Label: FAMOTIDINE for suspension


-
Contains inactivated NDC Code(s)
NDC Code(s): 40032-500-11 - Packager: Novel Laboratories, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated January 8, 2013
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
The active ingredient in Famotidine for Oral Suspension is a histamine H2-receptor antagonist. Famotidine is N-(aminosulfonyl)-3-[[[2-[(diaminomethylene)amino]-4- thiazolyl]methyl]thio]propanimidamide. The empirical formula of famotidine is C8H15N7O2S3 and its molecular weight is 337.43. Its structural formula is:
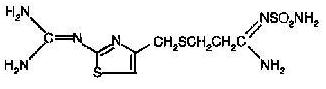
Famotidine is a white to pale yellow crystalline compound that is freely soluble in glacial acetic acid, slightly soluble in methanol, very slightly soluble in water, and practically insoluble in ethanol.Each 5 mL of the oral suspension when prepared as directed contains 40 mg of famotidine and the following inactive ingredients: anhydrous citric acid, flavors (cherry, banana, and mint), microcrystalline cellulose and carboxymethylcellulose sodium, confectioner’s sugar, corn starch, colloidal silicon-dioxide and xanthan gum. Added as preservatives are sodium benzoate and sodium methylparaben.
-
CLINICAL PHARMACOLOGY IN ADULTS
GI Effects
Famotidine is a competitive inhibitor of histamine H2-receptors. The primary clinically important pharmacologic activity of Famotidine is inhibition of gastric secretion. Both the acid concentration and volume of gastric secretion are suppressed by Famotidine, while changes in pepsin secretion are proportional to volume output.
In normal volunteers and hypersecretors, Famotidine inhibited basal and nocturnal gastric secretion, as well as secretion stimulated by food and pentagastrin. After oral administration, the onset of the antisecretory effect occurred within one hour; the maximum effect was dose-dependent, occurring within one to three hours. Duration of inhibition of secretion by doses of 20 and 40 mg was 10 to 12 hours.
Single evening oral doses of 20 and 40 mg inhibited basal and nocturnal acid secretion in all subjects; mean nocturnal gastric acid secretion was inhibited by 86% and 94%, respectively, for a period of at least 10 hours. The same doses given in the morning suppressed food-stimulated acid secretion in all subjects. The mean suppression was 76% and 84%, respectively, 3 to 5 hours after administration, and 25% and 30%, respectively, 8 to 10 hours after administration. In some subjects who received the 20-mg dose, however, the antisecretory effect was dissipated within 6 to 8 hours. There was no cumulative effect with repeated doses. The nocturnal intragastric pH was raised by evening doses of 20 and 40 mg of Famotidine to mean values of 5.0 and 6.4, respectively. When Famotidine was given after breakfast, the basal daytime interdigestive pH at 3 and 8 hours after 20 or 40 mg of Famotidine was raised to about 5.
Famotidine had little or no effect on fasting or postprandial serum gastrin levels. Gastric emptying and exocrine pancreatic function were not affected by Famotidine.
Other Effects
Systemic effects of Famotidine in the CNS, cardiovascular, respiratory or endocrine systems were not noted in clinical pharmacology studies. Also, no antiandrogenic effects were noted. (See ADVERSE REACTIONS.) Serum hormone levels, including prolactin, cortisol, thyroxine (T4), and testosterone, were not altered after treatment with Famotidine.
Pharmacokinetics
Famotidine is incompletely absorbed. The bioavailability of oral doses is 40 to 45%. Famotidine Tablets and Famotidine for Oral Suspension are bioequivalent. Bioavailability may be slightly increased by food, or slightly decreased by antacids; however, these effects are of no clinical consequence. Famotidine undergoes minimal first-pass metabolism. After oral doses, peak plasma levels occur in 1 to 3 hours. Plasma levels after multiple doses are similar to those after single doses. Fifteen to 20% of Famotidine in plasma is protein bound. Famotidine has an elimination half-life of 2.5 to 3.5 hours. Famotidine is eliminated by renal (65 to 70%) and metabolic (30 to 35%) routes. Renal clearance is 250 to 450 mL/min, indicating some tubular excretion. Twenty-five to 30% of an oral dose and 65 to 70% of an intravenous dose are recovered in the urine as unchanged compound. The only metabolite identified in man is the S-oxide.
