464] and Study 3 [NCT01335477]). These were randomized, double-blind, placebo-controlled studies comparing treatment with OFEV 150 mg twice daily to placebo for 52 weeks.
Study 2 and Study 3 were identical in design. Study 1 was very similar in design. Patients were randomized in a 3:2 ratio (1:1 for Study 1) to either OFEV 150 mg or placebo twice daily for 52 weeks. Study 1 also included other treatment arms (50 mg daily, 50 mg twice daily, and 100 mg twice daily) that are not further discussed. The primary endpoint was the annual rate of decline in Forced Vital Capacity (FVC). Time to first acute IPF exacerbation was a key secondary endpoint in Study 2 and Study 3 and a secondary endpoint in Study 1. Change from baseline in FVC percent predicted and survival were additional secondary endpoints in all studies.
Patients were required to have a diagnosis of IPF (ATS/ERS/JRS/ALAT criteria) for less than 5 years. Diagnoses were centrally adjudicated based on radiologic and, if applicable, histopathologic confirmation. Patients were required to be greater than or equal to 40 years of age with an FVC greater than or equal to 50% of predicted and a carbon monoxide diffusing capacity (DLCO, corrected for hemoglobin) 30% to 79% of predicted. Patients with relevant airways obstruction (i.e., pre-bronchodilator FEV1/FVC less than 0.7) or, in the opinion of the investigator, likely to receive a lung transplant during the studies were excluded (being listed for lung transplant was acceptable for inclusion). Patients with greater than 1.5 times ULN of ALT, AST, or bilirubin, patients with a known risk or predisposition to bleeding, patients receiving a full dose of anticoagulation treatment, and patients with a recent history of myocardial infarction or stroke were excluded from the studies. Patients were also excluded if they received other investigational therapy, azathioprine, cyclophosphamide, or cyclosporine A within 8 weeks of entry into this trial, or n-acetyl cysteine and prednisone (greater than 15 mg/day or equivalent) within 2 weeks. The majority of patients were Caucasian (60%) or Asian (30%) and male (79%). Patients had a mean age of 67 years and a mean FVC percent predicted of 80%.
Annual Rate of Decline in FVC
A statistically significant reduction in the annual rate of decline of FVC (in mL) was demonstrated in patients receiving OFEV compared to patients receiving placebo based on the random coefficient regression model, adjusted for gender, height, and age. The treatment effect on FVC was consistent in all 3 studies. See Table 3 for individual study results.
Table 3 Annual Rate of Decline in FVC (mL) in Study 1, Study 2, and Study 3a Study 1 Study 2 Study 3 OFEV
150 mg
twice dailyPlacebo OFEV
150 mg
twice dailyPlacebo OFEV
150 mg
twice dailyPlacebo aRandomized set in Study 1; treated set in Study 2 and Study 3
bEstimated based on a random coefficient regression modelNumber of analyzed patients 84 83 309 204 329 219 Ratea of decline over 52 weeks -60 -191 -115 -240 -114 -207 Comparison vs placebo Differenceb 131 125 94 95% CI (27, 235) (78, 173) (45, 143) Figure 1 displays the change from baseline over time in both treatment groups for Study 2. When the mean observed FVC change from baseline was plotted over time, the curves diverged at all timepoints through Week 52. Similar plots were seen for Study 1 and Study 3.
Figure 1 Mean (SEM) Observed FVC Change from Baseline (mL) Over Time in Study 2
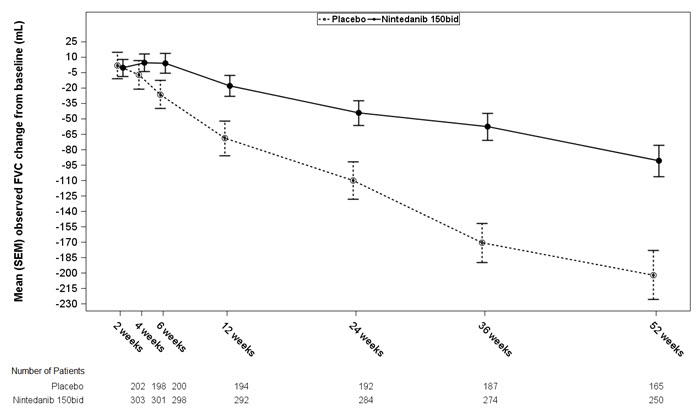
bid = twice daily
Change from Baseline in Percent Predicted Forced Vital Capacity
Figure 2 presents the cumulative distribution for all cut-offs for the change from baseline in FVC percent predicted at Week 52 for Study 2. For all categorical declines in lung function, the proportion of patients declining was lower on OFEV than on placebo. Study 3 showed similar results.
