Label: ANAGRELIDE HYDROCHLORIDE capsule

-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-5385-0, 54868-5385-1, 54868-5443-2 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 0172-5240, 0172-5241
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated April 27, 2012
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Anagrelide hydrochloride is an off white powder that is very slightly soluble in water and sparingly soluble in dimethyl sulfoxide and in dimethylformamide. Anagrelide hydrochloride is a platelet-reducing agent with a chemical name of 6,7-dichloro-1,5-dihydroimidazo[2,1-b] quinazolin-2(3H)-one monohydrochloride monohydrate, and it has the following structural formula:
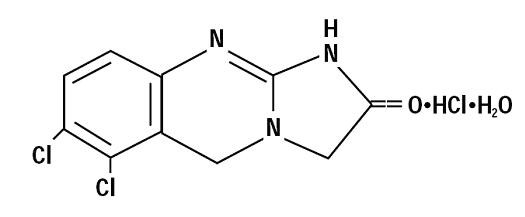
C10H7Cl2N3O·HCl·H2O M.W. 310.55
Each anagrelide hydrochloride capsule, for oral administration, contains either 0.5 mg or 1 mg of anagrelide base (as anagrelide hydrochloride) and has the following inactive ingredients: black iron oxide, crospovidone, D&C yellow #10 aluminum lake, FD&C blue #1/brilliant blue aluminum lake, FD&C blue #2/indigo carmine aluminum lake, FD&C red #40/allura red aluminum lake, gelatin, lactose anhydrous, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, propylene glycol, shellac glaze and titanium dioxide.
-
CLINICAL PHARMACOLOGY
The mechanism by which anagrelide reduces blood platelet count is still under investigation. Studies in patients support a hypothesis of dose-related reduction in platelet production resulting from a decrease in megakaryocyte hypermaturation. In blood withdrawn from normal volunteers treated with anagrelide, a disruption was found in the postmitotic phase of megakaryocyte development and a reduction in megakaryocyte size and ploidy. At therapeutic doses, anagrelide does not produce significant changes in white cell counts or coagulation parameters, and may have a small, but clinically insignificant effect on red cell parameters. Anagrelide inhibits cyclic AMP phosphodiesterase III (PDEIII). PDEIII inhibitors can also inhibit platelet aggregation. However, significant inhibition of platelet aggregation is observed only at doses of anagrelide higher than those required to reduce platelet count.
Following oral administration of 14C-anagrelide in people, more than 70% of radioactivity was recovered in urine. Based on limited data, there appears to be a trend toward dose linearity between doses of 0.5 mg and 2 mg. At fasting and at a dose of 0.5 mg of anagrelide, the plasma half-life is 1.3 hours. The available plasma concentration time data at steady state in patients showed that anagrelide does not accumulate in plasma after repeated administration.
Two major metabolites have been identified (RL603 and 3-hydroxy anagrelide).
There were no apparent differences between patient groups (pediatric versus adult patients) for tmax and for t1/2 for anagrelide, 3-hydroxy anagrelide, or RL603.
Pharmacokinetic data obtained from healthy volunteers comparing the pharmacokinetics of anagrelide in the fed and fasted states showed that administration of a 1 mg dose of anagrelide with food decreased the Cmax by 14%, but increased the AUC by 20%.
Pharmacokinetic (PK) data from pediatric (age range 7 to 14 years) and adult (age range 16 to 86 years) patients with thrombocythemia secondary to a myeloproliferative disorder (MPD), indicate that dose- and body weight-normalized exposure, Cmax and AUCτ, of anagrelide were lower in the pediatric patients compared to the adult patients (Cmax 48%, AUCτ 55%).
Pharmacokinetic data from fasting elderly patients with ET (age range 65 to 75 years) compared to fasting adult patients (age range 22 to 50 years) indicate that the Cmax and AUC of anagrelide were 36% and 61% higher respectively in elderly patients, but that the Cmax and AUC of the active metabolite, 3-hydroxy anagrelide, were 42% and 37% lower respectively in the elderly patients.
A pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with severe renal impairment (creatinine clearance < 30 mL/min) showed no significant effects on the pharmacokinetics of anagrelide.
A pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with moderate hepatic impairment showed an 8 fold increase in total exposure (AUC) to anagrelide.
