BUDEPRION SR- bupropion hydrochloride tablet, extended release
Cardinal Health
----------
BUDEPRION SR®
(buPROPion HCl Extended-Release Tablets)
"Patient Information" enclosed.
Suicidality and Antidepressant Drugs
Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Anyone considering the use of BUDEPRION SR® or any other antidepressant in a child, adolescent, or young adult must balance this risk with the clinical need. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction in risk with antidepressants compared to placebo in adults aged 65 and older. Depression and certain other psychiatric disorders are themselves associated with increases in the risk of suicide. Patients of all ages who are started on antidepressant therapy should be monitored appropriately and observed closely for clinical worsening, suicidality, or unusual changes in behavior. Families and caregivers should be advised of the need for close observation and communication with the prescriber. BUDEPRION SR® is not approved for use in pediatric patients. (See WARNINGS, Clinical Worsening and Suicide Risk , PRECAUTIONS, Information for Patients, and PRECAUTIONS, Pediatric Use.)
DESCRIPTION
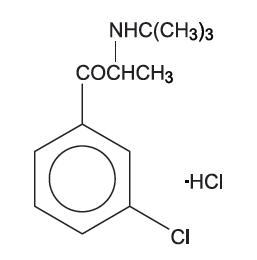
BUDEPRION SR® (bupropion hydrochloride), an antidepressant of the aminoketone class, is chemically unrelated to tricyclic, tetracyclic, selective serotonin re-uptake inhibitor, or other known antidepressant agents. Its structure closely resembles that of diethylpropion; it is related to phenylethylamines. It is designated as (±)-1-(3-chlorophenyl)-2-[(1,1-dimethylethyl)amino]-1-propanone hydrochloride. The molecular weight is 276.2. The molecular formula is C13H18CINO•HCI. Bupropion hydrochloride powder is white, crystalline, and highly soluble in water. It has a bitter taste and produces the sensation of local anesthesia on the oral mucosa. The structural formula is:
BUDEPRION SR® is supplied for oral administration as 100 mg and 150 mg, film-coated, extended-release tablets. Each tablet contains the labeled amount of bupropion hydrochloride and the inactive ingredients: colloidal silicon dioxide, hydroxypropyl cellulose, magnesium stearate, and microcrystalline cellulose. The 100 mg tablet also contains FD&C red # 40, FD&C yellow # 5, hypromellose, iron oxide yellow, macrogol, polydextrose, titanium dioxide and triacetin. The 150 mg tablet also contains hypromellose, iron oxide yellow, macrogol, polydextrose, titanium dioxide and triacetin.
This product meets USP Drug Release Test #3.
CLINICAL PHARMACOLOGY
Pharmacodynamics: Bupropion is a relatively weak inhibitor of the neuronal uptake of norepinephrine and dopamine, and does not inhibit monoamine oxidase or the reuptake of serotonin. While the mechanism of action of bupropion, as with other antidepressants, is unknown, it is presumed that this action is mediated by noradrenergic and/or dopaminergic mechanisms.
Pharmacokinetics: Bupropion is a racemic mixture. The pharmacological activity and pharmacokinetics of the individual enantiomers have not been studied. The mean elimination half-life (±SD) of bupropion after chronic dosing is 21 (±9) hours, and steady-state plasma concentrations of bupropion are reached within 8 days. In a study comparing chronic dosing with BUDEPRION SR® 150 mg twice daily to the immediate-release formulation of bupropion at 100 mg 3 times daily, peak plasma concentrations of bupropion at steady state for BUDEPRION SR® were approximately 85% of those achieved with the immediate-release formulation. There was equivalent for bupropion AUCs, as well as equivalence for both peak plasma concentration and AUCs for all 3 of the detectable bupropion metabolites. Thus, at steady state, BUDEPRION SR®, given twice daily, and the immediate-release formulation of bupropion, given 3 times daily, is essentially bioequivalent for both bupropion and the 3 quantitatively important metabolites.
Absorption: Following oral administration of BUDEPRION SR® to healthy volunteers, peak plasma concentrations of bupropion are achieved within 3 hours. Food increased Cmax and AUC of bupropion by 11% and 17%, respectively, indicating that there is no clinically significant food effect.
Distribution: In vitro tests show that bupropion is 84% bound to human plasma proteins at concentrations up to 200 mcg/mL. The extent of protein binding of the hydroxybupropion metabolite is similar to that for bupropion, whereas the extent of protein binding of the threohydrobupropion metabolite is about half that seen with bupropion.
Metabolism: Bupropion is extensively metabolized in humans. Three metabolites have been shown to be active: hydroxybupropion, which is formed via hydroxylation of the tert-butyl group of bupropion, and the amino-alcohol isomers threohydrobupropion and erythrohydrobupropion, which are formed via reduction of the carbonyl group. In vitro findings suggest that cytochrome P450IIB6 (CYP2B6) is the principal isoenzyme involved in the formation of hydroxybupropion, while cytochrome P450 isoenzymes are not involved in the formation of threohydrobupropion. Oxidation of the bupropion side chain results in the formation of a glycine conjugate of meta-chlorobenzoic acid, which is then excreted as the major urinary metabolite. The potency and toxicity of the metabolites relative to bupropion have not been fully characterized. However, it has been demonstrated in an antidepressant screening test in mice that hydroxybupropion is one half as potent as bupropion while threohydrobupropion and erthrohydrobupropion are 5-fold less potent than bupropion. This may be of clinical importance because the plasma concentrations of the metabolites are as high or higher than those of bupropion.
Because bupropion is extensively metabolized, there is the potential for drug-drug interactions, particularly with those agents that are metabolized by the cytochrome P450IIB6 (CYP2B6) isoenzyme. Although bupropion is not metabolized by cytochrome P450IID6 (CYP2D6), there is the potential for drug-drug interactions when bupropion is co-administered with drugs metabolized by this isoenzyme (see PRECAUTIONS: Drug Interactions).
Following a single dose in humans, peak plasma concentrations of hydroxybupropion occur approximately 6 hours after administration of BUDEPRION SR®. Peak plasma concentrations of hydroxybupropion are approximately 10 times the peak level of the parent drug at steady state. The elimination half-life of hydroxybupropion is approximately 20 (±5) hours, and its AUC at steady state is about 17 times that of bupropion. The times to peak concentrations for the erythrohydrobupropion and threohydrobupropion metabolites are similar to that of the hydroxybupropion metabolite. However, their elimination half-lives are longer, 33 (±10) and 37 (±13) hours, respectively, and steady-state AUCs are 1.5 and 7 times that of bupropion, respectively.
Bupropion and its metabolites exhibit linear kinetics following chronic administration of 300 to 450 mg/day.
Elimination: Following oral administration of 200 mg of 14C-bupropion in humans, 87% and 10% of the radioactive dose were recovered in the urine and feces, respectively. However, the fraction of the oral dose of bupropion excreted unchanged was only 0.5%, a finding consistent with the extensive metabolism of bupropion.
Population Subgroups: Factors or conditions altering metabolic capacity (e.g., liver disease, congestive heart failure [CHF], age, concomitant medications, etc.) or elimination may be expected to influence the degree and extent of accumulation of the active metabolites of bupropion. The elimination of the major metabolites of bupropion may be affected by reduced renal or hepatic function because they are moderately polar compounds and are likely to undergo further metabolism or conjugation in the liver prior to urinary excretion.
Hepatic: The effect of hepatic impairment on the pharmacokinetics of bupropion was characterized in 2 single-dose studies, one in patients with alcoholic liver disease and one in patients with mild to severe cirrhosis. The first study showed that the half-life of hydroxybupropion was significantly longer in 8 patients with alcoholic liver disease than in 8 healthy volunteers (32±14 hours versus 21±5 hours, respectively). Although not statistically significant, the AUCs for bupropion and hydroxybupropion were more variable and tended to be greater (by 53% to 57%) in patients with alcoholic liver disease. The differences in half-life for bupropion and the other metabolites in the 2 patient groups were minimal.
