PRISTIQ EXTENDED-RELEASE- desvenlafaxine succinate tablet, extended release
PD-Rx Pharmaceuticals, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use PRISTIQ safely and effectively. See full prescribing information for PRISTIQ.
PRISTIQ ® (desvenlafaxine) Extended-Release Tablets, for oral use Initial U.S. Approval: 2008 WARNING: SUICIDAL THOUGHTS AND BEHAVIORSSee full prescribing information for complete boxed warning.RECENT MAJOR CHANGESINDICATIONS AND USAGEPRISTIQ is a serotonin and norepinephrine reuptake inhibitor (SNRI) indicated for the treatment of adults with major depressive disorder (MDD) ( 1). DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥5% and twice the rate of placebo in the 50 or 100 mg dose groups) were: nausea, dizziness, insomnia, hyperhidrosis, constipation, somnolence, decreased appetite, anxiety, and specific male sexual function disorders ( 6.1). To report SUSPECTED ADVERSE REACTIONS, contact Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc., at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 11/2021 |
FULL PRESCRIBING INFORMATION
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term studies. These studies did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in patients over age 24; there was a reduction in risk with antidepressant use in patients aged 65 and older [see Warnings and Precautions (5.1)] .
In patients of all ages who are started on antidepressant therapy, monitor closely for worsening, and for emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber [see Warnings and Precautions (5.1)] .
PRISTIQ is not approved for use in pediatric patients [see Use in Specific Populations (8.4)] .
1 INDICATIONS AND USAGE
PRISTIQ is indicated for the treatment of adults with major depressive disorder (MDD) [see Clinical Studies (14)] .
2 DOSAGE AND ADMINISTRATION
2.1 General Instructions for Use
The recommended dose for PRISTIQ is 50 mg once daily, with or without food. The 50 mg dose is both a starting dose and the therapeutic dose. PRISTIQ should be taken at approximately the same time each day. Tablets must be swallowed whole with fluid and not divided, crushed, chewed, or dissolved.
In clinical studies, doses of 10 mg to 400 mg per day were studied. In clinical studies, doses of 50 mg to 400 mg per day were shown to be effective, although no additional benefit was demonstrated at doses greater than 50 mg per day and adverse reactions and discontinuations were more frequent at higher doses.
The 25 mg per day dose is intended for a gradual reduction in dose when discontinuing treatment. When discontinuing therapy, gradual dose reduction is recommended whenever possible to minimize discontinuation symptoms [see Dosage and Administration (2.5) and Warnings and Precautions (5.7)] .
2.2 Dosage Recommendations for Patients with Renal Impairment
The maximum recommended dose in patients with moderate renal impairment (24-hr creatinine clearance [Cl Cr] = 30 to 50 mL/min, Cockcroft-Gault [C-G]) is 50 mg per day. The maximum recommended dose in patients with severe renal impairment (Cl Cr 15 to 29 mL/min, C-G) or end-stage renal disease (ESRD, Cl Cr < 15 mL/min, C-G) is 25 mg every day or 50 mg every other day. Supplemental doses should not be given to patients after dialysis [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)] .
2.3 Dosage Recommendations for Patients with Hepatic Impairment
The recommended dose in patients with moderate to severe hepatic impairment (Child-Pugh score 7 to 15) is 50 mg per day. Dose escalation above 100 mg per day is not recommended [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)] .
2.4 Maintenance/Continuation/Extended Treatment
It is generally agreed that acute episodes of major depressive disorder require several months or longer of sustained pharmacologic therapy. Longer-term efficacy of PRISTIQ (50–400 mg) was established in two maintenance trials [see Clinical Studies (14)] . Patients should be periodically reassessed to determine the need for continued treatment.
2.5 Discontinuing PRISTIQ
Adverse reactions may occur upon discontinuation of PRISTIQ [see Warnings and Precautions (5.7)]. Gradually reduce the dosage rather than stopping PRISTIQ abruptly when discontinuing therapy with PRISTIQ. In some patients, discontinuation may need to occur over a period of several months.
2.6 Switching Patients From Other Antidepressants to PRISTIQ
Discontinuation symptoms have been reported when switching patients from other antidepressants, including venlafaxine, to PRISTIQ. Tapering of the initial antidepressant may be necessary to minimize discontinuation symptoms.
2.7 Switching Patients to or from a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders
At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with PRISTIQ. Conversely, at least 7 days should be allowed after stopping PRISTIQ before starting an MAOI intended to treat psychiatric disorders [see Contraindications (4)].
2.8 Use of PRISTIQ with other MAOIs such as Linezolid or Methylene Blue
Do not start PRISTIQ in a patient who is being treated with linezolid or intravenous methylene blue because there is increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered [see Contraindications (4)] .
In some cases, a patient already receiving PRISTIQ therapy may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue treatment are judged to outweigh the risks of serotonin syndrome in a particular patient, PRISTIQ should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for 7 days or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with PRISTIQ may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue [see Warnings and Precautions (5.2)] .
The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with PRISTIQ is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use [see Warnings and Precautions (5.2)] .
3 DOSAGE FORMS AND STRENGTHS
- 25 mg Tablet: tan, square pyramid tablet debossed with "W" over "25" on the flat side
- 50 mg Tablet: light pink, square pyramid tablet debossed with "W" over "50" on the flat side
- 100 mg Tablet: reddish-orange, square pyramid tablet debossed with "W" over "100" on the flat side
4 CONTRAINDICATIONS
- Hypersensitivity to desvenlafaxine succinate, venlafaxine hydrochloride or to any excipients in the PRISTIQ formulation. Angioedema has been reported in patients treated with PRISTIQ [see Adverse Reactions (6.1)] .
- The use of MAOIs intended to treat psychiatric disorders with PRISTIQ or within 7 days of stopping treatment with PRISTIQ is contraindicated because of an increased risk of serotonin syndrome. The use of PRISTIQ within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated [see Dosage and Administration (2.7) and Warnings and Precautions (5.2)].
- Starting PRISTIQ in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration (2.8) and Warnings and Precautions (5.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Pediatric and Young Adult Patients
Patients with MDD, both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment. Pooled analyses of short-term placebo-controlled studies of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18 to 24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled studies in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term studies of 9 antidepressant drugs in over 4,400 patients. The pooled analyses of placebo-controlled studies in adults with MDD or other psychiatric disorders included a total of 295 short-term studies (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug vs. placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1,000 patients treated) are provided in Table 1.
| Age Range | Drug-Placebo Difference in Number of Cases of Suicidality per 1,000 Patients Treated |
|---|---|
| Increases Compared to Placebo | |
| <18 | 14 additional cases |
| 18 to 24 | 5 additional cases |
| Decreases Compared to Placebo | |
| 25 to 64 | 1 fewer case |
| ≥65 | 6 fewer cases |
No suicides occurred in any of the pediatric studies. There were suicides in the adult studies, but the number was not sufficient to reach any conclusion about drug effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms.
If the decision has been made to discontinue treatment, medication should be tapered, as rapidly as is feasible, but with recognition that abrupt discontinuation can be associated with certain symptoms [see Dosage and Administration (2.4), Warnings and Precautions (5.7)] .
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to healthcare providers. Such monitoring should include daily observation by families and caregivers .
Prescriptions for PRISTIQ should be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose.
Screening Patients for Bipolar Disorder
A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled studies) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that PRISTIQ is not approved for use in treating bipolar depression.
5.2 Serotonin Syndrome
Serotonin-norepinephrine reuptake inhibitors (SNRIs) and selective-serotonin reuptake inhibitors (SSRIs), including PRISTIQ, can precipitate serotonin syndrome, a potentially life-threatening condition. The risk is increased with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, amphetamines, and St. John's Wort) and with drugs that impair metabolism of serotonin, i.e., MAOIs [see Contraindications (4), Drug Interactions (7.1)] . Serotonin syndrome can also occur when these drugs are used alone.