CLINICAL STUDIES
A total of 942 patients with myeloproliferative disorders including 551 patients with Essential Thrombocythemia (ET), 117 patients with Polycythemia Vera (PV), 178 patients with Chronic Myelogenous Leukemia (CML), and 96 patients with other myeloproliferative disorders (OMPD), were treated with anagrelide in three clinical trials. Patients with OMPD included 87 patients who had Myeloid Metaplasia with Myelofibrosis (MMM), and 9 patients who had unknown myeloproliferative disorders.
Clinical Studies
Patients with ET, PV, CML, or MMM were diagnosed based on the following criteria:
ET:
- Platelet count ≥ 900,000/µL on two determinations
- Profound megakaryocytic hyperplasia in bone marrow
- Absence of Philadelphia chromosome
- Normal red cell mass
- Normal serum iron and ferritin and normal marrow iron stores
CML:
- Persistent granulocyte count ≥ 50,000/µL without evidence of infection
- Absolute basophil count ≥ 100/µL
- Evidence for hyperplasia of the granulocytic line in the bone marrow
- Philadelphia chromosome is present
- Leukocyte alkaline phosphatase ≤ lower limit of the laboratory normal range
PV†:
- A1 Increased red cell mass
- A2 Normal arterial oxygen saturation
- A3 Splenomegaly
- B1 Platelet count ≥ 400,000/µL, in absence of iron deficiency or bleeding
- B2 Leukocytosis (≥ 12,000/µL, in the absence of infection)
- B3 Elevated leukocyte alkaline phosphatase
- B4 Elevated Serum B12
†Diagnosis positive if A1, A2, and A3 present; or, if no splenomegaly, diagnosis is positive if A1 and A2 are present with any two of B1, B2, or B3.
MMM:
- Myelofibrotic (hypocellular, fibrotic) bone marrow
- Prominent megakaryocytic metaplasia in bone marrow
- Splenomegaly
- Moderate to severe normo-chromic normocytic anemia
- White cell count may be variable; (80,000 to 100,000/µL)
- Increased platelet count
- Variable red cell mass; teardrop poikilocytes
- Normal to high leukocyte alkaline phosphatase
- Absence of Philadelphia chromosome
Patients were enrolled in clinical trials if their platelet count was ≥ 900,000/µL on two occasions or ≥ 650,000/µL on two occasions with documentation of symptoms associated with thrombocythemia. The mean duration of anagrelide therapy for ET, PV, CML, and OMPD patients was 65, 67, 40, and 44 weeks, respectively; 23% of patients received treatment for 2 years. Patients were treated with anagrelide starting at doses of 0.5 to 2 mg every 6 hours. The dose was increased if the platelet count was still high, but to no more than 12 mg each day. Efficacy was defined as reduction of platelet count to or near physiologic levels (150,000 to 400,000/µL). The criteria for defining subjects as "responders" were reduction in platelets for at least 4 weeks to ≤600,000/µL, or by at least 50% from baseline value. Subjects treated for less than 4 weeks were not considered evaluable. The results are depicted graphically below:
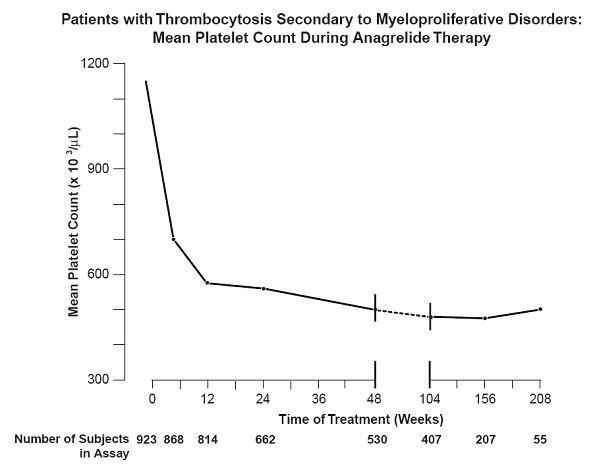
* x103/µL
** Nine hundred and forty-two subjects with myeloproliferative disorders were enrolled in three research studies. Of these, 923 had platelet counts over the duration of the studies.
Time on Treatment Weeks Years Baseline 4 12 24 48 2 3 4 Mean* 1131 683 575 526 484 460 437 457 N 923 ** 868 814 662 530 407 207 55 Anagrelide was effective in phlebotomized patients as well as in patients treated with other concomitant therapies including hydroxyurea, aspirin, interferon, radioactive phosphorus, and alkylating agents.