The second study showed no statistically significant differences in the pharmacokinetics of bupropion and its active metabolites in 9 patients with mild to moderate hepatic cirrhosis compared to 8 healthy volunteers. However, more variability was observed in some of the pharmacokinetic parameters for bupropion (AUC, Cmax, and Tmax) and its active metabolites (t1/2) in patients with mild to moderate hepatic cirrhosis. In addition, in patients with severe hepatic cirrhosis, the bupropion Cmax and AUC were substantially increased (mean difference: by approximately 70% and 3-fold, respectively) and more variable when compared to values in healthy volunteers; the mean bupropion half-life was also longer (29 hours in patients with severe hepatic cirrhosis vs. 19 hours in healthy subjects). For the metabolite hydroxybupropion, the mean Cmax was approximately 69% lower. For the combined amino-alcohol isomers threohydrobupropion and erythrohydrobupropion, the mean Cmax was approximately 31% lower. The mean AUC increased by about 11/2 fold for hydroxybupropion and about 21/2 fold for threo/erythrohydrobupropion. The median Tmax was observed 19 hours later for hydroxybupropion and 31 hours later for threo/erythrohydrobupropion. The mean half-lives for hydroxybupropion and threo/erythrohydrobupropion were increased 5- and 2-fold, respectively, in patients with severe hepatic cirrhosis compared to healthy volunteers (see WARNINGS, PRECAUTIONS, and DOSAGE AND ADMINISTRATION).
Renal: There is limited information on pharmacokinetics of bupropion in patients with renal impairment. An inter-study comparison between normal subjects and patients with end-stage renal failure demonstrated that the parent drug Cmax and AUC values were comparable in the 2 groups, whereas the hydroxybupropion and threohydrobupropion metabolites had a 2.3- and 2.8-fold increase, respectively, in AUC for patients with end-stage renal failure. The elimination of the major metabolites or bupropion may be reduced by impaired renal function (see PRECAUTIONS, Renal Impairment).
Left Ventricular Dysfunction: During a chronic dosing study with bupropion in 14 depressed patients with left ventricular dysfunction (history of CHF or an enlarged heart on x-ray), no apparent effect on the pharmacokinetics of bupropion or its metabolites was revealed, compared to healthy normal volunteers.
Age: The effects of age on the pharmacokinetics of bupropion and its metabolites have not been fully characterized, but an exploration of steady-state bupropion concentrations from several depression efficacy studies involving patients dosed in a range of 300 to 750 mg/day, on a three times daily schedule, revealed no relationship between age (18 to 83 years) and plasma concentration of bupropion. A single-dose pharmacokinetic study demonstrated that the disposition of bupropion and its metabolites in elderly subjects was similar to that of younger subjects. These data suggest there is no prominent effect of age on bupropion concentration; however, another pharmacokinetic study, single and multiple dose, had suggested that the elderly are at increased risk for accumulation of bupropion and its metabolites (see PRECAUTIONS: Geriatric Use).
Gender: A single-dose study involving 12 healthy male and 12 healthy female volunteers revealed no sex-related differences in the pharmacokinetic parameters of bupropion.
Smokers: The effects of cigarette smoking on the pharmacokinetics of bupropion were studied in 34 healthy male and female volunteers; 17 were chronic cigarette smokers and 17 were nonsmokers. Following oral administration of a single 150 mg dose of bupropion, there was no statistically significant difference in Cmax, half-life, Tmax, AUC, or clearance of bupropion or its active metabolites between smokers and nonsmokers.
CLINICAL TRIALS
The efficacy of the immediate-release formulation of bupropion as a treatment for depression was established in two 4-week, placebo-controlled trials in adult inpatients with depression and in one 6-week, placebo-controlled trial in adult outpatients with depression. In the first study, patients were titrated in a bupropion dose range of 300 to 600 mg/day on a three times daily schedule; 78% of patients received maximum doses of 450 mg/day or less. This trial demonstrated the effectiveness of the immediate-release formulation of bupropion on the Hamilton Depression Rating Scale (HDRS) total score, the depressed mood item (item 1) from the scale, and the Clinical Global Impressions (CGI) severity score. A second study included two fixed doses of the immediate-release formulation of bupropion (300 and 450 mg/day) and placebo. This trial demonstrated the effectiveness of the immediate-release formulation of bupropion, but only at the 450 mg/day dose; the results were positive for the HDRS total score and the CGI severity score, but not for HDRS item 1. In the third study, outpatients received 300 mg/day of the immediate-release formulation of bupropion. This study demonstrated the effectiveness of the immediate-release formulation of bupropion on the HDRS total score, HDRS item 1, the Montgomery-Asberg Depression Rating Scale, the CGI severity score, and the CGI improvement score.
Although there are not as yet independent trials demonstrating the antidepressant effectiveness of the extended-release formulation of bupropion, studies have demonstrated the bioequivalence of the immediate-release and extended-release forms of bupropion under steady-state conditions, i.e., bupropion extended-release 150 mg twice daily was shown to be bioequivalent to 100 mg three times daily of the immediate-release formulation of bupropion, with regard to both rate and extent of absorption, for parent drug and metabolites.
In a longer-term study, outpatients meeting DSM-IV criteria for major depressive disorder, recurrent type, who had responded during an 8-week open trial on BUDEPRION SR® (150 mg twice daily) were randomized to continuation of their same BUDEPRION SR® dose or placebo, for up to 44 weeks of observation for relapse. Response during the open phase was defined as CGI Improvement score of 1 (very much improved) or 2 (much improved) for each of the final three weeks. Relapse during the double-blind phase was defined as the investigator’s judgment that drug treatment was needed for worsening depressive symptoms. Patients receiving continued BUDEPRION SR® treatment experienced significantly lower relapse rates over the subsequent 44 weeks compared to those receiving placebo.
INDICATIONS AND USAGE
BUDEPRION SR® is indicated for the treatment of major depressive disorder. The efficacy of bupropion in the treatment of major depressive episode was established in two 4-week controlled trials of depressed inpatients and in one 6-week controlled trial of depressed outpatients whose diagnoses corresponded most closely to the Major Depression category of the APA Diagnostic and Statistical Manual (DSM) (see CLINICAL PHARMACOLOGY).
A major depressive episode (DSM-IV) implies the presence of 1) depressed mood or 2) loss of interest or pleasure; in addition, at least five of the following symptoms have been present during the same 2-week period and represent a change from previous functioning: depressed mood, markedly diminished interest or pleasure in usual activities, significant change in weight and/or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, increased fatigue, feelings of guilt or worthlessness, slowed thinking or impaired concentration, a suicide attempt or suicidal ideation.
The efficacy of BUDEPRION SR® in maintaining an antidepressant response for up to 44 weeks following 8 weeks of acute treatment was demonstrated in a placebo controlled trial (see CLINICAL PHARMACOLOGY). Nevertheless, the physician who elects to use BUDEPRION SR® for extended periods should periodically reevaluate the long-term usefulness of the drug for the individual patient.
CONTRAINDICATIONS
BUDEPRION SR® is contraindicated in patients with a seizure disorder.
BUDEPRION SR® is contraindicated in patients treated with ZYBAN® [bupropion hydrochloride extended-release tablets (SR)]; WELLBUTRIN® (bupropion hydrochloride tablets), the immediate-release formulation; WELLBUTRIN XL® [bupropion hydrochloride extended-release tablets (XL)], the extended-release formulation; or any other medications that contain bupropion because the incidence of seizure is dose dependent.
BUDEPRION SR® is contraindicated in patients with a current or prior diagnosis of bulimia or anorexia nervosa because of a higher incidence of seizures noted in patients treated for bulimia with the immediate-release formulation of bupropion. BUDEPRION SR® is contraindicated in patients undergoing abrupt discontinuation of alcohol or sedatives (including benzodiazepines).
The concurrent administration of BUDEPRION SR® and a monoamine oxidase (MAO) inhibitor is contraindicated. At least 14 days should elapse between discontinuation of an MAO inhibitor and initiation of treatment with BUDEPRION SR®.
BUDEPRION SR® is contraindicated in patients who have shown an allergic response to bupropion or the other ingredients that make up BUDEPRION SR®.
WARNINGS
Clinical Worsening and Suicide Risk :
Patients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment. Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18-24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled trials in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug vs placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1000 patients treated) are provided in Table 1.
| Age Range | Drug-Placebo Difference in Number of Cases of Suicidality per 1,000 Patients Treated |
|---|---|
|
Increases Compared to Placebo |
|
|
< 18 |
14 additional cases |
|
18 to 24 |
5 additional cases |
|
Decreases Compared to Placebo |
|
|
25 to 64 |
1 fewer case |
|
≥ 65 |
6 fewer cases |
No suicides occurred in any of the pediatric trials. There were suicides in the adult trials, but the number was not sufficient to reach any conclusion about drug effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, andunusual changes in behavior, especially during the initial few months of a courseof drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient’s presenting symptoms.
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric,should be alerted about the need to monitor patients for the emergence of agitation,irritability, unusual changes in behavior, and the other symptoms described above,as well as the emergence of suicidality, and to report such symptoms immediatelyto health care providers. Such monitoring should include daily observation by familiesand caregivers. Prescriptions for BUDEPRION SR® should be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose.
Screening Patients for Bipolar Disorder:A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled trials) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that BUDEPRION SR® is not approved for use in treating bipolar depression.
