Label: FASLODEX- fulvestrant injection

- NDC Code(s): 0310-0720-10
- Packager: AstraZeneca Pharmaceuticals LP
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 25, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FASLODEX safely and effectively. See full prescribing information for FASLODEX.
FASLODEX® (fulvestrant) injection, for intramuscular use
Initial U.S. Approval: 2002INDICATIONS AND USAGE
FASLODEX is an estrogen receptor antagonist indicated for the treatment of:
- •
- Hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer in postmenopausal women not previously treated with endocrine therapy. (1)
- •
- HR-positive advanced breast cancer in postmenopausal women with disease progression following endocrine therapy. (1)
- •
- HR-positive, HER2-negative advanced or metastatic breast cancer in postmenopausal women in combination with ribociclib, as initial endocrine based therapy or following disease progression on endocrine therapy. (1)
- •
- HR-positive, HER2-negative advanced or metastatic breast cancer in combination with palbociclib or abemaciclib in women with disease progression after endocrine therapy. (1)
DOSAGE AND ADMINISTRATION
- •
- FASLODEX 500 mg should be administered intramuscularly into the buttocks (gluteal area) slowly (1 - 2 minutes per injection) as two 5 mL injections, one in each buttock, on Days 1, 15, 29, and once monthly thereafter. (2.1, 14)
- •
- A dose of 250 mg is recommended in patients with moderate hepatic impairment to be administered intramuscularly into the buttock (gluteal area) slowly (1 - 2 minutes) as one 5 mL injection on Days 1, 15, 29, and once monthly thereafter. (2.2, 5.2, 8.6)
DOSAGE FORMS AND STRENGTHS
FASLODEX, an injection for intramuscular administration, is supplied as 250 mg/5 mL fulvestrant. (3)
CONTRAINDICATIONS
- •
- Hypersensitivity. (4)
WARNINGS AND PRECAUTIONS
- •
- Risk of Bleeding: Use with caution in patients with bleeding diatheses, thrombocytopenia, or anticoagulant use. (5.1)
- •
- Increased Exposure in Patients with Hepatic Impairment: Use a 250 mg dose for patients with moderate hepatic impairment. (2.2, 5.2, 8.6)
- •
- Injection Site Reaction: Use caution while administering FASLODEX at the dorsogluteal injection site due to the proximity of the underlying sciatic nerve. (5.3)
- •
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.4, 8.1, 8.3)
- •
- Immunoassay Measurement of Serum Estradiol: FASLODEX can interfere with estradiol measurement by immunoassay, resulting in falsely elevated estradiol levels. (5.5)
ADVERSE REACTIONS
- •
- The most common adverse reactions occurring in ≥5% of patients receiving FASLODEX 500 mg were: injection site pain, nausea, bone pain, arthralgia, headache, back pain, fatigue, pain in extremity, hot flash, vomiting, anorexia, asthenia, musculoskeletal pain, cough, dyspnea, and constipation. (6.1)
- •
- Increased hepatic enzymes (ALT, AST, ALP) occurred in >15% of FASLODEX patients and were not dose-dependent. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- There are no known drug-drug interactions. (7)
USE IN SPECIFIC POPULATIONS
- •
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Dose Modification
2.3 Administration Technique
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Bleeding
5.2 Increased Exposure in Patients with Hepatic Impairment
5.3 Injection Site Reaction
5.4 Embryo-Fetal Toxicity
5.5 Immunoassay Measurement of Serum Estradiol
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Monotherapy
FASLODEX is indicated for the treatment of:
- •
- Hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer in postmenopausal women not previously treated with endocrine therapy, or
- •
- HR-positive advanced breast cancer in postmenopausal women with disease progression following endocrine therapy.
Combination Therapy
FASLODEX is indicated for the treatment of:
- •
- HR-positive, HER2-negative advanced or metastatic breast cancer in postmenopausal women in combination with ribociclib as initial endocrine based therapy or following disease progression on endocrine therapy.
- •
- HR-positive, HER2-negative advanced or metastatic breast cancer in combination with palbociclib or abemaciclib in women with disease progression after endocrine therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
Monotherapy
The recommended dose of FASLODEX is 500 mg to be administered intramuscularly into the buttocks (gluteal area) slowly (1 - 2 minutes per injection) as two 5 mL injections, one in each buttock, on Days 1, 15, 29, and once monthly thereafter [see Clinical Studies (14)].
Combination Therapy
When FASLODEX is used in combination with palbociclib, abemaciclib, or ribociclib, the recommended dose of FASLODEX is 500 mg to be administered intramuscularly into the buttocks (gluteal area) slowly (1 - 2 minutes per injection) as two 5 mL injections, one in each buttock, on Days 1, 15, 29, and once monthly thereafter.
When FASLODEX is used in combination with palbociclib, the recommended dose of palbociclib is a 125 mg capsule taken orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a complete cycle of 28 days. Palbociclib should be taken with food. Refer to the Full Prescribing Information for palbociclib.
When FASLODEX is used in combination with abemaciclib, the recommended dose of abemaciclib is 150 mg orally, twice daily. Abemaciclib may be taken with or without food. Refer to the Full Prescribing Information for abemaciclib.
When FASLODEX is used in combination with ribociclib, the recommended dose of ribociclib is 600 mg taken orally, once daily for 21 consecutive days followed by 7 days off treatment resulting in a complete cycle of 28 days. Ribociclib can be taken with or without food. Refer to the Full Prescribing Information for ribociclib.
Pre/perimenopausal women treated with the combination of FASLODEX plus palbociclib, abemaciclib, or ribociclib, should be treated with luteinizing hormone-releasing hormone (LHRH) agonists according to current clinical practice standards [see Clinical Studies (14)].
2.2 Dose Modification
Monotherapy
Hepatic Impairment:
A dose of 250 mg is recommended for patients with moderate hepatic impairment (Child-Pugh class B) to be administered intramuscularly into the buttock (gluteal area) slowly (1 - 2 minutes) as one 5 mL injection on Days 1, 15, 29, and once monthly thereafter.
FASLODEX has not been evaluated in patients with severe hepatic impairment (Child-Pugh class C) [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
Combination Therapy
When FASLODEX is used in combination with palbociclib, abemaciclib, or ribociclib, refer to monotherapy dose modification instructions for FASLODEX.
Refer to the Full Prescribing Information of co-administered palbociclib, abemaciclib, or ribociclib for dose modification guidelines in the event of toxicities, for use with concomitant medications, and other relevant safety information.
2.3 Administration Technique
Administer the injection according to the local guidelines for performing large volume intramuscular injections.
NOTE: Due to the proximity of the underlying sciatic nerve, caution should be taken if administering FASLODEX at the dorsogluteal injection site [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
The proper method of administration of FASLODEX for intramuscular use is described in the following instructions.
For each single-dose prefilled syringe:
- 1.
- Remove glass syringe barrel from tray and check that it is not damaged.
- 2.
- Remove perforated patient record label from syringe.
- 3.
- Inspect drug product in glass syringe for any visible particulate matter or discoloration prior to use. Discard if particulate matter or discoloration is present.
- 4.
- Peel open the safety needle (SafetyGlide™) outer packaging.
- 5.
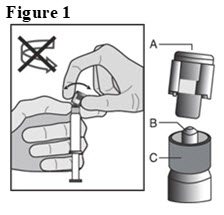
- Hold the syringe upright on the ribbed part (C). With the other hand, take hold of the cap (A) and carefully tilt cap back and forth (DO NOT TWIST CAP) until the cap disconnects for removal (see Figure 1).
- 6.
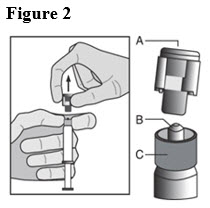
- Pull the cap (A) off in a straight upward direction. DO NOT TOUCH THE STERILE SYRINGE TIP (Luer-Lok) (B) (see Figure 2).
- 7.
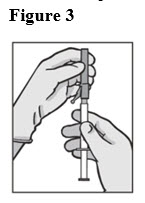
- Attach the safety needle to the syringe tip (Luer-Lok). Twist needle until firmly seated (see Figure 3). Confirm that the needle is locked to the Luer connector before moving or tilting the syringe out of the vertical plane to avoid spillage of syringe contents.
- For Administration:
- 8.
- Pull shield straight off needle to avoid damaging needle point.
- 9.
- Remove needle sheath.
- 10.
- Expel excess gas from the syringe (a small gas bubble may remain).
- 11.
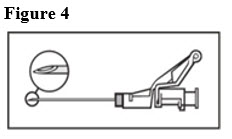
- Administer intramuscularly slowly (1-2 minutes/injection) into the buttock (gluteal area). For user convenience, the needle ‘bevel up’ position is orientated to the lever arm, as shown in Figure 4.
- 12.
- After injection, immediately activate the lever arm to deploy the needle shielding by applying a single-finger stroke to the activation assisted lever arm to push the lever arm completely forward. Listen for a click. Confirm that the needle shielding has completely covered the needle (see Figure 5).
- NOTE: Activate away from self and others.
- 13.
- Discard the empty syringe into an approved sharps collector in accordance with applicable regulations and institutional policy.
- 14.
- Repeat steps 1 through 13 for second syringe.
How To Use FASLODEX
For the 2 x 5 mL syringe package, the contents of both syringes must be injected to receive the 500 mg recommended dose.
SAFETYGLIDE™ INSTRUCTIONS FROM BECTON DICKINSON
SafetyGlide™ is a trademark of Becton Dickinson and Company.
Important Administration Information
To help avoid HIV (AIDS), HBV (Hepatitis), and other infectious diseases due to accidental needlesticks, contaminated needles should not be recapped or removed, unless there is no alternative or that such action is required by a specific medical procedure. Hands must remain behind the needle at all times during use and disposal.
Do not autoclave SafetyGlide™ Needle before use.
Becton Dickinson guarantees the contents of their unopened or undamaged packages to be sterile, non-toxic, and non-pyrogenic.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
FASLODEX is contraindicated in patients with a known hypersensitivity to the drug or to any of its components. Hypersensitivity reactions, including urticaria and angioedema, have been reported in association with FASLODEX [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Bleeding
Because FASLODEX is administered intramuscularly, it should be used with caution in patients with bleeding diatheses, thrombocytopenia, or anticoagulant use.
5.2 Increased Exposure in Patients with Hepatic Impairment
The safety and pharmacokinetics of FASLODEX were evaluated in a study in seven subjects with moderate hepatic impairment (Child-Pugh class B) and seven subjects with normal hepatic function. Exposure was increased in patients with moderate hepatic impairment, therefore, a dose of 250 mg is recommended [see Dosage and Administration (2.2)].
FASLODEX has not been studied in patients with severe hepatic impairment (Child-Pugh class C) [see Use in Specific Populations (8.6)].
5.3 Injection Site Reaction
Injection site related events including sciatica, neuralgia, neuropathic pain, and peripheral neuropathy have been reported with FASLODEX injection. Caution should be taken while administering FASLODEX at the dorsogluteal injection site due to the proximity of the underlying sciatic nerve [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.4 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, FASLODEX can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of fulvestrant to pregnant rats and rabbits during organogenesis resulted in embryo-fetal toxicity at daily doses that are significantly less than the maximum recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with FASLODEX and for one year after the last dose [see Use in Specific Populations (8.1), (8.3) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- •
- Risk of Bleeding [see Warnings and Precautions (5.1)]
- •
- Increased Exposure in Patients with Hepatic Impairment [see Warnings and Precautions (5.2)]
- •
- Injection Site Reaction [see Warnings and Precautions (5.3)]
- •
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
Monotherapy
Comparison of FASLODEX 500 mg and FASLODEX 250 mg (CONFIRM)
The following adverse reactions (ARs) were calculated based on the safety analysis of CONFIRM comparing the administration of FASLODEX 500 mg intramuscularly once a month with FASLODEX 250 mg intramuscularly once a month. The most frequently reported adverse reactions in the FASLODEX 500 mg group were injection site pain (11.6% of patients), nausea (9.7% of patients), and bone pain (9.4% of patients); the most frequently reported adverse reactions in the FASLODEX 250 mg group were nausea (13.6% of patients), back pain (10.7% of patients), and injection site pain (9.1% of patients).