Serotonin syndrome signs and symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
The concomitant use of PRISTIQ with MAOIs is contraindicated. In addition, do not initiate PRISTIQ in a patient being treated with MAOIs such as linezolid or intravenous methylene blue. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection). If it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking PRISTIQ, discontinue PRISTIQ before initiating treatment with the MAOI [see Contraindications (4), Drug Interactions (7.1)] .
Monitor all patients taking PRISTIQ for the emergence of serotonin syndrome. Discontinue treatment with PRISTIQ and any concomitant serotonergic agents immediately if the above symptoms occur, and initiate supportive symptomatic treatment. If concomitant use of PRISTIQ with other serotonergic drugs is clinically warranted, inform patients of the increased risk for serotonin syndrome and monitor for symptoms.
5.3 Elevated Blood Pressure
Patients receiving PRISTIQ should have regular monitoring of blood pressure since increases in blood pressure were observed in clinical studies [see Adverse Reactions (6.1)] . Pre-existing hypertension should be controlled before initiating treatment with PRISTIQ. Caution should be exercised in treating patients with pre-existing hypertension, cardiovascular, or cerebrovascular conditions that might be compromised by increases in blood pressure. Cases of elevated blood pressure requiring immediate treatment have been reported with PRISTIQ.
Sustained blood pressure increases could have adverse consequences. For patients who experience a sustained increase in blood pressure while receiving PRISTIQ, either dose reduction or discontinuation should be considered [see Adverse Reactions (6.1)] .
5.4 Increased Risk of Bleeding
Drugs that interfere with serotonin reuptake inhibition, including PRISTIQ, may increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs, warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Bleeding events related to SSRIs and SNRIs have ranged from ecchymosis, hematoma, epistaxis, and petechiae to life-threatening hemorrhages. Inform patients about the risk of bleeding associated with the concomitant use of PRISTIQ and antiplatelet agents or anticoagulants. For patients taking warfarin, carefully monitor coagulation indices when initiating, titrating, or discontinuing PRISTIQ.
5.5 Angle Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including PRISTIQ may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy. Avoid use of antidepressants, including PRISTIQ, in patients with untreated anatomically narrow angles.
5.6 Activation of Mania/Hypomania
During all MDD phase 2 and phase 3 studies, mania was reported for approximately 0.02% of patients treated with PRISTIQ. Activation of mania/hypomania has also been reported in a small proportion of patients with major affective disorder who were treated with other marketed antidepressants. As with all antidepressants, PRISTIQ should be used cautiously in patients with a history or family history of mania or hypomania.
5.7 Discontinuation Syndrome
Adverse reactions after discontinuation of serotonergic antidepressants, particularly after abrupt discontinuation, include: nausea, sweating, dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesia, such as electric shock sensations), tremor, anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures [see Adverse Reactions (6.1)].
There have been postmarketing reports of serious discontinuation symptoms with PRISTIQ, which can be protracted and severe. Completed suicide, suicidal thoughts, and severe aggression (including hostility, rage, and homicidal ideation) have been observed in patients during reduction in PRISTIQ dosage, including during discontinuation. Other postmarketing reports describe visual changes (such as blurred vision or trouble focusing) and increased blood pressure after stopping or reducing the dose of PRISTIQ .
Patients should be monitored when discontinuing treatment with PRISTIQ. A gradual reduction in the dose, rather than abrupt cessation, is recommended. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, then resuming the previously prescribed dose may be considered . Subsequently, the healthcare provider may continue decreasing the dose, but at a more gradual rate. In some patients, discontinuation may need to occur over a period of several months [see Dosage and Administration (2.5)] .
5.8 Seizure
Cases of seizure have been reported in pre-marketing clinical studies with PRISTIQ. PRISTIQ has not been systematically evaluated in patients with a seizure disorder. Patients with a history of seizures were excluded from pre-marketing clinical studies. PRISTIQ should be prescribed with caution in patients with a seizure disorder.
5.9 Hyponatremia
Hyponatremia may occur as a result of treatment with SSRIs and SNRIs, including PRISTIQ. In many cases, this hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported. Elderly patients may be at greater risk of developing hyponatremia with SSRIs and SNRIs. Also, patients taking diuretics or who are otherwise volume depleted can be at greater risk [see Use in Specific Populations (8.5) and Clinical Pharmacology (12.3)] . Discontinuation of PRISTIQ should be considered in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted.
Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which can lead to falls. Signs and symptoms associated with more severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death.
5.10 Interstitial Lung Disease and Eosinophilic Pneumonia
Interstitial lung disease and eosinophilic pneumonia associated with venlafaxine (the parent drug of PRISTIQ) therapy have been rarely reported. The possibility of these adverse events should be considered in patients treated with PRISTIQ who present with progressive dyspnea, cough, or chest discomfort. Such patients should undergo a prompt medical evaluation, and discontinuation of PRISTIQ should be considered.
5.11 Sexual Dysfunction
Use of SNRIs, including PRISTIQ, may cause symptoms of sexual dysfunction [see Adverse Reactions (6.1)] . In male patients, SNRI use may result in ejaculatory delay or failure, decreased libido, and erectile dysfunction. In female patients, SNRI use may result in decreased libido and delayed or absent orgasm.
It is important for prescribers to inquire about sexual function prior to initiation of PRISTIQ and to inquire specifically about changes in sexual function during treatment, because sexual function may not be spontaneously reported. When evaluating changes in sexual function, obtaining a detailed history (including timing of symptom onset) is important because sexual symptoms may have other causes, including the underlying psychiatric disorder. Discuss potential management strategies to support patients in making informed decisions about treatment.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label.
- Hypersensitivity [see Contraindications (4)]
- Suicidal Thoughts and Behaviors in Pediatric and Young Adult Patients [see Warnings and Precautions (5.1)]
- Serotonin Syndrome [see Warnings and Precautions (5.2)]
- Elevated Blood Pressure [see Warnings and Precautions (5.3)]
- Increased Risk of Bleeding [see Warnings and Precautions (5.4)]
- Angle Closure Glaucoma [see Warnings and Precautions (5.5)]
- Activation of Mania/Hypomania [see Warnings and Precautions (5.6)]
- Discontinuation Syndrome [see Warnings and Precautions (5.7)]
- Seizure [see Warnings and Precautions (5.8)]
- Hyponatremia [see Warnings and Precautions (5.9)]
- Interstitial Lung Disease and Eosinophilic Pneumonia [see Warnings and Precautions (5.10)]
- Sexual Dysfunction [see Warnings and Precautions (5.11)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
Patient Exposure
PRISTIQ was evaluated for safety in 8,394 patients diagnosed with major depressive disorder who participated in multiple-dose pre-marketing studies, representing 2,784 patient-years of exposure. Of the total 8,394 patients exposed to at least one dose of PRISTIQ; 2,116 were exposed to PRISTIQ for 6 months, representing 1,658 patient-years of exposure, and 421 were exposed for one year, representing 416 patient-years of exposure.
Adverse Reactions Reported as Reasons for Discontinuation of Treatment
In the pre-marketing pooled 8-week placebo-controlled studies in patients with MDD, 1,834 patients were exposed to PRISTIQ (50 to 400 mg). Of the 1,834 patients, 12% discontinued treatment due to an adverse reaction, compared with 3% of the 1,116 placebo-treated patients. At the recommended dose of 50 mg, the discontinuation rate due to an adverse reaction for PRISTIQ (4.1%) was similar to the rate for placebo (3.8%). For the 100 mg dose of PRISTIQ the discontinuation rate due to an adverse reaction was 8.7%.