Patients should be made aware that BUDEPRION SR® contains the same active ingredient found in ZYBAN®, used as an aid to smoking cessation treatment, andthat BUDEPRION SR® should not be used in combination with ZYBAN®, or any othermedications that contain bupropion, such as WELLBUTRIN® (bupropion hydrochloride tablets), the immediate-release formulation or WELLBUTRIN XL® [bupropion hydrochloride extended-release tablets (XL)], the extended-release formulation.
Seizures: Bupropion is associated with a dose-related risk of seizures. The risk of seizures is also related to patient factors, clinical situations, and concomitantmedications, which must be considered in selection of patients for therapy withBUDEPRION SR®.
BUDEPRION SR® should be discontinued and not restarted in patients who experiencea seizure while on treatment.
• Dose: At doses of BUDEPRION SR® up to a dose of 300 mg/day, the incidenceof seizure is approximately 0.1% (1/1000) and increases to approximately 0.4%(4/1000) at the maximum recommended dose of 400 mg/day.Data for the immediate-release formulation of bupropion revealed a seizure incidenceof approximately 0.4% (i.e., 13 of 3200 patients followed prospectively) inpatients treated at doses in a range of 300 to 450 mg/day. The 450 mg/day upperlimit of this dose range is close to the currently recommended maximum dose of400 mg/day for BUDEPRION SR®. This seizure incidence (0.4%) may exceed thatof other marketed antidepressants and BUDEPRION SR® up to 300 mg/day by asmuch as 4-fold. This relative risk is only an approximate estimate because nodirect comparative studies have been conducted.
Additional data accumulated for the immediate-release formulation of bupropionsuggested that the estimated seizure incidence increases almost tenfold between 450and 600 mg/day, which is twice the usual adult dose and one and one-half the maximumrecommended daily dose (400 mg) of BUDEPRION SR®. This disproportionateincrease in seizure incidence with dose incrementation calls for caution in dosing.
Data for BUDEPRION SR® revealed a seizure incidence of approximately 0.1%(i.e., 3 of 3100 patients followed prospectively) in patients treated at doses ina range of 100 to 300 mg/day. It is not possible to know if the lower seizure incidenceobserved in this study involving the sustained-release formulation ofbupropion resulted from the different formulation or the lower dose used.However, as noted above, the immediate-release and sustained-release formulationsare bioequivalent with regard to both rate and extent of absorption duringsteady state (the most pertinent condition to estimating seizure incidence),since most observed seizures occur under steady-state conditions.
• Patient factors: Predisposing factors that may increase the risk of seizure withbupropion use include history of head trauma or prior seizure, central nervoussystem (CNS) tumor, the presence of severe hepatic cirrhosis and concomitantmedications that lower seizure threshold.
• Clinical situations: Circumstances associated with an increased seizure riskinclude, among others, excessive use of alcohol or sedatives (including benzodiazepines);addiction to opiates, cocaine, or stimulants; use of over-the-counterstimulants and anorectics; and diabetes treated with oral hypoglycemics or insulin.
• Concomitant medications: Many medications (e.g., antipsychotics, antidepressants,theophylline, systemic steroids) are known to lower seizure threshold.
Recommendations for Reducing the Risk of Seizure: Retrospective analysis of clinical experience gained during the development of bupropion suggests that therisk of seizure may be minimized if
• The total daily dose of BUDEPRION SR® does not exceed 400 mg.
• The daily dose is administered twice daily, and
• The rate of incrementation of dose is gradual.
• No single dose should exceed 200 mg to avoid high peak concentrations of bupropion and/or its metabolites.
BUDEPRION SR® should be administered with extreme caution to patients with ahistory of seizure, cranial trauma, or other predisposition(s) toward seizure, orpatients treated with other agents (e.g. antipsychotics, other antidepressants,theophylline, systemic steroids, etc.) that lower seizure threshold.
Hepatic Impairment: BUDEPRION SR® should be used with extreme caution in patientswith severe hepatic cirrhosis. In these patients a reduced frequency and/or dose isrequired, as peak bupropion levels, as well as AUC, levels are substantially increasedand accumulation is likely to occur in such patients to a greater extent than usual. Thedose should not exceed 100 mg every day or 150 mg every other day in these patients(see CLINICAL PHARMACOLOGY, PRECAUTIONS, and DOSAGE AND ADMINISTRATION).
Potential for Hepatotoxicity:In rats receiving large doses of bupropion chronically, there was an increase in incidence of hepatic hyperplastic nodules and hepatocellular hypertrophy. In dogs receiving large doses of bupropion chronically, various histologic changes were seen in the liver, and laboratory tests suggesting mild hepatocellular injury were noted.
PRECAUTIONS
General
Agitation and Insomnia: Patients in placebo-controlled trials with BUDEPRION SR® experienced agitation, anxiety, and insomnia as shown in Table 2.
| Adverse Event Term | Bupropion hydrochloride extended-release
Tablets (SR) 300 mg/day (n=376) | Bupropion hydrochloride extended-release
Tablets (SR) 400 mg/day (n=114) | Placebo
(n=385) |
|---|---|---|---|
|
Agitation |
3% |
9% |
2% |
|
Anxiety |
5% |
6% |
3% |
|
Insomnia |
11% |
16% |
6% |
In clinical studies, these symptoms were sometimes of sufficient magnitude to require treatment with sedative/hypnotic drugs.
Symptoms were sufficiently severe to require discontinuation of treatment in 1% and 2.6% of patients treated with 300 and 400 mg/day, respectively, of BUDEPRION SR® and 0.8% of patients treated with placebo.
Psychosis, Confusion, and Other Neuropsychiatric Phenomena: Depressed patients treated with an immediate-release formulation of bupropion or with BUDEPRION SR® have been reported to show a variety of neuropsychiatric signs and symptoms, including delusions, hallucinations, psychosis, concentration disturbance, paranoia, and confusion. In some cases, these symptoms abated upon dose reduction and/or withdrawal of treatment.
Activation of Psychosis and/or Mania: Antidepressants can precipitate manic episodes in bipolar disorder patients during the depressed phase of their illness and may activate latent psychosis in other susceptible patients. BUDEPRION SR® is expected to pose similar risks.
Altered Appetite and Weight: In placebo-controlled studies, patients experienced weight gain or weight loss as shown in Table 3.
| Weight Change | Bupropion hydrochloride extended-release
Tablets (SR) 300 mg/day (n=339) | Bupropion hydrochloride extended-release
Tablets (SR) 400 mg/day (n=112) | Placebo
(n=347) |
|---|---|---|---|
|
Gain > 5 lbs |
3% |
2% |
4% |
|
Lost > 5 lbs |
14% |
19% |
6% |
In studies conducted with the immediate-release formulation of bupropion, 35% of patients receiving tricyclic antidepressants gained weight, compared to 9% of patients treated with the immediate-release formulation of bupropion. If weight loss is a major presenting sign of a patient’s depressive illness, the anorectic and/or weight-reducing potential of BUDEPRION SR® should be considered.
Allergic Reactions
Allergic Reactions: Anaphylactoid/anaphylactic reactions characterized by symptoms such as pruritus, urticaria, angioedema, and dyspnea requiring medical treatment have been reported in clinical trials with bupropion. In addition, there have been rare spontaneous postmarketing reports of erythema multiforme, Stevens-Johnson syndrome, and anaphylactic shock associated with bupropion. A patient should stop taking BUDEPRION SR® and consult a doctor if experiencing allergic or anaphylactoid/anaphylactic reactions (e.g., skin rash, pruritus, hives, chest pain, edema, and shortness of breath) during treatment.
Arthralgia, myalgia, and fever with rash and other symptoms suggestive of delayed hypersensitivity have been reported in association with bupropion. These symptoms may resemble serum sickness.
The 100 mg strength of this product contains FD&C Yellow No. 5 (tartrazine) which may cause allergic type reactions (including bronchial asthma) in certain susceptible persons. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin sensitivity.
Cardiovascular Effects: In clinical practice, hypertension, in some cases severe, requiring acute treatment, has been reported in patients receiving bupropion alone and in combination with nicotine replacement therapy. These events have been observed in both patients with and without evidence of preexisting hypertension.
Data from a comparative study of the extended-release bupropion (ZYBAN® Sustained- Release Tablets), nicotine transdermal system (NTS), the combination sustained-release bupropion plus NTS, and placebo as an aid to smoking cessation suggest a higher incidence of treatment-emergent hypertension in patients treated with the combination of sustained-release bupropion and NTS. In this study, 6.1% of patients treated with the combination of sustained-released bupropion and NTS had treatment-emergent hypertension compared to 2.5%, 1.6%, and 3.1% of patients treated with sustained-release bupropion, NTS, and placebo, respectively. The majority of these patients had evidence of preexisting hypertension. Three patients (1.2%) treated with the combination of ZYBAN® and NTS and one patient (0.4%) treated with NTS had study medication discontinued due to hypertension compared to none of the patients treated with ZYBAN® or placebo. Monitoring of blood pressure is recommended in patients who receive the combination of bupropion and nicotine replacement.