Table 1 lists adverse reactions reported with an incidence of 5% or greater, regardless of assessed causality, from CONFIRM.
Table 1: Adverse Reactions in CONFIRM (≥5% in Either Treatment Group) - *
- Including more severe injection site related sciatica, neuralgia, neuropathic pain, and peripheral neuropathy.
Adverse Reactions
FASLODEX 500 mg
N=361
%
FASLODEX 250 mg
N=374
%
Body as a Whole
Injection Site Pain*
12
9
Headache
8
7
Back Pain
8
11
Fatigue
8
6
Pain in Extremity
7
7
Asthenia
6
6
Vascular System
Hot Flash
7
6
Digestive System
Nausea
10
14
Vomiting
6
6
Anorexia
6
4
Constipation
5
4
Musculoskeletal System
Bone Pain
9
8
Arthralgia
8
8
Musculoskeletal Pain
6
3
Respiratory System
Cough
5
5
Dyspnea
4
5
In the pooled safety population (N=1127) from clinical trials comparing FASLODEX 500 mg to FASLODEX 250 mg, post-baseline increases of ≥1 CTC grade in either AST, ALT, or alkaline phosphatase were observed in >15% of patients receiving FASLODEX. Grade 3-4 increases were observed in 1-2% of patients. The incidence and severity of increased hepatic enzymes (ALT, AST, ALP) did not differ between the 250 mg and the 500 mg FASLODEX arms.
Comparison of FASLODEX 500 mg and Anastrozole 1 mg (FALCON)
The safety of FASLODEX 500 mg versus anastrozole 1 mg was evaluated in FALCON. The data described below reflect exposure to FASLODEX in 228 out of 460 patients with HR-positive advanced breast cancer in postmenopausal women not previously treated with endocrine therapy who received at least one (1) dose of treatment in FALCON.
Permanent discontinuation associated with an adverse reaction occurred in 4 of 228 (1.8%) patients receiving FASLODEX and in 3 of 232 (1.3%) patients receiving anastrozole. Adverse reactions leading to discontinuation for those patients receiving FASLODEX included drug hypersensitivity (0.9%), injection site hypersensitivity (0.4%), and elevated liver enzymes (0.4%).
The most common adverse reactions (≥10%) of any grade reported in patients in the FASLODEX arm were arthralgia, hot flash, fatigue, and nausea.
Adverse reactions reported in patients who received FASLODEX in FALCON at an incidence of ≥5% in either treatment arm are listed in Table 2, and laboratory abnormalities are listed in Table 3.
Table 2: Adverse Reactions in FALCON Adverse Reactions
FASLODEX 500 mg
N=228
Anastrozole 1 mg
N=232
All Grades
%
Grade 3 or 4
%
All Grades
%
Grade 3 or 4
%
Vascular Disorders
Hot flash
11
0
10
0
Gastrointestinal Disorders
Nausea
11
0
10
<1
Diarrhea
6
0
6
<1
Musculoskeletal and Connective Tissue Disorders
Arthralgia
17
0
10
0
Myalgia
7
0
3
0
Pain in extremity
6
0
4
0
Back pain
9
<1
6
0
General Disorders and Administration Site Conditions
Fatigue
11
<1
7
<1
Table 3: Laboratory Abnormalities in FALCON* - *
- In FALCON, post-baseline increases of ≥1 CTC grade in either AST, ALT, or alkaline phosphatase were observed in >10% of patients receiving FASLODEX. Grade 3-4 increases were observed in 1%-3% of patients.
Laboratory Parameters
FASLODEX 500 mg
N=228
Anastrozole 1 mg
N=232
All Grades
%
Grade 3 or 4
%
All Grades
%
Grade 3 or 4
%
Alanine aminotransferase increased (ALT)
7
1
3
0
Aspartate aminotransferase increased (AST)
5
1
3
<1
Comparison of FASLODEX 250 mg and Anastrozole 1 mg in Combined Trials (Studies 0020 and 0021)
The most commonly reported adverse reactions in the FASLODEX and anastrozole treatment groups were gastrointestinal symptoms (including nausea, vomiting, constipation, diarrhea, and abdominal pain), headache, back pain, vasodilatation (hot flashes), and pharyngitis.
Injection site reactions with mild transient pain and inflammation were seen with FASLODEX and occurred in 7% of patients given the single 5 mL injection (Study 0020) and in 27% of patients given the 2 x 2.5 mL injections (Study 0021) in the two clinical trials that compared FASLODEX 250 mg and anastrozole 1 mg.
Table 4 lists adverse reactions reported with an incidence of 5% or greater, regardless of assessed causality, from the two controlled clinical trials comparing the administration of FASLODEX 250 mg intramuscularly once a month with anastrozole 1 mg orally once a day.
Table 4: Adverse Reactions in Studies 0020 and 0021 (≥5% from Combined Data) Adverse Reactions FASLODEX 250 mg
N=423
%Anastrozole 1 mg
N=423
%- *
- Including more severe injection site related sciatica, neuralgia, neuropathic pain, and peripheral neuropathy. All patients on FASLODEX received injections, but only those anastrozole patients who were in Study 0021 received placebo injections.
Body as a Whole
68
68
Asthenia
23
27
Pain
19
20
Headache
15
17
Back Pain
14
13
Abdominal Pain
12
12
Injection Site Pain*
11
7
Pelvic Pain
10
9
Chest Pain
7
5
Flu Syndrome
7
6
Fever
6
6
Accidental Injury
5
6
Cardiovascular System
30
28
Vasodilatation
18
17
Digestive System
52
48
Nausea
26
25
Vomiting
13
12
Constipation
13
11
Diarrhea
12
13
Anorexia
9
11
Hemic and Lymphatic Systems
14
14
Anemia
5
5
Metabolic and Nutritional Disorders
18
18
Peripheral Edema
9
10
Musculoskeletal System
26
28
Bone Pain
16
14
Arthritis
3
6
Nervous System
34
34
Dizziness
7
7
Insomnia
7
9
Paresthesia
6
8
Depression
6
7
Anxiety
5
4
Respiratory System
39
34
Pharyngitis
16
12
Dyspnea
15
12
Cough Increased
10
10
Skin and Appendages
22
23
Rash
7
8
Sweating
5
5
Urogenital System
18
15
Urinary Tract Infection
6
4
Combination Therapy
Combination Therapy with Palbociclib (PALOMA-3)
The safety of FASLODEX 500 mg plus palbociclib 125 mg/day versus FASLODEX plus placebo was evaluated in PALOMA-3. The data described below reflect exposure to FASLODEX plus palbociclib in 345 out of 517 patients with HR-positive, HER2-negative advanced or metastatic breast cancer who received at least 1 dose of treatment in PALOMA-3. The median duration of treatment for FASLODEX plus palbociclib was 10.8 months while the median duration of treatment for FASLODEX plus placebo arm was 4.8 months.
No dose reduction was allowed for FASLODEX in PALOMA-3. Dose reductions of palbociclib due to an adverse reaction of any grade occurred in 36% of patients receiving FASLODEX plus palbociclib.
Permanent discontinuation associated with an adverse reaction occurred in 19 of 345 (6%) patients receiving FASLODEX plus palbociclib, and in 6 of 172 (3%) patients receiving FASLODEX plus placebo. Adverse reactions leading to discontinuation for those patients receiving FASLODEX plus palbociclib included fatigue (0.6%), infections (0.6%), and thrombocytopenia (0.6%).
The most common adverse reactions (≥10%) of any grade reported in patients in the FASLODEX plus palbociclib arm by descending frequency were neutropenia, leukopenia, infections, fatigue, nausea, anemia, stomatitis, diarrhea, thrombocytopenia, vomiting, alopecia, rash, decreased appetite, and pyrexia.
The most frequently reported Grade ≥3 adverse reactions (≥5%) in patients receiving FASLODEX plus palbociclib in descending frequency were neutropenia and leukopenia.
Adverse reactions (≥10%) reported in patients who received FASLODEX plus palbociclib or FASLODEX plus placebo in PALOMA-3 are listed in Table 5, and laboratory abnormalities are listed in Table 6.
Table 5: Adverse Reactions (≥10%) in PALOMA-3 Adverse Reactions FASLODEX plus Palbociclib
N=345FASLODEX plus Placebo
N=172All Grades
%Grade 3
%Grade 4
%All Grades
%Grade 3
%Grade 4
%- *
- Infections includes all reported preferred terms (PTs) that are part of the System Organ Class Infections and infestations.
- †
- Most common infections (≥1%) include: nasopharyngitis, upper respiratory infection, urinary tract infection, influenza, bronchitis, rhinitis, conjunctivitis, pneumonia, sinusitis, cystitis, oral herpes, respiratory tract infection, gastroenteritis, tooth infection, pharyngitis, eye infection, herpes simplex, paronychia.
- ‡
- Stomatitis includes: aphthous stomatitis, cheilitis, glossitis, glossodynia, mouth ulceration, mucosal inflammation, oral pain, oropharyngeal discomfort, oropharyngeal pain, stomatitis.
- §
- Grade 1 events – 17%; Grade 2 events – 1%.
- ¶
- Grade 1 events – 6%.
- #
- Rash includes: rash, rash maculo-papular, rash pruritic, rash erythematous, rash papular, dermatitis, dermatitis acneiform, toxic skin eruption.
Infections and Infestations
Infections*
47†
3
1
31
3
0
Blood and Lymphatic System Disorders
Neutropenia
83
55
11
4
1
0
Leukopenia
53
30
1
5
1
1
Anemia
30
4
0
13
2
0
Thrombocytopenia
23
2
1
0
0
0
Metabolism and Nutrition Disorders
Decreased appetite
16
1
0
8
1
0
Gastrointestinal Disorders
Nausea
34
0
0
28
1
0
Stomatitis‡
28
1
0
13
0
0
Diarrhea
24
0
0
19
1
0
Vomiting
19
1
0
15
1
0
Skin and Subcutaneous Tissue Disorders
Alopecia
18§
N/A
N/A
6¶
N/A
N/A
Rash#
17
1
0
6
0
0
General Disorders and Administration Site Conditions
Fatigue
41
2
0
29
1
0
Pyrexia
13
<1
0
5
0
0
Grading according to CTCAE v.4.0.
CTCAE=Common Terminology Criteria for Adverse Events; N=number of patients; N/A=not applicable.
Additional adverse reactions occurring at an overall incidence of <10.0% of patients receiving FASLODEX plus palbociclib in PALOMA-3 included asthenia (7.5%), aspartate aminotransferase increased (7.5%), dysgeusia (6.7%), epistaxis (6.7%), lacrimation increased (6.4%), dry skin (6.1%), alanine aminotransferase increased (5.8%), vision blurred (5.8%), dry eye (3.8%), and febrile neutropenia (0.9%).
Table 6: Laboratory Abnormalities in PALOMA-3 Laboratory Parameters FASLODEX plus Palbociclib
N=345FASLODEX plus Placebo
N=172All Grades
%Grade 3
%Grade 4
%All Grades
%Grade 3
%Grade 4
%WBC decreased
99
45
1
26
0
1
Neutrophils decreased
96
56
11
14
0
1
Anemia
78
3
0
40
2
0
Platelets decreased
62
2
1
10
0
0
Aspartate aminotransferase increased
43
4
0
48
4
0
Alanine aminotransferase increased
36
2
0
34
0
0
N=number of patients; WBC=white blood cells.
Combination Therapy with Abemaciclib (MONARCH 2)
The safety of FASLODEX (500 mg) plus abemaciclib (150 mg twice daily) versus FASLODEX plus placebo was evaluated in MONARCH 2. The data described below reflect exposure to FASLODEX in 664 patients with HR-positive, HER2-negative advanced breast cancer who received at least one dose of FASLODEX plus abemaciclib or placebo in MONARCH 2.
Median duration of treatment was 12 months for patients receiving FASLODEX plus abemaciclib and 8 months for patients receiving FASLODEX plus placebo.