- *
- Missing data for change from baseline at Week 52 in percent predicted FVC (due to death, lost to follow-up or censoring before 52 weeks) was imputed using the worst decline from baseline at Week 52 observed among all patients with available data, regardless of treatment.
bid = twice daily
Figure 2 Cumulative Distribution of Patients by Change in Percent Predicted FVC from Baseline to Week 52 (Study 2).* The vertical lines indicate ≥0% decline or ≥10% decline. 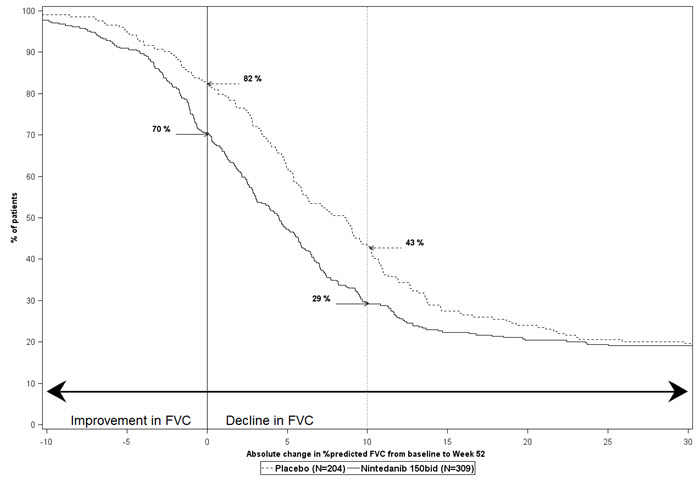
Time to First Acute IPF Exacerbation
Acute IPF exacerbation was defined as unexplained worsening or development of dyspnea within 30 days, new diffuse pulmonary infiltrates on chest x-ray, and/or new high-resolution CT parenchymal abnormalities with no pneumothorax or pleural effusion, and exclusion of alternative causes. Acute IPF exacerbation was adjudicated in Study 2 and Study 3. In Study 1 (investigator-reported) and Study 3 (adjudicated), the risk of first acute IPF exacerbation over 52 weeks was significantly reduced in patients receiving OFEV compared to placebo (hazard ratio [HR]: 0.16, 95% CI: 0.04, 0.71) and (HR: 0.20, 95% CI: 0.07, 0.56), respectively. In Study 2 (adjudicated), there was no difference between the treatment groups (HR: 0.55, 95% CI: 0.20, 1.54).
Survival
Survival was evaluated for OFEV compared to placebo in Study 2 and Study 3 as an exploratory analysis to support the primary endpoint (FVC). All-cause mortality was assessed over the study duration and available follow-up period, irrespective of cause of death and whether patients continued treatment. All-cause mortality did not show a statistically significant difference (See Figure 3).
Figure 3 Kaplan-Meier Estimates of All-Cause Mortality at Vital Status – End of Study: Study 2 and Study 3
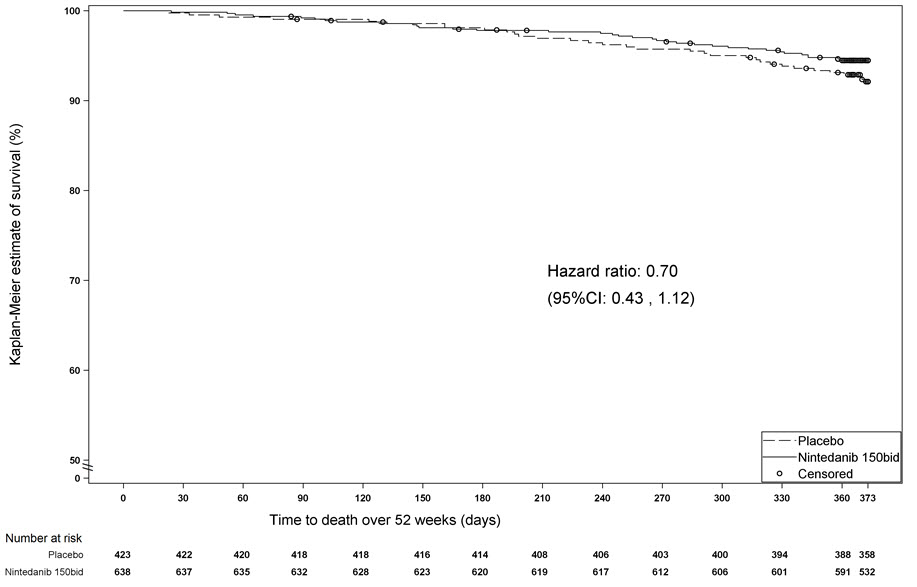
bid = twice daily
14.2 Chronic Fibrosing Interstitial Lung Diseases with a Progressive Phenotype
The clinical efficacy of OFEV has been studied in patients with chronic fibrosing ILDs with a progressive phenotype in a randomized, double-blind, placebo-controlled phase 3 trial (Study 5 [NCT02999178]). A total of 663 patients were randomized in a 1:1 ratio to receive either OFEV 150 mg twice daily or matching placebo for at least 52 weeks. Randomization was stratified based on high resolution computed tomography (HRCT) fibrotic pattern as assessed by central readers: 412 patients with UIP-like HRCT pattern and 251 patients with other HRCT fibrotic patterns were randomized. There were 2 co-primary populations defined for the analyses in this trial: all patients (the overall population) and patients with HRCT with UIP-like HRCT fibrotic pattern.
The primary endpoint was the annual rate of decline in FVC (in mL) over 52 weeks. Other endpoints included time to first acute ILD exacerbation and time to death.