There is no clinical experience establishing the safety of BUDEPRION SR® in patients with a recent history of myocardial infarction or unstable heart disease. Therefore, care should be exercised if it is used in these groups. Bupropion was well tolerated in depressed patients who had previously developed orthostatic hypotension while receiving tricyclic antidepressants, and was also generally well tolerated in a group of 36 depressed inpatients with stable congestive heart failure (CHF). However, bupropion was associated with a rise in supine blood pressure in the study of patients with CHF, resulting in discontinuation of treatment in two patients for exacerbation of baseline hypertension.
Hepatic Impairment: BUDEPRION SR® should be used with extreme caution in patients with severe hepatic cirrhosis. In these patients, a reduced frequency and/or dose is required. BUDEPRION SR® should be used with caution in patients with hepatic impairment (including mild to moderate hepatic cirrhosis) and reduced frequency and/or dose should be considered in patients with mild to moderate hepatic cirrhosis.
All patients with hepatic impairment should be closely monitored for possible adverse effects that could indicate high drug and metabolite levels (see CLINICAL PHARMACOLOGY, WARNINGS, and DOSAGE AND ADMINISTRATION).
Renal Impairment: There is limited information on pharmacokinetics of bupropion in patients with renal impairment. An inter-study comparison between normal subjects and patients with end-stage renal failure demonstrated that the parent drug Cmax and AUC values were comparable in the 2 groups, whereas the hydroxybupropion and threohydrobupropion metabolites had a 2.3- and 2.8-fold increase, respectively, in AUC for patients with end-stage renal failure. Bupropion is extensively metabolized in the liver to active metabolites, which are further metabolized and subsequently excreted by the kidneys. BUDEPRION SR® should be used with caution in patients with renal impairment and a reduced frequency of dosing should be considered as bupropion and the metabolites of bupropion may accumulate in such patients to a greater extent than usual. The patients should be closely monitored for possible adverse effects that could indicate high drug or metabolite levels.
Information for Patients: Prescribers or other health professionals should inform patients, their families, and their caregivers about the benefits and risks associated with treatment with BUDEPRION SR® and should counsel them in its appropriate use. A patient Medication Guide about “Antidepressant Medicines, Depression and other Serious Mental Illness, and Suicidal Thoughts or Actions” is available for BUDEPRION SR®. The prescriber or health professional should instruct patients, their families, and their caregivers to read the Medication Guide and Patient Package Insert, and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and Patient Package Insert, and to obtain answers to any questions they may have. The complete text of the Medication Guide and Patient Package Insert are provided in tear-off leaflets at the end of this document.
Patients should be advised of the following issues and asked to alert their prescriber if these occur while taking BUDEPRION SR®.
Clinical Worsening and Suicide Risk: Patients, their families, and their caregivers should be encouraged to be alert to the emergence of anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, mania, other unusual changes in behavior, worsening of depression, and suicidal ideation, especially early during antidepressant treatment and when the dose is adjusted up or down. Families and caregivers of patients should be advised to look for the emergence of such symptoms on a day-to-day basis, since changes may be abrupt. Such symptoms should be reported to the patient’s prescriber or health professional, especially if they are severe, abrupt in onset, or were not part of the patient’s presenting symptoms. Symptoms such as these may be associated with an increased risk for suicidal thinking and behavior and indicate a need for very close monitoring and possibly changes in the medication.
Patients should be made aware that BUDEPRION SR® contains the same active ingredient found in ZYBAN®, used as an aid to smoking cessation treatment, and that BUDEPRION SR® should not be used in combination with ZYBAN®, or any other medications that contain bupropion, such as WELLBUTRIN® (bupropion hydrochloride), the immediate release formulation or WELLBUTRIN XL® [bupropion hydrochloride extended-release tablets (XL)] the extended-release formulation.
As dose is increased during initial titration to doses above 150 mg/day, patients should be instructed to take BUDEPRION SR® in two divided doses, preferably with at least 8 hours between successive doses, to minimize the risk of seizures.
Patients should be told that BUDEPRION SR® should be discontinued and not restarted if they experience a seizure while on treatment.
Patients should be told that any CNS-active drug like BUDEPRION SR® may impair their ability to perform tasks requiring judgment or motor and cognitive skills. Consequently, until they are reasonably certain that BUDEPRION SR® does not adversely affect their performance, they should refrain from driving an automobile or operating complex, hazardous machinery.
Patients should be told that the excessive use or abrupt discontinuation of alcohol or sedatives (including benzodiazepines) may alter the seizure threshold. Some patients have reported lower alcohol tolerance during treatment with BUDEPRION SR®. Patients should be advised that the consumption of alcohol should be minimized or avoided.
Patients should be advised to inform their physicians if they are taking or plan to take any prescription or over-the-counter drugs. Concern is warranted because BUDEPRION SR® and other drugs may affect each other’s metabolism.
Patients should be advised to notify their physicians if they become pregnant or intend to become pregnant during therapy.
Patients should be advised to swallow BUDEPRION SR® whole so that the release rate is not altered. Do not chew, divide, or crush tablets.
Drug Interactions: Few systemic data have been collected on the metabolism of bupropion following concomitant administration with other drugs or, alternatively, the effect of concomitant administration of bupropion on the metabolism of other drugs. Because bupropion is extensively metabolized, the coadministration of other drugs may affect its clinical activity. In vitro studies indicate that bupropion is primarily metabolized to hydroxybupropion by the CYP2B6 isoenzyme. Therefore, the potential exists for a drug interaction between BUDEPRION SR® and drugs that affect the CYP2B6 isoenzyme (e.g., orphenadrine, thiotepa, and cyclophosphamide). In addition, in vitro studies suggest that paroxetine, sertraline, norfluoxetine, and fluvoxamine as well as nelfinavir, ritonavir, and efavirenz inhibit the hydroxylation of bupropion. No clinical studies have been performed to evaluate this finding. The threohydrobupropion metabolite of bupropion does not appear to be produced by the cytochrome P450 isoenzymes. The effects of concomitant administration of cimetidine on the pharmacokinetics of bupropion and its active metabolites were studied in 24 healthy young male volunteers. Following oral administration of two 150 mg BUDEPRION SR® with and without 800 mg of cimetidine, the pharmacokinetics of bupropion and hydroxybupropion were unaffected. However, there were 16% and 32% increases in the AUC and Cmax, respectively, of the combined moieties of threohydrobupropion and erythrohydrobupropion.
While not systematically studied, certain drugs may induce the metabolism of bupropion (e.g., carbamazepine, phenobarbital, phenytoin).
Multiple oral doses of bupropion had no statistically significant effects on the single dose pharmacokinetics of lamotrigine in 12 healthy volunteers and was associated with a slight increase in the AUC (15%) of lamotrigine glucuronide.
Animal data indicated that bupropion may be an inducer of drug-metabolizing enzymes in humans. In one study, following chronic administration of bupropion, 100 mg 3 times daily to 8 healthy male volunteers for 14 days, there was no evidence of induction of its own metabolism. Nevertheless, there may be the potential for clinically important alterations of blood levels of coadministered drugs.
Drug Metabolized By Cytochrome P450IID6 (CYP2D6): Many drugs, including most antidepressants (SSRIs, many tricyclics), beta-blockers, antiarrhythmics, and antipsychotics are metabolized by the CYP2D6 isoenzyme. Although bupropion is not metabolized by this isoenzyme, bupropion and hydroxybupropion are inhibitors of CYP2D6 isoenzyme in vitro. In a study of 15 male subjects (ages 19 to 35 years) who were extensive metabolizers of the CYP2D6 isoenzyme, daily doses of bupropion given as 150 mg twice daily followed by a single dose of 50 mg desipramine increased the Cmax, AUC, and t1/2 of desipramine by an average of approximately 2-, 5- and 2-fold, respectively. The effect was present for at least 7 days after the last dose of bupropion. Concomitant use of bupropion with other drugs metabolized by CYP2D6 has not been formally studied.