Dose reductions due to an adverse reaction occurred in 43% of patients receiving FASLODEX plus abemaciclib. Adverse reactions leading to dose reductions ≥5% of patients were diarrhea and neutropenia. Abemaciclib dose reduction due to diarrhea of any grade occurred in 19% of patients receiving FASLODEX plus abemaciclib compared to 0.4% of patients receiving FASLODEX plus placebo. Abemaciclib dose reductions due to neutropenia of any grade occurred in 10% of patients receiving FASLODEX plus abemaciclib compared to no patients receiving FASLODEX plus placebo.
Permanent study treatment discontinuation due to an adverse event was reported in 9% of patients receiving FASLODEX plus abemaciclib and in 3% of patients receiving FASLODEX plus placebo. Adverse reactions leading to permanent discontinuation for patients receiving FASLODEX plus abemaciclib were infection (2%), diarrhea (1%), hepatotoxicity (1%), fatigue (0.7%), nausea (0.2%), abdominal pain (0.2%), acute kidney injury (0.2%), and cerebral infarction (0.2%).
Deaths during treatment or during the 30-day follow up, regardless of causality, were reported in 18 cases (4%) of FASLODEX plus abemaciclib treated patients versus 10 cases (5%) of FASLODEX plus placebo treated patients. Causes of death for patients receiving FASLODEX plus abemaciclib included: 7 (2%) patient deaths due to underlying disease, 4 (0.9%) due to sepsis, 2 (0.5%) due to pneumonitis, 2 (0.5%) due to hepatotoxicity, and one (0.2%) due to cerebral infarction.
The most common adverse reactions reported (≥20%) in the FASLODEX plus abemaciclib arm were diarrhea, fatigue, neutropenia, nausea, infections, abdominal pain, anemia, leukopenia, decreased appetite, vomiting, and headache (Table 7). The most frequently reported (≥5%) Grade 3 or 4 adverse reactions were neutropenia, diarrhea, leukopenia, anemia, and infections.
Table 7: Adverse Reactions ≥10% of Patients Receiving FASLODEX Plus Abemaciclib and ≥2% Higher Than FASLODEX Plus Placebo in MONARCH 2 - *
- Includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal discomfort, abdominal tenderness.
- †
- Includes upper respiratory tract infection, urinary tract infection, lung infection, pharyngitis, conjunctivitis, sinusitis, vaginal infection, sepsis.
- ‡
- Includes neutropenia, neutrophil count decreased.
- §
- Includes anemia, hematocrit decreased, hemoglobin decreased, red blood cell count decreased.
- ¶
- Includes leukopenia, white blood cell count decreased.
- #
- Includes platelet count decreased, thrombocytopenia.
- Þ
- Includes asthenia, fatigue.
Adverse Reactions
FASLODEX plus Abemaciclib
N=441
FASLODEX plus Placebo
N=223
All Grades
%
Grade
3
%
Grade 4
%
All Grades
%
Grade
3
%
Grade 4
%
Gastrointestinal Disorders
Diarrhea
86
13
0
25
<1
0
Nausea
45
3
0
23
1
0
Abdominal pain*
35
2
0
16
1
0
Vomiting
26
<1
0
10
2
0
Stomatitis
15
<1
0
10
0
0
Infections and Infestations
Infections†
43
5
<1
25
3
<1
Blood and Lymphatic System Disorders
Neutropenia‡
46
24
3
4
1
<1
Anemia§
29
7
<1
4
1
0
Leukopenia¶
28
9
<1
2
0
0
Thrombocytopenia#
16
2
1
3
0
<1
General Disorders and Administration Site Conditions
FatigueÞ
46
3
0
32
<1
0
Edema peripheral
12
0
0
7
0
0
Pyrexia
11
<1
<1
6
<1
0
Metabolism and Nutrition Disorders
Decreased appetite
27
1
0
12
<1
0
Respiratory, Thoracic, and Mediastinal Disorders
Cough
13
0
0
11
0
0
Skin and Subcutaneous Tissue Disorders
Alopecia
16
0
0
2
0
0
Pruritus
13
0
0
6
0
0
Rash
11
1
0
4
0
0
Nervous System Disorders
Headache
20
1
0
15
<1
0
Dysgeusia
18
0
0
3
0
0
Dizziness
12
1
0
6
0
0
Investigations
Alanine aminotransferase increased
13
4
<1
5
2
0
Aspartate aminotransferase increased
12
2
0
7
3
0
Creatinine increased
12
<1
0
<1
0
0
Weight decreased
10
<1
0
2
<1
0
Additional adverse reactions in MONARCH 2 include venous thromboembolic events (deep vein thrombosis, pulmonary embolism, cerebral venous sinus thrombosis, subclavian vein thrombosis, axillary vein thrombosis, and DVT inferior vena cava), which were reported in 5% of patients treated with FASLODEX plus abemaciclib as compared to 0.9% of patients treated with FASLODEX plus placebo.
Table 8: Laboratory Abnormalities ≥10% in Patients Receiving FASLODEX Plus Abemaciclib and ≥2% Higher Than FASLODEX Plus Placebo in MONARCH 2 Laboratory Parameters Fulvestrant plus Abemaciclib
N=441Fulvestrant plus Placebo
N=223All Grades
%Grade 3
%Grade 4
%All Grades
%Grade 3
%Grade 4
%Creatinine increased
98
1
0
74
0
0
White blood cell decreased
90
23
<1
33
<1
0
Neutrophil count decreased
87
29
4
30
4
<1
Anemia
84
3
0
33
<1
0
Lymphocyte count decreased
63
12
<1
32
2
0
Platelet count decreased
53
<1
1
15
0
0
Alanine aminotransferase increased
41
4
<1
32
1
0
Aspartate aminotransferase increased
37
4
0
25
4
<1
Combination Therapy with Ribociclib (MONALEESA-3)
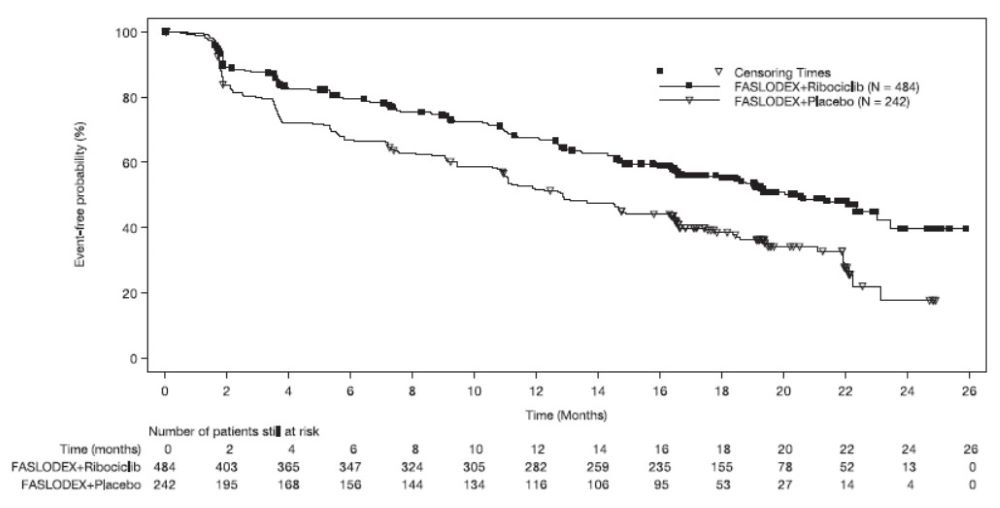
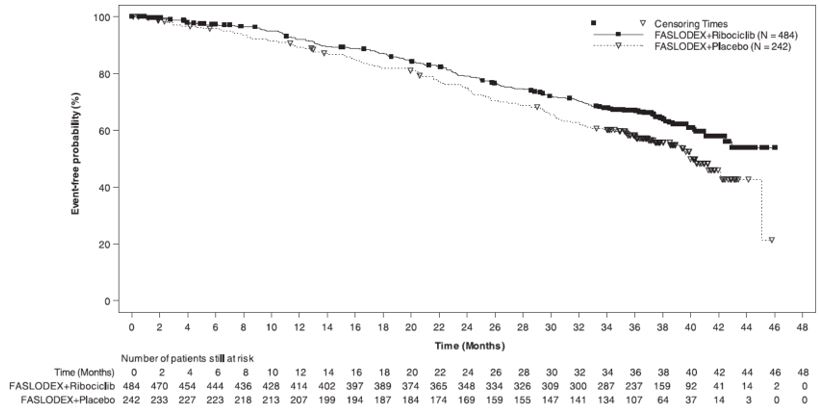
The safety of FASLODEX 500 mg plus ribociclib 600 mg versus FASLODEX plus placebo was evaluated in MONALEESA-3. The data described below reflect exposure to FASLODEX plus ribociclib in 483 out of 724 postmenopausal patients with HR-positive, HER2-negative advanced or metastatic breast cancer for initial endocrine based therapy or after disease progression on endocrine therapy who received at least one dose of FASLODEX plus ribociclib or placebo in MONALEESA-3. Median duration of treatment was 15.8 months for FASLODEX plus ribociclib and 12 months for FASLODEX plus placebo.
Dose reductions due to adverse reactions occurred in 32% of patients receiving FASLODEX plus ribociclib and in 3% of patients receiving FASLODEX plus placebo. Among patients receiving FASLODEX plus ribociclib, 8% were reported to have permanently discontinued both FASLODEX plus ribociclib, and 9% were reported to have discontinued ribociclib alone due to ARs. Among patients receiving FASLODEX plus placebo, 4% were reported to have permanently discontinued both FASLODEX and placebo and 2% were reported to have discontinued placebo alone due to ARs.
Adverse reactions leading to treatment discontinuation of FASLODEX plus ribociclib (as compared to FASLODEX plus placebo) were ALT increased (5% vs. 0%), AST increased (3% vs. 0.6%), and vomiting (1% vs. 0%).
The most common adverse reactions (reported at a frequency ≥20% on the FASLODEX plus ribociclib arm and ≥2% higher than FASLODEX plus placebo) were neutropenia, infections, leukopenia, cough, nausea, diarrhea, vomiting, constipation, pruritus, and rash. The most frequently reported Grade 3/4 adverse reactions (reported at a frequency ≥5%) in patients receiving FASLODEX plus ribociclib in descending frequency were neutropenia, leukopenia, infections, and abnormal liver function tests.
Adverse reactions and laboratory abnormalities occurring in patients in MONALEESA-3 are listed in Table 9 and Table 10, respectively.
Table 9: Adverse Reactions Occurring in ≥10% and ≥2% higher than FASLODEX plus Placebo Arm in MONALEESA-3 (All Grades) - *
- Infections; urinary tract infections; respiratory tract infections; gastroenteritis; sepsis (<1%).
Adverse Reactions
FASLODEX plus Ribociclib
N=483
FASLODEX plus Placebo
N=241
All Grades
%
Grade 3
%
Grade 4
%
All Grades
%
Grade 3
%
Grade 4
%
Infections and Infestations
Infections*
42
5
0
30
2
0
Blood and Lymphatic System Disorders
Neutropenia
69
46
7
2
0
0
Leukopenia
27
12
<1
<1
0
0
Anemia
17
3
0
5
2
0
Metabolism and Nutrition Disorders
Decreased appetite
16
<1
0
13
0
0
Nervous System Disorders
Dizziness
13
<1
0
8
0
0
Respiratory, Thoracic, and Mediastinal Disorders
Cough
22
0
0
15
0
0
Dyspnea
15
1
<1
12
2
0
Gastrointestinal Disorders
Nausea
45
1
0
28
<1
0
Diarrhea
29
<1
0
20
<1
0
Vomiting
27
1
0
13
0
0
Constipation
25
<1
0
12
0
0
Abdominal pain
17
1
0
13
<1
0
Skin and Subcutaneous Tissue Disorders
Alopecia
19
0
0
5
0
0
Pruritus
20
<1
0
7
0
0
Rash
23
<1
0
7
0
0
General Disorders and Administration Site Conditions
Edema peripheral
15
0
0
7
0
0
Pyrexia
11
<1
0
7
0
0
Investigations
Alanine aminotransferase increased
15
7
2
5
<1
0
Aspartate aminotransferase increased
13
5
1
5
<1
0
Grading according to CTCAE 4.03.