There is a close relationship between creatinine clearance values and the elimination half-life of Famotidine. In patients with severe renal insufficiency, i.e., creatinine clearance less than 10 mL/min, the elimination half-life of Famotidine may exceed 20 hours and adjustment of dose or dosing intervals in moderate and severe renal insufficiency may be necessary (see PRECAUTIONS, DOSAGE AND ADMINISTRATION).
In elderly patients, there are no clinically significant age-related changes in the pharmacokinetics of Famotidine. However, in elderly patients with decreased renal function, the clearance of the drug may be decreased (see PRECAUTIONS, Geriatric Use).
Duodenal Ulcer
In a U.S. multicenter, double-blind study in outpatients with endoscopically confirmed duodenal ulcer, orally administered Famotidine was compared to placebo. As shown in Table 1, 70% of patients treated with Famotidine 40 mg h.s. were healed by week 4.
Table 1 Outpatients with Endoscopically Confirmed Healed Duodenal Ulcers Famotidine
40 mg h.s.
(N = 89)Famotidine
20 mg b.i.d.
(N = 84)Placebo
h.s
(N = 97)** Statistically significantly different than placebo (p<0.001) Week 2 **32% **38% 17% Week 4 **70% **67% 31%
Patients not healed by week 4 were continued in the study. By week 8, 83% of patients treated with Famotidine had healed versus 45% of patients treated with placebo. The incidence of ulcer healing with Famotidine was significantly higher than with placebo at each time point based on proportion of endoscopically confirmed healed ulcers.In this study, time to relief of daytime and nocturnal pain was significantly shorter for patients receiving Famotidine than for patients receiving placebo; patients receiving Famotidine also took less antacid than the patients receiving placebo.
Long-Term Maintenance
Treatment of Duodenal Ulcers
Famotidine, 20 mg p.o. h.s., was compared to placebo h.s. as maintenance therapy in two double-blind, multicenter studies of patients with endoscopically confirmed healed duodenal ulcers. In the U.S. study the observed ulcer incidence within 12 months in patients treated with placebo was 2.4 times greater than in the patients treated with Famotidine. The 89 patients treated with Famotidine had a cumulative observed ulcer incidence of 23.4% compared to an observed ulcer incidence of 56.6% in the 89 patients receiving placebo (p<0.01). These results were confirmed in an international study where the cumulative observed ulcer incidence within 12 months in the 307 patients treated with Famotidine was 35.7%, compared to an incidence of 75.5% in the 325 patients treated with placebo (p<0.01).
Gastric Ulcer
In both a U.S. and an international multicenter, double-blind study in patients with endoscopically confirmed active benign gastric ulcer, orally administered Famotidine, 40 mg h.s., was compared to placebo h.s. Antacids were permitted during the studies, but consumption was not significantly different between the Famotidine and placebo groups. As shown in Table 2, the incidence of ulcer healing (dropouts counted as unhealed) with Famotidine was statistically significantly better than placebo at weeks 6 and 8 in the U.S. study, and at weeks 4, 6 and 8 in the international study, based on the number of ulcers that healed, confirmed by endoscopy.
Table 2 Patients with Endoscopically Confirmed Healed Gastric Ulcers U.S. Study International Study ***,† Statistically significantly better than placebo (p≤0.05, p≤0.01 respectively) Famotidine40 mg h.s.(N=74) Placeboh.s.(N=75) Famotidine40 mg h.s.(N=149) Placeboh.s.(N=145) Week 4 45% 39% †47% 31% Week 6 †66% 44% †65% 46% Week 8 ***78% 64% †80% 54% Time to complete relief of daytime and nighttime pain was statistically significantly shorter for patients receiving Famotidine than for patients receiving placebo; however, in neither study was there a statistically significant difference in the proportion of patients whose pain was relieved by the end of the study (week 8).
Gastroesophageal Reflux Disease (GERD)
Orally administered Famotidine was compared to placebo in a U.S. study that enrolled patients with symptoms of GERD and without endoscopic evidence of erosion or ulceration of the esophagus. Famotidine 20 mg b.i.d. was statistically significantly superior to 40 mg h.s. and to placebo in providing a successful symptomatic outcome, defined as moderate or excellent improvement of symptoms (Table 3).
Table 3 % Successful Symptomatic Outcome Famotidine20 mg b.i.d.(N=154) Famotidine40 mg h.s.(N=149) Placebo(N=73) †† p≤0.01 vs Placebo Week 6 82†† 69 62 By two weeks of treatment symptomatic success was observed in a greater percentage of patients taking Famotidine 20 mg b.i.d. compared to placebo (p≤0.01).
