VAPRISOL DEXTROSE IN PLASTIC CONTAINER- conivaptan hydrochloride injection, solution
Cumberland Pharmaceuticals Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use VAPRISOL safely and effectively. See full prescribing information for VAPRISOL.
VAPRISOL® (conivaptan hydrochloride) injection, for intravenous use Initial U.S. Approval: 2005 INDICATIONS AND USAGEVAPRISOL® is a vasopressin receptor antagonist indicated to raise serum sodium in hospitalized patients with euvolemic and hypervolemic hyponatremia (1).
Limitations of Use:
It has not been established that raising serum sodium with VAPRISOL provides a symptomatic benefit to patients (1). DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥ 10%) are infusion site reactions (including phlebitis), pyrexia, hypokalemia, headache and orthostatic hypotension (6). To report SUSPECTED ADVERSE REACTIONS, contact Cumberland Pharmaceuticals Inc. at 1-887-484-2700 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 9/2017 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
VAPRISOL® is indicated to raise serum sodium in hospitalized patients with euvolemic and hypervolemic hyponatremia.
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
VAPRISOL is for intravenous use only.
VAPRISOL is for use in hospitalized patients only.
Administer VAPRISOL through large veins and change of the infusion site every 24 hours to minimize the risk of vascular irritation [see Warnings and Precaution (5.4)].
Initiate with a loading dose of 20 mg VAPRISOL administered intravenously over 30 minutes.
Follow the loading dose with 20 mg VAPRISOL administered in a continuous intravenous infusion over 24 hours. After the initial day of treatment, administer VAPRISOL for an additional 1 to 3 days in a continuous infusion of 20 mg/day. If serum sodium is not rising at the desired rate, VAPRISOL may be titrated upward to a maximum dose of 40 mg daily, administered in a continuous intravenous infusion over 24 hours.
The total duration of infusion of VAPRISOL (after the loading dose) should not exceed four days.
Patients receiving VAPRISOL must have frequent monitoring of serum sodium and volume status [see Warnings and Precautions (5.2, 5.3)].
2.2 Preparation, Compatibility and Stability
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulate matter, discoloration or cloudiness is observed, the drug solution should not be used.
VAPRISOL is supplied ready-to-use; no further dilution of this preparation is necessary.
VAPRISOL is compatible with 5% Dextrose Injection. VAPRISOL is physically and chemically compatible with 0.9% Sodium Chloride Injection for up to 48 hours when the two solutions are co-administered via a Y-site connection at a flow rate for VAPRISOL of 4.2 mL/hour and at flow rates for 0.9% Sodium Chloride Injection of either 2.1 mL/hour or 6.3 mL/hour.
VAPRISOL is incompatible with both Lactated Ringer's Injection and furosemide injection when these products are mixed in the same container; therefore, do not combine VAPRISOL with these products in the same intravenous line or container.
Do not combine VAPRISOL with any other product in the same intravenous line or container.
Do not use plastic containers in series connections. Such use could result in air embolism due to residual air being drawn from the primary container before administration of the fluid from the secondary container is completed.
Do not remove container from overwrap until ready for use. The overwrap is a moisture and light barrier. The inner container maintains the sterility of the product.
Tear overwrap down side at slit and remove solution container. Some opacity of the plastic due to moisture absorption during the sterilization process may be observed. This is normal and does not affect the solution quality or safety. The opacity will diminish gradually. After removing overwrap, check for minute leaks by squeezing inner container firmly. If leaks are found, discard solution as sterility may be impaired. Do not use if the solution is cloudy or a precipitate is present.
Preparation for Administration:
- Suspend container from eyelet support.
- Remove protector from outlet port at bottom of container.
- Attach administration set. Refer to complete directions accompanying set.
2.3 Hepatic Impairment
In patients with moderate (Child-Pugh Class B) and severe (Child-Pugh Class C) hepatic impairment, initiate VAPRISOL with a loading dose of 10 mg over 30 minutes followed by 10 mg per day as a continuous infusion for 2 to 4 days. If serum sodium is not rising at the desired rate, VAPRISOL may be titrated upward to 20 mg per day [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
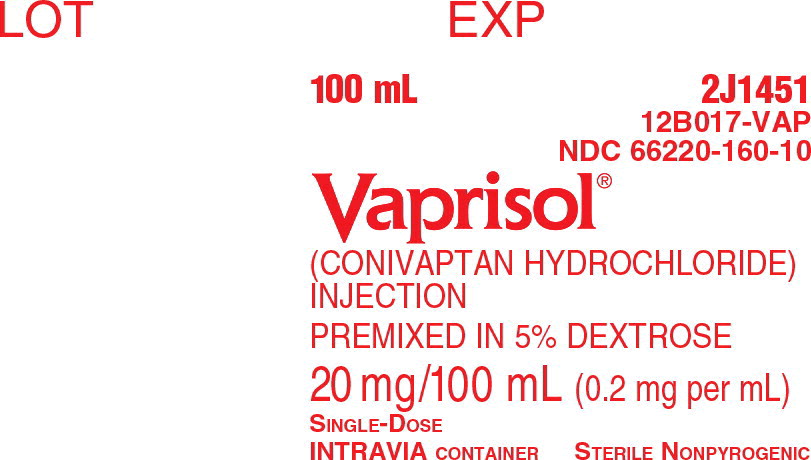
Intravenous injection solution: conivaptan hydrochloride 20 mg/100 mL premixed in 5% Dextrose in flexible plastic containers.