The most common adverse reactions leading to discontinuation in at least 2% and at a rate greater than placebo of the PRISTIQ treated patients in the short-term studies, up to 8 weeks, were: nausea (4%); dizziness, headache and vomiting (2% each). In a longer-term study, up to 9 months, the most common was vomiting (2%).
Common Adverse Reactions in Placebo-Controlled MDD Studies
The most commonly observed adverse reactions in PRISTIQ treated MDD patients in pre-marketing pooled 8-week, placebo-controlled, fixed-dose studies (incidence ≥ 5% and at least twice the rate of placebo in the 50 or 100 mg dose groups) were: nausea, dizziness, insomnia, hyperhidrosis, constipation, somnolence, decreased appetite, anxiety, and specific male sexual function disorders.
Table 2 shows the incidence of common adverse reactions that occurred in ≥ 2% of PRISTIQ treated MDD patients and twice the rate of placebo at any dose in the pre-marketing pooled 8-week, placebo-controlled, fixed dose clinical studies.
| Percentage of Patients Reporting Reaction | |||||
|---|---|---|---|---|---|
| PRISTIQ | |||||
| System Organ Class
Preferred Term | Placebo
(n=636) | 50 mg
(n=317) | 100 mg
(n=424) | 200 mg
(n=307) | 400 mg
(n=317) |
| Cardiac disorders | |||||
| Blood pressure increased | 1 | 1 | 1 | 2 | 2 |
| Gastrointestinal disorders | |||||
| Nausea | 10 | 22 | 26 | 36 | 41 |
| Dry mouth | 9 | 11 | 17 | 21 | 25 |
| Constipation | 4 | 9 | 9 | 10 | 14 |
| Vomiting | 3 | 3 | 4 | 6 | 9 |
| General disorders and administration site conditions | |||||
| Fatigue | 4 | 7 | 7 | 10 | 11 |
| Chills | 1 | 1 | <1 | 3 | 4 |
| Feeling jittery | 1 | 1 | 2 | 3 | 3 |
| Metabolism and nutrition disorders | |||||
| Decreased appetite | 2 | 5 | 8 | 10 | 10 |
| Nervous system disorders | |||||
| Dizziness | 5 | 13 | 10 | 15 | 16 |
| Somnolence | 4 | 4 | 9 | 12 | 12 |
| Tremor | 2 | 2 | 3 | 9 | 9 |
| Disturbance in attention | <1 | <1 | 1 | 2 | 1 |
| Psychiatric disorders | |||||
| Insomnia | 6 | 9 | 12 | 14 | 15 |
| Anxiety | 2 | 3 | 5 | 4 | 4 |
| Nervousness | 1 | <1 | 1 | 2 | 2 |
| Abnormal dreams | 1 | 2 | 3 | 2 | 4 |
| Renal and urinary disorders | |||||
| Urinary hesitation | 0 | <1 | 1 | 2 | 2 |
| Respiratory, thoracic and mediastinal disorders | |||||
| Yawning | <1 | 1 | 1 | 4 | 3 |
| Skin and subcutaneous tissue disorders | |||||
| Hyperhidrosis | 4 | 10 | 11 | 18 | 21 |
| Special Senses | |||||
| Vision blurred | 1 | 3 | 4 | 4 | 4 |
| Mydriasis | <1 | 2 | 2 | 6 | 6 |
| Vertigo | 1 | 2 | 1 | 5 | 3 |
| Tinnitus | 1 | 2 | 1 | 1 | 2 |
| Dysgeusia | 1 | 1 | 1 | 1 | 2 |
| Vascular disorders | |||||
| Hot flush | <1 | 1 | 1 | 2 | 2 |
Sexual Function Adverse Reactions
Table 3 shows the incidence of sexual function adverse reactions that occurred in ≥ 2% of PRISTIQ treated MDD patients in any fixed-dose group (pre-marketing pooled 8-week, placebo-controlled, fixed -dose, clinical studies) [s ee Warnings and Precautions (5.11)] .
| PRISTIQ | |||||
| Placebo
(n=239) | 50 mg
(n=108) | 100 mg
(n=157) | 200 mg
(n=131) | 400 mg
(n=154) |
|
| Men only | |||||
| Anorgasmia | 0 | 0 | 3 | 5 | 8 |
| Libido decreased | 1 | 4 | 5 | 6 | 3 |
| Orgasm abnormal | 0 | 0 | 1 | 2 | 3 |
| Ejaculation delayed | <1 | 1 | 5 | 7 | 6 |
| Erectile dysfunction | 1 | 3 | 6 | 8 | 11 |
| Ejaculation disorder | 0 | 0 | 1 | 2 | 5 |
| Ejaculation failure | 0 | 1 | 0 | 2 | 2 |
| Sexual dysfunction | 0 | 1 | 0 | 0 | 2 |
| PRISTIQ | |||||
| Placebo
(n=397) | 50 mg
(n=209) | 100 mg
(n=267) | 200 mg
(n=176) | 400 mg
(n=163) |
|
| Women only | |||||
| Anorgasmia | 0 | 1 | 1 | 0 | 3 |
Other Adverse Reactions Observed in Premarketing and Postmarketing Clinical Studies
Other infrequent adverse reactions, not described elsewhere in the label, occurring at an incidence of < 2% in MDD patients treated with PRISTIQ were:
Cardiac disorders – Tachycardia.
General disorders and administration site conditions – Asthenia.
Investigations – Weight increased, liver function test abnormal, blood prolactin increased.
Musculoskeletal and connective tissue disorders – Musculoskeletal stiffness.
Nervous system disorders –Syncope, convulsion, dystonia.
Psychiatric disorders – Depersonalization, bruxism.
Renal and urinary disorders – Urinary retention.
Skin and subcutaneous tissue disorders – Rash, alopecia, photosensitivity reaction, angioedema.
In clinical studies, there were uncommon reports of ischemic cardiac adverse reactions, including myocardial ischemia, myocardial infarction, and coronary occlusion requiring revascularization; these patients had multiple underlying cardiac risk factors. More patients experienced these events during PRISTIQ treatment as compared to placebo.
Laboratory, ECG and Vital Sign Changes Observed in MDD Clinical Studies
The following changes were observed in pre-marketing placebo-controlled, short-term MDD studies with PRISTIQ.
Lipids
Elevations in fasting serum total cholesterol, LDL (low density lipoproteins) cholesterol, and triglycerides occurred in the controlled studies. Some of these abnormalities were considered potentially clinically significant.