Patients with a clinical diagnosis of a chronic fibrosing ILD were selected if they had relevant fibrosis (greater than 10% fibrotic features) on HRCT and presented with clinical signs of progression (defined as FVC decline ≥10%, FVC decline ≥ 5% and <10% with worsening symptoms or imaging, or worsening symptoms and worsening imaging all in the 24 months prior to screening). Patients were required to have an FVC greater than or equal to 45% of predicted and a DLCO 30% to less than 80% of predicted. Patients were required to have progressed despite management deemed appropriate in clinical practice by investigators for the patient's relevant ILD.
Patients with IPF, relevant airways obstruction (i.e., pre-bronchodilator FEV1/FVC less than 0.7), or significant pulmonary hypertension were excluded from the trial. Patients with greater than 1.5 times ULN of ALT, AST, or bilirubin, patients with a known risk or predisposition to bleeding, patients receiving a full dose of anticoagulation treatment, and patients with a recent history of myocardial infarction or stroke were excluded. Patients were also excluded if they received other investigational therapy, azathioprine, cyclosporine, mycophenolate mofetil, tacrolimus, oral corticosteroids greater than 20 mg/day, or the combination of oral corticosteroids + azathioprine + n-acetylcysteine within 4 weeks of randomization, cyclophosphamide within 8 weeks prior to randomization, rituximab within 6 months, or previous treatment with nintedanib or pirfenidone.
The majority of patients were Caucasian (74%) or Asian (25%). Patients were mostly male (54%) and had a mean age of 66 years and a mean FVC percent predicted of 69%, and 49% were never-smokers. The underlying clinical ILD diagnoses in groups represented in the trial were hypersensitivity pneumonitis (26%), autoimmune ILDs (26%), idiopathic nonspecific interstitial pneumonia (19%), unclassifiable idiopathic interstitial pneumonia (17%), and other ILDs (12%).
Annual Rate of Decline in FVC
There was a statistically significant reduction in the annual rate of decline in FVC (in mL) over 52 weeks in patients receiving OFEV compared to patients receiving placebo. The annual rate of decline in FVC (in mL) over 52 weeks was significantly reduced by 107 mL in patients receiving OFEV compared to patients receiving placebo. Results in the subpopulations of patients with HRCT with UIP-like fibrotic pattern and patients with other fibrotic patterns (Other HRCT) are included with the overall population in Table 4.
Table 4 Annual Rate of Decline in FVC (mL) in Study 5 Overall UIP-like Subpopulation Other HRCT Subpopulation OFEV Placebo OFEV Placebo OFEV Placebo aBased on a random coefficient regression model with fixed categorical effects of treatment, HRCT pattern, fixed continuous effects of time, baseline FVC (mL), and including treatment by time and baseline by time interactions Number of analyzed patients 331 331 206 206 125 125 Adjusted annual rate of decline over 52 weeks -81 -188 -83 -211 -79 -154 Comparison vs placebo differencea 107 128 75* 95% CI (65, 148) (71, 186) (16, 135)* *Comparison based on the Other HRCT subpopulation was not included in the multiple testing procedure. Values shown here are for descriptive purposes. A post-hoc exploratory analysis by ILD diagnosis was performed and is shown in Figure 4. Treatment response across ILD diagnoses was consistent for FVC.
Figure 4 Annual Rate of Decline in FVC (mL) over 52 Weeks based on Underlying ILD Diagnosis in Study 5*
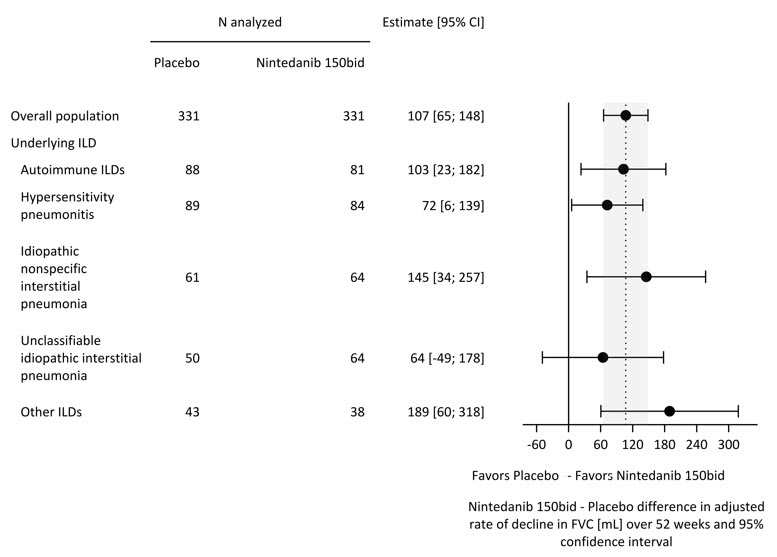
ILD = interstitial lung disease; Autoimmune ILDs: includes rheumatoid arthritis-associated ILD, mixed connective tissue disease, systemic sclerosis-associated ILD, and other terms; Other ILDs: includes fibrosing ILDs not categorized under autoimmune ILDs, hypersensitivity pneumonitis, idiopathic nonspecific interstitial pneumonia, or unclassifiable idiopathic interstitial pneumonia. The three most common ILDs in this category are exposure-related ILD, sarcoidosis, and pleuro-parenchymal fibroelastosis.