CTCAE=Common Terminology Criteria for Adverse Events; N=number of patients
Additional adverse reactions in MONALEESA-3 for patients receiving FASLODEX plus ribociclib included asthenia (14%), dyspepsia (10%), thrombocytopenia (9%), dry skin (8%), dysgeusia (7%), electrocardiogram QT prolonged (6%), dry mouth (5%), vertigo (5%), dry eye (5%), lacrimation increased (4%), erythema (4%), hypocalcemia (4%), blood bilirubin increased (1%), and syncope (1%).
Table 10: Laboratory Abnormalities Occurring in ≥10% of Patients in MONALEESA-3 Laboratory parameters FASLODEX plus Ribociclib
N=483FASLODEX plus Placebo
N=241All Grades
%Grade 3
%Grade 4
%All Grades
%Grade 3
%Grade 4
%Hematology
Leukocyte count decreased
95
25
<1
26
<1
0
Neutrophil count decreased
92
46
7
21
<1
0
Hemoglobin decreased
60
4
0
35
3
0
Lymphocyte count decreased
69
14
1
35
4
<1
Platelet count decreased
33
<1
1
11
0
0
Chemistry
Creatinine increased
65
<1
<1
33
<1
0
Gamma-glutamyl transferase increased
52
6
1
49
8
2
Aspartate aminotransferase increased
49
5
2
43
3
0
Alanine aminotransferase increased
44
8
3
37
2
0
Glucose serum decreased
23
0
0
18
0
0
Phosphorous decreased
18
5
0
8
<1
0
Albumin decreased
12
0
0
8
0
0
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of FASLODEX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
For FASLODEX 250 mg, other adverse reactions reported as drug-related and seen infrequently (<1%) include thromboembolic phenomena, myalgia, vertigo, leukopenia, and hypersensitivity reactions, including angioedema and urticaria.
Vaginal bleeding has been reported infrequently (<1%), mainly in patients during the first 6 weeks after changing from existing hormonal therapy to treatment with FASLODEX. If bleeding persists, further evaluation should be considered.
Elevation of bilirubin, elevation of gamma GT, hepatitis, and liver failure have been reported infrequently (<1%).
-
7 DRUG INTERACTIONS
There are no known drug-drug interactions. Although, fulvestrant is metabolized by CYP 3A4 in vitro, drug interactions studies with ketoconazole or rifampin did not alter fulvestrant pharmacokinetics. Dose adjustment is not needed in patients co-prescribed CYP 3A4 inhibitors or inducers [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, FASLODEX can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of fulvestrant to pregnant rats and rabbits during organogenesis caused embryo-fetal toxicity, including skeletal malformations and fetal loss, at daily doses that were 6% and 30% of the maximum recommended human dose based on mg/m2, respectively [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Administration of fulvestrant to rats prior to and up to implantation caused embryonic loss at daily doses that were 0.6% of the daily maximum recommended human dose based on mg/m2. When fulvestrant was administered to pregnant rats during the period of organogenesis, intramuscular doses ≥0.1 mg/kg/day (6% of the human recommended dose based on mg/m2) caused effects on embryo-fetal development consistent with its antiestrogenic activity. Fulvestrant caused an increased incidence of fetal abnormalities in rats (tarsal flexure of the hind paw at 2 mg/kg/day; equivalent to the human dose based on mg/m2) and non-ossification of the odontoid and ventral tubercle of the first cervical vertebra at doses ≥0.1 mg/kg/day. Fulvestrant administered at 2 mg/kg/day caused fetal loss.
When administered to pregnant rabbits during the period of organogenesis, fulvestrant caused pregnancy loss at an intramuscular dose of 1 mg/kg/day (equivalent to the human dose based on mg/m2). Further, at 0.25 mg/kg/day (30% the human dose based on mg/m2), fulvestrant caused increases in placental weight and post-implantation loss in rabbits. Fulvestrant was associated with an increased incidence of fetal variations in rabbits (backwards displacement of the pelvic girdle, and 27 pre-sacral vertebrae at 0.25 mg/kg/day; 30% the human dose based on mg/m2) when administered during the period of organogenesis.
8.2 Lactation
Risk Summary
There is no information regarding the presence of fulvestrant in human milk, nor of its effects on milk production or breastfed infant. Fulvestrant can be detected in rat milk [see Data]. Because of the potential for serious adverse reactions in breastfed infants from FASLODEX, advise a lactating woman not to breastfeed during treatment with FASLODEX and for one year after the final dose.
Data
Levels of fulvestrant were approximately 12-fold higher in milk than in plasma after exposure of lactating rats to a dose of 2 mg/kg. Drug exposure in rodent pups from fulvestrant-treated lactating dams was estimated as 10% of the administered dose. In a study in rats of fulvestrant at 10 mg/kg given twice or 15 mg/kg given once (less than the recommended human dose based on mg/m2) during lactation, offspring survival was slightly reduced.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential within seven days prior to initiating FASLODEX.
Contraception
Females
FASLODEX can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment and for one year after the last dose.
Infertility
Based on animal studies, FASLODEX may impair fertility in females and males of reproductive potential. The effects of fulvestrant on fertility were reversible in female rats [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. A multi-center, single-arm, open-label, study of fulvestrant was conducted in 30 girls with McCune-Albright Syndrome (MAS) associated with Progressive Precocious Puberty (PPP). The median age at informed consent was 6 years old (range: 1 to 8).
The first 10 patients initially received fulvestrant 2 mg/kg. Based on PK data from the first 6 patients, all 10 patients receiving 2 mg/kg were escalated to a dose of 4 mg/kg and all other patients received 4 mg/kg from study entry.
Baseline measurements for vaginal bleeding days, bone age, growth velocity, and Tanner staging for at least 6 months prior to study entry were provided retrospectively by the parent, guardian, or local consultant. All measurements during the study period were collected prospectively. Patients’ baseline characteristics included the following: a mean ± SD chronological age of 5.9 ± 1.8 years; a mean rate of bone age advancement (change in bone age in years divided by change in chronological age in years) of 2.0 ± 1.03; and a mean growth velocity z-score of 2.4 ± 3.26.
Twenty-nine of 30 patients completed the 12-month study period. The following results were observed: 35% (95% CI: 16%, 57%) of the 23 patients with baseline vaginal bleeding experienced a complete cessation of vaginal bleeding on-treatment (month 0 to 12); a reduction in the rate of bone age advancement during the 12-month study period compared to baseline (mean change=-0.9 [95% CI: -1.4, -0.4]); and a reduction in mean growth velocity Z-score on-treatment compared to baseline (mean change=-1.1 [95% CI: -2.7, 0.4]). There were no clinically meaningful changes in median Tanner stage (breast or pubic), mean uterine volume, or mean ovarian volume, or predicted adult height (PAH) on-treatment compared to baseline. The effect of FASLODEX on bone mineral density in children has not been studied and is not known.
Eight patients (27%) experienced adverse reactions that were considered possibly related to FASLODEX. These included injection site reactions (inflammation, pain, hematoma, pruritus, rash), abdominal pain, contusion, tachycardia, hot flash, extremity pain, and vomiting. Nine (30%) patients reported an SAE, none of which were considered related to FASLODEX. No patients discontinued study treatment due to an AE and no patients died.
Pharmacokinetics
The pharmacokinetics of fulvestrant was characterized using a population pharmacokinetic analysis with sparse samples per patient obtained from 30 female pediatric patients aged 1 to 8 years with PPP associated with MAS. Pharmacokinetic data from 294 postmenopausal women with breast cancer who received 125 or 250 mg monthly dosing regimen were also included in the analysis.
In these pediatric patients receiving 4 mg/kg monthly intramuscular dose of fulvestrant, the geometric mean (SD) CL/F was 444 (165) mL/min which was 32% lower than adults. The geometric mean (SD) steady state trough concentration (Cmin,ss) and AUCss was 4.19 (0.87) ng/mL and 3680 (1020) ng*hr/mL, respectively.
8.5 Geriatric Use
For FASLODEX 250 mg, when tumor response was considered by age, objective responses were seen in 22% and 24% of patients under 65 years of age and in 11% and 16% of patients 65 years of age and older, who were treated with FASLODEX in Study 0021 and Study 0020, respectively.
8.6 Hepatic Impairment
FASLODEX is metabolized primarily in the liver.
The pharmacokinetics of fulvestrant were evaluated after a single dose of 100 mg in subjects with mild and moderate hepatic impairment and normal hepatic function (n=7 subjects/group), using a shorter-acting intramuscular injection formulation. Subjects with mild hepatic impairment (Child-Pugh class A) had comparable mean AUC and clearance values to those with normal hepatic function. In subjects with moderate hepatic impairment (Child-Pugh class B), the average AUC of fulvestrant increased by 70% compared to patients with normal hepatic function. AUC was positively correlated with total bilirubin concentration (p=0.012). FASLODEX has not been studied in patients with severe hepatic impairment (Child-Pugh class C).
A dose of FASLODEX 250 mg is recommended in patients with moderate hepatic impairment (Child-Pugh class B) [see Dosage and Administration (2.2) and Warnings and Precautions (5.2)].
8.7 Renal Impairment
Negligible amounts of fulvestrant are eliminated in urine; therefore, a study in patients with renal impairment was not conducted. In the advanced breast cancer trials, fulvestrant concentrations in women with estimated creatinine clearance as low as 30 mL/min were similar to women with normal creatinine.
-
10 OVERDOSAGE
Human experience of overdose with FASLODEX is limited. There are isolated reports of overdose with FASLODEX in humans. No adverse reactions were seen in healthy male and female volunteers who received intravenous fulvestrant, which resulted in peak plasma concentrations at the end of the infusion, that were approximately 10 to 15 times those seen after intramuscular injection. The potential toxicity of fulvestrant at these or higher concentrations in cancer patients who may have additional comorbidities is unknown. There is no specific treatment in the event of fulvestrant overdose, and symptoms of overdose are not established. In the event of an overdose, healthcare practitioners should follow general supportive measures and should treat symptomatically.
-
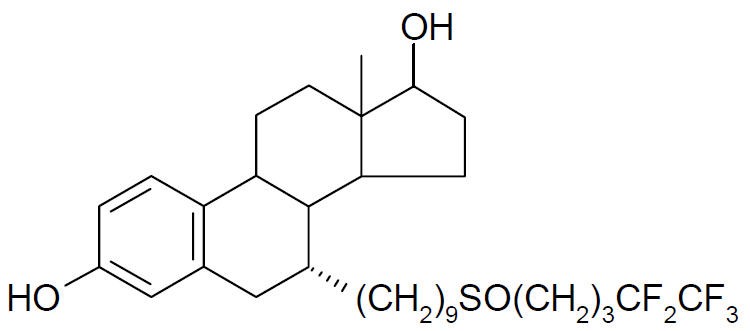
11 DESCRIPTION
FASLODEX® (fulvestrant) injection for intramuscular administration is an estrogen receptor antagonist. The chemical name is 7-alpha-[9-(4,4,5,5,5-penta fluoropentylsulphinyl) nonyl]estra-1,3,5-(10)- triene-3,17-beta-diol. The molecular formula is C32H47F5O3S and its structural formula is:
Fulvestrant is a white powder with a molecular weight of 606.77. The solution for injection is a clear, colorless to yellow, viscous liquid.
Each injection contains as inactive ingredients: 10% w/v Alcohol, USP, 10% w/v Benzyl Alcohol, NF, and 15% w/v Benzyl Benzoate, USP, as co-solvents, and made up to 100% w/v with Castor Oil, USP as a co-solvent and release rate modifier.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Many breast cancers have estrogen receptors (ER) and the growth of these tumors can be stimulated by estrogen. Fulvestrant is an estrogen receptor antagonist that binds to the estrogen receptor in a competitive manner with affinity comparable to that of estradiol and downregulates the ER protein in human breast cancer cells.