-
INDICATIONS AND USAGE
Anagrelide hydrochloride capsules are indicated for the treatment of patients with thrombocythemia, secondary to myeloproliferative disorders, to reduce the elevated platelet count and the risk of thrombosis and to ameliorate associated symptoms including thrombo-hemorrhagic events (see CLINICAL STUDIES, DOSAGE AND ADMINISTRATION).
-
CONTRAINDICATIONS
Anagrelide is contraindicated in patients with severe hepatic impairment. Exposure to anagrelide is increased 8 fold in patients with moderate hepatic impairment (see CLINICAL PHARMACOLOGY). Use of anagrelide in patients with severe hepatic impairment has not been studied (see also WARNINGS, Hepatic).
-
WARNINGS
Cardiovascular
Anagrelide should be used with caution in patients with known or suspected heart disease, and only if the potential benefits of therapy outweigh the potential risks. Because of the positive inotropic effects and side effects of anagrelide, a pre-treatment cardiovascular examination is recommended along with careful monitoring during treatment. In humans, therapeutic doses of anagrelide may cause cardiovascular effects, including vasodilation, tachycardia, palpitations, and congestive heart failure.
Hepatic
Exposure to anagrelide is increased 8 fold in patients with moderate hepatic impairment (see CLINICAL PHARMACOLOGY). Use of anagrelide in patients with severe hepatic impairment has not been studied. The potential risks and benefits of anagrelide therapy in a patient with mild and moderate impairment of hepatic function should be assessed before treatment is commenced. In patients with moderate hepatic impairment, dose reduction is required and patients should be carefully monitored for cardiovascular effects (see DOSAGE AND ADMINISTRATION for specific dosing recommendations).
Interstitial Lung Diseases
Interstitial lung diseases (including allergic alveolitis, eosinophilic pneumonia and interstitial pneumonitis) have been reported to be associated with the use of anagrelide in postmarketing reports. Most cases presented with progressive dyspnea with lung infiltrations. The time of onset ranged from 1 week to several years after initiating anagrelide. In most cases, the symptoms improved after discontinuation of anagrelide (see ADVERSE REACTIONS).
-
PRECAUTIONS
Laboratory Tests
Anagrelide therapy requires close clinical supervision of the patient. While the platelet count is being lowered (usually during the first two weeks of treatment), blood counts (hemoglobin, white blood cells) and renal function (serum creatinine, BUN) should be monitored. Cases of clinically significant hepatotoxicity (including symptomatic ALT and AST elevations and elevations greater than three times the ULN) have been reported in postmarketing surveillance. Measure liver function tests (ALT, AST) before initiating anagrelide treatment and during therapy.
In 9 subjects receiving a single 5 mg dose of anagrelide, standing blood pressure fell an average of 22/15 mm Hg, usually accompanied by dizziness. Only minimal changes in blood pressure were observed following a dose of 2 mg.
Cessation of Anagrelide Treatment
In general, interruption of anagrelide treatment is followed by an increase in platelet count. After sudden stoppage of anagrelide therapy, the increase in platelet count can be observed within four days.
Drug Interactions
Limited PK and/or PD studies investigating possible interactions between anagrelide and other medicinal products have been conducted. In vivo interaction studies in humans have demonstrated that digoxin and warfarin do not affect the PK properties of anagrelide, nor does anagrelide affect the PK properties of digoxin or warfarin.
In two clinical interaction studies in healthy subjects, coadministration of single-dose anagrelide 1 mg and aspirin 900 mg or repeat-dose anagrelide 1 mg once daily and aspirin 75 mg once daily showed greater ex vivo anti-platelet aggregation effects than administration of aspirin alone. Coadministered anagrelide 1 mg and aspirin 900 mg single-doses had no effect on bleeding time, prothrombin time (PT) or activated partial thromboplastin time (aPTT).
The potential risks and benefits of concomitant use of anagrelide with aspirin should be assessed, particularly in patients with a high risk profile for hemorrhage, before treatment is commenced.
Drug interaction studies have not been conducted with the other common medications used concomitantly with anagrelide in clinical trials which were acetaminophen, furosemide, iron, ranitidine, hydroxyurea, and allopurinol.