Therefore, coadministration of bupropion with drugs that are metabolized by CYP2D6 isoenzyme including certain antidepressants (e.g., nortriptyline, imipramine, desipramine, paroxetine, fluoxetine, sertraline), antipsychotics (e.g., haloperidol, risperidone, thioridazine), beta-blockers (e.g., metoprolol), and Type 1C antiarrhythmics (e.g., propafenone, flecainide), should be approached with caution and should be initiated at the lower end of the dose range of the concomitant medication. If bupropion is added to the treatment regimen of a patient already receiving a drug metabolized by CYP2D6, the need to decrease the dose of the original medication should be considered, particularly for those concomitant medications with a narrow therapeutic index.
MAO Inhibitors: Studies in animals demonstrate that the acute toxicity of bupropion is enhanced by the MAO inhibitor phenelzine (see CONTRAINDICATIONS).
Levodopa and Amantadine: Limited clinical data suggest a higher incidence of adverse experiences in patients receiving bupropion concurrently with either levodopa or amantadine. Administration of BUDEPRION SR® to patients receiving either levodopa or amantadine concurrently should be undertaken with caution, using small initial doses and gradual dose increases.
Drugs that Lower Seizure Threshold: Concurrent administration of BUDEPRION SR® and agents (e.g., antipsychotics, other antidepressants, theophylline, systemic steroids, etc.) that lower seizure threshold should be undertaken only with extreme caution (see WARNINGS). Low initial dosing and gradual dose increases should be employed.
Alcohol: In postmarketing experience, there have been rare reports of adverse neuropsychiatric events or reduced alcohol tolerance in patients who were drinking alcohol during treatment with BUDEPRION SR®. The consumption of alcohol during treatment with BUDEPRION SR® should be minimized or avoided (also see CONTRAINDICATIONS).
Carcinogenesis, Mutagenesis, Impairment of Fertility: Lifetime carcinogenicity studies were performed in rats and mice at doses up to 300 and 150 mg/kg per day, respectively. These doses are approximately 7 and 2 times the maximum recommended human dose (MRHD), respectively, on a mg/m2 basis. In the rat study there was an increase in nodular proliferative lesions of the liver at doses of 100 to 300 mg/kg per day (approximately 2 to 7 times the MRHD on a mg/m2 basis); lower doses were not tested. The question of whether or not such lesions may be precursors of neoplasms of the liver is currently unresolved. Similar liver lesions were not seen in the mouse study, and no increase in malignant tumors of the liver and other organs was seen in either study.
Bupropion produced a positive response (2 to 3 times control mutation rate) in 2 of 5 strains in the Ames bacterial mutagenicity test and an increase in chromosomal aberrations in 1 of 3 in vivo rat bone marrow cytogenetic studies.
A fertility study in rats at doses up to 300 mg/kg revealed no evidence of impaired fertility.
Pregnancy
Teratogenic Effects
Pregnancy Category C. In studies conducted in rats and rabbits, bupropion was administered orally at doses up to 450 and 150 mg/kg/day, respectively (approximately 11 and 7 times the maximum recommended human dose [MRHD], respectively, on a mg/m2 basis), during the period of organogenesis. No clear evidence of teratogenic activity was found in either species; however, in rabbits, slightly increased incidences of fetal malformations and skeletal variations were observed at the lowest dose tested (25 mg/kg/day, approximately equal to MRHD on a mg/m2 basis) and greater. Decreased fetal weights were seen at 50 mg/kg and greater.
When rats were administered bupropion at oral doses of up to 300 mg/kg/day (approximately 7 times the MRHD on a mg/m2 basis) prior to mating and throughout pregnancy and lactation, there were no apparent adverse effects on offspring development.
One study has been conducted in pregnant women. This retrospective, managed-care database study assessed the risk of congenital malformations overall, and cardiovascular malformations specifically, following exposure to bupropion in the first trimester compared to the risk of these malformations following exposure to other antidepressants in the first trimester and bupropion outside of the first trimester. This study included 7005 infants with antidepressant exposure during pregnancy, 1213 of whom were exposed to bupropion in the first trimester. The study showed no greater risk for congenital malformations overall, or cardiovascular malformations specifically, following first trimester bupropion exposure compared to exposure to all other antidepressants in the first trimester, or bupropion outside of the first trimester. The results of this study have not been corroborated. BUDEPRION SR® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: Like many other drugs, bupropion and its metabolites are secreted in human milk. Because of the potential for serious adverse reactions in nursing infants from BUDEPRION SR®, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use: Safety and effectiveness in the pediatric population have not been established (see BOX WARNING and WARNINGS-Clinical Worsening and Suicide Risk). Anyone considering the use of BUDEPRION SR® in a child or adolescent must balance the potential risks with the clinical need.
Geriatric Use: Of the approximately 6000 patients who participated in clinical trials with bupropion sustained-release tablets (depression and smoking cessation studies), 275 were 65 and over and 47 were 75 and over. In addition, several hundred patients 65 and over participated in clinical trials using the immediate-release formulation of bupropion (depression studies). No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
A single-dose pharmacokinetic study demonstrated that the disposition of bupropion and its metabolites in elderly subjects was similar to that of younger subjects; however, another pharmacokinetic study, single and multiple dose, has suggested that the elderly are at increased risk for accumulation of bupropion and its metabolites (see CLINICAL PHARMACOLOGY).
Bupropion is extensively metabolized in the liver to active metabolites, which are further metabolized and excreted by the kidneys. The risk of toxic reaction to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see PRECAUTIONS: Renal Impairment and DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
(See also WARNINGS and PRECAUTIONS)
The information included under the Incidence in Controlled Trials subsection of ADVERSE REACTIONS is based primarily on data from controlled clinical trials with BUDEPRION SR®. Information on additional adverse events associated with the sustained-release formulation of bupropion in smoking cessation trials, as well as the immediate-release formulation of bupropion, is included in a separate section (see Other Events Observed During the Clinical Development and Postmarketing Experience of Bupropion).
Incidence in Controlled Trials With BUDEPRION SR®:
Adverse Events Associated With Discontinuation of Treatment Among Patients Treated With BUDEPRION SR®:
In placebo-controlled clinical trials, 9% and 11% of patients treated with 300 and 400 mg/day, respectively, of BUDEPRION SR® and 4% of patients treated with placebo discontinued treatment due to adverse events. The specific adverse events in these trials that led to discontinuation in at least 1% of patients treated with either 300 or 400 mg/day of BUDEPRION SR® and at a rate at least twice the placebo rate are listed in Table 4.
| Adverse Event Term | Bupropion HCl ER
Tablets (SR) 300 mg/day (n=376) | Bupropion HCl ER
Tablets (SR) 400 mg/day (n=114) | Placebo
(n=385) |
|---|---|---|---|
|
Rash |
2.4% |
0.9% |
0.0% |
|
Nausea |
0.8% |
1.8% |
0.3% |
|
Agitation |
0.3% |
1.8% |
0.3% |
|
Migraine |
0.0% |
1.8% |
0.3% |
Adverse Events Occurring at an Incidence of 1% or More Among Patients Treated WithBUDEPRION SR®:
Table 5 enumerates treatment-emergent adverse events that occurred among patients treated with 300 and 400 mg/day of BUDEPRION SR® and with placebo in placebo-controlled trials. Events that occurred in either the 300- or 400-mg/day group at an incidence of 1% or more and were more frequent than in the placebo group are included. Reported adverse events were classified using a COSTART-based Dictionary.
Accurate estimates of the incidence of adverse events associated with the use of any drug are difficult to obtain. Estimates are influenced by drug dose, detection technique, setting, physician judgments, etc. The figures cited cannot be used to predict precisely the incidence of untoward events in the course of usual medical practice where patient characteristics and other factors differ from those that prevailed in the clinical trials. These incidence figures also cannot be compared with those obtained from other clinical studies involving related drug products as each group of drug trials is conducted under a different set of conditions.