In vitro studies demonstrated that fulvestrant is a reversible inhibitor of the growth of tamoxifen-resistant, as well as estrogen-sensitive human breast cancer (MCF-7) cell lines. In in vivo tumor studies, fulvestrant delayed the establishment of tumors from xenografts of human breast cancer MCF-7 cells in nude mice. Fulvestrant inhibited the growth of established MCF-7 xenografts and of tamoxifen-resistant breast tumor xenografts.
Fulvestrant showed no agonist-type effects in in vivo uterotrophic assays in immature or ovariectomized mice and rats. In in vivo studies in immature rats and ovariectomized monkeys, fulvestrant blocked the uterotrophic action of estradiol. In postmenopausal women, the absence of changes in plasma concentrations of FSH and LH in response to fulvestrant treatment (250 mg monthly) suggests no peripheral steroidal effects.
12.2 Pharmacodynamics
In a clinical study in postmenopausal women with primary breast cancer treated with single doses of FASLODEX 15-22 days prior to surgery, there was evidence of increasing down-regulation of ER with increasing dose. This was associated with a dose-related decrease in the expression of the progesterone receptor, an estrogen-regulated protein. These effects on the ER pathway were also associated with a decrease in Ki67 labeling index, a marker of cell proliferation.
12.3 Pharmacokinetics
Absorption:
The single dose and multiple dose PK parameters for the 500 mg dosing regimen with an additional dose (AD) at Day 15 are reported in Table 11. The additional dose of FASLODEX given two weeks after the initial dose allows for steady state concentrations to be reached within the first month of dosing.
Table 11: Summary of Fulvestrant Pharmacokinetic Parameters [gMean (CV%)] in Postmenopausal Advanced Breast Cancer Patients after Intramuscular Administration 500 mg + AD Dosing Regimen Cmax
(ng/mL)Cmin
(ng/mL)AUC
(ng.hr/mL)500 mg + AD*
Single dose
25.1 (35.3)
16.3 (25.9)
11400 (33.4)
Multiple dose steady state†
28.0 (27.9)
12.2 (21.7)
13100 (23.4)
Distribution:
The apparent volume of distribution at steady state is approximately 3 to 5 L/kg. This suggests that distribution is largely extravascular. Fulvestrant is highly (99%) bound to plasma proteins; VLDL, LDL, and HDL lipoprotein fractions appear to be the major binding components. The role of sex hormone-binding globulin, if any, could not be determined.
Metabolism:
Biotransformation and disposition of fulvestrant in humans have been determined following intramuscular and intravenous administration of 14C-labeled fulvestrant. Metabolism of fulvestrant appears to involve combinations of a number of possible biotransformation pathways analogous to those of endogenous steroids, including oxidation, aromatic hydroxylation, conjugation with glucuronic acid and/or sulphate at the 2, 3, and 17 positions of the steroid nucleus, and oxidation of the side chain sulphoxide. Identified metabolites are either less active or exhibit similar activity to fulvestrant in antiestrogen models.
Studies using human liver preparations and recombinant human enzymes indicate that cytochrome P-450 3A4 (CYP 3A4) is the only P-450 isoenzyme involved in the oxidation of fulvestrant; however, the relative contribution of P-450 and non-P-450 routes in vivo is unknown.
Excretion:
Fulvestrant was rapidly cleared by the hepatobiliary route with excretion primarily via the feces (approximately 90%). Renal elimination was negligible (less than 1%). After an intramuscular injection of 250 mg, the clearance (Mean ± SD) was 690 ± 226 mL/min with an apparent half-life about 40 days.
Special Populations:
Geriatric:
In patients with breast cancer, there was no difference in fulvestrant pharmacokinetic profile related to age (range 33 to 89 years).
Gender:
Following administration of a single intravenous dose, there were no pharmacokinetic differences between men and women or between premenopausal and postmenopausal women. Similarly, there were no differences between men and postmenopausal women after intramuscular administration.
Race:
In the advanced breast cancer treatment trials, the potential for pharmacokinetic differences due to race have been evaluated in 294 women including 87.4% Caucasian, 7.8% Black, and 4.4% Hispanic. No differences in fulvestrant plasma pharmacokinetics were observed among these groups. In a separate trial, pharmacokinetic data from postmenopausal ethnic Japanese women were similar to those obtained in non-Japanese patients.
Drug-Drug Interactions:
There are no known drug-drug interactions. Fulvestrant does not significantly inhibit any of the major CYP isoenzymes, including CYP 1A2, 2C9, 2C19, 2D6, and 3A4 in vitro, and studies of co-administration of fulvestrant with midazolam indicate that therapeutic doses of fulvestrant have no inhibitory effects on CYP 3A4 or alter blood levels of drug metabolized by that enzyme. Although fulvestrant is partly metabolized by CYP 3A4, a clinical study with rifampin, an inducer of CYP 3A4, showed no effect on the pharmacokinetics of fulvestrant. Also, results from a healthy volunteer study with ketoconazole, a potent inhibitor of CYP 3A4, indicated that ketoconazole had no effect on the pharmacokinetics of fulvestrant and dosage adjustment is not necessary in patients co-prescribed CYP 3A4 inhibitors or inducers [see Drug Interactions (7)]. Data from a clinical trial in patients with breast cancer showed that there was no clinically relevant drug interaction when fulvestrant is co-administered with palbociclib, abemaciclib, or ribociclib.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenesis studies were conducted in rats and mice. Positive findings were observed in both species. Rats were treated at intramuscular doses of 15 mg/kg/30 days, 10 mg/rat/30 days, and 10 mg/rat/15 days.
These doses correspond to 0.9-, 1.5-, and 3-fold (in females) and 0.8-, 0.8-, and 2-fold (in males) the systemic exposure [AUC0-30 days] achieved in women receiving the recommended dose of 500 mg/month. An increased incidence of benign ovarian granulosa cell tumors and testicular Leydig cell tumors was evident, in females dosed at 10 mg/rat/15 days and males dosed at 15 mg/rat/30 days, respectively. Mice were treated at oral doses of 0, 20, 150, and 500 mg/kg/day. These doses correspond to 0-, 0.8-, 8.4-, and 18-fold (in females) and 0.8-, 7.1-, and 11.9-fold (in males), the systemic exposure (AUC0-30 days) achieved in women receiving the recommended dose of 500 mg/month. There was an increased incidence of sex cord stromal tumors (both benign and malignant) in the ovary of mice at doses of 150 and 500 mg/kg/day. Induction of such tumors is consistent with the pharmacology-related endocrine feedback alterations in gonadotropin levels caused by an antiestrogen.
Fulvestrant was not mutagenic or clastogenic in multiple in vitro tests with and without the addition of a mammalian liver metabolic activation factor (bacterial mutation assay in strains of Salmonella typhimurium and Escherichia coli, in vitro cytogenetics study in human lymphocytes, mammalian cell mutation assay in mouse lymphoma cells, and in vivo micronucleus test in rat).
In female rats, fulvestrant administered at doses ≥0.01 mg/kg/day (0.6% the human recommended dose based on body surface area [BSA in mg/m2]), for 2 weeks prior to and for 1 week following mating, caused a reduction in fertility and embryonic survival. No adverse effects on female fertility and embryonic survival were evident in female animals dosed at 0.001 mg/kg/day (0.06% the human dose based on BSA in mg/m2). Restoration of female fertility to values similar to controls was evident following a 29-day withdrawal period after dosing at 2 mg/kg/day (equivalent to the human dose based on BSA in mg/m2). The effects of fulvestrant on the fertility of female rats appear to be consistent with its antiestrogenic activity. The potential effects of fulvestrant on the fertility of male animals were not studied, but in a 6-month toxicology study, male rats treated with intramuscular doses of 15 mg/kg/30 days, 10 mg/rat/30 days, or 10 mg/rat/15 days fulvestrant showed a loss of spermatozoa from the seminiferous tubules, seminiferous tubular atrophy, and degenerative changes in the epididymides. Changes in the testes and epididymides had not recovered 20 weeks after cessation of dosing. These fulvestrant doses correspond to 1.3-, 1.2-, and 3.5-fold the systemic exposure [AUC0-30 days] achieved in women receiving the recommended dose of 500 mg/month.
-
14 CLINICAL STUDIES
The efficacy of FASLODEX 500 mg versus FASLODEX 250 mg was compared in CONFIRM. The efficacy of FASLODEX 250 mg was compared to 1 mg anastrozole in Studies 0020 and 0021. The efficacy of FASLODEX 500 mg was compared to 1 mg anastrozole in FALCON. The efficacy of FASLODEX 500 mg in combination with palbociclib 125 mg was compared to FASLODEX 500 mg plus placebo in PALOMA-3. The efficacy of FASLODEX 500 mg in combination with abemaciclib 150 mg was compared to FASLODEX 500 mg plus placebo in MONARCH 2. The efficacy of FASLODEX 500 mg in combination with ribociclib 600 mg was compared to FASLODEX 500 mg plus placebo in MONALEESA-3.
Monotherapy
Comparison of FASLODEX 500 mg and FASLODEX 250 mg (CONFIRM)
A randomized, double-blind, controlled clinical trial (CONFIRM, NCT00099437) was completed in 736 postmenopausal women with advanced breast cancer who had disease recurrence on or after adjuvant endocrine therapy or progression following endocrine therapy for advanced disease. This trial compared the efficacy and safety of FASLODEX 500 mg (n=362) with FASLODEX 250 mg (n=374).
FASLODEX 500 mg was administered as two 5 mL injections each containing FASLODEX 250 mg/5 mL, one in each buttock, on Days 1, 15, 29, and every 28 (+/- 3) days thereafter. FASLODEX 250 mg was administered as two 5 mL injections (one containing FASLODEX 250 mg/5 mL injection plus one placebo injection), one in each buttock, on Days 1, 15 (2 placebo injections only), 29, and every 28 (+/- 3) days thereafter.
The median age of study participants was 61 years. All patients had ER+ advanced breast cancer. Approximately 30% of subjects had no measurable disease. Approximately 55% of patients had visceral disease.