Symptomatic improvement and healing of endoscopically verified erosion and ulceration were studied in two additional trials. Healing was defined as complete resolution of all erosions or ulcerations visible with endoscopy. The U.S. study comparing Famotidine 40 mg p.o. b.i.d. to placebo and Famotidine 20 mg p.o. b.i.d. showed a significantly greater percentage of healing for Famotidine 40 mg b.i.d. at weeks 6 and 12 (Table 4).
Table 4 % Endoscopic Healing - U.S. Study Famotidine40 mg b.i.d.(N=127) Famotidine20 mg b.i.d.(N=125) Placebo(N=66) ††† p≤0.01 vs Placebo
‡ p≤0.05 vs Famotidine 20 mg b.i.d.
‡‡ p≤0.01 vs Famotidine 20 mg b.i.d.Week 6 48†††,‡‡ 32 18 Week 12 69†††,‡ 54††† 29 As compared to placebo, patients who received Famotidine had faster relief of daytime and nighttime heartburn and a greater percentage of patients experienced complete relief of nighttime heartburn. These differences were statistically significant.
In the international study, when Famotidine 40 mg p.o. b.i.d. was compared to ranitidine 150 mg p.o. b.i.d., a statistically significantly greater percentage of healing was observed with Famotidine 40 mg b.i.d. at week 12 (Table 5). There was, however, no significant difference among treatments in symptom relief.
Table 5 % Endoscopic Healing - International Study Famotidine40 mg b.i.d.(N=175) Famotidine20 mg b.i.d.(N=93) Ranitidine150 mg b.i.d.(N=172) ‡‡‡ p≤0.05 vs Ranitidine 150 mg b.i.d. Week 6 48 52 42 Week 12 71‡‡‡ 68 60 Pathological Hypersecretory Conditions (e.g., Zollinger-Ellison Syndrome, Multiple Endocrine Adenomas)
In studies of patients with pathological hypersecretory conditions such as Zollinger-Ellison Syndrome with or without multiple endocrine adenomas, Famotidine significantly inhibited gastric acid secretion and controlled associated symptoms. Orally administered doses from 20 to 160 mg q 6 h maintained basal acid secretion below 10 mEq/hr; initial doses were titrated to the individual patient need and subsequent adjustments were necessary with time in some patients. Famotidine was well tolerated at these high dose levels for prolonged periods (greater than 12 months) in eight patients, and there were no cases reported of gynecomastia, increased prolactin levels, or impotence which were considered to be due to the drug.
CLINICAL PHARMACOLOGY IN PEDIATRIC PATIENTS
Table 6 presents pharmacokinetic data from clinical trials and a published study in pediatric patients (<1 year of age; N=27) given famotidine I.V. 0.5 mg/kg and from published studies of small numbers of pediatric patients (1 to 15 years of age) given famotidine intravenously. Areas under the curve (AUCs) are normalized to a dose of 0.5 mg/kg I.V. for pediatric patients 1 to 15 years of age and compared with an extrapolated 40 mg intravenous dose in adults (extrapolation based on results obtained with a 20 mg I.V. adult dose).
Table 6 Pharmacokinetic Parametersa of Intravenous Famotidine Age(N=number ofpatients) Area Underthe Curve (AUC)(ng hr/mL) TotalClearance (Cl)(L/hr/kg) Volume ofDistribution (Vd)(L/kg) EliminationHalf-life (T1/2)(hours) aValues are presented as means ±SD unless indicated otherwise.
bMean value only.
cSingle center study.
dMulticenter study.0-1 monthc (N=10) NA 0.13 ± 0.06 1.4 ± 0.4 10.5 ± 5.4 0-3 monthsd (N=6) 2688 ± 847 0.21 ± 0.06 1.8 ± 0.3 8.1 ± 3.5 >3–12 monthsd (N=11) 1160 ± 474 0.49 ± 0.17 2.3 ± 0.7 4.5 ± 1.1 1-11 yrs (N=20) 1089 ± 834 0.54 ± 0.34 2.07 ± 1.49 3.38 ± 2.60 11-15 yrs (N=6) 1140 ± 320 0.48 ± 0.14 1.5 ± 0.4 2.3 ± 0.4 Adult (N=16) 1726b 0.39 ± 0.14 1.3 ± 0.2 2.83 ± 0.99 Plasma clearance is reduced and elimination half-life is prolonged in pediatric patients 0 to 3 months of age compared to older pediatric patients. The pharmacokinetic parameters for pediatric patients, ages >3 months to 15 years, are comparable to those obtained for adults.