The percentage of patients who exceeded a predetermined threshold value is shown in Table 4.
| PRISTIQ | |||||
|---|---|---|---|---|---|
| Placebo | 50 mg | 100 mg | 200 mg | 400 mg | |
| Total Cholesterol
*(Increase of ≥ 50 mg/dl and an absolute value of ≥ 261 mg/dl) | 2 | 3 | 4 | 4 | 10 |
| LDL Cholesterol
*(Increase ≥ 50 mg/dl and an absolute value of ≥ 190 mg/dl) | 0 | 1 | 0 | 1 | 2 |
| Triglycerides, fasting
*(Fasting: ≥ 327 mg/dl) | 3 | 2 | 1 | 4 | 6 |
Proteinuria
Proteinuria, greater than or equal to trace, was observed in the pre-marketing fixed-dose controlled studies (see Table 5). This proteinuria was not associated with increases in BUN or creatinine and was generally transient.
| PRISTIQ | |||||
|---|---|---|---|---|---|
| Placebo | 50 mg | 100 mg | 200 mg | 400 mg | |
| Proteinuria | 4 | 6 | 8 | 5 | 7 |
Vital Sign Changes
Table 6 summarizes the changes that were observed in placebo-controlled, short-term, pre-marketing studies with PRISTIQ in patients with MDD (doses 50 to 400 mg).
| PRISTIQ | |||||
|---|---|---|---|---|---|
| Placebo | 50 mg | 100 mg | 200 mg | 400 mg | |
| Blood pressure | |||||
| Supine systolic bp (mm Hg) | -1.4 | 1.2 | 2.0 | 2.5 | 2.1 |
| Supine diastolic bp (mm Hg) | -0.6 | 0.7 | 0.8 | 1.8 | 2.3 |
| Pulse rate | |||||
| Supine pulse (bpm) | -0.3 | 1.3 | 1.3 | 0.9 | 4.1 |
| Weight (kg) | 0.0 | -0.4 | -0.6 | -0.9 | -1.1 |
Treatment with PRISTIQ at all doses from 50 mg per day to 400 mg per day in controlled studies was associated with sustained hypertension, defined as treatment-emergent supine diastolic blood pressure (SDBP) ≥90 mm Hg and ≥10 mm Hg above baseline for 3 consecutive on-therapy visits (see Table 7). Analyses of patients in PRISTIQ pre-marketing short-term controlled studies who met criteria for sustained hypertension revealed a consistent increase in the proportion of patients who developed sustained hypertension. This was seen at all doses with a suggestion of a higher rate at 400 mg per day.
| Treatment Group | Proportion of Patients with Sustained Hypertension |
|---|---|
| Placebo | 0.5% |
| PRISTIQ 50 mg per day | 1.3% |
| PRISTIQ 100 mg per day | 0.7% |
| PRISTIQ 200 mg per day | 1.1% |
| PRISTIQ 400 mg per day | 2.3% |
Orthostatic Hypotension
In the pre-marketing short-term, placebo-controlled clinical studies with doses of 50 to 400 mg, systolic orthostatic hypotension (decrease ≥30 mm Hg from supine to standing position) occurred more frequently in patients ≥65 years of age receiving PRISTIQ (8%, 7/87) versus placebo (2.5%, 1/40), compared to patients <65 years of age receiving PRISTIQ (0.9%, 18/1,937) versus placebo (0.7%, 8/1,218).
6.2 Postmarketing Experience
The following adverse reaction has been identified during post-approval use of PRISTIQ. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Skin and subcutaneous tissue disorders – Stevens-Johnson syndrome
Gastrointestinal disorders – Pancreatitis acute
Cardiovascular system – Takotsubo cardiomyopathy
7 DRUG INTERACTIONS
7.1 Drugs Having Clinically Important Interactions with PRISTIQ
| Monoamine Oxidase Inhibitors (MAOI) | |
| Clinical Impact | The concomitant use of SSRIs and SNRIs including PRISTIQ with MAOIs increases the risk of serotonin syndrome. |
| Intervention | Concomitant use of PRISTIQ is contraindicated:
|
| Examples | selegiline, tranylcypromine, isocarboxazid, phenelzine, linezolid, methylene blue |
| Other Serotonergic Drugs | |
| Clinical Impact | Concomitant use of PRISTIQ with other serotonergic drugs increases the risk of serotonin syndrome. |
| Intervention | Monitor for symptoms of serotonin syndrome when PRISTIQ is used concomitantly with other drugs that may affect the serotonergic neurotransmitter systems. If serotonin syndrome occurs, consider discontinuation of PRISTIQ and/or concomitant serotonergic drugs [see Warnings and Precautions (5.2)]. |
| Examples | other SNRIs, SSRIs, triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, amphetamines, tryptophan, and St. John's Wort |
| Drugs that Interfere with Hemostasis | |
| Clinical Impact | Concomitant use of PRISTIQ with an antiplatelet or anticoagulant drug may potentiate the risk of bleeding. This may be due to the effect of PRISTIQ on the release of serotonin by platelets. |
| Intervention | Closely monitor for bleeding for patients receiving an antiplatelet or anticoagulant drug when PRISTIQ is initiated or discontinued [see Warnings and Precautions (5.4)] . |
| Examples | NSAIDs, aspirin, and warfarin |
| Drugs that are Primarily Metabolized by CYP2D6 | |
| Clinical Impact | Concomitant use of PRISTIQ increases C max and AUC of a drug primarily metabolized by CYP2D6 which may increase the risk of toxicity of the CYP2D6 substrate drug [see Clinical Pharmacology (12.3)] . |
| Intervention | Original dose should be taken when co-administered with PRISTIQ 100 mg or lower. Reduce the dose of these drugs by up to one-half if co-administered with 400 mg of PRISTIQ. |
| Examples | desipramine, atomoxetine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine |
7.2 Drugs Having No Clinically Important Interactions with PRISTIQ
Based on pharmacokinetic studies, no dosage adjustment is required for drugs that are mainly metabolized by CYP3A4 (e.g., midazolam), or for drugs that are metabolized by both CYP2D6 and CYP3A4 (e.g., tamoxifen, aripiprazole), when administered concomitantly with PRISTIQ [see Clinical Pharmacology (12.3)].
7.3 Alcohol
A clinical study has shown that PRISTIQ does not increase the impairment of mental and motor skills caused by ethanol. However, as with all CNS-active drugs, patients should be advised to avoid alcohol consumption while taking PRISTIQ.
7.4 Drug-Laboratory Test Interactions
False-positive urine immunoassay screening tests for phencyclidine (PCP) and amphetamine have been reported in patients taking desvenlafaxine. This is due to lack of specificity of the screening tests. False positive test results may be expected for several days following discontinuation of desvenlafaxine therapy. Confirmatory tests, such as gas chromatography/mass spectrometry, will distinguish desvenlafaxine from PCP and amphetamine.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Antidepressants at 1-844-405-6185.
Risk Summary
There are no published studies on PRISTIQ in pregnant women; however published epidemiologic studies of pregnant women exposed to venlafaxine, the parent compound, have not reported a clear association with adverse developmental outcomes (see Data) . There are risks associated with untreated depression in pregnancy and with exposure to SNRIs and SSRIs, including PRISTIQ, during pregnancy ( see Clinical Considerations).
In reproductive developmental studies in rats and rabbits treated with desvenlafaxine succinate, there was no evidence of teratogenicity at a plasma exposure (AUC) that is up to 19-times (rats) and 0.5-times (rabbits) the exposure at an adult human dose of 100 mg per day. However, fetotoxicity and pup deaths were observed in rats at 4.5-times the AUC exposure observed with an adult human dose of 100 mg per day.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
A prospective longitudinal study of 201 women with a history of major depression who were euthymic at the beginning of pregnancy, showed that women who discontinued antidepressant medication during pregnancy were more likely to experience a relapse of major depression than women who continued antidepressant medication.
Maternal Adverse Reactions
Exposure to SNRIs in mid to late pregnancy may increase the risk for preeclampsia, and exposure to SNRIs near delivery may increase the risk for postpartum hemorrhage.
Fetal/Neonatal Adverse Reactions
Exposure to SNRIs or SSRIs in late pregnancy may lead to an increased risk for neonatal complications requiring prolonged hospitalization, respiratory support, and tube feeding. Monitor neonates who were exposed to PRISTIQ in the third trimester of pregnancy for drug discontinuation syndrome (see Data).
Data
Human Data
Published epidemiological studies of pregnant women exposed to the parent compound venlafaxine have not reported a clear association with major birth defects or miscarriage. Methodological limitations of these observational studies include possible exposure and outcome misclassification, lack of adequate controls, adjustment for confounders, and confirmatory studies; therefore, these studies cannot establish or exclude any drug-associated risk during pregnancy.