Anagrelide is metabolized at least in part by CYP1A2. It is known that CYP1A2 is inhibited by several medicinal products, including fluvoxamine, and such medicinal products could theoretically adversely influence the clearance of anagrelide. Anagrelide demonstrates some limited inhibitory activity towards CYP1A2 which may present a theoretical potential for interaction with other coadministered medicinal products sharing that clearance mechanism e.g., theophylline.
Anagrelide is an inhibitor of cyclic AMP PDE III. The effects of medicinal products with similar properties such as inotropes milrinone, enoximone, amrinone, olprinone and cilostazol may be exacerbated by anagrelide.
There is a single case report which suggests that sucralfate may interfere with anagrelide absorption.
Food has no clinically significant effect on the bioavailability of anagrelide.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a two year rat carcinogenicity study a higher incidence of uterine adenocarcinoma, relative to controls, was observed in females receiving 30 mg/kg/day (at least 174 times human AUC exposure after a 1 mg twice daily dose). Adrenal phaeochromocytomas were increased relative to controls in males receiving 3 mg/kg/day and above, and in females receiving 10 mg/kg/day and above (at least 10 and 18 times respectively human AUC exposure after a 1 mg twice daily dose). Anagrelide hydrochloride was not genotoxic in the Ames test, the mouse lymphoma cell (L5178Y, TK+/-) forward mutation test, the human lymphocyte chromosome aberration test, or the mouse micronucleus test. Anagrelide hydrochloride at oral doses up to 240 mg/kg/day (1,440 mg/m2/day, 195 times the recommended maximum human dose based on body surface area) was found to have no effect on fertility and reproductive performance of male rats. However, in female rats, at oral doses of 60 mg/kg/day (360 mg/m2/day, 49 times the recommended maximum human dose based on body surface area) or higher, it disrupted implantation when administered in early pregnancy and retarded or blocked parturition when administered in late pregnancy.
Pregnancy
Pregnancy Category C
Teratogenic Effects
Teratology studies have been performed in pregnant rats at oral doses up to 900 mg/kg/day (5,400 mg/m2/day, 730 times the recommended maximum human dose based on body surface area) and in pregnant rabbits at oral doses up to 20 mg/kg/day (240 mg/m2/day, 32 times the recommended maximum human dose based on body surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to anagrelide hydrochloride.
Nonteratogenic Effects
A fertility and reproductive performance study performed in female rats revealed that anagrelide hydrochloride at oral doses of 60 mg/kg/day (360 mg/m2/day, 49 times the recommended maximum human dose based on body surface area) or higher disrupted implantation and exerted adverse effect on embryo/fetal survival.
A perinatal and postnatal study performed in female rats revealed that anagrelide hydrochloride at oral doses of 60 mg/kg/day (360 mg/m2/day, 49 times the recommended maximum human dose based on body surface area) or higher produced delay or blockage of parturition, deaths of non-delivering pregnant dams and their fully developed fetuses, and increased mortality in the pups born.
There are however no adequate and well-controlled studies with anagrelide hydrochloride in pregnant women. Because animal reproduction studies are not always predictive of human response, anagrelide hydrochloride should be used during pregnancy only if clearly needed.
Nonclinical Toxicology
In the 2-year rat study, a significant increase in non-neoplastic lesions were observed in anagrelide treated males and females in the adrenal (medullary hyperplasia), heart (myocardial hypertrophy and chamber distension), kidney (hydronephrosis, tubular dilation and urothelial hyperplasia) and bone (femur enostosis). Vascular effects were observed in tissues of the pancreas (arteritis/periarteritis, intimal proliferation and medial hypertrophy), kidney (arteritis/periarteritis, intimal proliferation and medial hypertrophy), sciatic nerve (vascular mineralization), and testes (tubular atrophy and vascular infarct) in anagrelide treated males.