Bioavailability studies of 8 pediatric patients (11 to 15 years of age) showed a mean oral bioavailability of 0.5 compared to adult values of 0.42 to 0.49. Oral doses of 0.5 mg/kg achieved AUCs of 645 ± 249 ng-hr/mL and 580 ± 60 ng-hr/mL in pediatric patients <1 year of age (N=5) and in pediatric patients 11 to 15 years of age, respectively, compared to 482 ± 181 ng-hr/mL in adults treated with 40 mg orally.
Pharmacodynamics of famotidine were evaluated in 5 pediatric patients 2 to 13 years of age using the sigmoid Emax model. These data suggest that the relationship between serum concentration of famotidine and gastric acid suppression is similar to that observed in one study of adults (Table 7).
Table 7 Pharmacodynamics of famotidine using the sigmoid Emax model EC50 (ng/mL)* *Serum concentration of famotidine associated with 50% maximum gastric acid reduction. Values are presented as means ± SD. Pediatric Patients 26 ± 13 Data from one study a) healthy adult subjects 26.5 ± 10.3 b) adult patients with upper GI bleeding 18.7 ± 10.8
Five published studies (Table 8) examined the effect of famotidine on gastric pH and duration of acid suppression in pediatric patients. While each study had a different design, acid suppression data over time are summarized as follows:Table 8 Dosage Route Effecta Number of Patients (age range) aValues reported in published literature.
bMeans ± SD.
cMean (95% confidence interval).0.5 mg/kg, single dose I.V. gastric pH >4 for 19.5 hours
(17.3, 21.8)c11 (5-19 days) 0.3 mg/kg, single dose I.V. gastric pH >3.5 for8.7 ± 4.7b hours 6 (2-7 years) 0.4-0.8 mg/kg I.V. gastric pH >4 for 6-9 hours 18 (2-69 months) 0.5 mg/kg, single dose I.V. a >2 pH unit increase abovebaseline in gastric pH for>8 hours 9 (2-13 years) 0.5 mg/kg b.i.d. I.V. gastric pH >5 for 13.5± 1.8b hours 4 (6-15 years) 0.5 mg/kg b.i.d. oral gastric pH >5 for 5.0± 1.1b hours 4 (11-15 years) The duration of effect of famotidine I.V. 0.5 mg/kg on gastric pH and acid suppression was shown in one study to be longer in pediatric patients <1 month of age than in older pediatric patients. This longer duration of gastric acid suppression is consistent with the decreased clearance in pediatric patients <3 months of age (see Table 6).
-
INDICATIONS AND USAGE
Famotidine for Oral Suspension is indicated in:
- Short-term treatment of active duodenal ulcer. Most adult patients heal within 4 weeks; there is rarely reason to use Famotidine at full dosage for longer than 6 to 8 weeks. Studies have not assessed the safety of famotidine in uncomplicated active duodenal ulcer for periods of more than eight weeks.
- Maintenance therapy for duodenal ulcer patients at reduced dosage after healing of an active ulcer. Controlled studies in adults have not extended beyond one year.
- Short-term treatment of active benign gastric ulcer. Most adult patients heal within 6 weeks. Studies have not assessed the safety or efficacy of famotidine in uncomplicated active benign gastric ulcer for periods of more than 8 weeks.
- Short-term treatment of gastroesophageal reflux disease (GERD). Famotidine is indicated for short-term treatment of patients with symptoms of GERD (see CLINICAL PHARMACOLOGY IN ADULTS, Clinical Studies).
Famotidine is also indicated for the short-term treatment of esophagitis due to GERD including erosive or ulcerative disease diagnosed by endoscopy (see CLINICAL PHARMACOLOGY IN ADULTS, Clinical Studies). - Treatment of pathological hypersecretory conditions (e.g., Zollinger-Ellison Syndrome, multiple endocrine adenomas) (see CLINICAL PHARMACOLOGY IN ADULTS, Clinical Studies).
- CONTRAINDICATIONS
-
PRECAUTIONS
General
Symptomatic response to therapy with Famotidine does not preclude the presence of gastric malignancy.
Patients with Moderate or Severe Renal Insufficiency
Since CNS adverse effects have been reported in patients with moderate and severe renal insufficiency, longer intervals between doses or lower doses may need to be used in patients with moderate (creatinine clearance <50 mL/min) or severe (creatinine clearance <10 mL/min) renal insufficiency to adjust for the longer elimination half-life of famotidine (see CLINICAL PHARMACOLOGY IN ADULTS and DOSAGE AND ADMINISTRATION ).