Retrospective cohort studies based on claims data have shown an association between venlafaxine use and preeclampsia, compared to depressed women who did not take an antidepressant during pregnancy. One study that assessed venlafaxine exposure in the second trimester or first half of the third trimester and preeclampsia showed an increased risk compared to unexposed depressed women [adjusted (adj) RR 1.57, 95% CI 1.29–1.91]. Preeclampsia was observed at venlafaxine doses equal to or greater than 75 mg/day and a duration of treatment >30 days. Another study that assessed venlafaxine exposure in gestational weeks 10–20 and preeclampsia showed an increased risk at doses equal to or greater than 150 mg/day. Available data are limited by possible outcome misclassification and possible confounding due to depression severity and other confounders.
Retrospective cohort studies based on claims data have suggested an association between venlafaxine use near the time of delivery or through delivery and postpartum hemorrhage. One study showed an increased risk for postpartum hemorrhage when venlafaxine exposure occurred through delivery, compared to unexposed depressed women [adj RR 2.24 (95% CI 1.69–2.97)]. There was no increased risk in women who were exposed to venlafaxine earlier in pregnancy. Limitations of this study include possible confounding due to depression severity and other confounders. Another study showed an increased risk for postpartum hemorrhage when SNRI exposure occurred for at least 15 days in the last month of pregnancy or through delivery, compared to unexposed women (adj RR 1.64–1.76). The results of this study may be confounded by the effects of depression.
Neonates exposed to SNRIs or SSRIs, late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremor, jitteriness, irritability, and constant crying. These features are consistent with either a direct toxic effect of SSRIs and SNRIs or, possibly, a drug discontinuation syndrome. It should be noted that, in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions (5.2)] .
Animal Data
When desvenlafaxine succinate was administered orally to pregnant rats and rabbits during the period of organogenesis at doses up to 300 mg/kg/day and 75 mg/kg/day, respectively, no teratogenic effects were observed. These doses were associated with a plasma exposure (AUC) 19 times (rats) and 0.5 times (rabbits) the AUC exposure at an adult human dose of 100 mg per day. However, fetal weights were decreased and skeletal ossification was delayed in rats in association with maternal toxicity at the highest dose, with an AUC exposure at the no-effect dose that is 4.5-times the AUC exposure at an adult human dose of 100 mg per day.
When desvenlafaxine succinate was administered orally to pregnant rats throughout gestation and lactation, there was a decrease in pup weights and an increase in pup deaths during the first four days of lactation at the highest dose of 300 mg/kg/day. The cause of these deaths is not known. The AUC exposure at the no-effect dose for rat pup mortality was 4.5-times the AUC exposure at an adult human dose of 100 mg per day. Post-weaning growth and reproductive performance of the progeny were not affected by maternal treatment with desvenlafaxine succinate at exposures 19 times the AUC exposure at an adult human dose of 100 mg per day.
8.2 Lactation
Risk Summary
Available limited data from published literature show low levels of desvenlafaxine in human milk, and have not shown adverse reactions in breastfed infants (see Data) . There are no data on the effects of desvenlafaxine on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRISTIQ and any potential adverse effects on the breastfed child from PRISTIQ or from the underlying maternal condition.
Data
A lactation study was conducted in 10 breastfeeding women (at a mean of 4.3 months post-partum) who were being treated with a 50–150 mg daily dose of desvenlafaxine for postpartum depression. Sampling was performed at steady state (up to 8 samples) over a 24 hour dosing period, and included foremilk and hindmilk. The mean relative infant dose was calculated to be 6.8% (range of 5.5–8.1%). No adverse reactions were seen in the infants.
8.4 Pediatric Use
The safety and effectiveness of PRISTIQ have not been established in pediatric patients for the treatment of MDD.
Efficacy was not demonstrated in two adequate and well controlled, 8-week, randomized, double-blind, placebo-controlled, parallel group studies conducted in 587 patients (7 to 17 years of age) for the treatment of MDD.
Antidepressants, such as PRISTIQ, increase the risk of suicidal thoughts and behaviors in pediatric patients [see the Boxed Warning and Warnings and Precautions (5.1)] .
PRISTIQ was associated with a decrease in body weight in placebo-controlled trials in pediatric patients with MDD. The incidence of weight loss (≥3.5% of baseline weight) was 22%, 14%, and 7% for patients treated with low dose PRISTIQ, high dose PRISTIQ, and placebo, respectively.
The risks associated with longer term PRISTIQ use were assessed in 6-month, open-label extension studies in pediatric patients (7 to 17 years of age) with MDD. Pediatric patients (7 to 17 years of age) had mean changes in weight that approximated expected changes, based on data from age- and sex-matched peers.
In clinical trials, when compared to adult patients receiving the same dose of PRISTIQ, exposure to desvenlafaxine was similar in adolescent patients 12 to 17 years of age, and was about 30% higher in pediatric patients 7 to 11 years of age.
Juvenile Animal Studies
In a juvenile animal study, male and female rats were treated with desvenlafaxine (75, 225 and 675 mg/kg/day) starting on postnatal day (PND) 22 through 112. Behavioral deficits (longer time immobile in a motor activity test, longer time swimming in a straight channel test, and lack of habituation in an acoustic startle test) were observed in males and females but were reversed after a recovery period. A No Adverse Effect Level (NOAEL) was not identified for these deficits. The Low Adverse Effect Level (LOAEL) was 75 mg/kg/day which was associated with plasma exposure (AUC) twice the levels measured with a pediatric dose of 100 mg/day.
In a second juvenile animal study, male and female rats were administered desvenlafaxine (75, 225 or 675 mg/kg/day) for 8–9 weeks starting on PND 22 and were mated with naïve counterparts. Delays in sexual maturation and decreased fertility, number of implantation sites and total live embryos were observed in treated females at all doses. The LOAEL for these findings is 75 mg/kg/day which was associated with an AUC twice the levels measured with a pediatric dose of 100 mg/day. These findings were reversed at the end of a 4-week recovery period. The relevance of these findings to humans is not known.
8.5 Geriatric Use
Of the 4,158 patients in pre-marketing clinical studies with PRISTIQ, 6% were 65 years of age or older. No overall differences in safety or efficacy were observed between these patients and younger patients; however, in the short-term placebo-controlled studies, there was a higher incidence of systolic orthostatic hypotension in patients ≥65 years of age compared to patients <65 years of age treated with PRISTIQ [see Adverse Reactions (6.1)] . For elderly patients, possible reduced renal clearance of PRISTIQ should be considered when determining dose [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)] .
SSRIs and SNRIs, including PRISTIQ, have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse event [see Warnings and Precautions (5.9)] .
8.6 Renal Impairment
Adjust the maximum recommended dosage in patients with moderate or severe renal impairment (CLcr 15 to 50 mL/min, C-G), or end-stage renal disease (CLcr < 15 mL/min, C-G) [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)] .
8.7 Hepatic Impairment
Adjust the maximum recommended dosage in patients with moderate to severe hepatic impairment (Child-Pugh score 7 to 15) [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)] .
10 OVERDOSAGE
10.1 Human Experience with Overdosage
There is limited clinical trial experience with desvenlafaxine succinate overdosage in humans. However, desvenlafaxine (PRISTIQ) is the major active metabolite of venlafaxine. Overdose experience reported with venlafaxine (the parent drug of PRISTIQ) is presented below; the identical information can be found in the Overdosage section of the venlafaxine package insert.
In postmarketing experience, overdose with venlafaxine (the parent drug of PRISTIQ) has occurred predominantly in combination with alcohol and/or other drugs. The most commonly reported events in overdosage include tachycardia, changes in level of consciousness (ranging from somnolence to coma), mydriasis, seizures, and vomiting. Electrocardiogram changes (e.g., prolongation of QT interval, bundle branch block, QRS prolongation), sinus and ventricular tachycardia, bradycardia, hypotension, rhabdomyolysis, vertigo, liver necrosis, serotonin syndrome, and death have been reported.