4 CONTRAINDICATIONS
4.2 Coadministration with Potent CYP3A Inhibitors
The coadministration of VAPRISOL with potent CYP3A inhibitors, such as ketoconazole, itraconazole, clarithromycin, ritonavir, and indinavir, is contraindicated [see Drug Interactions (7.1)].
4.3 Anuric Patients
In patients unable to make urine, no benefit can be expected [see Clinical Pharmacology (12.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Hyponatremia Associated with Heart Failure
The amount of safety data on the use of VAPRISOL in patients with hypervolemic hyponatremia associated with heart failure is limited. VAPRISOL should be used to raise serum sodium in such patients only after consideration of other treatment options [see Adverse Reactions (6.1)].
5.2 Overly Rapid Correction of Serum Sodium
Osmotic demyelination syndrome is a risk associated with overly rapid correction of hyponatremia (i.e., > 12 mEq/L/24 hours). Osmotic demyelination results in dysarthria, mutism, dysphagia, lethargy, affective changes, spastic quadriparesis, seizures, coma or death. In susceptible patients, including those with severe malnutrition, alcoholism or advanced liver disease, use slower rates of correction. In controlled clinical trials of VAPRISOL, about 9% of patients who received VAPRISOL in doses of 20-40 mg/day IV had rises of serum sodium >12 mEq/L/24 hours, but none of these patients had evidence of osmotic demyelination or permanent neurologic sequelae. Serum sodium concentration and neurologic status should be monitored appropriately during VAPRISOL administration, and VAPRISOL administration should be discontinued if the patient develops an undesirably rapid rate of rise of serum sodium. If the serum sodium concentration continues to rise, VAPRISOL should not be resumed. If hyponatremia persists or recurs (after initial discontinuation of VAPRISOL for an undesirably rapid rate of rise of serum sodium concentration), and the patient has had no evidence of neurologic sequelae of rapid rise in serum sodium, VAPRISOL may be resumed at a reduced dose [see Dosage and Administration (2.1)].
5.3 Hypovolemia or Hypotension
For patients who develop hypovolemia or hypotension while receiving VAPRISOL, VAPRISOL should be discontinued, and volume status and vital signs should be frequently monitored. Once the patient is again euvolemic and is no longer hypotensive, VAPRISOL may be resumed at a reduced dose if the patient remains hyponatremic.
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in labeling:
- Osmotic demyelination syndrome [see Warnings and Precautions (5.2)]
- Infusion site reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse event information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
The most common adverse reactions reported with VAPRISOL administration were infusion site reactions. In studies in patients and healthy volunteers, infusion site reactions occurred in 73% and 63% of subjects treated with VAPRISOL 20 mg/day and 40 mg/day, respectively, compared to 4% in the placebo group. Infusion site reactions were the most common type of adverse event leading to discontinuation of VAPRISOL. Discontinuations from treatment due to infusion site reactions were more common among VAPRISOL-treated patients (3%) than among placebo-treated patients (0%). Some serious infusion site reactions did occur [see Dosage and Administration (2.1) and Warnings and Precautions (5.4)].
The adverse reactions presented in Table 1 are derived from 72 healthy volunteers and 243 patients with euvolemic or hypervolemic hyponatremia who received VAPRISOL 20 mg IV as a loading dose followed by 40 mg/day IV for 2 to 4 days, from 37 patients with euvolemic or hypervolemic hyponatremia who received VAPRISOL 20 mg IV as a loading dose followed by 20 mg/day IV for 2 to 4 days in an open-label study, and from 40 healthy volunteers and 29 patients with euvolemic or hypervolemic hyponatremia who received placebo. The adverse reactions occurred in at least 5% of patients treated with VAPRISOL and at a higher incidence for VAPRISOL-treated patients than for placebo-treated patients.