Information for Patients
The patient should be instructed to shake the oral suspension vigorously for 5 to 10 seconds prior to each use. Unused constituted oral suspension should be discarded after 30 days.
Drug Interactions
No drug interactions have been identified. Studies with famotidine in man, in animal models, and in vitro have shown no significant interference with the disposition of compounds metabolized by the hepatic microsomal enzymes, e.g., cytochrome P450 system. Compounds tested in man include warfarin, theophylline, phenytoin, diazepam, aminopyrine and antipyrine. Indocyanine green as an index of hepatic drug extraction has been tested and no significant effects have been found.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 106 week study in rats and a 92-week study in mice given oral doses of up to 2000 mg/kg/day (approximately 2500 times the recommended human dose for active duodenal ulcer), there was no evidence of carcinogenic potential for Famotidine.
Famotidine was negative in the microbial mutagen test (Ames test) using Salmonella typhimurium and Escherichia coli with or without rat liver enzyme activation at concentrations up to 10,000 mcg/plate. In in vivo studies in mice, with a micronucleus test and a chromosomal aberration test, no evidence of a mutagenic effect was observed.
In studies with rats given oral doses of up to 2000 mg/kg/day or intravenous doses of up to 200 mg/kg/day, fertility and reproductive performance were not affected.
Pregnancy
Pregnancy Category B
Reproductive studies have been performed in rats and rabbits at oral doses of up to 2000 and 500 mg/kg/day, respectively, and in both species at I.V. doses of up to 200 mg/kg/day, and have revealed no significant evidence of impaired fertility or harm to the fetus due to Famotidine. While no direct fetotoxic effects have been observed, sporadic abortions occurring only in mothers displaying marked decreased food intake were seen in some rabbits at oral doses of 200 mg/kg/day (250 times the usual human dose) or higher. There are, however, no adequate or well-controlled studies in pregnant women. Because animal reproductive studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Studies performed in lactating rats have shown that famotidine is secreted into breast milk. Transient growth depression was observed in young rats suckling from mothers treated with maternotoxic doses of at least 600 times the usual human dose. Famotidine is detectable in human milk. Because of the potential for serious adverse reactions in nursing infants from Famotidine, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Patients <1 year of age
Use of Famotidine in pediatric patients <1 year of age is supported by evidence from adequate and well controlled studies of Famotidine in adults, and by the following studies in pediatric patients <1 year of age.
Two pharmacokinetic studies in pediatric patients <1 year of age (N=48) demonstrated that clearance of famotidine in patients >3 months to 1 year of age is similar to that seen in older pediatric patients (1 to 15 years of age) and adults. In contrast, pediatric patients 0 to 3 months of age had famotidine clearance values that were 2- to 4-fold less than those in older pediatric patients and adults. These studies also show that the mean bioavailability in pediatric patients <1 year of age after oral dosing is similar to older pediatric patients and adults. Pharmacodynamic data in pediatric patients 0 to 3 months of age suggest that the duration of acid suppression is longer compared with older pediatric patients, consistent with the longer famotidine half-life in pediatric patients 0 to 3 months of age. (See CLINICAL PHARMACOLOGY IN PEDIATRIC PATIENTS, Pharmacokinetics and Pharmacodynamics)
In a double-blind, randomized, treatment-withdrawal study, 35 pediatric patients <1 year of age who were diagnosed as having gastroesophageal reflux disease were treated for up to 4 weeks with famotidine oral suspension (0.5 mg/kg/dose or 1 mg/kg/dose). Although an intravenous famotidine formulation was available, no patients were treated with intravenous famotidine in this study. Also, caregivers were instructed to provide conservative treatment including thickened feedings. Enrolled patients were diagnosed primarily by history of vomiting (spitting up) and irritability (fussiness). The famotidine dosing regimen was once daily for patients <3 months of age and twice daily for patients ≥3 months of age. After 4 weeks of treatment, patients were randomly withdrawn from the treatment and followed an additional 4 weeks for adverse events and symptomatology. Patients were evaluated for vomiting (spitting up), irritability (fussiness) and global assessments of improvement. The study patients ranged in age at entry from 1.3 to 10.5 months (mean 5.6 ± 2.9 months), 57% were female, 91% were white and 6% were black. Most patients (27/35) continued into the treatment-withdrawal phase of the study. Two patients discontinued famotidine due to adverse events. Most patients improved during the initial treatment phase of the study. Results of the treatment-withdrawal phase were difficult to interpret because of small numbers of patients. Of the 35 patients enrolled in the study, agitation was observed in 5 patients on famotidine that resolved when the medication was discontinued; agitation was not observed in patients on placebo (see ADVERSE REACTIONS, Pediatric Patients).