Published retrospective studies report that venlafaxine overdosage may be associated with an increased risk of fatal outcomes compared to that observed with SSRI antidepressant products, but lower than that for tricyclic antidepressants. Epidemiological studies have shown that venlafaxine-treated patients have a higher pre-existing burden of suicide risk factors than SSRI-treated patients. The extent to which the finding of an increased risk of fatal outcomes can be attributed to the toxicity of venlafaxine in overdosage, as opposed to some characteristic(s) of venlafaxine-treated patients, is not clear.
11 DESCRIPTION
PRISTIQ is an extended-release tablet for oral administration that contains desvenlafaxine succinate, a structurally novel SNRI for the treatment of MDD. Desvenlafaxine (O-desmethylvenlafaxine) is the major active metabolite of the antidepressant venlafaxine, a medication used to treat major depressive disorder.
Desvenlafaxine is designated RS-4-[2-dimethylamino-1-(1-hydroxycyclohexyl)ethyl]phenol and has the empirical formula of C 16H 25NO 2 (free base) and C 16H 25NO 2∙C 4H 6O 4∙H 2O (succinate monohydrate). Desvenlafaxine succinate monohydrate has a molecular weight of 399.48. The structural formula is shown below.
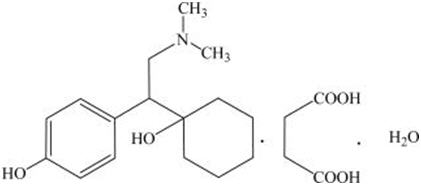
Desvenlafaxine succinate is a white to off-white powder that is soluble in water. The solubility of desvenlafaxine succinate is pH dependent. Its octanol:aqueous system (at pH 7.0) partition coefficient is 0.21.
PRISTIQ is formulated as an extended-release tablet for once-a-day oral administration.
Each tablet contains 38 mg, 76 mg or 152 mg of desvenlafaxine succinate equivalent to 25 mg, 50 mg or 100 mg of desvenlafaxine, respectively.
Inactive ingredients for the 25 mg tablet consist of hypromellose, microcrystalline cellulose, talc, magnesium stearate, a film coating which consists of polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, and iron oxides.
Inactive ingredients for the 50 mg tablet consist of hypromellose, microcrystalline cellulose, talc, magnesium stearate and film coating, which consists of polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, and iron oxides.
Inactive ingredients for the 100 mg tablet consist of hypromellose, microcrystalline cellulose, talc, magnesium stearate and film coating, which consists of polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, iron oxide and FD&C yellow #6.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The exact mechanism of the antidepressant action of desvenlafaxine is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of their reuptake. Non-clinical studies have shown that desvenlafaxine is a potent and selective SNRI.
12.2 Pharmacodynamics
Desvenlafaxine lacked significant affinity for numerous receptors, including muscarinic-cholinergic, H 1-histaminergic, or α 1-adrenergic receptors in vitro. Desvenlafaxine also lacked monoamine oxidase (MAO) inhibitory activity.
ECG changes
Electrocardiograms were obtained from 1,492 desvenlafaxine treated patients with major depressive disorder and 984 placebo-treated patients in clinical studies lasting up to 8 weeks. No clinically relevant differences were observed between desvenlafaxine treated and placebo-treated patients for QT, QTc, PR, and QRS intervals. In a thorough QTc study with prospectively determined criteria, desvenlafaxine did not cause QT prolongation. No difference was observed between placebo and desvenlafaxine treatments for the QRS interval.
12.3 Pharmacokinetics
The single-dose pharmacokinetics of desvenlafaxine are linear and dose-proportional in a dose range of 50 to 600 mg (1 to 12 times the recommended approved dosage) per day. With once-daily dosing, steady-state plasma concentrations are achieved within approximately 4 to 5 days. At steady-state, multiple-dose accumulation of desvenlafaxine is linear and predictable from the single-dose pharmacokinetic profile.
Distribution
Steady-state volume of distribution of desvenlafaxine is 3.4 L/kg. Plasma protein binding of desvenlafaxine is 30% and is independent of drug concentration.
Elimination
Metabolism
Desvenlafaxine is primarily metabolized by conjugation (mediated by UGT isoforms) and, to a minor extent, through oxidative metabolism. CYP3A4 mediates the oxidative metabolism (N-demethylation) of desvenlafaxine. The CYP2D6 metabolic pathway is not involved. The pharmacokinetics of desvenlafaxine was similar in subjects with CYP2D6 poor and extensive metabolizer phenotype.
Specific Populations
No clinically significant differences in the exposures of desvenlafaxine were observed based on ethnicity (White, Black, Hispanic).
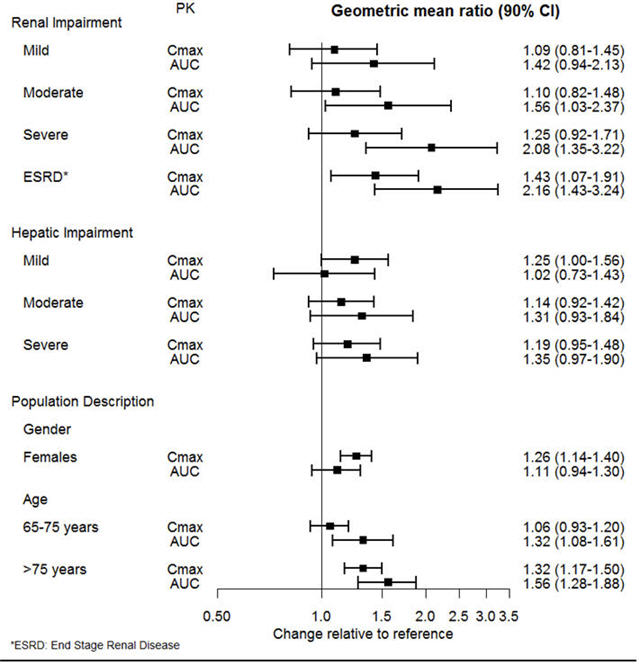
The effect of intrinsic patient factors on the pharmacokinetics of desvenlafaxine is presented in Figure 1.
| Figure 1 Impact of Intrinsic Factors (Renal, Hepatic Impairment and Population Description) on Desvenlafaxine Pharmacokinetics |
|
|
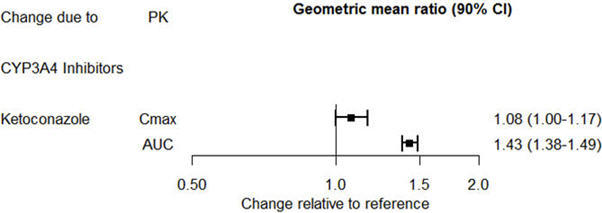
Drug Interaction Studies
Clinical Studies
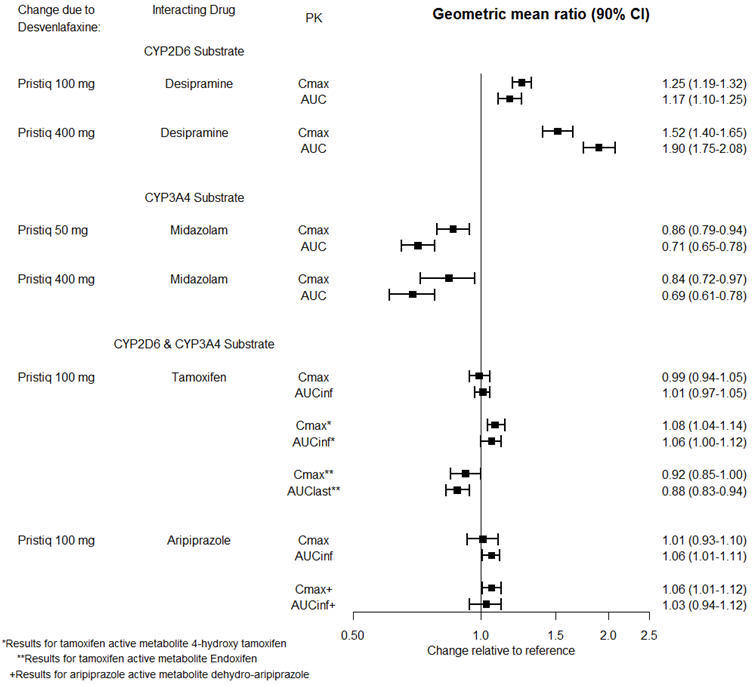
In Vitro Studies
Based on in vitro data, drugs that inhibit CYP isozymes 1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19, and 2E1 are not expected to have significant impact on the pharmacokinetic profile of desvenlafaxine.
