CLORPRES- clonidine hydrochloride and chlorthalidone tablet
Mylan Bertek Pharmaceuticals Inc.
----------
CLORPRES®
(Clonidine Hydrochloride and Chlorthalidone)
TABLETS, USP
DESCRIPTION
CLORPRES® is a combination of clonidine hydrochloride (a centrally acting antihypertensive agent) and chlorthalidone (a diuretic). CLORPRES® is available as tablets for oral administration in three dosage strengths: 0.1 mg/15 mg, 0.2 mg/15 mg and 0.3 mg/15 mg of clonidine hydrochloride/chlorthalidone, respectively.
The inactive ingredients are ammonium chloride, colloidal silicon dioxide, croscarmellose sodium (Type A), magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, D&C yellow #10.
Clonidine Hydrochloride
Clonidine hydrochloride is an imidazoline derivative and exists as a mesomeric compound. The chemical name is 2-[(2,6-dichlorophenyl)imino]imidazoline monohydrochloride. The following are the structural formula, molecular formula and molecular weight:
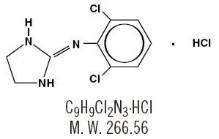
Clonidine hydrochloride is an odorless, bitter, white crystalline substance soluble in water and alcohol.
Chlorthalidone
Chlorthalidone is a monosulfamyl diuretic that differs chemically from thiazide diuretics in that a double ring system is incorporated in its structure. It is 2-chloro-5-(1-hydroxy-3-oxo-1-isoindolinyl) benzenesulfonamide with the following structural formula, molecular formula and molecular weight:
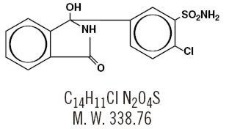
Chlorthalidone is practically insoluble in water, in ether and in chloroform; soluble in methanol; slightly soluble in alcohol.
CLINICAL PHARMACOLOGY
CLORPRES®
Clorpres produces a more pronounced antihypertensive response than occurs after either clonidine hydrochloride or chlorthalidone alone in equivalent doses.
Clonidine Hydrochloride
Clonidine hydrochloride acts relatively rapidly. The patient's blood pressure declines within 30 to 60 minutes after an oral dose, the maximum decrease occurring within 2 to 4 hours. The plasma level of clonidine hydrochloride peaks in approximately 3 to 5 hours and the plasma half-life ranges from 12 to 16 hours. The half-life increases up to 41 hours in patients with severe impairment of renal function. Following oral administration about 40 to 60% of the absorbed dose is recovered in the urine as unchanged drug in 24 hours. About 50% of the absorbed dose is metabolized in the liver.
Clonidine stimulates alpha-adrenoreceptors in the brain stem, resulting in reduced sympathetic outflow from the central nervous system and a decrease in peripheral resistance, renal vascular resistance, heart rate, and blood pressure. Renal blood flow and glomerular filtration rate remain essentially unchanged. Normal postural reflexes are intact and therefore orthostatic symptoms are mild and infrequent.
Acute studies with clonidine hydrochloride in humans have demonstrated a moderate reduction (15 to 20%) of cardiac output in the supine position with no change in the peripheral resistance; at a 45° tilt there is a smaller reduction in cardiac output and a decrease of peripheral resistance. During long-term therapy, cardiac output tends to return to control values, while peripheral resistance remains decreased. Slowing of the pulse rate has been observed in most patients given clonidine but the drug does not alter normal hemodynamic response to exercise.
Other studies in patients have provided evidence of a reduction in plasma renin activity and in the excretion of aldosterone and catecholamines, but the exact relationship of these pharmacologic actions to the antihypertensive effect has not been fully elucidated.
Clonidine acutely stimulates growth hormone release in both children and adults, but does not produce a chronic elevation of growth hormone with long-term use.
Tolerance may develop in some patients, necessitating a reevaluation of therapy.
Chlorthalidone
Chlorthalidone is a long-acting oral diuretic with antihypertensive activity. Its diuretic action commences a mean of 2.6 hours after dosing and continues for up to 72 hours. The drug produces diuresis with increased excretion of sodium and chloride. The diuretic effects of chlorthalidone and the benzothiadiazine (thiazide) diuretics appear to arise from similar mechanisms and the maximal effect of chlorthalidone and the thiazides appears to be similar. The site of action appears to be the distal convoluted tubule of the nephron. The diuretic effects of chlorthalidone lead to decreased extracellular fluid volume, plasma volume, cardiac output, total exchangeable sodium, glomerular filtration rate, and renal plasma flow. Although the mechanism of action of chlorthalidone and related drugs is not wholly clear, sodium and water depletion appear to provide a basis for its antihypertensive effect. Like the thiazide diuretics, chlorthalidone produces dose-related reductions in serum potassium levels, elevations in serum uric acid and blood glucose, and it can lead to decreased sodium and chloride levels.