| Term | Placebo (N=69)
N (%) | 20 mg (N=37)
N (%) | 40 mg (N=315)
N (%) |
|---|---|---|---|
|
Adapted from MedDRA version 6.0 |
|||
| Blood and lymphatic system disorders | |||
| Anemia NOS | 2 (3%) | 2 (5%) | 18 (6%) |
| Cardiac disorders | |||
| Atrial fibrillation | 0 (0%) | 2 (5%) | 7 (2%) |
| Gastrointestinal disorders | |||
| Constipation | 2 (3%) | 3 (8%) | 20 (6%) |
| Diarrhea NOS | 0 (0%) | 0 (0%) | 23 (7%) |
| Nausea | 3 (4%) | 1 (3%) | 17 (5%) |
| Vomiting NOS | 0 (0%) | 2 (5%) | 23 (7%) |
| General disorders and administration site conditions | |||
| Edema peripheral | 1 (1%) | 1 (3%) | 24 (8%) |
| Infusion site erythema | 0 (0%) | 0 (0%) | 18 (6%) |
| Infusion site pain | 1 (1%) | 0 (0%) | 16 (5%) |
| Infusion site phlebitis | 1 (1%) | 19 (51%) | 102 (32%) |
| Infusion site reaction | 0 (0%) | 8 (22%) | 61 (19%) |
| Pyrexia | 0 (0%) | 4 (11%) | 15 (5%) |
| Thirst | 1 (1%) | 1 (3%) | 19 (6%) |
| Infections and infestations | |||
| Pneumonia NOS | 0 (0%) | 2 (5%) | 7 (2%) |
| Urinary tract infection NOS | 2 (3%) | 2 (5%) | 14 (4%) |
| Injury, poisoning and procedural complications | |||
| Post procedural diarrhea | 0 (0%) | 2 (5%) | 0 (0%) |
| Investigations | |||
| Electrocardiogram ST segment depression | 0 (0%) | 2 (5%) | 0 (0%) |
| Metabolism and nutrition disorders | |||
| Hypokalemia | 2 (3%) | 8 (22%) | 30 (10%) |
| Hypomagnesemia | 0 (0%) | 2 (5%) | 6 (2%) |
| Hyponatremia | 1 (1%) | 3 (8%) | 20 (6%) |
| Nervous system disorders | |||
| Headache | 2 (3%) | 3 (8%) | 32 (10%) |
| Psychiatric disorders | |||
| Confusional state | 2 (3%) | 0 (0%) | 16 (5%) |
| Insomnia | 0 (0%) | 2 (5%) | 12 (4%) |
| Respiratory, thoracic and mediastinal disorders | |||
| Pharyngolaryngeal pain | 3 (4%) | 2 (5%) | 3 (1%) |
| Skin and subcutaneous tissue disorders | |||
| Pruritus | 0 (0%) | 2 (5%) | 2 (1%) |
| Vascular disorders | |||
| Hypertension NOS | 0 (0%) | 3 (8%) | 20 (6%) |
| Hypotension NOS | 2 (3%) | 3 (8%) | 16 (5%) |
| Orthostatic hypotension | 0 (0%) | 5 (14%) | 18 (6%) |
Although a dose of 80 mg/day of VAPRISOL was also studied, it was associated with a higher incidence of infusion site reactions and a higher rate of discontinuation for adverse events than was the 40 mg/day VAPRISOL dose. The maximum recommended daily dose of VAPRISOL (after the loading dose) is 40 mg/day.
Heart failure with hypervolemic hyponatremia
In clinical trials where VAPRISOL was administered to 79 hypervolemic hyponatremic patients with underlying heart failure and intravenous placebo administered to 10 patients, adverse cardiac failure events, atrial dysrhythmias, and sepsis occurred more frequently among patients treated with VAPRISOL (32%, 5% and 8% respectively) than among patients treated with placebo (20%, 0% and 0% respectively) [see Warnings and Precautions (5.1)].
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors and Substrates
Conivaptan is a sensitive substrate of CYP3A. Coadministration with strong CYP3A inhibitors (e.g. ketoconazole, itraconazole, clarithromycin, ritonavir and indinavir) increases conivaptan exposure and is contraindicated [see Contraindications (4.2) and Clinical Pharmacology (12.3)].
Coadministration with CYP3A substrates results in increased exposure of the other drug. Avoid concomitant use with drugs eliminated primarily by CYP3A-mediated metabolism. Subsequent treatment with CYP3A substrates may be initiated no sooner than 1 week after the infusion of VAPRISOL is completed [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data with VAPRISOL in pregnant women to inform a drug-associated risk for major birth defects and miscarriage.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major malformations and miscarriage in clinically recognized pregnancies is 2-4%, and 15-20%, respectively.
Data
Animal Data
When pregnant rats were given intravenous conivaptan hydrochloride up to 2.5 mg/kg/day on gestation days 7 through 17 (systemic exposures less than human therapeutic exposure based on AUC comparisons), no significant fetal or maternal effects were noted. However, when the same doses were administered to pregnant rats from gestation day 7 through lactation day 20 (weaning), the pups showed decreased neonatal viability and weaning indices, decreased body weight, and delayed reflex and physical development (including sexual maturation). These effects occurred only at the highest dose administered (2.5 mg/kg/day). No maternal adverse effects of conivaptan were seen in this study. When pregnant rabbits were administered intravenous doses of conivaptan hydrochloride up to 12 mg/kg/day on gestation days 6 through 18 (at about twice the human therapeutic exposure), there were no fetal or maternal findings.