Five women became pregnant while on anagrelide treatment at doses of 1 to 4 mg/day. Treatment was stopped as soon as it was realized that they were pregnant. All delivered normal, healthy babies. There are no adequate and well-controlled studies in pregnant women. Anagrelide hydrochloride should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Anagrelide is not recommended in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential harm to the fetus. Women of child-bearing potential should be instructed that they must not be pregnant and that they should use contraception while taking anagrelide. Anagrelide may cause fetal harm when administered to a pregnant woman.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reaction in nursing infants from anagrelide hydrochloride, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Myeloproliferative disorders are uncommon in pediatric patients and limited data are available in this population. An open label safety and PK/PD study (see CLINICAL PHARMACOLOGY) was conducted in 17 pediatric patients 7 to 14 years of age (8 patients 7 to 11 years of age and 9 patients 11 to 14 years of age, mean age of 11 years; 8 males and 9 females) with thrombocythemia secondary to ET as compared to 18 adult patients (mean age of 63 years, 9 males and 9 females). Prior to entry on to the study, 16 of 17 pediatric patients and 13 of 18 adult patients had received anagrelide treatment for an average of 2 years. The median starting total daily dose, determined by retrospective chart review, for pediatric and adult ET patients who had received anagrelide prior to study entry was 1 mg for each of the three age groups (7 to 11 and 11 to 14 year old patients and adults). The starting dose for 6 anagrelide-naive patients at study entry was 0.5 mg once daily. At study completion, the median total daily maintenance doses were similar across age groups, median of 1.75 mg for patients of 7 to 11 years of age, 2 mg in patients 11 to 14 years of age, and 1.5 mg for adults.
The study evaluated the pharmacokinetic (PK) and pharmacodynamic (PD) profile of anagrelide, including platelet counts (see CLINICAL PHARMACOLOGY).
The frequency of adverse events observed in pediatric patients was similar to adult patients. The most common adverse events observed in pediatric patients were fever, epistaxis, headache, and fatigue during a 3 months treatment of anagrelide in the study. Adverse events that had been reported in these pediatric patients prior to the study and were considered to be related to anagrelide treatment based on retrospective review were palpitation, headache, nausea, vomiting, abdominal pain, back pain, anorexia, fatigue, and muscle cramps. Episodes of increased pulse rate and decreased systolic or diastolic blood pressure beyond the normal ranges in the absence of clinical symptoms were observed in some patients. Reported AEs were consistent with the known pharmacological profile of anagrelide and the underlying disease. There were no apparent trends or differences in the types of adverse events observed between the pediatric patients compared with those of the adult patients. No overall difference in dosing and safety were observed between pediatric and adult patients.
In another open-label study, anagrelide had been used successfully in 12 pediatric patients (age range 6.8 to 17.4 years; 6 male and 6 female), including 8 patients with ET, 2 patients with CML, 1 patient with PV, and 1 patient with OMPD. Patients were started on therapy with 0.5 mg q.i.d. up to a maximum daily dose of 10 mg. The median duration of treatment was 18.1 months with a range of 3.1 to 92 months. Three patients received treatment for greater than three years. Other adverse events reported in spontaneous reports and literature reviews include anemia, cutaneous photosensitivity and elevated leukocyte count.
Geriatric Use
Of the total number of subjects in clinical studies of anagrelide, 42.1% were 65 years and over, while 14.9% were 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in response between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
ADVERSE REACTIONS
Analysis of the adverse events in a population consisting of 942 patients in 3 clinical studies diagnosed with myeloproliferative diseases of varying etiology (ET: 551; PV: 117; OMPD: 274) has shown that all disease groups have the same adverse event profile. While most reported adverse events during anagrelide therapy have been mild in intensity and have decreased in frequency with continued therapy, serious adverse events were reported in these patients. These include the following: congestive heart failure, myocardial infarction, cardiomyopathy, cardiomegaly, complete heart block, atrial fibrillation, cerebrovascular accident, pericarditis, pericardial effusion, pleural effusion, pulmonary infiltrates, pulmonary fibrosis, pulmonary hypertension, pancreatitis, gastric/duodenal ulceration, and seizure.
Of the 942 patients treated with anagrelide for a mean duration of approximately 65 weeks, 161 (17%) were discontinued from the study because of adverse events or abnormal laboratory test results. The most common adverse events for treatment discontinuation were headache, diarrhea, edema, palpitation, and abdominal pain. Overall, the occurrence rate of all adverse events was 17.9 per 1,000 treatment days. The occurrence rate of adverse events increased at higher dosages of anagrelide.