The mean plasma half-life of chlorthalidone is about 40 to 60 hours. It is eliminated primarily as unchanged drug in the urine. Non-renal routes of elimination have yet to be clarified. In the blood, approximately 75% of the drug is bound to plasma proteins.
INDICATIONS AND USAGE
CLORPRES® (clonidine hydrochloride USP/chlorthalidone USP) is indicated in the treatment of hypertension. This fixed combination drug is not indicated for initial therapy of hypertension. Hypertension requires therapy titrated to the individual patient. If the fixed combination represents the dosage so determined, its use may be more convenient in patient management. The treatment of hypertension is not static, but must be reevaluated as conditions in each patient warrant.
WARNINGS
Chlorthalidone should be used with caution in severe renal disease. In patients with renal disease, chlorthalidone or related drugs may precipitate azotemia. Cumulative effects of the drug may develop in patients with impaired renal function. Chlorthalidone should be used with caution in patients with impaired hepatic function or progressive liver disease, because minor alterations of fluid and electrolyte balance may precipitate hepatic coma.
Sensitivity reactions may occur in patients with a history of allergy or bronchial asthma.
The possibility of exacerbation or activation of systemic lupus erythematosus has been reported with thiazide diuretics which are structurally related to chlorthalidone. However, systemic lupus erythematosus has not been reported following chlorthalidone administration.
PRECAUTIONS
Clonidine Hydrochloride
General
In patients who have developed localized contact sensitization to transdermal clonidine, substitution of oral clonidine hydrochloride therapy may be associated with the development of a generalized skin rash.
In patients who develop an allergic reaction from transdermal clonidine that extends beyond the local patch site (such as generalized skin rash, urticaria, or angioedema), oral clonidine hydrochloride substitution may elicit a similar reaction.
As with all antihypertensive therapy, clonidine hydrochloride should be used with caution in patients with severe coronary insufficiency, recent myocardial infarction, cerebrovascular disease or chronic renal failure.
Withdrawal
Patients should be instructed not to discontinue therapy without consulting their physician. Sudden cessation of clonidine treatment has resulted in subjective symptoms such as nervousness, agitation and headache, accompanied or followed by a rapid rise in blood pressure and elevated catecholamine concentrations in the plasma, but such occurrences have usually been associated with previous administration of high oral doses (exceeding 1.2 mg/day) and/or with continuation of concomitant beta-blocker therapy. Rare instances of hypertensive encephalopathy and death have been reported. When discontinuing therapy with clonidine hydrochloride, the physician should reduce the dose gradually over 2 to 4 days to avoid withdrawal symptomatology.
An excessive rise in blood pressure following clonidine hydrochloride discontinuance can be reversed by administration of oral clonidine or by intravenous phentolamine. If therapy is to be discontinued in patients receiving beta-blockers and clonidine concurrently, beta-blockers should be discontinued several days before the gradual withdrawal of clonidine hydrochloride.
Perioperative Use
Administration of clonidine hydrochloride should be continued to within four hours of surgery and resumed as soon as possible thereafter. The blood pressure should be carefully monitored and appropriate measures instituted to control it as necessary.
Information for Patients
Patients who engage in potentially hazardous activities, such as operating machinery or driving, should be advised of a potential sedative effect of clonidine. Patients should be cautioned against interruption of clonidine hydrochloride therapy without a physician's advice.
Drug Interactions
If a patient receiving clonidine hydrochloride is also taking tricyclic antidepressants, the effect of clonidine may be reduced, thus necessitating an increase in dosage. Clonidine hydrochloride may enhance the CNS-depressive effects of alcohol, barbiturates or other sedatives. Amitriptyline in combination with clonidine enhances the manifestation of corneal lesions in rats (see Ocular Toxicity).
Ocular Toxicity
In several studies, oral clonidine hydrochloride produced a dose-dependent increase in the incidence and severity of spontaneously occurring retinal degeneration in albino rats treated for six months or longer. Tissue distribution studies in dogs and monkeys revealed that clonidine hydrochloride was concentrated in the choroid of the eye. In view of the retinal degeneration observed in rats, eye examinations were performed in 908 patients prior to the start of clonidine hydrochloride therapy, who were then examined periodically thereafter. In 353 of these 908 patients, examinations were performed for periods of 24 months or longer. Except for some dryness of the eyes, no drug-related abnormal ophthalmologic findings were recorded and clonidine hydrochloride did not alter retinal function as shown by specialized tests such as the electroretinogram and macular dazzle.