Desvenlafaxine does not inhibit CYP1A2, 2A6, 2C8, 2C9, 2C19 CYP2D6, or CYP3A4 isozymes. Desvenlafaxine does not induce CYP3A4 either.
Desvenlafaxine is not a substrate or an inhibitor for the P-glycoprotein (P-gp) transporter.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Desvenlafaxine succinate administered by oral gavage to mice and rats for 2 years did not increase the incidence of tumors in either study.
Mice received desvenlafaxine succinate at dosages up to 500/300 mg/kg/day (dosage lowered after 45 weeks of dosing). The AUC exposure at 300 mg/kg/day dose is estimated at 10 times the AUC exposure at an adult human dose of 100 mg per day.
Rats received desvenlafaxine succinate at dosages up to 300 mg/kg/day (males) or 500 mg/kg/day (females). The AUC exposure at the highest dose is estimated at 11 (males) or 26 (females) times the AUC exposure at an adult human dose of 100 mg per day.
Mutagenesis
Desvenlafaxine was not mutagenic in the in vitro bacterial mutation assay (Ames test) and was not clastogenic in an in vitro chromosome aberration assay in cultured CHO cells, an in vivo mouse micronucleus assay, or an in vivo chromosome aberration assay in rats. Additionally, desvenlafaxine was not genotoxic in the in vitro CHO mammalian cell forward mutation assay and was negative in the in vitro BALB/c-3T3 mouse embryo cell transformation assay.
Impairment of Fertility
When desvenlafaxine succinate was administered orally to male and female rats, fertility was reduced at the high dose of 300 mg/kg/day, which is 10 (males) and 19 (females) times the AUC exposure at an adult human dose of 100 mg per day. There was no effect on fertility at 100 mg/kg/day, which is 3 (males) or 5 (females) times the AUC exposure at an adult human dose of 100 mg per day. These studies did not address reversibility of the effect on fertility. The relevance of these findings to humans is not known.
14 CLINICAL STUDIES
Major Depressive Disorder
The efficacy of PRISTIQ as a treatment for depression was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose studies (at doses of 50 mg per day to 400 mg per day) in adult outpatients who met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria for major depressive disorder. In the first study, patients received 100 mg (n = 114), 200 mg (n = 116), or 400 mg (n = 113) of PRISTIQ once daily, or placebo (n = 118). In a second study, patients received either 200 mg (n = 121) or 400 mg (n = 124) of PRISTIQ once daily, or placebo (n = 124). In two additional studies, patients received 50 mg (n = 150 and n = 164) or 100 mg (n = 147 and n = 158) of PRISTIQ once daily, or placebo (n = 150 and n = 161).
PRISTIQ showed superiority over placebo as measured by improvement in the 17-item Hamilton Rating Scale for Depression (HAM-D 17) total score in four studies and overall improvement, as measured by the Clinical Global Impressions Scale - Improvement (CGI-I), in three of the four studies. In studies directly comparing 50 mg per day and 100 mg per day there was no suggestion of a greater effect with the higher dose and adverse reactions and discontinuations were more frequent at higher doses [see Dosage and Administration (2.1)] .
| PRISTIQ | ||||||
|---|---|---|---|---|---|---|
| Study No. | Primary Endpoint: HAM-D 17 | Placebo | 50 mg/day | 100 mg/day | 200 mg/day | 400 mg/day |
| 1 | Baseline Score (SD *) | 23.1 (2.5) | 23.2 (2.5) | 22.9 (2.4) | 23.0 (2.2) | |
| Difference from Placebo (95% CI †) | -2.9
‡
(-5.1, -0.8) | -2.0 | -3.1
‡
(-5.2, -0.9) |
|||
| 2 | Baseline Score (SD *) | 25.3 (3.3) | 24.8 (2.9) | 25.2 (3.2) | ||
| Difference from Placebo (95% CI †) | -3.3
‡
(-5.3, -1.2) | -2.8
‡
(-4.8, -0.7) |
||||
| 3 | Baseline Score (SD *) | 23.0 (2.6) | 23.4 (2.6) | 23.4 (2.6) | ||
| Difference from Placebo (95% CI †) | -1.9
‡
(-3.5, -0.3) | -1.5 | ||||
| 4 | Baseline Score (SD *) | 24.3 (2.6) | 24.3 (2.4) | 24.4 (2.7) | ||
| Difference from Placebo (95% CI †) | -2.5
‡
(-4.1, -0.9) | -3.0
‡
(-4.7, -1.4) | ||||
Analyses of the relationships between treatment outcome and age and treatment outcome and gender did not suggest any differential responsiveness on the basis of these patient characteristics. There was insufficient information to determine the effect of race on outcome in these studies.
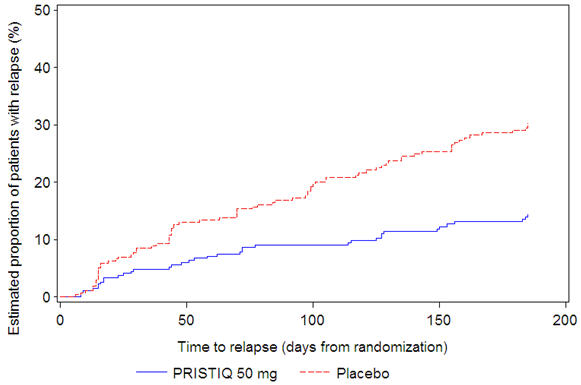
In a longer-term trial (Study 5), adult outpatients meeting DSM-IV criteria for major depressive disorder, who responded to 8 weeks of open-label acute treatment with 50 mg per day desvenlafaxine and subsequently remained stable for 12 weeks on desvenlafaxine, were assigned randomly in a double-blind manner to remain on active treatment or switch to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D 17 total score of ≤ 11 and CGI-I ≤ 2 at the day 56 evaluation; stability was defined as HAM-D 17 total score of ≤ 11 and CGI-I ≤ 2 at week 20 and not having a HAM-D 17 total score of ≥ 16 or a CGI-I score ≥ 4 at any office visit. Relapse during the double-blind phase was defined as follows: (1) a HAM-D 17 total score of ≥ 16 at any office visit, (2) discontinuation for unsatisfactory efficacy response, (3) hospitalized for depression, (4) suicide attempt, or (5) suicide. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse compared with placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 14% with desvenlafaxine treatment versus 30% with placebo.