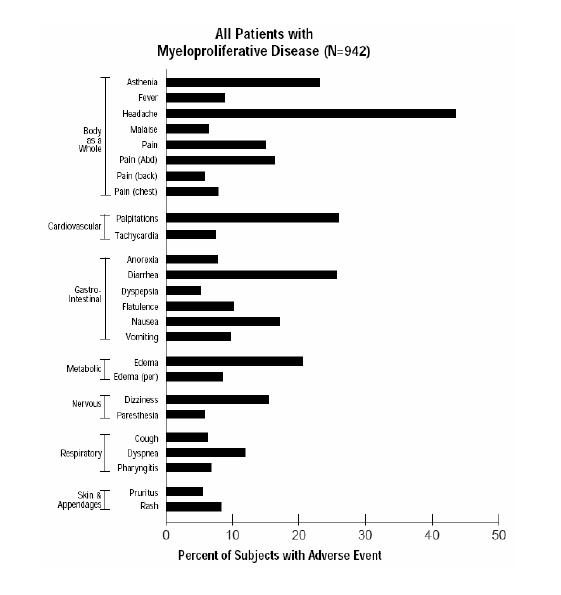
The most frequently reported adverse reactions to anagrelide (in 5% or greater of 942 patients with myeloproliferative disease) in clinical trials were:
Headache…………………………………….. 43.5% Palpitations…………………………………... 26.1% Diarrhea……………………………………… 25.7% Asthenia……………………………………… 23.1% Edema, other…………………………………. 20.6% Nausea……………………………………….. 17.1% Abdominal pain……………………………… 16.4% Dizziness…………………………………….. 15.4% Pain, other……………………………………. 15% Dyspnea……………………………………… 11.9% Flatulence……………………………………. 10.2% Vomiting……………………………………... 9.7% Fever…………………………………………. 8.9% Peripheral edema……………………………. 8.5% Rash, including urticaria…………………….. 8.3% Chest pain……………………………………. 7.8% Anorexia……………………………………... 7.7% Tachycardia………………………………….. 7.5% Pharyngitis…………………………………… 6.8% Malaise………………………………………. 6.4% Cough………………………………………... 6.3% Paresthesia…………………………………… 5.9% Back pain…………………………………….. 5.9% Pruritus………………………………………. 5.5% Dyspepsia……………………………………. 5.2% Adverse events with an incidence of 1% to < 5% included:
Cardiovascular System
Arrhythmia, hemorrhage, hypertension, cardiovascular disease, angina pectoris, heart failure, postural hypotension, thrombosis, vasodilatation, migraine, syncope
Digestive System
Constipation, GI distress, GI hemorrhage, gastritis, melena, aphthous stomatitis, eructation
Hemic and Lymphatic System
Anemia, thrombocytopenia, ecchymosis, lymphadenopathy
Platelet counts below 100,000/µL occurred in 84 patients (ET: 35; PV: 9; OMPD: 40), reduction below 50,000/µL occurred in 44 patients (ET: 7; PV: 6; OMPD: 31) while on anagrelide therapy. Thrombocytopenia promptly recovered upon discontinuation of anagrelide.
Hepatic System
Elevated liver enzymes were observed in 3 patients (ET: 2; OMPD: 1) during anagrelide therapy.
Respiratory System
Rhinitis, epistaxis, respiratory disease, sinusitis, pneumonia, bronchitis, asthma
Urogenital System
Dysuria, hematuria
Renal abnormalities occurred in 15 patients (ET: 10; PV: 4; OMPD: 1). Six ET, 4 PV and 1 with OMPD experienced renal failure (approximately 1%) while on anagrelide treatment; in 4 cases, the renal failure was considered to be possibly related to anagrelide treatment. The remaining 11 were found to have pre-existing renal impairment. Doses ranged from 1.5 to 6 mg/day, with exposure periods of 2 to 12 months. No dose adjustment was required because of renal insufficiency.
The adverse event profile for patients in three clinical trials on anagrelide therapy (in 5% or greater of 942 patients with myeloproliferative diseases) is shown in the following bar graph:
Postmarketing Reports
Cases of interstitial lung diseases (including allergic alveolitis, eosinophilic pneumonia and interstitial pneumonitis), tubulointerstitial nephritis and clinically significant hepatotoxicity have been reported (see WARNINGS, Interstitial Lung Diseases and PRECAUTIONS, Laboratory Tests).
-
OVERDOSAGE
Acute Toxicity and Symptoms
Single oral doses of anagrelide hydrochloride at 2,500, 1,500 and 200 mg/kg in mice, rats and monkeys, respectively, were not lethal. Symptoms of acute toxicity were: decreased motor activity in mice and rats and softened stools and decreased appetite in monkeys.