Rat fetal tissue levels were < 10% of maternal plasma concentrations while placental levels were 2.2-fold higher than maternal plasma concentrations. Conivaptan that is taken up by fetal tissue is slowly cleared, suggesting that fetal accumulation is possible.
Conivaptan hydrochloride delayed delivery in rats dosed at 10 mg/kg/day by oral gavage (systemic exposure equivalent to the human therapeutic exposure based on AUC comparison).
8.2 Lactation
Risk Summary
There is no information regarding conivaptan or its metabolites in human milk, the effects of conivaptan on the breastfed infant, or the effects of conivaptan on milk production. Conivaptan is present in rat milk; however, due to species-specific differences in lactation physiology, the clinical relevance of these data are not clear [see Data]. Because of the potential for serious adverse reactions, including electrolyte abnormalities (e.g., hypernatremia), hypotension, and volume depletion in breastfed infants, advise a woman not to breastfeed during treatment with VAPRISOL.
8.3 Females and Males of Reproductive Potential
Infertility
Females
Based on findings of decreased fertility in female rats, conivaptan may impair fertility in females of reproductive potential. It is not known whether these effects on fertility are reversible [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
In clinical studies of VAPRISOL administered as a 20 mg IV loading dose followed by 20 mg/day or 40 mg/day IV for 2 to 4 days, 89% (20 mg/day regimen) and 60% (40 mg/day regimen) of participants were greater than or equal to 65 years of age and 60% (20 mg/day regimen) and 40% (40 mg/day regimen) were greater than or equal to 75 years of age. In general, the adverse event profile in elderly patients was similar to that seen in the general study population.
8.6 Use in Patients with Hepatic Impairment
No clinically relevant increase in exposure was observed in subjects with mild hepatic impairment; therefore no dose adjustment of VAPRISOL is necessary. The systemic exposure to unbound conivaptan doubled in subjects with moderate and severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Use in Patients with Renal Impairment
No clinically relevant increase in exposure was observed in subjects with mild and moderate renal impairment (CLcr 30 – 80 mL/min). No dose adjustment of VAPRISOL is necessary.
Because of the high incidence of infusion site phlebitis (which can reduce vascular access sites) and unlikely benefit, use in patients with severe renal impairment (CLcr<30 mL/min) is not recommended [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Although no data on overdosage in humans are available, VAPRISOL has been administered as a 20 mg loading dose on Day 1 followed by continuous infusion of 80 mg/day for 4 days in hyponatremia patients and up to 120 mg/day for 2 days in CHF patients. No new toxicities were identified at these higher doses, but adverse events related to the pharmacologic activity of VAPRISOL, e.g. hypotension and thirst, occurred more frequently at these higher doses.
In case of overdose, based on expected exaggerated pharmacological activity, symptomatic treatment with frequent monitoring of vital signs and close observation of the patient is recommended.
11 DESCRIPTION
Conivaptan hydrochloride is chemically [1,1'-biphenyl]-2-carboxamide, N-[4-[(4,5-dihydro-2-methylimidazo[4,5-d][1]benzazepin-6(1H)-yl)carbonyl]phenyl]-, monohydrochloride, having a molecular weight of 535.04 and molecular formula C32H26N4O2∙HCl. The structural formula of conivaptan hydrochloride is:
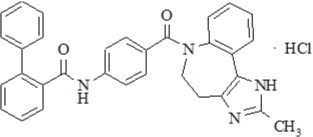
Conivaptan hydrochloride is a white to off-white or pale orange-white powder that is very slightly soluble in water (0.15 mg/mL at 23° C). Conivaptan hydrochloride injection is supplied as a sterile premixed solution with dextrose in a flexible plastic container.
Each container contains a clear, colorless, sterile, non-pyrogenic solution of conivaptan hydrochloride in dextrose injection for intravenous use. Each 100 mL, single-use premixed INTRAVIA Container contains 20 mg of conivaptan hydrochloride and 5 g of Dextrose Hydrous, USP. Lactic Acid, USP is added for pH adjustment to pH 3.4 to 3.8. The flexible plastic container is fabricated from a specially designed multilayer plastic (PL 2408). Solutions in contact with the plastic container leach out certain of the chemical components from the plastic in very small amounts; however, biological testing was supportive of the safety of the plastic container materials. The flexible container has a foil overwrap. Water can permeate the plastic into the overwrap, but the amount is insufficient to affect the premixed solution significantly.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Conivaptan hydrochloride is a dual arginine vasopressin (AVP) antagonist with nanomolar affinity for human V1A and V2 receptors in vitro. The level of AVP in circulating blood is critical for the regulation of water and electrolyte balance and is usually elevated in both euvolemic and hypervolemic hyponatremia. The AVP effect is mediated through V2 receptors, which are functionally coupled to aquaporin channels in the apical membrane of the collecting ducts of the kidney. These receptors help to maintain plasma osmolality within the normal range. The predominant pharmacodynamic effect of conivaptan hydrochloride in the treatment of hyponatremia is through its V2 antagonism of AVP in the renal collecting ducts, an effect that results in aquaresis, or excretion of free water.