| Figure 4. Estimated Proportion of Relapses vs. Number of Days since Randomization (Study 5) |
|
|
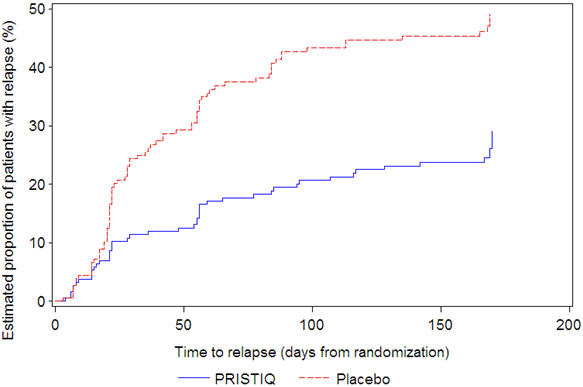
In another longer-term trial (Study 6), adult outpatients meeting DSM-IV criteria for major depressive disorder and who responded to 12 weeks of acute treatment with desvenlafaxine were assigned randomly to the same dose (200 or 400 mg per day) they had received during acute treatment or to placebo for up to 26 weeks of observation for relapse. Response during the open-label phase was defined as a HAM-D 17 total score of ≤ 11 at the day 84 evaluation. Relapse during the double-blind phase was defined as follows: (1) a HAM-D 17 total score of ≥ 16 at any office visit, (2) a CGI-I score of ≥ 6 (versus day 84) at any office visit, or (3) discontinuation from the trial due to unsatisfactory response. Patients receiving continued desvenlafaxine treatment experienced statistically significantly longer time to relapse over the subsequent 26 weeks compared with those receiving placebo. At 26 weeks, the Kaplan-Meier estimated proportion of relapse was 29% with desvenlafaxine treatment versus 49% with placebo.
| Figure 5. Estimated Proportion of Relapses vs. Number of Days since Randomization (Study 6) |
|
|
In a postmarketing study, the efficacy of PRISTIQ at a dose lower than 50 mg per day was evaluated in an 8-week, multicenter, randomized, double-blind, placebo-controlled, fixed-dose study in adult outpatients with Major Depressive Disorder. The treatment arms were 25 mg (n=232), 50 mg (n=236), and placebo (n=231). The 50 mg dose was superior to placebo, as measured by the mean change from baseline on the HAMD-17. The 25 mg dose was not superior to placebo.
16 HOW SUPPLIED/STORAGE AND HANDLING
PRISTIQ ® (desvenlafaxine) extended-release tablets are available as follows:
50 mg, light pink, square pyramid tablet debossed with "W" (over) "50" on the flat side
Each tablet contains 76 mg of desvenlafaxine succinate equivalent to 50 mg of desvenlafaxine, respectively.
NDC 43063-262-30, bottle of 30 tablets
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Suicidal Thoughts and Behaviors
Advise patients and caregivers to look for the emergence of suicidality, especially early during treatment and when the dose is adjusted up or down, and instruct them to report such symptoms to the healthcare provider [see Boxed Warning and Warnings and Precautions (5.1)] .
Concomitant Medication
Advise patients taking PRISTIQ not to use concomitantly other products containing desvenlafaxine or venlafaxine. Healthcare professionals should instruct patients not to take PRISTIQ with an MAOI or within 14 days of stopping an MAOI and to allow 7 days after stopping PRISTIQ before starting an MAOI [see Contraindications (4)] .
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of PRISTIQ with other serotonergic agents (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, amphetamines, tryptophan, buspirone, and St. John's Wort supplements) [see Warnings and Precautions (5.2)] .
Elevated Blood Pressure
Advise patients that they should have regular monitoring of blood pressure when taking PRISTIQ [see Warnings and Precautions (5.3)] .
Increased Risk of Bleeding
Inform patients about the concomitant use of PRISTIQ with NSAIDs, aspirin, other antiplatelet drugs, warfarin, or other coagulants because the combined use of has been associated with an increased risk of bleeding. Advise patients to inform their healthcare providers if they are taking or planning to take any prescription or over-the-counter medications that increase the risk of bleeding [see Warnings and Precautions (5.4)] .
Activation of Mania/Hypomania
Advise patients, their families and caregivers to observe for signs of activation of mania/hypomania [see Warnings and Precautions (5.6)] .
Discontinuation Syndrome
Advise patients not to abruptly stop taking PRISTIQ without talking first with their healthcare professional. Patients should be aware that discontinuation effects may occur when stopping PRISTIQ, and a dose of 25 mg per day is available for discontinuing therapy [see Warnings and Precautions (5.7) and Adverse Reactions (6.1)] .
Sexual Dysfunction
Advise patients that use of PRISTIQ may cause symptoms of sexual dysfunction in both male and female patients. Inform patients that they should discuss any changes in sexual function and potential management strategies with their healthcare provider [see Warnings and Precautions (5.11)].
Switching Patients from Other Antidepressants to PRISTIQ
Discontinuation symptoms have been reported when switching patients from other antidepressants, including venlafaxine, to PRISTIQ. Tapering of the initial antidepressant may be necessary to minimize discontinuation symptoms.
Interference with Cognitive and Motor Performance
Caution patients about operating hazardous machinery, including automobiles, until they are reasonably certain that PRISTIQ therapy does not adversely affect their ability to engage in such activities.
Allergic Reactions
Advise patients to notify their physician if they develop allergic phenomena such as rash, hives, swelling, or difficulty breathing.
Pregnancy
Advise patients to notify their physician if they become pregnant or intend to become pregnant during therapy. Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to PRISTIQ during pregnancy [see Use in Specific Populations (8.1)] .
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 11/2021 | |||
| MEDICATION GUIDE
PRISTIQ ® (pris- TEEK ) (desvenlafaxine) extended-release tablets |
||||
| What is the most important information I should know about PRISTIQ?
PRISTIQ can cause serious side effects, including:
|
||||
|
|
|||
What is PRISTIQ?
|
||||
Do not take PRISTIQ if you:
|
||||
Before taking PRISTIQ tell your healthcare provider about all your medical conditions, including if you:
|
||||
|
||||
PRISTIQ and other medicines may affect each other causing possible serious side effects. PRISTIQ may affect the way other medicines work and other medicines may affect the way PRISTIQ works. Especially tell your healthcare provider if you take:
Do not start or stop any other medicines during treatment with PRISTIQ without talking to your healthcare provider first. Stopping PRISTIQ suddenly may cause you to have serious side effects. See, " What are the possible side effects of PRISTIQ?" Know the medicines you take. Keep a list of them to show to your healthcare providers when you get a new medicine. |
||||
How should I take PRISTIQ?
|
||||
What should I avoid while taking PRISTIQ?
|
||||
| What are the possible side effects of PRISTIQ?
PRISTIQ can cause serious side effects, including:
|
||||
|
|
|||
|
||||
|
|
|
||
|
||||
|
|
|||
| These are not all the possible side effects of PRISTIQ.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
How should I store PRISTIQ?
|
||||
| General Information about the safe and effective use of PRISTIQ
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take PRISTIQ for a condition for which it was not prescribed. Do not give PRISTIQ to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about PRISTIQ that is written for healthcare professionals. |
||||
| What are the ingredients in PRISTIQ?
Active ingredient: desvenlafaxine Inactive ingredients:
This product's label may have been updated. For current full prescribing information, please visit www.pfizer.com. LAB-0539-13.0 For more information, go to www.pristiq.com or call 1-888-PRISTIQ (774-7847). |
||||
| PRISTIQ
EXTENDED-RELEASE
desvenlafaxine succinate tablet, extended release |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - PD-Rx Pharmaceuticals, Inc. (156893695) |
| Registrant - PD-Rx Pharmaceuticals, Inc. (156893695) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| PD-Rx Pharmaceuticals, Inc. | 156893695 | repack(43063-262) | |