These studies suggest that a starting dose of 0.5 mg/kg/dose of famotidine oral suspension may be of benefit for the treatment of GERD for up to 4 weeks once daily in patients <3 months of age and twice daily in patients 3 months to <1 year of age; the safety and benefit of famotidine treatment beyond 4 weeks have not been established. Famotidine should be considered for the treatment of GERD only if conservative measures (e.g., thickened feedings) are used concurrently and if the potential benefit outweighs the risk.
Pediatric Patients 1 to 16 years of age
Use of Famotidine in pediatric patients 1 to 16 years of age is supported by evidence from adequate and well-controlled studies of Famotidine in adults, and by the following studies in pediatric patients: In published studies in small numbers of pediatric patients 1 to 15 years of age, clearance of famotidine was similar to that seen in adults. In pediatric patients 11 to 15 years of age, oral doses of 0.5 mg/kg were associated with a mean area under the curve (AUC) similar to that seen in adults treated orally with 40 mg. Similarly, in pediatric patients 1 to 15 years of age, intravenous doses of 0.5 mg/kg were associated with a mean AUC similar to that seen in adults treated intravenously with 40 mg. Limited published studies also suggest that the relationship between serum concentration and acid suppression is similar in pediatric patients 1 to 15 years of age as compared with adults. These studies suggest a starting dose for pediatric patients 1 to 16 years of age as follows:
Peptic ulcer - 0.5 mg/kg/day p.o. at bedtime or divided b.i.d. up to 40 mg/day.
Gastroesophageal Reflux Disease with or without esophagitis including erosions and ulcerations - 1.0 mg/kg/day p.o. divided b.i.d. up to 40 mg b.i.d.
While published uncontrolled studies suggest effectiveness of famotidine in the treatment of gastroesophageal reflux disease and peptic ulcer, data in pediatric patients are insufficient to establish percent response with dose and duration of therapy. Therefore, treatment duration (initially based on adult duration recommendations) and dose should be individualized based on clinical response and/or pH determination (gastric or esophageal) and endoscopy. Published uncontrolled clinical studies in pediatric patients have employed doses up to 1 mg/kg/day for peptic ulcer and 2 mg/kg/day for GERD with or without esophagitis including erosions and ulcerations.
Of the 4,966 subjects in clinical studies who were treated with famotidine, 488 subjects (9.8%) were 65 and older, and 88 subjects (1.7%) were greater than 75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. However, greater sensitivity of some older individuals cannot be ruled out.
No dosage adjustment is required based on age (see CLINICAL PHARMACOLOGY IN ADULTS, Pharmacokinetics). This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Dosage adjustment in the case of moderate or severe renal impairment is necessary (see PRECAUTIONS, Patients with Moderate or Severe Renal Insufficiency and DOSAGE AND ADMINISTRATION, Dosage Adjustment for Patients with Moderate or Severe Renal Insufficiency).
-
ADVERSE REACTIONS
The adverse reactions listed below have been reported during domestic and international clinical trials in approximately 2500 patients. In those controlled clinical trials in which Famotidine Tablets were compared to placebo, the incidence of adverse experiences in the group which received Famotidine Tablets, 40 mg at bedtime, was similar to that in the placebo group.
The following adverse reactions have been reported to occur in more than 1% of patients on therapy with Famotidine in controlled clinical trials, and may be causally related to the drug: headache (4.7%), dizziness (1.3%), constipation (1.2%) and diarrhea (1.7%).
The following other adverse reactions have been reported infrequently in clinical trials or since the drug was marketed. The relationship to therapy with Famotidine has been unclear in many cases. Within each category the adverse reactions are listed in order of decreasing severity:
Body as a Whole: fever, asthenia, fatigue
Cardiovascular: arrhythmia, AV block, palpitation
Gastrointestinal: cholestatic jaundice, liver enzyme abnormalities, vomiting, nausea, abdominal discomfort, anorexia, dry mouth
Hematologic: rare cases of agranulocytosis, pancytopenia, leukopenia, thrombocytopenia
Hypersensitivity: anaphylaxis, angioedema, orbital or facial edema, urticaria, rash, conjunctival injection
Musculoskeletal: musculoskeletal pain including muscle cramps, arthralgia
Nervous System/Psychiatric: grand mal seizure; psychic disturbances, which were reversible in cases for which follow-up was obtained, including hallucinations, confusion, agitation, depression, anxiety, decreased libido; paresthesia; insomnia; somnolence. Convulsions, in patients with impaired renal function, have been reported very rarely.