12.2 Pharmacodynamics
The pharmacodynamic effects of conivaptan hydrochloride include increased free water excretion (i.e., effective water clearance [EWC]) generally accompanied by increased net fluid loss, increased urine output, and decreased urine osmolality. Studies in animal models of hyponatremia showed that conivaptan hydrochloride prevented the occurrence of hyponatremia-related physical signs in rats with the syndrome of inappropriate antidiuretic hormone secretion.
Electrophysiology
The effect of VAPRISOL 40 mg IV and 80 mg IV on the QT interval was evaluated after the first dose (Day 1) and at the last day during treatment (Day 4) in a randomized, single-blind, parallel group, placebo- and positive-controlled (moxifloxacin 400 mg IV) study in healthy male and female volunteers aged 18 to 45 years. Digital ECGs were obtained at baseline and on Days 1 and 4. Moxifloxacin elicited placebo-corrected changes from baseline in individualized QT correction (QTcI) of +7 to +10 msec on Days 1 and 4, respectively, indicating that the study had assay sensitivity. The placebo-corrected changes from baseline in QTcI in the VAPRISOL 40 mg and 80 mg dose groups on Day 1 were -3.5 msec and -2.9 msec, respectively, and -2.1 msec for both dose groups on Day 4. The results suggest that conivaptan has no clinically significant effect on cardiac repolarization.
12.3 Pharmacokinetics
The pharmacokinetics of conivaptan have been characterized in healthy subjects, specific populations and patients following both oral and intravenous dosing regimens. The pharmacokinetics of conivaptan following intravenous infusion (40 mg/day to 80 mg/day) and oral administration are non-linear, and inhibition by conivaptan of its own metabolism seems to be the major factor for the non-linearity. The intersubject variability of conivaptan pharmacokinetics is high (94% CV in CL).
The pharmacokinetics of conivaptan and its metabolites were characterized in healthy male subjects administered conivaptan hydrochloride as a 20 mg loading dose (infused over 30 minutes) followed by a continuous infusion of 40 mg/day for 3 days. Mean Cmax for conivaptan was 619 ng/mL and occurred at the end of the loading dose. Plasma concentrations reached a minimum at approximately 12 hours after start of the loading dose, then gradually increased over the duration of the infusion to a mean concentration of 188 ng/mL at the end of the infusion. The mean terminal elimination half-life after conivaptan infusion was 5.0 hours, and the mean clearance was 253.3 mL/min.
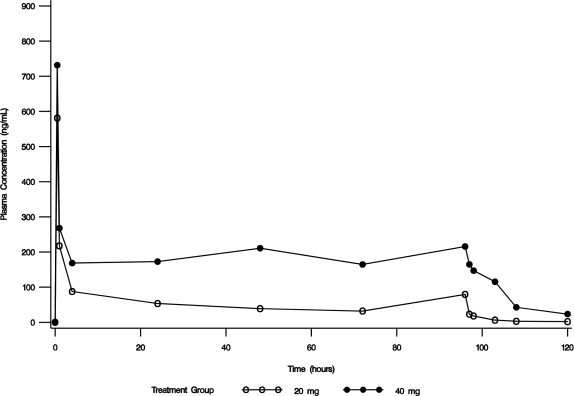
In an open-label safety and efficacy study, the pharmacokinetics of conivaptan were characterized in hypervolemic or euvolemic hyponatremia patients (ages 20 - 92 years) receiving conivaptan hydrochloride as a 20 mg loading dose (infused over 30 minutes) followed by a continuous infusion of 20 or 40 mg/day for 4 days. The median-plasma conivaptan concentrations are shown in Figure 1. The median (range) elimination half-life was 5.3 (3.3 - 9.3) or 8.1 (4.1 - 22.5) hours in the 20 mg/day or 40 mg/day group, respectively, based on data from rich PK sampling.
Figure 1. Median Plasma Concentration-Time Profiles from Rich PK Sampling Post 20 mg Loading Dose and 20 mg/day (open circle) or 40 mg/day (closed circle) Infusion for 4 Days
Distribution
Conivaptan is extensively bound to human plasma proteins, being 99% bound over the concentration range of approximately 10 to 1000 ng/mL.
Metabolism and Excretion
CYP3A was identified as the sole cytochrome P450 isozyme responsible for the metabolism of conivaptan. Four metabolites have been identified. The pharmacological activity of the metabolites at V1A and V2 receptors ranged from approximately 3-50% and 50-100% that of conivaptan, respectively. The combined exposure of the metabolites following intravenous administration of conivaptan is approximately 7% that of conivaptan and hence, their contribution to the clinical effect of conivaptan is minimal.