*These results are from a post-hoc exploratory analysis. Values shown here are for descriptive purposes.Figure 5 shows the change in FVC from baseline over time in the treatment groups. When the mean observed FVC change from baseline was plotted over time, the curves diverged at all timepoints through Week 52.
Figure 5 Mean (SEM) Observed FVC Change from Baseline (mL) Over 52 Weeks in Study 5
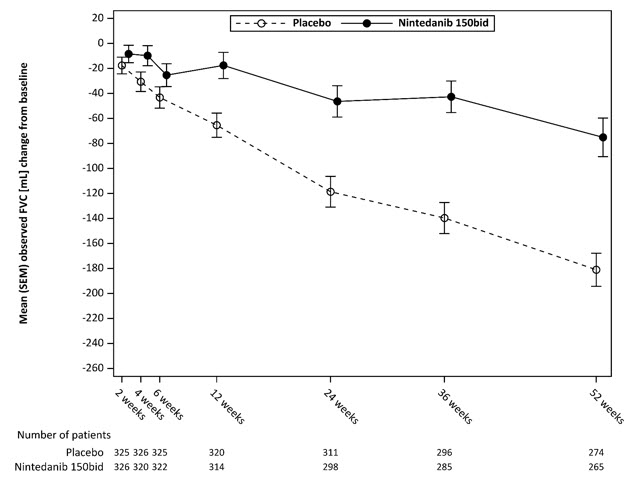
bid = twice daily
Percent Change from Baseline in Forced Vital Capacity
Figure 6 presents the percent change from baseline in FVC in mL at Week 52 for Study 5. For the majority of patients, the decline in lung function was less on OFEV than on placebo.
Figure 6 Histogram of the Percent Change in FVC (mL) from Baseline to Week 52 According to Treatment and Percent Increments or Decrements of 5 (Study 5)a
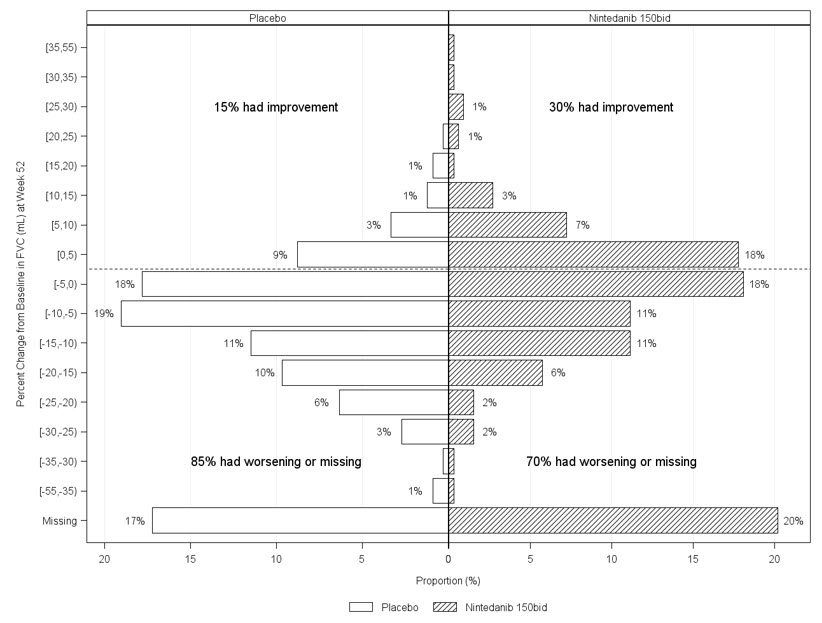
a Patients classified as having missing FVC data at Week 52 are those with no FVC assessment between Day 310 and Day 373.
bid = twice dailyTime to First Acute ILD Exacerbation
Acute ILD exacerbation was defined as unexplained worsening or development of dyspnea within 30 days, new diffuse pulmonary infiltrates on chest x-ray, and/or new HRCT parenchymal abnormalities with no pneumothorax or pleural effusion, and exclusion of alternative causes. Acute ILD exacerbations were not adjudicated.
The risk of first acute ILD exacerbation did not show a statistically significant difference between the OFEV group compared to placebo (52 week treatment period: HR 0.72, (95% CI: 0.38, 1.37); whole trial: HR 0.63 (95% CI: 0.37, 1.07)).
Survival
Survival was evaluated for OFEV compared to placebo in Study 5 to support the primary endpoint (FVC). All-cause mortality was assessed over the study duration and available follow-up period, irrespective of cause of death and whether patients continued treatment. All-cause mortality did not show a statistically significant difference (52 week treatment period: HR 0.94 (95% CI: 0.47, 1.86); whole trial: HR 0.78 (95% CI: 0.50, 1.21)).