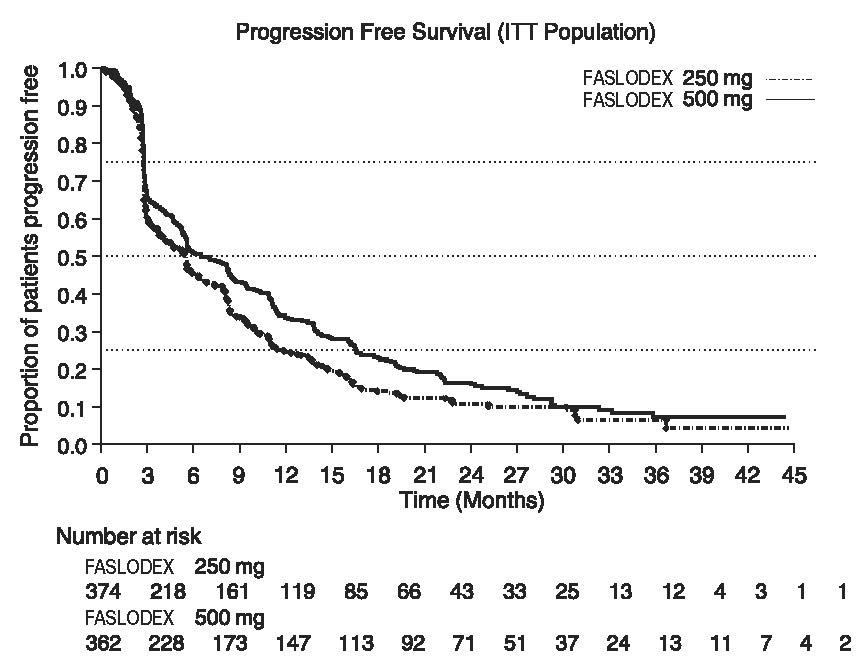
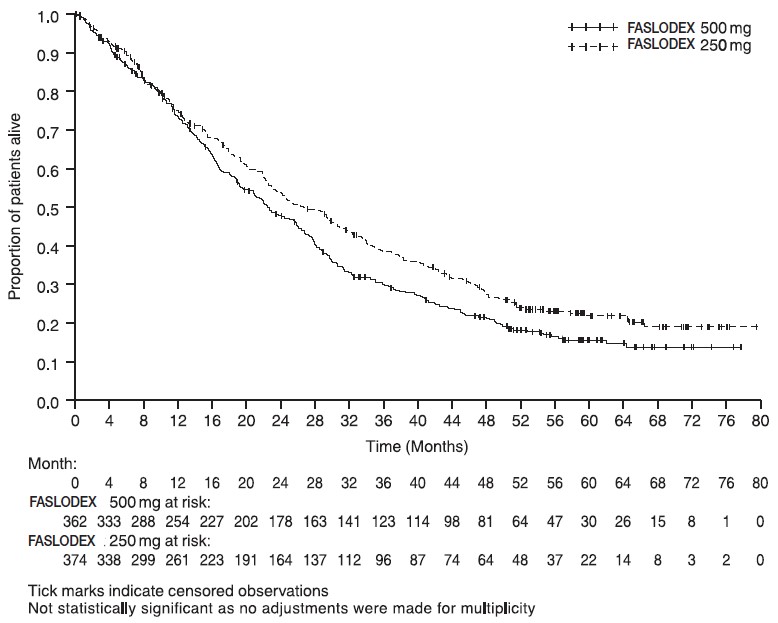
Results of CONFIRM are summarized in Table 12. The efficacy of FASLODEX 500 mg was compared to that of FASLODEX 250 mg. Figure 6 shows a Kaplan-Meier plot of the Progression Free Survival (PFS) data after a minimum follow-up duration of 18 months demonstrating statistically significant superiority of FASLODEX 500 mg vs. FASLODEX 250 mg. In the initial Overall Survival (OS) analysis after a minimum follow-up duration of 18 months, there was no statistically significant difference in OS between the two treatment groups. After a minimum follow-up duration of 50 months, an updated OS analysis was performed. Figure 7 shows a Kaplan-Meier plot of the updated OS data.
Table 12: Efficacy Results in CONFIRM (Intent-To-Treat (ITT) Population) - *
- PFS (Progression Free Survival)=the time between randomization and the earliest of progression or death from any cause. Minimum follow-up duration of 18 months.
- †
- Hazard Ratio <1 favors FASLODEX 500 mg.
- ‡
- CI=Confidence Interval
- §
- OS=Overall Survival
- ¶
- Minimum follow up duration of 50 months.
- #
- Not statistically significant as no adjustments were made for multiplicity.
- Þ
- ORR (Objective Response Rate), as defined as number (%) of patients with complete response or partial response, was analyzed in the evaluable patients with measurable disease at baseline (fulvestrant 500 mg N=240; fulvestrant 250 mg N=261). Minimum follow-up duration of 18 months.
Endpoint
FASLODEX 500 mg
(N=362)
FASLODEX 250 mg
(N=374)
PFS*
Median (months)
6.5
5.4
0.80 (0.68-0.94)
p-value
0.006
(% patients who died)
261 (72.1%)
293 (78.3%)
Median OS (months)
26.4
22.3
0.81 (0.69-0.96)
13.8% (9.7%, 18.8%)
(33/240)
14.6% (10.5%, 19.4%)
(38/261)
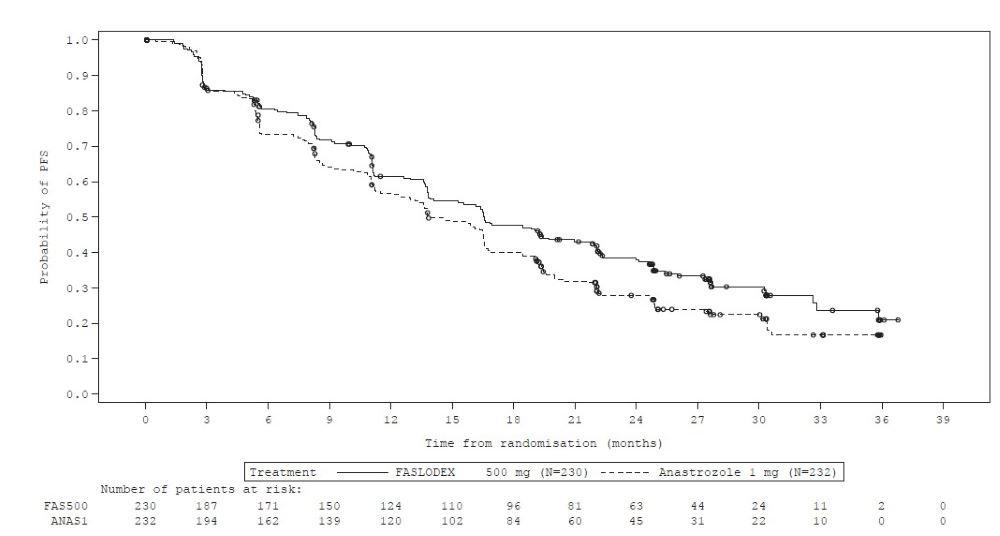
Figure 6 Kaplan-Meier PFS: CONFIRM ITT Population
Figure 7 Kaplan-Meier OS (Minimum Follow-up Duration of 50 Months): CONFIRM ITT Population
Comparison of FASLODEX 500 mg and Anastrozole 1 mg (FALCON)
A randomized, double-blind, double-dummy, multi-center study (FALCON, NCT01602380) of FASLODEX 500 mg versus anastrozole 1 mg was conducted in postmenopausal women with ER-positive and/or PgR-positive, HER2-negative locally advanced or metastatic breast cancer who had not previously been treated with any hormonal therapy. A total of 462 patients were randomized 1:1 to receive administration of FASLODEX 500 mg as an intramuscular injection on Days 1, 15, 29, and every 28 (+/- 3) days thereafter or daily administration of 1 mg of anastrozole orally. This study compared the efficacy and safety of FASLODEX 500 mg and anastrozole 1 mg.
Randomization was stratified by disease setting (locally advanced or metastatic), use of prior chemotherapy for advanced disease, and presence or absence of measurable disease.
The major efficacy outcome measure of the study was investigator-assessed progression-free survival (PFS) evaluated according to RECIST v.1.1 (Response Evaluation Criteria in Solid Tumors). Key secondary efficacy outcome measures included overall survival (OS), objective response rate (ORR), and duration of response (DoR).
Patients enrolled in this study had a median age of 63 years (range 36-90). The majority of patients (87%) had metastatic disease at baseline. Fifty-five percent (55%) of patients had visceral metastasis at baseline. A total of 17% of patients had received one prior chemotherapy regimen for advanced disease; 84% of patients had measurable disease. Sites of metastases were as follows: musculoskeletal 59%, lymph nodes 50%, respiratory 40%, liver (including gall bladder) 18%.
The efficacy results of FALCON are presented in Table 13 and Figure 8.
Table 13: Efficacy Results in FALCON (Investigator Assessment, ITT Population) - *
- Interim OS analysis with 61% of total number of events required for the final OS analysis.
FASLODEX
500 mg
N=230
Anastrozole
1 mg
N=232
Progression-Free Survival
Number of PFS Events (%)
143 (62.2%)
166 (71.6%)
Median PFS (months)
16.6
13.8
PFS Hazard Ratio (95% CI)
0.797 (0.637 - 0.999)
p-value
0.049
Overall Survival*
Number of OS Events
67 (29.1%)
75 (32.3%)
Median OS (months)
NR
NR
OS Hazard Ratio (95% CI)
0.874 (0.629 – 1.216)
Objective Response for Patients with Measurable Disease
N=193
N=196
Objective Response Rate (%, 95% CI)
46.1% (38.9%, 53.4%)
44.9% (37.8%, 52.1%)
Median DoR (months)
20.0
13.2
NR: Not reached
Figure 8 Kaplan-Meier Plot of Progression-Free Survival (Investigator Assessment, ITT Population) ─ FALCON
Comparison of FASLODEX 250 mg and Anastrozole 1 mg in Combined Data (Studies 0020 and 0021)
Efficacy of FASLODEX was established by comparison to the selective aromatase inhibitor anastrozole in two randomized, controlled clinical trials (one conducted in North America, Study 0021, NCT00635713; the other predominantly in Europe, Study 0020) in postmenopausal women with locally advanced or metastatic breast cancer. All patients had progressed after previous therapy with an antiestrogen or progestin for breast cancer in the adjuvant or advanced disease setting.
The median age of study participants was 64 years. 81.6% of patients had ER+ and/or PgR+ tumors. Patients with ER-/PgR- or unknown tumors were required to have demonstrated a prior response to endocrine therapy. Sites of metastases occurred as follows: visceral only 18.2%; viscera – liver involvement 23.0%; lung involvement 28.1%; bone only 19.7%; soft tissue only 5.2%; skin and soft tissue 18.7%.
In both trials, eligible patients with measurable and/or evaluable disease were randomized to receive either FASLODEX 250 mg intramuscularly once a month (28 days + 3 days) or anastrozole 1 mg orally once a day. All patients were assessed monthly for the first three months and every three months thereafter. Study 0021 was a double-blind, randomized trial in 400 postmenopausal women. Study 0020 was an open-label, randomized trial conducted in 451 postmenopausal women. Patients on the FASLODEX arm of Study 0021 received two separate injections (2 x 2.5 mL), whereas FASLODEX patients received a single injection (1 x 5 mL) in Study 0020. In both trials, patients were initially randomized to a 125 mg per month dose as well, but interim analysis showed a very low response rate, and low dose groups were dropped.
Results of the trials, after a minimum follow-up duration of 14.6 months, are summarized in Table 14. The effectiveness of FASLODEX 250 mg was determined by comparing Objective Response Rate (ORR) and Time to Progression (TTP) results to anastrozole 1 mg, the active control. The two studies ruled out (by one-sided 97.7% confidence limit) inferiority of FASLODEX to anastrozole of 6.3% and 1.4% in terms of ORR. There was no statistically significant difference in overall survival (OS) between the two treatment groups after a follow-up duration of 28.2 months in Study 0021 and 24.4 months in Study 0020.
Table 14: Efficacy Results in Studies 0020 and 0021 (Objective Response Rate (ORR) and Time to Progression (TTP)) Study 0021
(Double-Blind)Study 0020
(Open-Label)Endpoint FASLODEX
250 mg
N=206Anastrozole
1 mg
N=194FASLODEX
250 mg
N=222Anastrozole
1 mg
N=229Objective Tumor Response Number (%) of subjects with CR* + PR†
35 (17.0)
33 (17.0)
45 (20.3)
34 (14.9)
% Difference in Tumor Response Rate (FAS‡-ANA§) 2-sided 95.4% CI¶
0.0
(-6.3, 8.9)
5.4
(-1.4, 14.8)
Time to Progression (TTP)
Median TTP (days)
165
103
166
156
Hazard Ratio#
2-sided 95.4% CI
0.9
(0.7, 1.1)
1.0
(0.8, 1.2)
Stable Disease for ≥24 weeks (%)
26.7
19.1
24.3
30.1
Overall Survival (OS)
Died n (%)
Median Survival (days)
152 (73.8%)
844
149 (76.8%)
913
167 (75.2%)
803
173 (75.5%)
736
Hazard Ratio#
(2-sided 95% CI)
0.98
(0.78, 1.24)
0.97
(0.78, 1.21)
Combination Therapy
Patients with HR-positive, HER2-negative advanced or metastatic breast cancer who have had disease progression on or after prior adjuvant or metastatic endocrine therapy
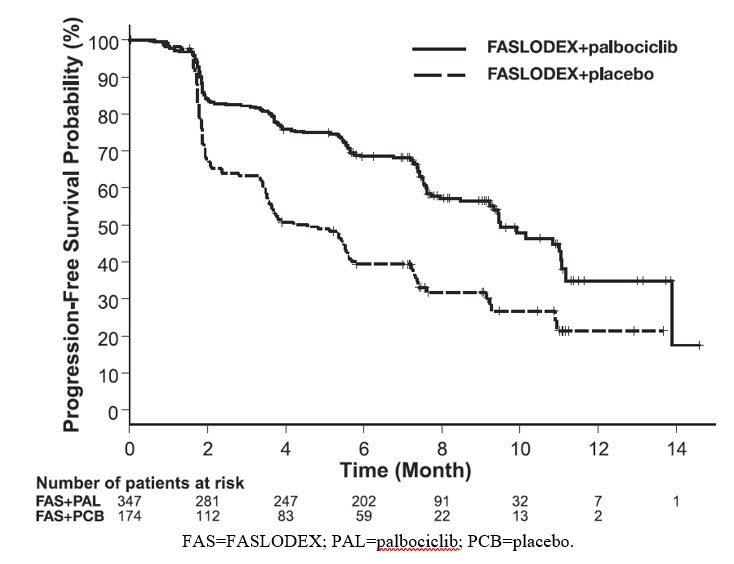
FASLODEX 500 mg in Combination with Palbociclib 125 mg (PALOMA-3)
PALOMA-3 (NCT-1942135) was an international, randomized, double-blind, parallel group, multi-center study of FASLODEX plus palbociclib versus FASLODEX plus placebo conducted in women with HR-positive, HER2-negative advanced breast cancer, regardless of their menopausal status, whose disease progressed on or after prior endocrine therapy.