After intravenous (10 mg) or oral (20 mg) administration of conivaptan hydrochloride in a mass balance study, approximately 83% of the dose was excreted in feces as total radioactivity and 12% in urine over several days of collection. Over the first 24 hours after dosing, approximately 1% of the intravenous dose was excreted in urine as intact conivaptan.
Specific Populations
Hepatic Impairment
In subjects with moderate and severe hepatic impairment, the area under the plasma concentration-time curve for unbound conivaptan was 2.3- to 2.5-fold the values observed in normal volunteers. The plasma protein binding of conivaptan decreased approximately 27% and 50%, respectively in patients with moderate and severe hepatic impairment. No clinically relevant increase in systemic exposure was observed in subjects with mild hepatic impairment [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)].
Renal Impairment
Mild and moderate renal impairment (CLcr 30 – 80 mL/min) do not affect exposure to VAPRISOL to a clinically relevant extent. Use in patients with severe renal impairment (CLcr < 30 mL/min) is not recommended [see Use in Specific Populations (8.7)].
Drug Interactions
CYP3A
Conivaptan is a sensitive substrate of CYP3A. The effect of ketoconazole, a potent CYP3A inhibitor, on the pharmacokinetics of intravenous conivaptan has not been evaluated. Coadministration of oral conivaptan hydrochloride 10 mg with ketoconazole 200 mg resulted in Cmax and AUC of conivaptan 4- and 11-fold, respectively, levels with conivaptan alone [see Contraindications (4.2) Drug Interactions (7.1)].
Conivaptan is a potent mechanism-based inhibitor of CYP3A. The effect of conivaptan on the pharmacokinetics of co-administered CYP3A substrates has been evaluated with the coadministration of conivaptan with midazolam, simvastatin, and amlodipine. VAPRISOL 40 mg/day increased the mean AUC values by approximately 100%-for 1 mg intravenous or by 200% for 2 mg oral doses of midazolam. VAPRISOL 30 mg/day tripled the AUC of simvastatin. Oral conivaptan hydrochloride 40 mg twice daily doubled the AUC and half-life of amlodipine.
Digoxin
Coadministration of a 0.5 mg dose of digoxin, a P-glycoprotein substrate, with oral conivaptan hydrochloride 40 mg twice daily resulted in a 30% reduction in clearance and 79% and 43% increases in digoxin Cmax and AUC values, respectively [see Drug Interactions (7.2)].
Warfarin
VAPRISOL (40 mg/day for 4 days) administered with a single 25 mg dose of warfarin, which undergoes major metabolism by CYP2C9 and minor metabolism by CYP3A, increased the mean S-warfarin AUC and S-warfarin Cmax by 14% and 17%, respectively. The corresponding prothrombin time and international normalized ratio values were unchanged.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Standard lifetime (104 week) carcinogenicity bioassays were conducted in mice and rats. Male and female mice were given oral doses of conivaptan hydrochloride up to 30 mg/kg/day and 10 mg/kg/day, respectively, by gavage. Male and female rats were given oral doses of up to 10 mg/kg/day and 30 mg/kg/day, respectively, by gavage. There was no increased incidence of tumors associated with exposure to conivaptan in either species. The 30 mg/kg/day dosage regimen in male mice and female rats was shown to result in a systemic exposure (AUC) about twice the human systemic exposure from an IV bolus of 20 mg on day 1 followed by IV infusion of 40 mg/day for 3 days. The 10 mg/kg/day dosage regimen in female mice and male rats was shown to result in about one-fourth and one-half the human therapeutic exposure, respectively.
Conivaptan was not genotoxic in the bacterial reverse mutation assay, the in vitro human peripheral blood lymphocyte chromosomal aberration assay, or in vivo rat micronucleus assay.
Fertility of male rats treated with conivaptan hydrochloride by IV bolus doses of up to 2.5 mg/kg/day for the 4 weeks preceding mating and throughout the mating period was unaffected. However, when female rats were given IV bolus conivaptan from 15 days before mating through gestation day 7, there was prolonged diestrus, decreased fertility (decreased numbers of corpora lutea and implantations) and increased post-implantation loss at 2.5 mg/kg/day (systemic exposure less than human exposure at the therapeutic dose).
14 CLINICAL STUDIES
14.1 Hyponatremia
The effect on serum sodium of VAPRISOL was demonstrated in a double-blind, placebo-controlled, randomized, multicenter study conducted in 84 patients with euvolemic (N=56) or hypervolemic (N=28) hyponatremia (serum sodium 115 -130 mEq/L) from a variety of underlying causes (malignant or nonmalignant diseases of the central nervous system, lung, or abdomen; congestive heart failure; hypertension; myocardial infarction; diabetes; osteoarthritis; or idiopathic). Study participants were randomized to receive either placebo IV (N=29), VAPRISOL 40 mg/day IV (N=29), or VAPRISOL 80 mg/day IV (N=26). Daily fluid intake was restricted to 2 liters. VAPRISOL or placebo was administered as a continuous infusion following a 30 minute IV loading dose on the first treatment day and patients were treated for 4 days. Serum or plasma sodium concentrations were assessed pre-dose (Hour 0) and at 4, 6, 10, and 24 hours post-dose on all treatment days.