In rats, clonidine hydrochloride in combination with amitriptyline produced corneal lesions within 5 days.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 132-week (fixed concentration) dietary administration study in rats, clonidine hydrochloride administered at 32 to 46 times the maximum recommended daily human oral dose was unassociated with evidence of carcinogenic potential.
Fertility of male or female rats was unaffected by clonidine hydrochloride doses as high as 150 mcg/kg or about 3 times the maximum recommended daily human oral dose (MRDHD). Fertility of female rats did, however, appear to be affected (in another experiment) at dose levels of 500 to 2000 mcg/kg or 10 to 40 times the MRDHD.
Usage in Pregnancy
Teratogenic Effect
Pregnancy Category C
Reproduction studies performed in rabbits at doses up to approximately 3 times the maximum recommended daily human dose (MRDHD) of clonidine hydrochloride have revealed no evidence of teratogenic or embryotoxic potential. In rats however, doses as low as 1/3 the MRDHD were associated with increased resorptions in a study in which dams were treated continuously from 2 months prior to mating. Increased resorptions were not associated with treatment at the same or at higher dose levels (up to 3 times the MRDHD) when dams were treated days 6 to 15 of gestation. Increased resorptions were observed at much higher levels (40 times the MRDHD) in rats and mice treated days 1 to 14 of gestation (lowest dose employed in that study was 500 mcg/kg). There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Chlorthalidone
General
Hypokalemia and other electrolyte abnormalities, including hyponatremia and hypochloremic alkalosis, are common in patients receiving chlorthalidone. These abnormalities are dose-related but may occur even at the lowest marketed doses of chlorthalidone. Serum electrolytes should be determined before initiating therapy and at periodic intervals during therapy. Serum and urine electrolyte determinations are particularly important when the patient is vomiting excessively or receiving parenteral fluids. All patients taking chlorthalidone should be observed for clinical signs of electrolyte imbalance, including dryness of mouth, thirst, weakness, lethargy, drowsiness, restlessness, muscle pains or cramps, muscular fatigue, hypotension, oliguria, tachycardia, palpitations and gastrointestinal disturbances, such as nausea and vomiting. Digitalis therapy may exaggerate metabolic effects of hypokalemia especially with reference to myocardial activity.
Any chloride deficit is generally mild and usually does not require specific treatment except under extraordinary circumstances (as in liver disease or renal disease). Dilutional hyponatremia may occur in edematous patients in hot weather: appropriate therapy is water restriction, rather than administration of salt, except in rare instances when the hyponatremia is life-threatening. In cases of actual salt depletion, appropriate replacement is the therapy of choice.
Uric Acid
Hyperuricemia may occur or frank gout may be precipitated in certain patients receiving chlorthalidone.
Other
Increases in serum glucose may occur and latent diabetes mellitus may become manifest during chlorthalidone therapy (see PRECAUTIONS: Chlorthalidone: Drug Interactions). Chlorthalidone and related drugs may decrease serum PBI levels without signs of thyroid disturbance.
Information for Patients
Patients should inform their doctor if they have: 1) had an allergic reaction to chlorthalidone or other diuretics or have asthma 2) kidney disease 3) liver disease 4) gout 5) systemic lupus erythematosus, or 6) been taking other drugs such as cortisone, digitalis, lithium carbonate, or drugs for diabetes.
Patients should be cautioned to contact their physician if they experience any of the following symptoms of potassium loss: excess thirst, tiredness, drowsiness, restlessness, muscle pains or cramps, nausea, vomiting or increased heart rate or pulse.
Patients should also be cautioned that taking alcohol can increase the chance of dizziness occurring.
Laboratory Tests
Periodic determination of serum electrolytes to detect possible electrolyte imbalance should be performed at appropriate intervals.
All patients receiving chlorthalidone should be observed for clinical signs of fluid or electrolyte imbalance: namely, hyponatremia, hypochloremic alkalosis and hypokalemia. Serum and urine electrolyte determinations are particularly important when the patient is vomiting excessively or receiving parenteral fluids.