A total of 521 pre/postmenopausal women were randomized 2:1 to FASLODEX plus palbociclib or FASLODEX plus placebo and stratified by documented sensitivity to prior hormonal therapy, menopausal status at study entry (pre/peri versus postmenopausal), and presence of visceral metastases. Palbociclib was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment. Fulvestrant 500 mg was administered as two 5 mL injections each containing fulvestrant 250 mg/5 mL, one in each buttock, on Days 1, 15, 29, and every 28 (+/- 3) days thereafter. Pre/perimenopausal women were enrolled in the study and received the LHRH agonist goserelin for at least 4 weeks prior to and for the duration of PALOMA-3.
Patients continued to receive assigned treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. The major efficacy outcome of the study was investigator-assessed PFS evaluated according to RECIST v.1.1.
Patients enrolled in this study had a median age of 57 years (range 29 to 88). The majority of patients on study were White (74%), all patients had an ECOG PS of 0 or 1, and 80% were postmenopausal. All patients had received prior systemic therapy and 75% of patients had received a previous chemotherapy regimen. Twenty-five percent of patients had received no prior therapy in the metastatic disease setting, 60% had visceral metastases, and 23% had bone only disease.
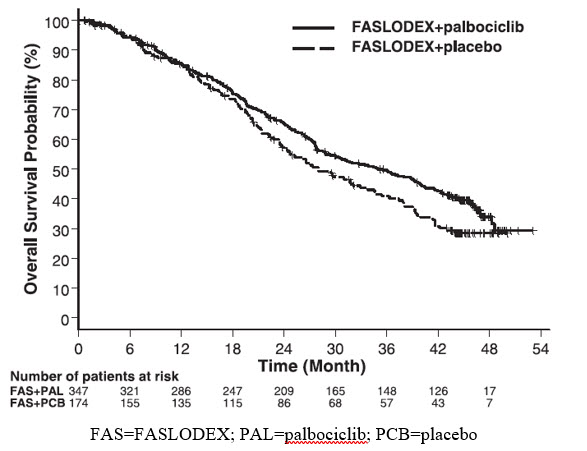
The results from the investigator-assessed PFS and final OS data from PALOMA-3 are summarized in Table 15. The relevant Kaplan-Meier plots are shown in Figures 9 and 10, respectively. Consistent PFS results were observed across patient subgroups of disease site, sensitivity to prior hormonal therapy, and menopausal status. After a median follow-up time of 45 months, the final OS results were not statistically significant.
Table 15: Efficacy Results in PALOMA-3 (Investigator Assessment, ITT Population) FASLODEX plus Palbociclib
FASLODEX plus Placebo
Progression-Free Survival for ITT
N=347
N=174
Number of PFS Events (%)
145 (41.8%)
114 (65.5%)
Median PFS (months) (95% CI)
9.5 (9.2-11.0)
4.6 (3.5-5.6)
Hazard Ratio (95% CI) and p-value
0.461 (0.360-0.591)
p <0.0001
Objective Response for Patients with Measurable Disease
N=267
N=138
Objective response rate* (%, 95% CI)
24.6 (19.6-30.2)
10.9 (6.2-17.3)
Overall Survival for ITT population
N=347
N=174
Number of OS events (%)
201 (57.9)
109 (62.6)
Median OS (months) (95% CI)
34.9 (28.8, 40.0)
28.0 (23.6, 34.6)
Hazard Ratio (95% CI) and p-value
N=number of patients; PFS=progression-free survival; CI=confidence interval; ITT=Intent-to-Treat; OS=overall survival.
Figure 9 Kaplan-Meier Plot of Progression-Free Survival (Investigator Assessment, ITT Population) – PALOMA-3
Figure 10 Kaplan-Meier Plot of Overall Survival (ITT Population) ─ PALOMA-3
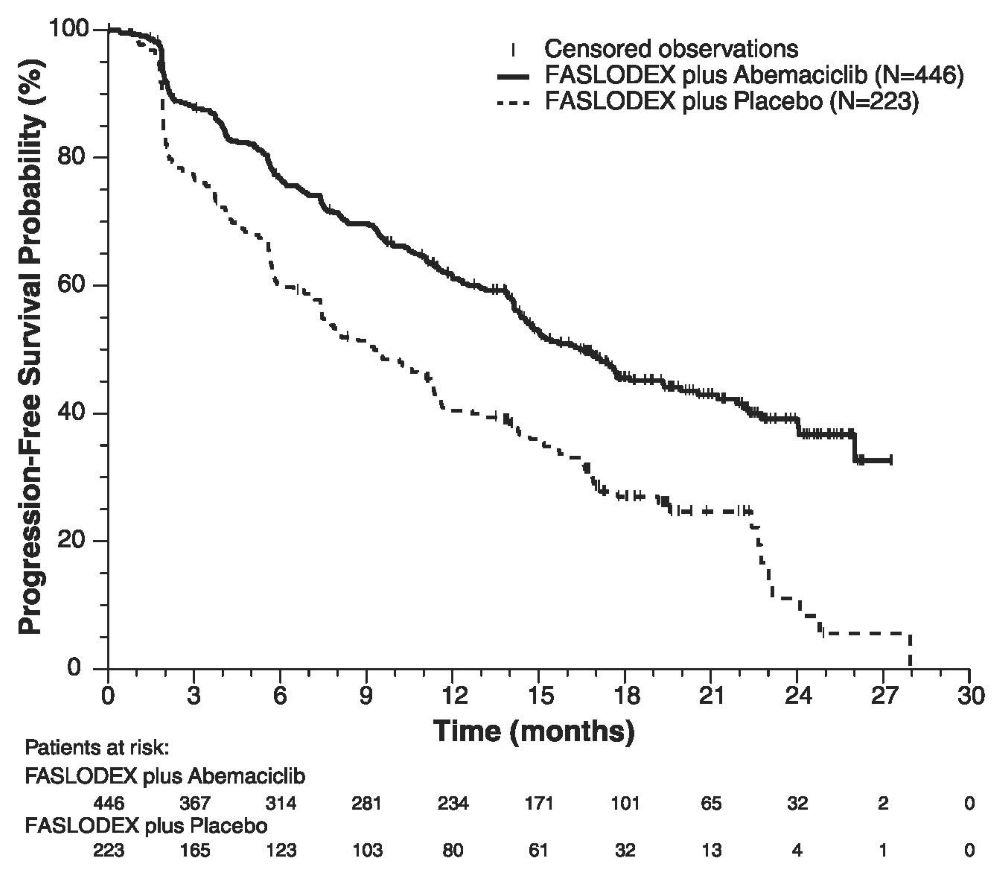
FASLODEX 500 mg in Combination with Abemaciclib 150 mg (MONARCH 2)
MONARCH 2 (NCT02107703) was a randomized, placebo-controlled, multi-center study conducted in women with HR-positive, HER2-negative metastatic breast cancer with disease progression following endocrine therapy treated with FASLODEX plus abemaciclib versus FASLODEX plus placebo. Randomization was stratified by disease site (visceral, bone only, or other) and by sensitivity to prior endocrine therapy (primary or secondary resistance). A total of 669 patients received intramuscular injection of FASLODEX 500 mg on Days 1 and 15 of cycle 1 and then on Day 1 of cycle 2 and beyond (28-day cycles), plus abemaciclib or placebo orally twice daily. Pre/perimenopausal women were enrolled in the study and received the gonadotropin-releasing hormone agonist goserelin for at least 4 weeks prior to and for the duration of MONARCH 2. Patients remained on continuous treatment until development of progressive disease or unmanageable toxicity.
Patient median age was 60 years (range, 32-91 years), and 37% of patients were older than 65. The majority were White (56%), and 99% of patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Twenty percent (20%) of patients had de novo metastatic disease, 27% had bone only disease, and 56% had visceral disease. Twenty-five percent (25%) of patients had primary endocrine therapy resistance. Seventeen percent (17%) of patients were pre- or perimenopausal.
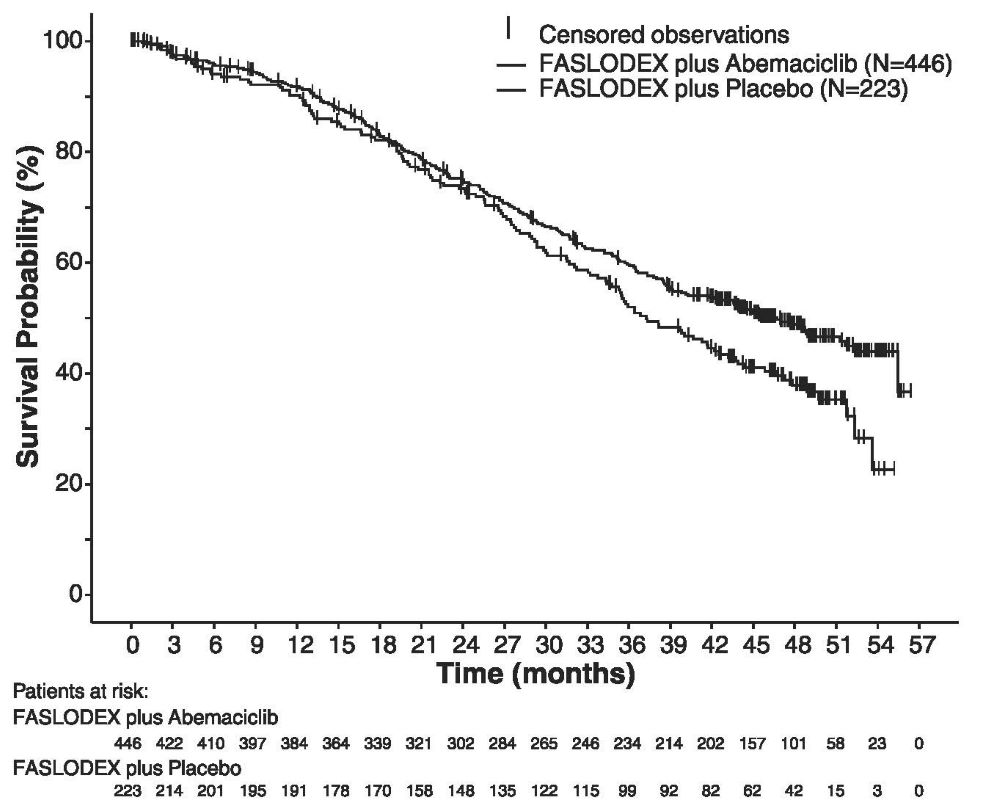
The efficacy results from the MONARCH 2 study are summarized in Table 16, Figure 11, and Figure 12. PFS assessment based on a blinded independent radiologic review was consistent with the investigator assessment. Consistent results were observed across patient stratification subgroups of disease site and endocrine therapy resistance for PFS and OS.