Mean serum sodium concentration was 123.3 mEq/L at study entry. The mean change in serum sodium concentration from baseline over the 4-day treatment period is shown in Figure 2.
Figure 2. Mean (SE) Change from Baseline in Sodium Concentrations with VAPRISOL 40 mg/day
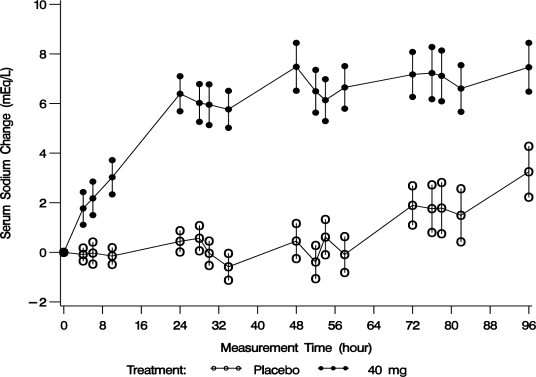
Following treatment with 40 mg/day of VAPRISOL, the mean change from baseline in serum sodium concentration at the end of 2 days of treatment with VAPRISOL was 5.3 mEq/L (mean concentration 128.6 mEq/L). At the end of the 4-day treatment period, the mean change from baseline was 6.5 mEq/L (mean concentration 129.8 mEq/L). In addition, after 2 days and 4 days of treatment with VAPRISOL, 41% (after 2 days) and 69% (after 4 days) of patients achieved a ≥ 6 mEq/L increase in serum sodium concentration or a normal serum sodium of ≥ 135 mEq/L. Although 80 mg/day was also studied, it was not significantly more effective than 40 mg/day and was associated with a higher incidence of infusion site reactions and a higher rate of discontinuations for adverse events [see Adverse Reactions (6.1)]. Additional efficacy data are summarized in Table 2.
| Efficacy Variable | Placebo (N=29) | VAPRISOL 40 mg/day ( N=29) | ||
|---|---|---|---|---|
|
*: P ≤ 0.001 vs placebo |
||||
|
‡: efficacy variables were assessed on Day 2 of a 4-day treatment period |
||||
| Day 2‡ | Day 4 | Day 2‡ | Day 4 | |
| Baseline adjusted serum Na+ AUC over duration of treatment (mEq·hr/L) Mean (SD) LS Mean ± SE |
6.2 (81.8) 3.8 ± 26.9 |
61.4 (242.3) 12.9 ± 61.2 |
205.9 (171.6) 205.6 ± 26.6* |
500.8 (365.5) 490.9 ± 56.8* |
| Number of patients (%) and median event time (h) from first dose of study medication to a confirmed ≥ 4 mEq/L increase from Baseline in serum Na+, [95% CI] | 2 (7%) Not estimable Not estimable | 9 (31%) Not estimable Not estimable | 22 (76%) 23.7* [10, 2] | 23 (79%) 23.7* [10, 2] |
| Serum Na+
(mEq/L)
Baseline mean (SD) Mean (SD) at end of treatment Change from Baseline to end of treatment Mean change (SD) LS Mean change ± SE | 124.3 (4.1) 124.5 (4.7) 0.2 (2.5) 0.1 ± 0.7 | 124.3 (4.1) 125.8 (4.9) 1.5 (4.6) 0.8 ± 0.8 | 123.3 (4.7) 128.6 (5.9) 5.3 (4.4) 5.2 ± 0.7* | 123.3 (4.7) 129.8 (4.8) 6.5 (4.4) 6.3 ± 0.7* |
| Number (%) of patients who obtained a confirmed ≥ 6 mEq/L increase from Baseline in serum Na+ or a normal serum Na+ concentration ≥ 135 mEq/L during treatment |
0 (0) |
6 (21%) |
12 (41%)* |
20 (69%)* |
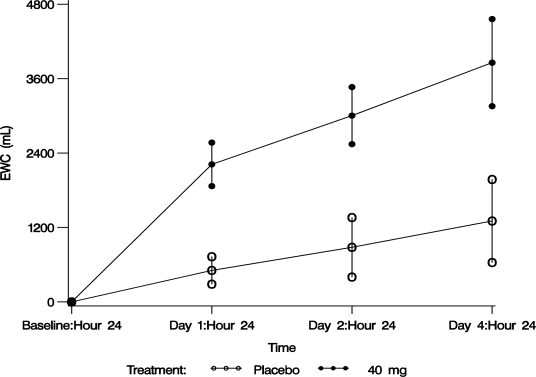
The aquaretic effect of VAPRISOL is shown in Figure 3. VAPRISOL produced a baseline-corrected cumulative increase in effective water clearance of over 3800 mL compared to approximately 1300 mL with placebo by Day 4.