Finally, it is important to emphasize that the tabulation does not reflect the relative severity and/or clinical importance of the events. A better perspective on the serious adverse events associated with the use of BUDEPRION SR® is provided in the WARNINGS and PRECAUTIONS sections.
| Body System/Adverse Event | Bupropion HCl ER Tablets (SR)
300 mg/day (n=376) | Bupropion HCl ER Tablets (SR)
400 mg/day (n=114) | Placebo
(n = 385) |
|---|---|---|---|
| t Incidence based on the number of female patients. -- Hyphen denotes adverse events occurring in greater than 0 but less than 0.5% of patients. |
|||
|
|||
|
Body (General) | |||
|
Headache |
26% |
25% |
23% |
|
Infection |
8% |
9% |
6% |
|
Abdominal pain |
3% |
9% |
2% |
|
Asthenia |
2% |
4% |
2% |
|
Chest pain |
3% |
4% |
1% |
|
Pain |
2% |
3% |
2% |
|
Fever |
1% |
2% |
-- |
|
Cardiovascular | |||
|
Palpitation |
2% |
6% |
2% |
|
Flushing |
1% |
4% |
-- |
|
Migraine |
1% |
4% |
1% |
|
Hot flashes |
1% |
3% |
1% |
|
Digestive | |||
|
Dry mouth |
17% |
24% |
7% |
|
Nausea |
13% |
18% |
8% |
|
Constipation |
10% |
5% |
7% |
|
Diarrhea |
5% |
7% |
6% |
|
Anorexia |
5% |
3% |
2% |
|
Vomiting |
4% |
2% |
2% |
|
Dysphagia |
0% |
2% |
0% |
|
Musculoskeletal | |||
|
Myalgia |
2% |
6% |
3% |
|
Arthralgia |
1% |
4% |
1% |
|
Arthritis |
0% |
2% |
0% |
|
Twitch |
1% |
2% |
-- |
|
Nervous system | |||
|
Insomnia |
11% |
16% |
6% |
|
Dizziness |
7% |
11% |
5% |
|
Agitation |
3% |
9% |
2% |
|
Anxiety |
5% |
6% |
3% |
|
Tremor |
6% |
3% |
1% |
|
Nervousness |
5% |
3% |
3% |
|
Somnolence |
2% |
3% |
2% |
|
Irritability |
3% |
2% |
2% |
|
Memory decreased |
-- |
3% |
1% |
|
Paresthesia |
1% |
2% |
1% |
|
CNS stimulation |
2% |
1% |
1% |
|
Respiratory | |||
|
Pharyngitis |
3% |
11% |
2% |
|
Sinusitis |
3% |
1% |
2% |
|
Increased cough |
1% |
2% |
1% |
|
Skin | |||
|
Sweating |
6% |
5% |
2% |
|
Rash |
5% |
4% |
1% |
|
Pruritus |
2% |
4% |
2% |
|
Urticaria |
2% |
1% |
0% |
|
Special senses | |||
|
Tinnitus |
6% |
6% |
2% |
|
Taste perversion |
2% |
4% |
-- |
|
Amblyopia |
3% |
2% |
2% |
|
Urogenital | |||
|
Urinary frequency |
2% |
5% |
2% |
|
Urinary urgency |
-- |
2% |
0% |
|
Vaginal hemorrhage # |
0% |
2% |
-- |
|
Urinary tract infection |
1% |
0% |
-- |
Incidence of Commonly Observed Adverse Events in Controlled Clinical Trials: Adverse events from Table 5 occurring in at least 5% of patients treated with BUDEPRION SR® and at a rate at least twice the placebo rate are listed below for the 300 and 400 mg/day dose groups.
BUDEPRION SR® 300 mg/day: Anorexia, dry mouth, rash, sweating, tinnitus, and tremor.
BUDEPRION SR® 400 mg/day: Abdominal pain, agitation, anxiety, dizziness, dry mouth, insomnia, myalgia, nausea, palpitation, pharyngitis, sweating, tinnitus, and urinary frequency.
Other Events Observed During the Clinical Development and PostmarketingExperience of Bupropion: In addition to the adverse events noted above, the following events have been reported in clinical trials and postmarketing experience with the sustained-release formulation of bupropion in depressed patients and in nondepressed smokers, as well as in clinical trials and postmarketing clinical experience with the immediate-release formulation of bupropion.
Adverse events for which frequencies are provided below occurred in clinical trials with the sustained-release formulation of bupropion. The frequencies represent the proportion of patients who experienced a treatment-emergent adverse event on at least one occasion in placebo-controlled studies for depression (n=987) or smoking cessation (n=1013), or patients who experienced an adverse event requiring discontinuation of treatment in an open-label surveillance study with BUDEPRION SR® (n=3100). All treatment-emergent adverse events are included except those listed in Tables 2 through 5, those events listed in other safety-related sections, those adverse events subsumed under COSTART terms that are either overly general or excessively specific so as to be uninformative, those events not reasonably associated with the use of the drug, and those events that were not serious and occurred in fewer than two patients. Events of major clinical importance are described in the WARNINGS and PRECAUTIONS sections of labeling.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions of frequency: Frequent adverse events are defined as those occurring in at least 1/100 patients. Infrequent adverse events are those occurring in 1/100 to 1/1000 patients, while rare events are those occurring in less than 1/1000 patients.
Adverse events for which frequencies are not provided occurred in clinical trials or postmarketing experience with bupropion. Only those adverse events not previously listed for sustained-release bupropion are included. The extent to which these events may be associated with BUDEPRION SR® is unknown.
Body (general): Infrequent were chills, facial edema, musculoskeletal chest pain, and photosensitivity. Rare was malaise. Also observed were arthralgia, myalgia, and fever with rash and other symptoms suggestive of delayed hypersensitivity. These symptoms may resemble serum sickness (see PRECAUTIONS).
Cardiovascular: Infrequent were postural hypotension, stroke, tachycardia, and vasodilation. Rare was syncope. Also observed were complete atrioventricular block, extrasystoles, hypotension, hypertension (in some cases severe, see PRECAUTIONS), myocardial infarction, phlebitis, and pulmonary embolism.
Digestive: Infrequent were abnormal liver function, bruxism, gastric reflux, gingivitis, glossitis, increased salivation, jaundice, mouth ulcers, stomatitis, and thirst. Rare was edema of tongue. Also observed were colitis, esophagitis, gastrointestinal hemorrhage, gum hemorrhage, hepatitis, intestinal perforation, liver damage, pancreatitis, and stomach ulcer.
Endocrine: Also observed were hyperglycemia, hypoglycemia, and syndrome of inappropriate antidiuretic hormone.
Hemic and Lymphatic: Infrequent was ecchymosis. Also observed were anemia, leukocytosis, leukopenia, lymphadenopathy, pancytopenia, and thrombocytopenia. Altered PT and/or INR, infrequently associated with hemorrhagic or thrombotic complications, were observed when bupropion was coadministered with warfarin.
Metabolic and Nutritional: Infrequent were edema and peripheral edema. Also observed was glycosuria.
Musculoskeletal: Infrequent were leg cramps. Also observed were muscle rigidity/fever/rhabdomyolysis and muscle weakness.
Nervous System: Infrequent were abnormal coordination, decreased libido, depersonalization, dysphoria, emotional lability, hostility, hyperkinesia, hypertonia, hypesthesia, suicidal ideation, and vertigo. Rare were amnesia, ataxia, derealization, and hypomania. Also observed were abnormal electroencephalogram (EEG), akinesia, aggression, aphasia, coma, delirium, delusion, dysarthria, dyskinesia, dystonia, euphoria, extrapyramidal syndrome, hallucinations, hypokinesia, increased libido, manic reaction, neuralgia, neuropathy, paranoid ideation, restlessness, and unmasking tardive dyskinesia.
Skin: Rare was maculopapular rash. Also observed were alopecia, angioedema, exfoliative dermatitis, and hirsutism.
DRUG ABUSE AND DEPENDENCE
Humans: Controlled clinical studies of bupropion (immediate-release formulation) conducted in normal volunteers, in subjects with a history of multiple drug abuse, and in depressed patients showed some increase in motor activity and agitation/excitement.
In a population of individuals experienced with drugs of abuse, a single dose of 400 mg of bupropion produced mild amphetamine-like activity as compared to placebo on the Morphine-Benzedrine Subscale of the Addiction Research Center Inventories (ARCI), and a score intermediate between placebo and amphetamine on the Liking Scale of the ARCI. These scales measure general feelings of euphoria and drug desirability.
Findings in clinical trials, however, are not known to reliably predict the abuse potential of drugs. Nonetheless, evidence from single-dose studies does suggest that the recommended daily dosage of bupropion when administered in divided doses is not likely to be especially reinforcing to amphetamine or stimulant abusers. However, higher doses that could not be tested because of the risk of seizure might be modestly attractive to those who abuse stimulant drugs.
Animals: Studies in rodents and primates have shown that bupropion exhibits some pharmacologic actions common to psychostimulants. In rodents, it has been shown to increase locomotor activity, elicit a mild stereotyped behavioral response, and increase rates of responding in several schedule-controlled behavior paradigms. In primate models to assess the positive reinforcing effects of psychoactive drugs, bupropion was self-administered intravenously. In rats, bupropion produced amphetamine-like and cocaine-like discriminative stimulus effects in drug discrimination paradigms used to characterize the subjective effects of psychoactive drugs.
OVERDOSAGE
Human Overdose Experience: Overdoses of up to 30 g or more of bupropion have been reported. Seizure was reported in approximately one third of all cases. Other serious reactions reported with overdoses of bupropion alone included hallucinations, loss of consciousness, sinus tachycardia, and ECG changes such as conduction disturbances or arrhythmias. Fever, muscle rigidity, rhabdomyolysis, hypotension, stupor, coma, and respiratory failure have been reported mainly when bupropion was part of multiple drug overdoses.