Drug Interactions
Chlorthalidone may add to or potentiate the action of other antihypertensive drugs. Insulin requirements in diabetic patients may be increased, decreased or unchanged. Higher dosage of oral hypoglycemic agents may be required. Chlorthalidone and related drugs may increase the responsiveness to tubocurarine. Chlorthalidone and related drugs may decrease arterial responsiveness to norepinephrine. This diminution is not sufficient to preclude effectiveness of the pressor agent for therapeutic use. Lithium renal clearance is reduced by chlorthalidone, increasing the risk of lithium toxicity.
Drug/Laboratory Test Interactions
Chlorthalidone and related drugs may decrease serum PBI levels without signs of thyroid disturbance.
Usage in Pregnancy
Teratogenic Effects
Pregnancy Category B
Reproduction studies have been performed in the rat and the rabbit at doses up to 420 times the human dose and have revealed no evidence of harm to the fetus due to chlorthalidone. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Non-Teratogenic Effects
Thiazides cross the placental barrier and appear in cord blood. The use of chlorthalidone and related drugs in pregnant women requires that the anticipated benefits of the drug be weighed against possible hazards to the fetus. These hazards include fetal or neonatal jaundice, thrombocytopenia, and possibly other adverse reactions that have occurred in the adult.
ADVERSE REACTIONS
CLORPRES® is generally well tolerated. Most adverse effects are mild and tend to diminish with continued therapy. The most frequent (which appear to be dose-related) are dry mouth, occurring in about 40 of 100 patients; drowsiness, about 33 in 100; dizziness, about 16 in 100; constipation and sedation, each about 10 in 100.
In addition to the reactions listed above, certain less frequent adverse experiences, which are shown below, have also been reported in patients receiving the component drugs of CLORPRES® but in many cases patients were receiving concomitant medication and a causal relationship has not been established:
Clonidine Hydrochloride
Gastrointestinal: Nausea and vomiting, about 5 in 100 patients; anorexia and malaise, each about 1 in 100; mild transient abnormalities in liver function tests, about 1 in 100; rare reports of hepatitis; parotitis, rarely.
Metabolic: Weight gain, about 1 in 100 patients; gynecomastia, about 1 in 1000, transient elevation of blood glucose or serum creatine phosphokinase, rarely.
Central Nervous System: Nervousness and agitation, about 3 in 100 patients; mental depression, about 1 in 100; headache, about 1 in 100; insomnia, about 5 in 1000. Vivid dreams or nightmares, other behavioral changes, restlessness, anxiety, visual and auditory hallucinations and delirium have been reported.
Cardiovascular: Orthostatic symptoms, about 3 in 100 patients; palpitations and tachycardia, and bradycardia, each about 5 in 1000. Raynaud's phenomenon, congestive heart failure, and electrocardiographic abnormalities, i.e., conduction disturbances and arrhythmias, have been reported rarely. Rare cases of sinus bradycardia and atrioventricular block have been reported, both with and without the use of concomitant digitalis.
Dermatological: Rash, about 1 in 100 patients; pruritus, about 7 in 1000; hives, angioneurotic edema and urticaria, about 5 in 1000, alopecia, about 2 in 1000.
Genitourinary: Decreased sexual activity, impotence and loss of libido, about 3 in 100 patients; nocturia, about 1 in 100; difficulty in micturition, about 2 in 1000; urinary retention, about 1 in 1000.
Other: Weakness, about 10 in 100 patients; fatigue, about 4 in 100; discontinuation syndrome, about 1 in 100; muscle or joint pain, about 6 in 1000 and cramps of the lower limbs, about 3 in 1000. Dryness, burning of the eyes, blurred vision, dryness of the nasal mucosa, pallor, weakly positive Coombs' test, increased sensitivity to alcohol and fever have been reported.
Chlorthalidone
Gastrointestinal: Anorexia, gastric irritation, nausea, vomiting, cramping, diarrhea, constipation, jaundice (intrahepatic cholestatic jaundice), pancreatitis.
Central Nervous System: Dizziness, vertigo, paresthesias, headache, xanthopsia.
Hematologic: Leukopenia, agranulocytosis, thrombocytopenia, aplastic anemia.
Dermatologic-Hypersensitivity: Purpura, photosensitivity, rash, urticaria, necrotizing angiitis (vasculitis) (cutaneous vasculitis), Lyell's syndrome (toxic epidermal necrolysis).
Cardiovascular: Orthostatic hypotension may occur and may be aggravated by alcohol, barbiturates or narcotics.
Other Adverse Reactions: Hyperglycemia, glycosuria, hyperuricemia, muscle spasm, weakness, restlessness, impotence.