Figure 3. Baseline-Corrected Mean (SE) Cumulative Effective Water Clearance (EWC)
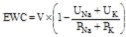 , where V is urine volume (mL/d), UNa is urine sodium concentration, UK is urine potassium concentration, PNa is plasma/serum sodium concentration, and PK is plasma/serum potassium concentration.
, where V is urine volume (mL/d), UNa is urine sodium concentration, UK is urine potassium concentration, PNa is plasma/serum sodium concentration, and PK is plasma/serum potassium concentration.
The effect on serum sodium of VAPRISOL (administered as a 20 or 40 mg/day IV continuous infusion for 4 days following a 30 minute IV infusion of a 20 mg loading dose on the first treatment day) was also evaluated in an open-label study of 251 patients with euvolemic or hypervolemic hyponatremia. The results are shown in Table 3.
| Primary Efficacy Endpoint | 20 mg/day
N=37 | 40 mg/day
N=214 |
|---|---|---|
| Baseline adjusted serum Na+ AUC over duration of treatment (mEq·hr/L) Mean (SD) | 753.8 (429.9) | 689.2 (417.3) |
| Secondary Efficacy Endpoints | ||
| Number of patients (%) and median event time (h) from first dose of study medication to a confirmed ≥ 4 mEq/L increase from Baseline in serum Na+, [95% CI] | 29 (78%) 23.8[12.0, 36.0] | 178 (83%) 24.4 [24.0, 35.8] |
| Total time (h) from first dose of study medication to end of treatment in which patients had a confirmed ≥ 4 mEq/L increase in serum Na+ from Baseline Mean (SD) | 60.6 (35.2) | 59.5 (33.2) |
| Serum Na+
(mEq/L)
Baseline mean (SD) Mean (SD) at end of treatment Mean Change (SD) from Baseline to End of Treatment Mean (SD) at Follow-up Day 11 Mean Change (SD) from Baseline to Follow-up Day 11 Mean (SD) at Follow-up Day 34 Mean Change (SD) from Baseline to Follow-up Day 34 |
122.5 (5.2) 131.8 (3.9) 9.4 (5.3) 129.9 (6.2) 7.1 (8.2) 134.3 (4.5) 11.5 (7.3) |
123.8 (4.6) 132.5 (4.6) 8.8 (5.4) 131.8 (5.8) 8.0 (6.5) 134.3 (5.2) 10.7 (6.7) |
| Number (%) of patients who obtained a confirmed ≥ 6 mEq/L increase from Baseline in serum Na+ or a normal serum Na+ concentration ≥135 mEq/L during treatment | 26 (70%) | 154 (72%) |
14.2 Heart Failure
The effectiveness of VAPRISOL for the treatment of congestive heart failure has not been established. In 10 Phase 2/pilot heart failure studies, VAPRISOL did not show statistically significant improvement for heart failure outcomes, including such measures as length of hospital stay, changes in categorized physical findings of heart failure, change in ejection fraction, change in exercise tolerance, change in functional status, or change in heart failure symptoms, compared to placebo. In these studies, the changes in the physical findings and heart failure symptoms were no worse in the VAPRISOL-treated group (N=818) compared to the placebo group (N=290) [see Indications and Usage (1)].
16 HOW SUPPLIED/STORAGE AND HANDLING
VAPRISOL (conivaptan hydrochloride) Injection is supplied as a single-use, premixed solution, containing 20 mg of conivaptan hydrochloride in 5% Dextrose in 100 mL INTRAVIA Plastic Containers.
- 1 container/carton (NDC 66220-160-10)
VAPRISOL in INTRAVIA Plastic Containers should be stored at 25°C (77°F); however, brief exposure up to 40°C (104°F) does not adversely affect the product. Avoid excessive heat. Protect from freezing. Protect from light until ready to use.
17 PATIENT COUNSELING INFORMATION
Inform patients about the common adverse effects of VAPRISOL including infusion site effects (edema, erythema, pain, and phlebitis), pyrexia, hypokalemia, headache, orthostatic hypotension and potential for overly rapid increase in serum sodium which can cause serious neurologic sequelae. Instruct patients to inform their healthcare provider if they develop any unusual symptoms, or if any known symptom persists or worsens, with special attention to potential manifestations of osmotic demyelination syndrome.
Ask patients about what other medications they are currently taking with VAPRISOL, including over-the-counter medications.
Lactation
Advise women not to breastfeed during treatment with VAPRISOL [see Use in Specific Populations (8.2)].
Marketed by:
Cumberland Pharmaceuticals Inc.
Nashville TN 37203
VAPRISOL is a registered trademark of Cumberland Pharmaceuticals Inc.
INTRAVIA is a registered trademark of Baxter International Inc.
US Patent Number 5,723,606
07-19-73-925

| VAPRISOL DEXTROSE IN PLASTIC CONTAINER
conivaptan hydrochloride injection, solution |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Cumberland Pharmaceuticals Inc. (069532880) |