Respiratory: bronchospasm, interstitial pneumonia
Skin: toxic epidermal necrolysis/Stevens Johnson syndrome (very rare), alopecia, acne, pruritus, dry skin, flushing
Special Senses: tinnitus, taste disorder
Other: rare cases of impotence and rare cases of gynecomastia have been reported; however, in controlled clinical trials, the incidences were not greater than those seen with placebo.
The adverse reactions reported for Famotidine Tablets may also occur with Famotidine for Oral Suspension.
In a clinical study in 35 pediatric patients <1 year of age with GERD symptoms [e.g., vomiting (spitting up), irritability (fussing)], agitation was observed in 5 patients on famotidine that resolved when the medication was discontinued.
-
OVERDOSAGE
The adverse reactions in overdose cases are similar to the adverse reactions encountered in normal clinical experience (see ADVERSE REACTIONS). Oral doses of up to 640 mg/day have been given to adult patients with pathological hypersecretory conditions with no serious adverse effects. In the event of overdosage, treatment should be symptomatic and supportive. Unabsorbed material should be removed from the gastrointestinal tract, the patient should be monitored, and supportive therapy should be employed.
The oral LD50 of famotidine in male and female rats and mice was greater than 3000 mg/kg and the minimum lethal acute oral dose in dogs exceeded 2000 mg/kg. Famotidine did not produce overt effects at high oral doses in mice, rats, cats and dogs, but induced significant anorexia and growth depression in rabbits starting with 200 mg/kg/day orally. The intravenous LD50 of famotidine for mice and rats ranged from 254 to 563 mg/kg and the minimum lethal single I.V. dose in dogs was approximately 300 mg/kg. Signs of acute intoxication in I.V. treated dogs were emesis, restlessness, pallor of mucous membranes or redness of mouth and ears, hypotension, tachycardia and collapse.
-
DOSAGE AND ADMINISTRATION
Duodenal Ulcer
Acute Therapy: The recommended adult oral dosage for active duodenal ulcer is 40 mg once a day at bedtime. Most patients heal within 4 weeks; there is rarely reason to use famotidine for oral suspension at full dosage for longer than 6 to 8 weeks. A regimen of 20 mg b.i.d. is also effective.
Maintenance Therapy: The recommended adult oral dose is 20 mg once a day at bedtime.
Benign Gastric Ulcer
Acute Therapy: The recommended adult oral dosage for active benign gastric ulcer is 40 mg once a day at bedtime.
Gastroesophageal Reflux Disease (GERD)
The recommended oral dosage for treatment of adult patients with symptoms of GERD is 20 mg b.i.d. for up to 6 weeks. The recommended oral dosage for the treatment of adult patients with esophagitis including erosions and ulcerations and accompanying symptoms due to GERD is 20 or 40 mg b.i.d. for up to 12 weeks (see CLINICAL PHARMACOLOGY IN ADULTS, Clinical Studies).
Dosage for Pediatric Patients <1year of age Gastroesophageal Reflux Disease (GERD)
See PRECAUTIONS, Pediatric Patients <1 year of age.
The studies described in PRECAUTIONS, Pediatric Patients < 1 year of age suggest the following starting doses in pediatric patients <1 year of age: Gastroesophageal Reflux Disease (GERD) - 0.5 mg/kg/dose of famotidine oral suspension for the treatment of GERD for up to 8 weeks once daily in patients <3 months of age and 0.5 mg/kg/dose twice daily in patients 3 months to <1 year of age. Patients should also be receiving conservative measures (e.g., thickened feedings). The use of intravenous famotidine in pediatric patients <1 year of age with GERD has not been adequately studied.
Dosage for Pediatric Patients 1 to 16 years of age
See PRECAUTIONS, Pediatric Patients 1 to 16 years of age.
The studies described in PRECAUTIONS, Pediatric Patients 1 to 16 years of age suggest the following starting doses in pediatric patients 1 to 16 years of age:
Peptic ulcer - 0.5 mg/kg/day p.o. at bedtime or divided b.i.d. up to 40 mg/day.
Gastroesophageal Reflux Disease with or without esophagitis including erosions and ulcerations - 1.0 mg/kg/day p.o. divided b.i.d. up to 40 mg b.i.d.