14.3 Systemic Sclerosis-Associated Interstitial Lung Disease
The clinical efficacy of nintedanib has been studied in patients with SSc-ILD in a randomized, double-blind, placebo-controlled phase 3 trial (Study 4 [NCT02597933]). A total of 580 patients were randomized in a 1:1 ratio to receive either OFEV 150 mg twice daily or matching placebo for at least 52 weeks, of which 576 patients were treated. Randomization was stratified by anti-topoisomerase antibody (ATA) status. Individual patients remained on blinded trial treatment for up to 100 weeks. The primary endpoint was the annual rate of decline in FVC over 52 weeks. The absolute change from baseline in the modified Rodnan skin score (mRSS) at Week 52 was a key secondary endpoint. Mortality over the whole trial was an additional secondary endpoint.
Patients were diagnosed with SSc-ILD based upon the 2013 American College of Rheumatology / European League Against Rheumatism classification criteria for SSc with onset of disease (first non-Raynaud symptom) of less than 7 years and greater than or equal to 10% fibrosis on a chest high resolution computed tomography (HRCT) scan conducted within the previous 12 months. Patients were required to have an FVC greater than or equal to 40% of predicted and a DLCO 30-89% of predicted. Patients with relevant airways obstruction (i.e., pre-bronchodilator FEV1/FVC less than 0.7) or previous or planned hematopoietic stem cell transplant were excluded from the trial. Patients with greater than 1.5 times ULN of ALT, AST, or bilirubin, patients with a known risk or predisposition to bleeding, patients receiving a full dose of anticoagulation treatment, and patients with a recent history of myocardial infarction or stroke were excluded from the study. Patients were excluded if they had significant pulmonary hypertension, more than three digital fingertip ulcers, a history of severe digital necrosis requiring hospitalization, or a history of scleroderma renal crisis. Patients were also excluded if they received other investigational therapy, azathioprine within 8 weeks prior to randomization, cyclophosphamide or cyclosporine A within 6 months prior to randomization, or previous treatment with nintedanib or pirfenidone.
The majority of patients were female (75%). Patients were mostly Caucasian (67%), Asian (25%), or Black (6%). The mean age was 54 years. Overall, 52% of patients had diffuse cutaneous systemic sclerosis (SSc) and 48% had limited cutaneous SSc. The mean time since first onset of a non-Raynaud symptom was 3.49 years. At baseline, 49% of patients were on stable therapy with mycophenolate.
Annual Rate of Decline in FVC
The annual rate of decline of FVC (in mL) over 52 weeks was significantly reduced by 41 mL in patients receiving OFEV compared to patients receiving placebo, corresponding to a relative treatment effect of 44%. See Table 5.
Table 5 Annual Rate of Decline in FVC (mL) in Study 4 OFEV
150 mg
twice dailyPlacebo aBased on a random coefficient regression model, adjusted for gender, height, age, ATA status, FVC at baseline, FVC at baseline-by-time Number of analyzed patients 287 288 Adjusted rate of decline over 52 weeks -52 -93 Comparison vs placebo Differencea 41 95% CI (3, 79) Figure 7 displays the change from baseline over time in both treatment groups. When the mean observed FVC change from baseline was plotted over time, the curves diverged at all timepoints through Week 52. Separation of the mean values is seen after 12 weeks of treatment.
Figure 7 Mean (SEM) Observed FVC Change from Baseline (mL) Over Time in Study 4
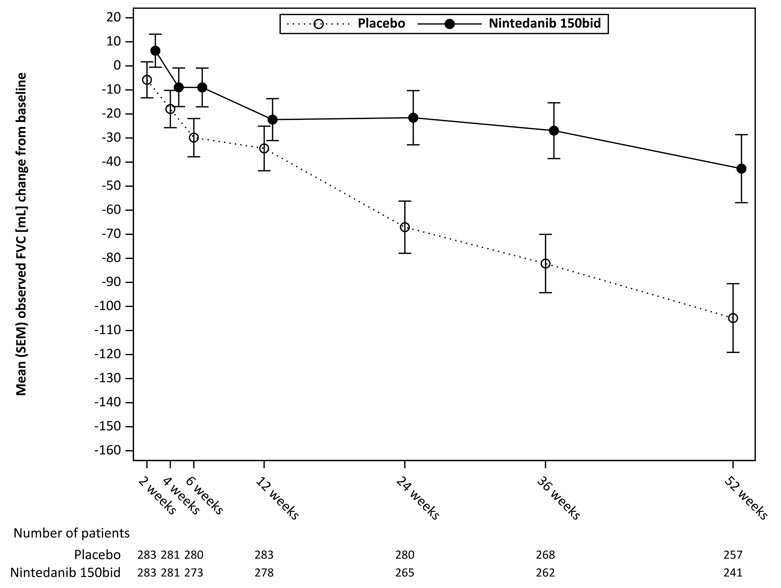
bid = twice daily
In two pre-specified subgroup efficacy analyses, the mean treatment difference in FVC decline at 52 weeks in patients were examined by region and mycophenolate use (Figure 8).