Although most patients recovered without sequelae, deaths associated with overdoses of bupropion alone have been reported in patients ingesting large doses of the drug. Multiple uncontrolled seizures, bradycardia, cardiac failure, and cardiac arrest prior to death were reported in these patients.
Overdosage Management: Ensure an adequate airway, oxygenation, and ventilation. Monitor cardiac rhythm and vital signs. EEG monitoring is also recommended for the first 48 hours post-ingestion. General supportive and symptomatic measures are also recommended. Induction of emesis is not recommended. Gastric lavage with a large-bore orogastric tube with appropriate airway protection, if needed, may be indicated if performed soon after ingestion or in symptomatic patients.
Activated charcoal should be administered. There is no experience with the use of forced diuresis, dialysis, hemoperfusion, or exchange transfusion in the management of bupropion overdoses. No specific antidotes for bupropion are known.
Due to the dose-related risk of seizures with BUDEPRION SR®, hospitalization following suspected overdose should be considered. Based on studies in animals, it is recommended that seizures be treated with intravenous benzodiazepine administration and other supportive measures, as appropriate.
In managing overdosage, consider the possibility of multiple drug involvement. The physician should consider contacting a poison control center for additional information on the treatment of any overdose. Telephone numbers for certified poison control centers are listed in the Physicians’ Desk Reference (PDR).
DOSAGE AND ADMINISTRATION
General Dosing Considerations: It is particularly important to administer BUDEPRION SR® in a manner most likely to minimize the risk of seizure (see WARNINGS). Gradual escalation in dosage is also important if agitation, motor restlessness, and insomnia, often seen during the initial days of treatment, are to be minimized. If necessary, these effects may be managed by temporary reduction of dose or the short-term administration of an intermediate to long-acting sedative hypnotic. A sedative hypnotic usually is not required beyond the first week of treatment. Insomnia may also be minimized by avoiding bedtime doses. If distressing, untoward effects supervene, dose escalation should be stopped. BUDEPRION SR® should be swallowed whole and not crushed, divided, or chewed.
Initial Treatment: The usual adult target dose for BUDEPRION SR® is 300 mg/day, given as 150 mg twice daily. Dosing with BUDEPRION SR® should begin at 150 mg/day given as a single daily dose in the morning. If the 150 mg initial dose is adequately tolerated, an increase to the 300 mg/day target dose, given as 150 mg twice daily, may be made as early as day 4 of dosing. There should be an interval of at least 8 hours between successive doses.
Increasing the Dosage Above 300 mg/day: As with other antidepressants, the full antidepressant effect of BUDEPRION SR® may not be evident until 4 weeks of treatment or longer. An increase in dosage to the maximum of 400 mg/day, given as 200 mg twice daily, may be considered for patients in whom no clinical improvement is noted after several weeks of treatment at 300 mg/day.
Maintenance Treatment: It is generally agreed that acute episodes of depression require several months or longer of sustained pharmacological therapy beyond response to the acute episode. In a study in which patients with major depressive disorder, recurrent type, who had responded during 8 weeks of acute treatment with BUDEPRION SR® were assigned randomly to placebo or to the same dose of BUDEPRION SR® (150 mg twice daily) during 44 weeks of maintenance treatment as they had received during the acute stabilization phase, longer-term efficacy was demonstrated (see CLINICAL TRIALS under CLINICAL PHARMACOLOGY). Based on these limited data, it is unknown whether or not the dose of BUDEPRION SR® needed for maintenance treatment is identical to the dose needed to achieve an initial response. Patients should be periodically reassessed to determine the need for maintenance treatment and the appropriate dose for such treatment.
Dosage Adjustment for Patients with Impaired Hepatic Function: BUDEPRION SR® should be used with extreme caution in patients with severe hepatic cirrhosis. The dose should not exceed 100 mg every day or 150 mg every other day in these patients. BUDEPRION SR® should be used with caution in patients with hepatic impairment (including mild to moderate hepatic cirrhosis) and a reduced frequency and/or dose should be considered in patients with mild to moderate hepatic cirrhosis (see CLINICAL PHARMACOLOGY, WARNINGS, and PRECAUTIONS).
HOW SUPPLIED
BUDEPRION SR®, 100 mg of bupropion hydrochloride, are yellow, round, convex, film-coated tablets, debossed with “G” on one side and “2442” on the other side.
Bottles of 60....................................................NDC 0093-5501-06
Bottles of 100..................................................NDC 0093-5501-01
BUDEPRION SR®, 150 mg of bupropion hydrochloride, are light yellow, round, convex, film-coated tablets debossed with “G” on one side and “2444” on the other side.
Bottles of 60....................................................NDC 0093-5502-06
Bottles of 100..................................................NDC 0093-5502-01
BUDEPRION SR® 100 mg and 150 mg tablets are available from Cardinal Health in unit dose packages of 100 tablets.
100 mg, unit dose package of 100 tablets, NDC 55154-8255-4
150 mg, unit dose package of 100 tablets, NDC 55154-8250-4
Manufactured By:
IMPAX Laboratories, Inc.
Hayward, California 94544 USA
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
ZYBAN®, WELLBUTRIN®, and WELLBUTRIN XL® are registered trademarks of GlaxoSmithKline.
Rev. 06/2007
514-03
IU37361050408
PHARMACIST - DETACH HERE AND GIVE MEDICATION GUIDE TO PATIENT.
MEDICATION GUIDE
Antidepressant Medicines, Depression and other Serious Mental Illnesses, and Suicidal Thoughts orActions
Read the Medication Guide that comes with you or your family member’s antidepressant medicine. This Medication Guide is only about the risk of suicidal thoughts and actions with antidepressant medicines. Talk to your or your familymember’s healthcare provider about:
• all risks and benefits of treatment with antidepressant medicines
• all treatment choices for depression or other serious mental illness
What is the most important information I should know about antidepressant medicines, depression and otherserious mental illnesses, and suicidal thoughts oractions?
1. Antidepressant medicines may increase suicidalthoughts or actions in some children, teenagers,and young adults within the first few months oftreatment.
2. Depression and other serious mental illnesses arethe most important causes of suicidal thoughts andactions. Some people may have a particularly highrisk of having suicidal thoughts or actions. These include people who have (or have a family history of) bipolar illness (also called manic-depressive illness) or suicidal thoughts or actions.
3. How can I watch for and try to prevent suicidalthoughts and actions in myself or a family member?
• Pay close attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings. This is very important when an antidepressant medicine is started or when the dose is changed.
• Call the healthcare provider right away to report new or sudden changes in mood, behavior, thoughts, or feelings.
• Keep all follow-up visits with the healthcare provider as scheduled. Call the healthcare provider between visits as needed, especially if you have concerns about symptoms.
Call a healthcare provider right away if you or your family member has any of the following symptoms,especially if they are new, worse, or worry you:
• thoughts about suicide or dying
• attempts to commit suicide
• new or worse depression
• new or worse anxiety
• feeling very agitated or restless
• panic attacks
• trouble sleeping (insomnia)
• new or worse irritability
• acting aggressive, being angry, or violent
• acting on dangerous impulses
• an extreme increase in activity and talking (mania)
• other unusual changes in behavior or mood
What else do I need to know about antidepressant
medicines?
• Never stop an antidepressant medicine without first talking to a healthcare provider. Stopping an antidepressant medicine suddenly can cause other symptoms.
• Antidepressants are medicines used to treatdepression and other illnesses. It is important to discuss all the risks of treating depression and also the risks of not treating it. Patients and their families or other caregivers should discuss all treatment choices with the healthcare provider, not just the use of antidepressants.
• Antidepressant medicines have other side effects. Talk to the healthcare provider about the side effects of the medicine prescribed for you or your family member.
• Antidepressant medicines can interact with othermedicines. Know all of the medicines that you or your family member takes. Keep a list of all medicines to show the healthcare provider. Do not start new medicines without first checking with your healthcare provider.
• Not all antidepressant medicines prescribed for children are FDA approved for use in children. Talk to your child’s healthcare provider for more information.
This Medication Guide has been approved by the U.S. Food and Drug Administration for all antidepressants.
Manufactured By:
IMPAX Laboratories, Inc.
Hayward, CA 94544 USA
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
Rev. 06/2007
514-03
IU37361050408
PHARMACIST - DETACH HERE AND GIVE LEAFLET TO PATIENT.