While published uncontrolled studies suggest effectiveness of famotidine in the treatment of gastroesophageal reflux disease and peptic ulcer, data in pediatric patients are insufficient to establish percent response with dose and duration of therapy. Therefore, treatment duration (initially based on adult duration recommendations) and dose should be individualized based on clinical response and/or pH determination (gastric or esophageal) and endoscopy. Published uncontrolled clinical studies in pediatric patients 1to 16 years of age have employed doses up to 1 mg/kg/day for peptic ulcer and 2 mg/kg/day for GERD with or without esophagitis including erosions and ulcerations.
Pathological Hypersecretory Conditions (e.g., Zollinger-Ellison Syndrome, Multiple Endocrine Adenomas)
The dosage of Famotidine in patients with pathological hypersecretory conditions varies with the individual patient. The recommended adult oral starting dose for pathological hypersecretory conditions is 20 mg q 6 h. In some patients, a higher starting dose may be required. Doses should be adjusted to individual patient needs and should continue as long as clinically indicated. Doses up to 160 mg q 6 h have been administered to some adult patients with severe Zollinger-Ellison Syndrome.
Oral Suspension
Famotidine for Oral Suspension may be substituted for Famotidine Tablets in any of the above indications. Each five mL contains 40 mg of famotidine after constitution of the powder with 46 mL of Purified Water as directed.
Directions for Preparing Famotidine for Oral Suspension
Prepare suspension at time of dispensing. Slowly add 46 mL of Purified Water. Shake vigorously for 5 to 10 seconds immediately after adding the water and immediately before use.
Stability of Famotidine for Oral Suspension
Unused constituted oral suspension should be discarded after 30 days.
Concomitant Use of Antacids
Antacids may be given concomitantly if needed.
Dosage Adjustment for Patients with Moderate or Severe Renal Insufficiency
In adult patients with moderate (creatinine clearance <50 mL/min) or severe (creatinine clearance <10 mL/min) renal insufficiency, the elimination half-life of Famotidine is increased. For patients with severe renal insufficiency, it may exceed 20 hours, reaching approximately 24 hours in anuric patients. Since CNS adverse effects have been reported in patients with moderate and severe renal insufficiency, to avoid excess accumulation of the drug in patients with moderate or severe renal insufficiency, the dose of Famotidine may be reduced to half the dose or the dosing interval may be prolonged to 36 to 48 hours as indicated by the patient’s clinical response.
Based on the comparison of pharmacokinetic parameters for Famotidine in adults and pediatric patients, dosage adjustment in pediatric patients with moderate or severe renal insufficiency should be considered.
-
HOW SUPPLIED
Famotidine for Oral Suspension is a white to off-white powder containing 400 mg of famotidine for constitution. When constituted to 50 mL as directed, Famotidine for Oral Suspension is a smooth, mobile, off-white, homogeneous suspension with a cherry-banana-mint flavor, containing 40 mg of famotidine per 5 mL.
NDC 40032-500-11, bottles containing 400 mg famotidine.
Storage
Preserve in well-closed, light resistant containers. Store at controlled room temperature.
Store Famotidine for Oral Suspension dry powder and suspension at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Suspension: Protect from freezing. Discard unused suspension after 30 days.
Manufactured by:
Novel Laboratories, Inc.
Somerset, NJ USA
PI5001100101Rev. 07/2012
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FAMOTIDINE
famotidine for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:40032-500 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FAMOTIDINE (UNII: 5QZO15J2Z8) (FAMOTIDINE - UNII:5QZO15J2Z8) FAMOTIDINE 40 mg in 5 mL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SUCROSE (UNII: C151H8M554) XANTHAN GUM (UNII: TTV12P4NEE) CARBOXYMETHYLCELLULOSE SODIUM (UNII: K679OBS311) STARCH, CORN (UNII: O8232NY3SJ) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM BENZOATE (UNII: OJ245FE5EU) METHYLPARABEN SODIUM (UNII: CR6K9C2NHK) Product Characteristics Color WHITE (Off-White) Score Shape Size Flavor CHERRY (Cherry-Banana-Mint) Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:40032-500-11 50 mL in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA201695 12/17/2012 Labeler - Novel Laboratories, Inc. (793518643) Registrant - Novel Labortaories, Inc. (793518643) Establishment Name Address ID/FEI Business Operations Novel Labortaories, Inc. 793518643 MANUFACTURE(40032-500) , ANALYSIS(40032-500)