Figure 8 Subgroup Analyses of the Mean Treatment Difference in FVC (mL) Decline at Week 52 by Region and Mycophenolate Use (Study 4)
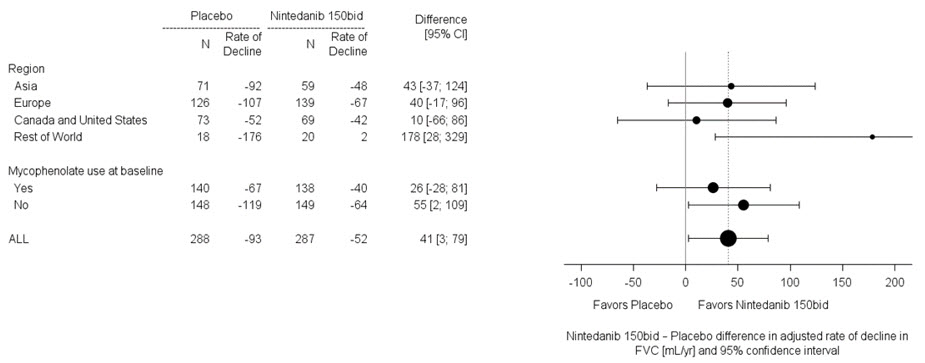
Percent Change from Baseline in Forced Vital Capacity
Figure 9 presents the percent change from baseline in FVC in mL at Week 52 for Study 4. For the majority of patients, the decline in lung function was less on OFEV than on placebo.
a Patients classified as having missing FVC data at Week 52 are those with no FVC assessment between Day 310 and Day 373. Figure 9 Histogram of the Percent Change in FVC (mL) from Baseline to Week 52 According to Treatment and Percent Increments or Decrements of 5 (Study 4)a 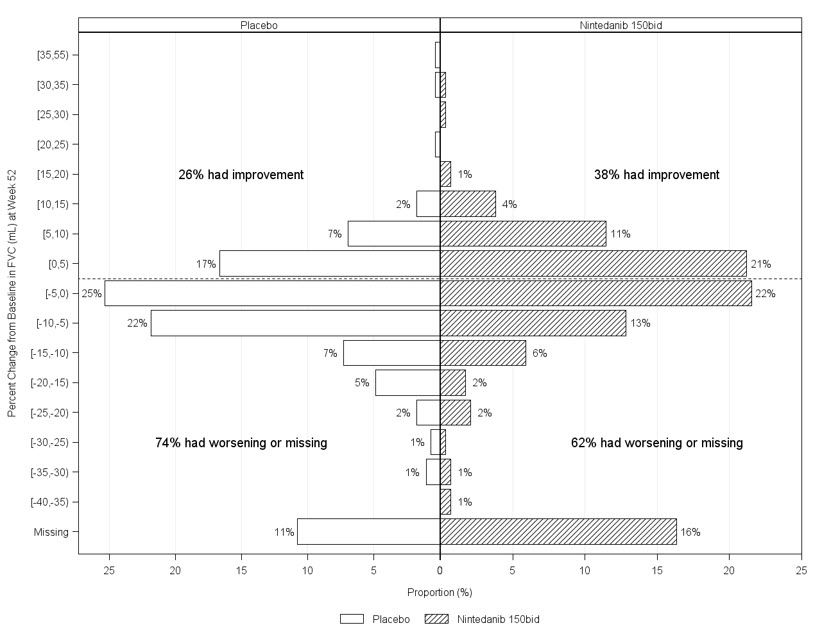
bid = twice daily
Modified Rodnan Skin Score
No benefit in mRSS was observed in patients receiving OFEV. The adjusted mean absolute change from baseline in mRSS at Week 52 was comparable between the OFEV group (-2.17 (95% CI: -2.69, -1.65)) and the placebo group (-1.96 (95% CI: -2.48, -1.45)). The adjusted mean difference between the treatment groups was -0.21 (95% CI: -0.94, 0.53).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
150 mg: brown, opaque, oblong, soft capsules imprinted in black with the Boehringer Ingelheim company symbol and "150". They are packaged in HDPE bottles with a child-resistant closure, available as follows:
Bottles of 60 NDC: 0597-0145-60 100 mg: peach, opaque, oblong, soft capsules imprinted in black with the Boehringer Ingelheim company symbol and "100". They are packaged in HDPE bottles with a child-resistant closure, available as follows:
Bottles of 60 NDC: 0597-0143-60 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Elevated Liver Enzymes and Drug-Induced Liver Injury
Advise patients that they will need to undergo liver function testing periodically. Advise patients to immediately report any symptoms of a liver problem (e.g., skin or the whites of eyes turn yellow, urine turns dark or brown (tea colored), pain on the right side of stomach, bleed or bruise more easily than normal, lethargy, loss of appetite) [see Warnings and Precautions (5.2)].