Table 16: Efficacy Results in MONARCH 2 (Intent-to-Treat Population) - *
- Stratified by disease site (visceral metastases vs. bone-only metastases vs. other) and endocrine therapy resistance (primary resistance vs. secondary resistance)
- †
- Data from a pre-specified interim analysis (77% of the number of events needed for the planned final analysis) with the p-value compared with the allocated alpha of 0.021.
- ‡
- Complete response + partial response.
FASLODEX plus Abemaciclib
FASLODEX plus Placebo
Progression-Free Survival
(Investigator Assessment)
N=446
N=223
Number of patients with an event (n, %)
222 (49.8)
157 (70.4)
Median (months, 95% CI)
16.4 (14.4, 19.3)
9.3 (7.4, 12.7)
Hazard ratio (95% CI)
0.553 (0.449, 0.681)
p-value*
p<0.0001
Overall Survival†
Number of deaths (n, %)
211 (47.3)
127 (57.0)
Median OS in months (95% CI)
46.7 (39.2, 52.2)
37.3 (34.4, 43.2)
Hazard ratio (95% CI)1
0.757 (0.606, 0.945)
p-value*
p=0.0137
Objective Response for Patients with Measurable Disease
N=318
N=164
Objective response rate‡ (n, %)
153 (48.1)
35 (21.3)
95% CI
42.6, 53.6
15.1, 27.6
Abbreviations: CI=confidence interval, OS=overall survival.
Figure 11 Kaplan-Meier Curves of Progression-Free Survival: FASLODEX Plus Abemaciclib versus FASLODEX plus Placebo (MONARCH 2)
Figure 12 Kaplan-Meier Curves of Overall Survival: FASLODEX plus Abemaciclib versus FASLODEX plus Placebo (MONARCH 2)
Postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer for initial endocrine based therapy or after disease progression on endocrine therapy
FASLODEX 500 mg in Combination with Ribociclib 600 mg (MONALEESA-3)
MONALEESA-3 (NCT 02422615) was a randomized double-blind, placebo-controlled study of FASLODEX plus ribociclib versus FASLODEX plus placebo conducted in postmenopausal women with hormone receptor positive, HER2-negative, advanced breast cancer who have received no or only one line of prior endocrine treatment.
A total of 726 patients were randomized in a 2:1 ratio to receive FASLODEX plus ribociclib or FASLODEX plus placebo and stratified according to the presence of liver and/or lung metastases and prior endocrine therapy for advanced or metastatic disease. Fulvestrant 500 mg was administered intramuscularly on Days 1, 15, 29, and once monthly thereafter, with either ribociclib 600 mg or placebo given orally once daily for 21 consecutive days followed by 7 days off until disease progression or unacceptable toxicity. The major efficacy outcome measure for the study was investigator-assessed progression-free survival (PFS) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
Patients enrolled in this study had a median age of 63 years (range 31 to 89). Of the patients enrolled, 47% were 65 years and older, including 14% age 75 years and older. The patients enrolled were primarily Caucasian (85%), Asian (9%), and Black (0.7%). Nearly all patients (99.7%) had an ECOG performance status of 0 or 1. First- and second-line patients were enrolled in this study (of which 19% had de novo metastatic disease). Forty-three percent (43%) of patients had received chemotherapy in the adjuvant vs. 13% in the neoadjuvant setting and 59% had received endocrine therapy in the adjuvant vs. 1% in the neoadjuvant setting prior to study entry. Twenty-one percent (21%) of patients had bone-only disease and 61% had visceral disease. Demographics and baseline disease characteristics were balanced and comparable between study arms.
The efficacy results from MONALEESA-3 are summarized in Table 17, Figure 13, and Figure 14. Consistent results were observed in stratification factor subgroups of disease site and prior endocrine treatment for advanced disease.
Table 17: Efficacy Results – MONALEESA-3 (Investigator Assessment, Intent-to-Treat Population) FASLODEX plus Ribociclib
FASLODEX plus Placebo
Progression-free survival*
N=484
N=242
Events (n, %)
210 (43.4%)
151 (62.4%)
Median (months, 95% CI)
20.5 (18.5, 23.5)
12.8 (10.9, 16.3)
Hazard Ratio (95% CI)
0.593 (0.480 to 0.732)
p-value†
<0.0001
Overall Survival
N=484
N=242
Events (n, %)
167 (34.5%)
108 (44.6%)
Median (months, 95% CI)
NR (42.5, NR)
40.0 (37.0, NR)
Hazard Ratio (95% CI)
0.724 (0.568, 0.924)
p-value†
0.00455
N=379
N=181
Patients with measurable disease (95% CI)
40.9 (35.9, 45.8)
28.7 (22.1, 35.3)
- Abbreviation: NR, not reached
Figure 13 Kaplan-Meier Progression Free Survival Curves – MONALEESA-3 (Intent-To-Treat Population, Investigator assessment)
Figure 14 Kaplan-Meier plot of Overall Survival – MONALEESA-3 (Intent -to-Treat Population)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
FASLODEX is supplied as two 5 mL clear neutral glass (Type 1) barrels, each containing 250 mg/5 mL of FASLODEX solution for intramuscular injection and fitted with a tamper evident closure.
NDC 0310–0720–10
The single-dose prefilled syringes are presented in a tray with polystyrene plunger rod and safety needles (SafetyGlide™) for connection to the barrel.
Discard each syringe after use. If a patient dose requires only one syringe, unused syringe should be stored as directed below.
Storage:
REFRIGERATE, 2°-8°C (36°-46°F). TO PROTECT FROM LIGHT, STORE IN THE ORIGINAL CARTON UNTIL TIME OF USE.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Monotherapy
Risk of Bleeding:
- •
- Because FASLODEX is administered intramuscularly, it should be used with caution in patients with bleeding disorders, decreased platelet count, or in patients receiving anticoagulants (for example, warfarin) [see Warnings and Precautions (5.1)].
Embryo-Fetal Toxicity:
- •
- Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with FASLODEX and for one year after the last dose. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1), (8.3)].
Lactation:
- •
- Advise women not to breastfeed during treatment with FASLODEX and for one year after the last dose [see Use in Specific Populations (8.2)].
Combination Therapy
When FASLODEX is used in combination with palbociclib, abemaciclib, or ribociclib, refer to the respective Full Prescribing Information for Patient Counseling Information.
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
FASLODEX is a registered trademark of the AstraZeneca group of companies
©AstraZeneca 2020
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
FASLODEX® (faz-lo-dex)
(fulvestrant)
injection
What is FASLODEX?
FASLODEX is a prescription medicine used to treat advanced breast cancer or breast cancer that has spread to other parts of the body (metastatic).
FASLODEX may be used alone, if you have gone through menopause, and your advanced breast cancer is:
- •
- hormone receptor (HR)-positive and human epidermal growth factor receptor 2 (HER2)-negative and has not been previously treated with endocrine therapy
or - •
- HR-positive and has progressed after endocrine therapy.
FASLODEX may be used in combination with ribociclib, if you have gone through menopause, and your advanced or metastatic breast cancer is HR-positive and HER2-negative, and has not been previously treated with endocrine therapy or has progressed after endocrine therapy.
FASLODEX may be used in combination with palbociclib or abemaciclib if your advanced or metastatic breast cancer is HR-positive and HER2-negative, and has progressed after endocrine therapy.
When FASLODEX is used in combination with palbociclib, abemaciclib, or ribociclib, also read the Patient Information for the prescribed product.
It is not known if FASLODEX is safe and effective in children.
It is not known if FASLODEX is safe and effective in people with severe liver problems.
Who should not receive FASLODEX?
Do not receive FASLODEX if you have had an allergic reaction to fulvestrant or any of the ingredients in FASLODEX. See the end of this leaflet for a list of the ingredients in FASLODEX.
Symptoms of an allergic reaction to FASLODEX may include:
- •
- itching or hives
- •
- swelling of your face, lips, tongue, or throat
- •
- trouble breathing
What should I tell my healthcare provider before receiving FASLODEX?
Before receiving FASLODEX, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have a low level of platelets in your blood or bleed easily.
- •
- have liver problems.
- •
- are pregnant or plan to become pregnant. FASLODEX can harm your unborn baby.
Females who are able to become pregnant:- •
- Your healthcare provider may perform a pregnancy test within 7 days before you start FASLODEX.
- •
- You should use effective birth control during treatment with FASLODEX and for one year after the last dose of FASLODEX.
- •
- Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with FASLODEX.
- •
- are breastfeeding or plan to breastfeed. It is not known if FASLODEX passes into your breast milk. Do not breastfeed during your treatment with FASLODEX and for one year after the final dose of FASLODEX. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. FASLODEX may affect the way other medicines work, and other medicines may affect how FASLODEX works.
Especially tell your healthcare provider if you take a blood thinner medicine.
How will I receive FASLODEX?
- •
- Your healthcare provider will give you FASLODEX by injection into the muscle of each buttock.
- •
- Your healthcare provider may change your dose of FASLODEX if needed.
What are the possible side effects of FASLODEX?
FASLODEX may cause serious side effects, including:
- •
-
Injection site related nerve damage. Call your healthcare provider if you develop any of the following symptoms in your legs following a FASLODEX injection:
- •
- numbness
- •
- tingling
- •
- weakness
The most common side effects of FASLODEX include:
- •
- injection site pain
- •
- nausea
- •
- muscle, joint, and bone pain
- •
- headache
- •
- back pain
- •
- tiredness
- •
- pain in arms, hands, legs, or feet
- •
- hot flashes
- •
- vomiting
- •
- loss of appetite
- •
- weakness
- •
- cough
- •
- shortness of breath
- •
- constipation
- •
- increased liver enzymes
- •
- diarrhea
FASLODEX may cause fertility problems in males and females. Talk to your healthcare provider if you plan to become pregnant.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects with FASLODEX. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of FASLODEX
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about FASLODEX that is written for health professionals.
What are the ingredients in FASLODEX?
Active ingredient: fulvestrant.
Inactive ingredients: alcohol, benzyl alcohol, benzyl benzoate, and castor oil.
SafetyGlide™ is a trademark of Becton Dickinson and Company.
FASLODEX is a trademark of the AstraZeneca group of companies.
© AstraZeneca 2020
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
Manufactured for:
AstraZeneca UK Limited
Macclesfield, Cheshire, England
By: Vetter Pharma-Fertigung GMBH & Co. KG
Ravensburg, Germany
For more information, go to www.FASLODEX.com or call 1-800-236-9933.
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 8/2020
-
Package/Label Display Panel – 250 mg/5 mL (50 mg/mL)
FASLODEX®
fulvestrant injection
250 mg/5 mL (50 mg/mL)
For Intramuscular Use Only
AstraZeneca
NDC 0310-0720-10
This carton contains a total of 500 mg fulvestrant in TWO single-dose prefilled syringes each containing 250 mg/5 mL, and two Safety Glide™ shielding intramuscular injection needles.
Discard each syringe after use.
Both single-dose prefilled syringes must be administered to receive the 500 mg dose.
REFRIGERATE, 2-8°C (36-46°F). TO PROTECT FROM LIGHT, STORE IN THE ORIGINAL CARTON UNTIL TIME OF USE.
Rx only
Contains 2 single-dose prefilled syringes.
-
INGREDIENTS AND APPEARANCE
FASLODEX
fulvestrant injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0310-0720 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FULVESTRANT (UNII: 22X328QOC4) (FULVESTRANT - UNII:22X328QOC4) FULVESTRANT 50 mg in 1 mL Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) BENZYL ALCOHOL (UNII: LKG8494WBH) BENZYL BENZOATE (UNII: N863NB338G) CASTOR OIL (UNII: D5340Y2I9G) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0310-0720-10 2 in 1 CARTON 11/01/2010 1 5 mL in 1 SYRINGE, GLASS; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021344 11/01/2010 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Registrant - AstraZeneca PLC (230790719)