Whenever adverse reactions are moderate or severe, chlorthalidone dosage should be reduced or therapy withdrawn.
OVERDOSAGE
Clonidine Hydrochloride
The signs and symptoms of clonidine hydrochloride overdosage include hypotension, bradycardia, lethargy, irritability, weakness, somnolence, diminished or absent reflexes, miosis, vomiting and hypoventilation. With large overdoses, reversible cardiac conduction defects or arrhythmias, apnea, seizures and transient hypertension have been reported. The oral LD50 of clonidine in rats was 465 mg/kg, and in mice 206 mg/kg.
The general treatment of clonidine hydrochloride overdosage may include intravenous fluids as indicated. Bradycardia can be treated with intravenous atropine sulfate and hypotension with dopamine infusion in addition to intravenous fluids. Hypertension, associated with overdosage, has been treated with intravenous furosemide or diazoxide or alpha-blocking agents such as phentolamine. Tolazoline, an alpha-blocker, in intravenous doses of 10 mg at 30-minute intervals, may reverse clonidine's effects if other efforts fail. Routine hemodialysis is of limited benefit, since a maximum of 5% of circulating clonidine is removed.
In a patient who ingested 100 mg clonidine hydrochloride, plasma clonidine levels were 60 ng/mL (one hour), 190 ng/mL (1.5 hours), 370 ng/mL (two hours) and 120 ng/mL (5.5 and 6.5 hours). This patient developed hypertension followed by hypotension, bradycardia, apnea, hallucinations, semicoma, and premature ventricular contractions. The patient fully recovered after intensive treatment.
Chlorthalidone
Symptoms of acute overdosage include nausea, weakness, dizziness and disturbances of electrolyte balance. The oral LD50 of the drug in the mouse and the rat is more than 25,000 mg/kg body weight. The minimum lethal dose (MLD) in humans has not been established. There is no specific antidote but gastric lavage is recommended, followed by supportive treatment. Where necessary, this may include intravenous dextrose-saline with potassium, administered with caution.
DOSAGE AND ADMINISTRATION
The dosage must be determined by individual titration. (See INDICATIONS AND USAGE.)
Chlorthalidone is usually initiated at a dose of 25 mg once daily and may be increased to 50 mg if the response is insufficient after a suitable trial.
Clonidine hydrochloride is usually initiated at a dose of 0.1 mg twice daily. Elderly patients may benefit from a lower initial dose. Further increments of 0.1 mg/day may be made if necessary until the desired response is achieved. The therapeutic doses most commonly employed have ranged from 0.2 to 0.6 mg per day in divided doses.
One CLORPRES® (clonidine hydrochloride/chlorthalidone) Tablet administered once or twice daily can be used to administer a minimum of 0.1 mg clonidine hydrochloride and 15 mg chlorthalidone to a maximum of 0.6 mg clonidine hydrochloride and 30 mg chlorthalidone.
HOW SUPPLIED
CLORPRES® (clonidine hydrochloride and chlorthalidone) Tablets, USP are available containing:
0.1 mg clonidine hydrochloride, USP and 15 mg chlorthalidone, USP
or
0.2 mg clonidine hydrochloride, USP and 15 mg chlorthalidone, USP
or
0.3 mg clonidine hydrochloride, USP and 15 mg chlorthalidone, USP
The 0.1 mg/15 mg product is a yellow, round, scored tablet debossed with M1. They are available as follows:
NDC 62794-001-01
bottles of 100 tablets
The 0.2 mg/15 mg product is a yellow, round, scored tablet debossed with M27. They are available as follows:
NDC 62794-027-01
bottles of 100 tablets
The 0.3 mg/15 mg product is a yellow, round, scored tablet debossed with M72. They are available as follows:
NDC 62794-072-01
bottles of 100 tablets
Dispense in tight, light-resistant container as defined in the USP using a child-resistant closure.
Keep this and all medication out of the reach of children.
Store at 20° to 25°C (68° to 77°F). [See USP for Controlled Room Temperature.]
Avoid excessive humidity.
BERTEK
PHARMACEUTICALS INC.
Morgantown,WV 26505
REVISED FEBRUARY 2008
BKCLCH:R4



| CLORPRES
clonidine hydrochloride and chlorthalidone tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| CLORPRES
clonidine hydrochloride and chlorthalidone tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| CLORPRES
clonidine hydrochloride and chlorthalidone tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Mylan Bertek Pharmaceuticals Inc. (191600790) |