Gastrointestinal Disorders
Inform patients that gastrointestinal disorders such as diarrhea, nausea, and vomiting were the most commonly reported gastrointestinal events occurring in patients who received OFEV. Advise patients that their healthcare provider may recommend hydration, antidiarrheal medications (e.g., loperamide), or anti-emetic medications to treat these side effects. Temporary dosage reductions or discontinuations may be required. Instruct patients to contact their healthcare provider at the first signs of diarrhea or for any severe or persistent diarrhea, nausea, or vomiting [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Counsel patients on pregnancy prevention and planning. Advise females of reproductive potential of the potential risk to a fetus and to avoid becoming pregnant while receiving treatment with OFEV. Advise females of reproductive potential to use highly effective contraception at initiation of, during treatment, and for at least 3 months after taking the last dose of OFEV. Advise women taking oral hormonal contraceptives who experience vomiting and/or diarrhea or other conditions where the drug absorption may be reduced to contact their doctor to discuss alternative highly effective contraception. Advise female patients to notify their doctor if they become pregnant or suspect they are pregnant during therapy with OFEV [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1, 8.3)].
Arterial Thromboembolic Events
Advise patients about the signs and symptoms of acute myocardial ischemia and other arterial thromboembolic events and the urgency to seek immediate medical care for these conditions [see Warnings and Precautions (5.5)].
Risk of Bleeding
Bleeding events have been reported. Advise patients to report unusual bleeding [see Warnings and Precautions (5.6)].
Gastrointestinal Perforation
Serious gastrointestinal perforation events have been reported. Advise patients to report signs and symptoms of gastrointestinal perforation [see Warnings and Precautions (5.7)].
Nephrotic Range Proteinuria
Nephrotic range proteinuria has been reported. Advise patients to report signs and symptoms of proteinuria (e.g., fluid retention, foamy urine) [see Warnings and Precautions (5.8)].
Lactation
Advise patients that breastfeeding is not recommended while taking OFEV [see Use in Specific Populations (8.2)].
Smokers
Encourage patients to stop smoking prior to treatment with OFEV and to avoid smoking when using OFEV [see Clinical Pharmacology (12.3)].
Administration
Instruct patients to take OFEV with food, to swallow OFEV capsules whole with liquid, and not to chew the capsules due to the bitter taste. Advise patients or caregivers not to open or crush OFEV capsules and to wash hands immediately and thoroughly if contact with the content of the capsule occurs. Advise patients to not make up for a missed dose [see Dosage and Administration (2.2)].
-
SPL UNCLASSIFIED SECTION
Distributed by:
Boehringer Ingelheim Pharmaceuticals, Inc.
Ridgefield, CT 06877 USALicensed from:
Boehringer Ingelheim International GmbHOFEV is a registered trademark of and used under license from Boehringer Ingelheim International GmbH.
Copyright © 2022 Boehringer Ingelheim International GmbH
ALL RIGHTS RESERVEDCOL9114EJ132022
SPL9116D -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: October 2022 Patient Information
OFEV® (OH-fev)
(nintedanib capsules)What is the most important information I should know about OFEV? - OFEV can cause birth defects or death to an unborn baby. Women should not become pregnant while taking OFEV. Women who are able to become pregnant should have a pregnancy test before starting treatment with OFEV.
- Women who are able to become pregnant should use highly effective birth control at the start of treatment, during treatment, and for at least 3 months after treatment. Talk with your doctor about what birth control method is right for you during this time.
- Birth control pills may not work as well in women having vomiting, diarrhea, or other problems reducing the drug absorption. If you have any of these problems, talk with your doctor about which highly effective birth control method is right for you.
- If you become pregnant or think you are pregnant while taking OFEV, tell your doctor right away.
What is OFEV? - OFEV is a prescription medicine used:
- to treat adults with a lung disease called idiopathic pulmonary fibrosis (IPF).
- to treat adults with a long-lasting (chronic) interstitial lung disease in which lung fibrosis continues to worsen (progress).
- to slow the rate of decline in lung function in adults with systemic sclerosis-associated interstitial lung disease (SSc-ILD) (also known as scleroderma-associated ILD).
- It is not known if OFEV is safe and effective in children.
What should I tell my doctor before taking OFEV?
Before you take OFEV, tell your doctor about all of your medical conditions, including if you:- have liver problems.
- have heart problems.
- have a history of blood clots.
- have a bleeding problem or a family history of a bleeding problem.
- have had recent surgery in your stomach (abdominal) area.
- are a smoker.
- are pregnant or plan to become pregnant. OFEV can harm your unborn baby. OFEV can cause birth defects or death to an unborn baby. See "What is the most important information I should know about OFEV?"
- are breastfeeding or plan to breastfeed. It is not known if OFEV passes into your breast milk. You should not breastfeed while taking OFEV.
How should I take OFEV? - Take OFEV exactly as your doctor tells you to take it.
- Your doctor will tell you how much OFEV to take and when to take it.
- Take OFEV with food. Swallow the OFEV capsules whole with a liquid.
- Do not chew, crush, or open OFEV capsules. If you or your caregiver accidently comes in contact with the content of the capsule, wash hands well right away.
- If you miss a dose of OFEV, take your next dose at your regular time. Do not take the missed dose.
- Do not take more than 300 mg of OFEV in 1 day.
- If you take too much OFEV, call your doctor or go to the nearest hospital emergency room right away.
- Your doctor should do certain blood tests before you start taking OFEV.
What are the possible side effects of OFEV?
OFEV may cause serious side effects, including:- See "What is the most important information I should know about OFEV?"