Patient Information
BUDEPRION SR®
(buPROPion HCI Extended-Release Tablets)
Read the Patient Information that comes with BUDEPRION SR® before you starttaking BUDEPRION SR® and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your doctor about your medical condition or your treatment.
BUDEPRION SR® has not been studied in children under the age of 18 and is not approved for use in children and teenagers.
What other important information should I know about BUDEPRION SR®?
There is a chance of having a seizure (convulsion, fit) with BUDEPRION SR®,especially in people:
• with certain medical problems.
• who take certain medicines.
The chance of having seizures increases with higher doses of BUDEPRION SR®.
For more information, see the sections “Who should not take BUDEPRION SR®?” and “What should I tell my doctor before using BUDEPRION SR®?” Tell your doctor about all of your medical conditions and all the medicines you take. Do nottake any other medicines while you are using BUDEPRION SR® unless yourdoctor has said it is okay to take them.
If you have a seizure while taking BUDEPRION SR®, stop taking the tablets and call your doctor right away. Do not take BUDEPRION SR® again if you have a seizure.
What is BUDEPRION SR®?
BUDEPRION SR® is a prescription medicine used to treat adults with a certain type of depression called major depressive disorder.
Who should not take BUDEPRION SR®?
Do not take BUDEPRION SR® if you
• have or had a seizure disorder or epilepsy
• are taking ZYBAN® (used to help people stop smoking) or any other medicines that contain bupropion hydrochloride, such as WELLBUTRIN® Tablets or WELLBUTRIN XL® [bupropion hydrochloride extended-release Tablets (XL)]. Bupropion is the same active ingredient that is in BUDEPRION SR®
• drink a lot of alcohol and abruptly stop drinking, or use medicines called sedatives (these make you sleepy) or benzodiazepines and you stop using them all of a sudden.
• have taken within the last 14 days medicine for depression called a monoamine oxidase inhibitor (MAOI), such as NARDIL® (phenelzine sulfate), PARNATE® (tranylcypromine sulfate), or MARPLAN® (isocarboxazid).
• have or had an eating disorder such as anorexia nervosa or bulimia.
• are allergic to the active ingredient in BUDEPRION SR®, bupropion, or to any of the inactive ingredients. See the end of this leaflet for a complete list of ingredients in BUDEPRION SR®.
What should I tell my doctor before using BUDEPRION SR®?
• Tell your doctor about your medical conditions. Tell your doctor if you:
• are pregnant or plan to become pregnant. It is not known if BUDEPRION SR® can harm your unborn baby.
• are breastfeeding. BUDEPRION SR® passes through your milk. It is not known if BUDEPRION SR® can harm your baby.
• have liver problems, especially cirrhosis of the liver.
• have kidney problems.
• have an eating disorder such as anorexia nervosa or bulimia.
• have had a head injury.
• have had a seizure (convulsion, fit).
• have a tumor in your nervous system (brain or spine).
• have had a heart attack, heart problems, or high blood pressure.
• are a diabetic taking insulin or other medicines to control your blood sugar.
• drink a lot of alcohol.
• abuse prescription medicines or street drugs.
• Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Many medicines increase your chances of having seizures or other serious side effects if you take them while you are using BUDEPRION SR®.
How should I take BUDEPRION SR®?
• Take BUDEPRION SR® exactly as prescribed by your doctor.
• Do not chew, cut, or crush BUDEPRION SR®. You must swallow the tablets whole.
Tell your doctor if you cannot swallow medicine tablets.
• Take BUDEPRION SR® at the same time each day.
• Take your doses of BUDEPRION SR® at least 8 hours apart.
• You may take BUDEPRION SR® with or without food.
• If you miss a dose, do not take an extra tablet to make up for the dose you forgot. Wait and take your next tablet at the regular time. This is very important. Too much BUDEPRION SR® can increase your chance of having a seizure.
• If you take too much BUDEPRION SR®, or overdose, call your local emergency room or poison control center right away.
• Do not take any other medicines while using BUDEPRION SR® unless yourdoctor has told you it is okay.
• It may take several weeks for you to feel that BUDEPRION SR® is working. Once you feel better, it is important to keep taking BUDEPRION SR® exactly as directed by your doctor. Call your doctor if you do not feel BUDEPRION SR® is working for you.
• Do not change your dose or stop taking BUDEPRION SR® without talking with your doctor first.
What should I avoid while taking BUDEPRION SR®?
• Do not drink a lot of alcohol while taking BUDEPRION SR®. If you usually drink a lot of alcohol, talk with your doctor before suddenly stopping. If you suddenly stop drinking alcohol, you may increase your chance of having seizures.
• Do not drive a car or use heavy machinery until you know how BUDEPRION SR ® affects you. BUDEPRION SR® can impair your ability to perform these tasks.
What are possible side effects of BUDEPRION SR®?
• Seizures. Some patients get seizures while taking BUDEPRION SR®. If you have a seizure while taking BUDEPRION SR®, stop taking the tablets and call your doctor right away. Do not take BUDEPRION SR® again if you have a seizure.
• Hypertension (high blood pressure). Some patients get high blood pressure, sometimes severe, while taking BUDEPRION SR®. The chance of high blood pressure may be increased if you also use nicotine replacement therapy (for example, a nicotine patch) to help you stop smoking.
• Severe allergic reactions: Stop taking BUDEPRION SR® and call your doctor right away if you get a rash, itching, hives, fever, swollen lymph glands, painful sores in the mouth or around the eyes, swelling of the lips or tongue, chest pain, or have trouble breathing. These could be signs of a serious allergic reaction.
• Unusual thoughts or behaviors. Some patients have unusual thoughts or behaviors while taking BUDEPRION SR®, including delusions (believe you are someone else), hallucinations (seeing or hearing things that are not there), paranoia (feeling that people are against you), or feeling confused. If this happens to you, call your doctor.
The most common side effects of BUDEPRION SR® are loss of appetite, dry mouth, skin rash, sweating, ringing in the ears, shakiness, stomach pain, agitation, anxiety, dizziness, trouble sleeping, muscle pain, nausea, fast heart beat, sore throat, and urinating more often.
If you have nausea, you may want to take your medicine with food. If you have trouble sleeping, do not take your medicine too close to bedtime.
Tell your doctor right away about any side effects that bother you.
These are not all the side effects of BUDEPRION SR®. For a complete list, ask your doctor or pharmacist.
How should I store BUDEPRION SR®?
• Store BUDEPRION SR® at room temperature. Store out of direct sunlight. Keep BUDEPRION SR® in its tightly closed bottle.
• BUDEPRION SR® may have an odor.
General Information about BUDEPRION SR®.
• Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use BUDEPRION SR® for a condition for which it was not prescribed. Do not give BUDEPRION SR® to other people, even if they have the same symptoms you have. It may harm them. Keep BUDEPRION SR® out of the reach of children.
This leaflet summarizes important information about BUDEPRION SR®. For more information, talk with your doctor. You can ask your doctor or pharmacist for information about BUDEPRION SR® that is written for health professionals.
What are the ingredients in BUDEPRION SR®?
Active ingredient: bupropion hydrochloride.
Inactive ingredients: colloidal silicon dioxide, hydroxypropyl cellulose, magnesium stearate, and microcrystalline cellulose. The 100 mg tablet also contains FD&C red # 40, FD&C yellow # 5, hypromellose, iron oxide yellow, macrogol, polydextrose, titanium dioxide and triacetin. The 150 mg tablet also contains hypromellose, iron oxide yellow, macrogol, polydextrose, titanium dioxide and triacetin.
Rx only
Manufactured By:
IMPAX Laboratories, Inc.
Hayward, CA 94544 USA
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
ZYBAN®, WELLBUTRIN®, and WELLBUTRIN XL® are registered trademarks of
GlaxoSmithKline.
Nardil® is a registered trademark of Parke Davis.
Parnate® is a registered trademark of GlaxoSmithKline.
Marplan® is a registered trademark of Oxford Pharmaceutical Services.
Rev. 06/2007
514-03
BUDEPRION SR® 100 mg and 150 mg tablets are available from Cardinal Health in
unit dose packages of 100 tablets.
100 mg, unit dose package of 100 tablets, NDC 55154-8255-4
150 mg, unit dose package of 100 tablets, NDC 55154-8250-4
Cardinal Health
Zanesville OH 43701
IU37361050408
PRINCIPAL DISPLAY PANEL - 100 mg Carton
Budeprion SR®
(bupropion HCl Extended-release)
100 mg
100 Tablets




| BUDEPRION
SR
bupropion hydrochloride tablet, extended release |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| BUDEPRION
SR
bupropion hydrochloride tablet, extended release |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Cardinal Health (188557102) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Cardinal Health | 188557102 | REPACK(55154-8255, 55154-8250) | |