There have been postmarketing case reports of intentional overdose with anagrelide hydrochloride. Reported symptoms include sinus tachycardia and vomiting. Symptoms resolved with conservative management. Platelet reduction from anagrelide therapy is dose-related; therefore, thrombocytopenia, which can potentially cause bleeding, is expected from overdosage. Should overdosage occur, cardiac and central nervous system toxicity can also be expected.
-
DOSAGE AND ADMINISTRATION
Treatment with anagrelide hydrochloride capsules should be initiated under close medical supervision. The recommended starting dosage of anagrelide hydrochloride capsules for adult patients is 0.5 mg q.i.d. or 1 mg b.i.d (2 capsules of 0.5 mg twice a day), which should be maintained for at least one week. Starting doses in pediatric patients have ranged from 0.5 mg per day to 0.5 mg q.i.d. As there are limited data on the appropriate starting dose for pediatric patients, an initial dose of 0.5 mg per day is recommended. In both adult and pediatric patients, dosage should then be adjusted to the lowest effective dosage required to reduce and maintain platelet count below 600,000/µL, and ideally to the normal range. The dosage should be increased by not more than 0.5 mg/day in any one week. Maintenance dosing is not expected to be different between adult and pediatric patients. Dosage should not exceed 10 mg/day or 2.5 mg in a single dose (see PRECAUTIONS).
There are no special requirements for dosing the geriatric population.
It is recommended that patients with moderate hepatic impairment start anagrelide therapy at a dose of 0.5 mg/day and be maintained for a minimum of one week with careful monitoring of cardiovascular effects. The dosage increment must not exceed more than 0.5 mg/day in any one week. The potential risks and benefits of anagrelide therapy in a patient with mild or moderate impairment of hepatic function should be assessed before treatment is commenced. Use of anagrelide in patients with severe hepatic impairment has not been studied. Use of anagrelide in patients with severe hepatic impairment is contraindicated (see CONTRAINDICATIONS).
To monitor the effect of anagrelide and prevent the occurrence of thrombocytopenia, platelet counts should be performed every two days during the first week of treatment and at least weekly thereafter until the maintenance dosage is reached.
Typically, platelet count begins to respond within 7 to 14 days at the proper dosage. The time to complete response, defined as platelet count ≤600,000/µL, ranged from 4 to 12 weeks. Most patients will experience an adequate response at a dose of 1.5 to 3 mg/day. Patients with known or suspected heart disease, renal insufficiency, or hepatic dysfunction should be monitored closely.
-
HOW SUPPLIED
Anagrelide Hydrochloride Capsules are available as light gray cap/white body hard gelatin capsules, spin printed in black ink
“5241” on the cap and "0.5 mg" on the body containing 0.5 mg of anagrelide base (as anagrelide hydrochloride) packaged in bottles of 30 capsules.
PHARMACIST: Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Manufactured In India By:
Cipla Ltd.
Goa, India
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
Rev. C 2/2011
ANAGRELIDE HYDROCHLORIDE CAPSULES
Relabeling and Repackaging by:
Physicians Total Care, Inc.
Tulsa, Oklahoma 74146
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ANAGRELIDE HYDROCHLORIDE
anagrelide hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5443(NDC:0172-5241) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Anagrelide Hydrochloride (UNII: VNS4435G39) (Anagrelide - UNII:K9X45X0051) Anagrelide 0.5 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE (UNII: 68401960MK) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) ALUMINUM OXIDE (UNII: LMI26O6933) FERROSOFERRIC OXIDE (UNII: XM0M87F357) GELATIN (UNII: 2G86QN327L) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE (UNII: FZ989GH94E) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color GRAY (light gray cap/white body) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code 5241;05mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5443-2 30 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076468 07/19/2007 ANAGRELIDE HYDROCHLORIDE
anagrelide hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5385(NDC:0172-5240) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Anagrelide Hydrochloride (UNII: VNS4435G39) (Anagrelide - UNII:K9X45X0051) Anagrelide 1 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE (UNII: 68401960MK) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C RED NO. 40 (UNII: WZB9127XOA) ALUMINUM OXIDE (UNII: LMI26O6933) FERROSOFERRIC OXIDE (UNII: XM0M87F357) GELATIN (UNII: 2G86QN327L) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE (UNII: FZ989GH94E) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color WHITE Score no score Shape CAPSULE Size 16mm Flavor Imprint Code 5240;1mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5385-0 30 in 1 BOTTLE 2 NDC:54868-5385-1 10 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076468 08/15/2005 06/30/2011 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel, repack