- liver problems. Call your doctor right away if you have unexplained symptoms such as yellowing of your skin or the white part of your eyes (jaundice), dark or brown (tea colored) urine, pain on the upper right side of your stomach area (abdomen), bleeding or bruising more easily than normal, feeling tired, or loss of appetite. Your doctor will do blood tests to check how well your liver is working before starting and during your treatment with OFEV.
- diarrhea, nausea, and vomiting. While you are taking OFEV, your doctor may recommend that you drink fluids or take medicine to treat these side effects. Tell your doctor if you have diarrhea, nausea, or vomiting or if these symptoms do not go away or become worse. Tell your doctor if you are taking over-the-counter laxatives, stool softeners, and other medicines or dietary supplements that can cause diarrhea.
- heart attack. Tell your doctor right away if you have symptoms of a heart problem. These symptoms may include chest pain or pressure, pain in your arms, back, neck or jaw, or shortness of breath.
- stroke. Tell your doctor right away if you have symptoms of a stroke. These symptoms may include numbness or weakness on 1 side of your body, trouble talking, headache, or dizziness.
- bleeding problems. OFEV may increase your chances of having bleeding problems. Tell your doctor if you have unusual bleeding, bruising, or wounds that do not heal. Tell your doctor if you are taking a blood thinner, including prescription blood thinners and over-the-counter aspirin.
- tear in your stomach or intestinal wall (perforation). OFEV may increase your chances of having a tear in your stomach or intestinal wall. Tell your doctor if you have pain or swelling in your stomach area.
- increased protein in your urine (proteinuria). OFEV may increase your chances of having protein in your urine. Tell your doctor if you have any signs and symptoms of protein in the urine such as foamy urine, swelling, including in your hands, arms, legs, or feet, or sudden weight gain.
These are not all the possible side effects of OFEV. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store OFEV? - Store OFEV at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep OFEV dry and protect from high heat.
General information about the safe and effective use of OFEV.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use OFEV for a condition for which it was not prescribed. Do not give OFEV to other people, even if they have the same symptoms you have. It may harm them. This Patient Information leaflet summarizes the most important information about OFEV. If you would like more information, talk to your doctor. You can ask your pharmacist or doctor for information about OFEV that is written for health professionals.
For more information about OFEV, including the current prescribing information, and patient support, go to www.ofev.com/support or call Boehringer Ingelheim Pharmaceuticals, Inc. at 1-800-542-6257, or scan the code to go to www.ofev.com/support.
What are the ingredients in OFEV?
Active ingredient: nintedanib
Inactive ingredients: Fill Material: triglycerides, hard fat, lecithin. Capsule Shell: gelatin, glycerol, titanium dioxide, red ferric oxide, yellow ferric oxide, black ink
Distributed by: Boehringer Ingelheim Pharmaceuticals, Inc. Ridgefield, CT 06877 USA
Licensed from: Boehringer Ingelheim International GmbH
OFEV is a registered trademark of and used under license from Boehringer Ingelheim International GmbH.
Copyright © 2022 Boehringer Ingelheim International GmbH, ALL RIGHTS RESERVED
COL9114EJ132022 - PRINCIPAL DISPLAY PANEL - 150 mg Capsule Bottle Carton
- PRINCIPAL DISPLAY PANEL - 100 mg Capsule Bottle Carton
-
INGREDIENTS AND APPEARANCE
OFEV
nintedanib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0597-0145 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NINTEDANIB ESYLATE (UNII: 42F62RTZ4G) (NINTEDANIB - UNII:G6HRD2P839) NINTEDANIB 150 mg Product Characteristics Color BROWN Score no score Shape OVAL Size 18mm Flavor Imprint Code 150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0597-0145-60 1 in 1 CARTON 10/17/2014 1 60 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205832 10/15/2014 OFEV
nintedanib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0597-0143 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NINTEDANIB ESYLATE (UNII: 42F62RTZ4G) (NINTEDANIB - UNII:G6HRD2P839) NINTEDANIB 100 mg Product Characteristics Color PINK (peach) Score no score Shape OVAL Size 16mm Flavor Imprint Code 100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0597-0143-60 1 in 1 CARTON 10/17/2014 1 60 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205832 10/15/2014 Labeler - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Registrant - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Establishment Name Address ID/FEI Business Operations Boehringer Ingelheim Pharma GmbH and Co. KG 551147440 API MANUFACTURE(0597-0143, 0597-0145) , PACK(0597-0143, 0597-0145) , LABEL(0597-0143, 0597-0145) Establishment Name Address ID/FEI Business Operations Catalent Germany Eberbach GmbH 318612223 MANUFACTURE(0597-0143, 0597-0145) Establishment Name Address ID/FEI Business Operations Sixarp, LLC - Praxis Packaging Solutions 016329513 PACK(0597-0143, 0597-0145) , LABEL(0597-0143, 0597-0145) Establishment Name Address ID/FEI Business Operations Bidachem S.p.a. 429232812 ANALYSIS(0597-0143, 0597-0145) , API MANUFACTURE(0597-0143, 0597-0145)

