Label: SPRYCEL- dasatinib tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-5759-0 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 0003-0524
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 1, 2012
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SPRYCEL safely and effectively. See full prescribing information for SPRYCEL.
SPRYCEL® (dasatinib) Tablet for Oral Use
Initial U.S. Approval: 2006RECENT MAJOR CHANGES
Warnings and Precautions,
Pulmonary Arterial Hypertension (5.6) 10/2011INDICATIONS AND USAGE
SPRYCEL is a kinase inhibitor indicated for the treatment of
- newly diagnosed adults with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase. The trial is ongoing and further data will be required to determine long-term outcome. (1, 14)
- adults with chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib. (1, 14)
- adults with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with resistance or intolerance to prior therapy. (1, 14)
DOSAGE AND ADMINISTRATION
- Chronic phase CML: 100 mg once daily. (2)
- Accelerated phase CML, myeloid or lymphoid blast phase CML, or Ph+ ALL: 140 mg once daily. (2)
Administered orally, with or without a meal. Tablets should not be crushed or cut. (2)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Myelosuppression: Severe thrombocytopenia, neutropenia, and anemia may occur and require dose interruption or reduction. Monitor complete blood counts regularly. (2.3, 5.1, 6.1)
- Bleeding Related Events (mostly associated with severe thrombocytopenia): CNS and gastrointestinal hemorrhages, including fatalities, have occurred. Severe hemorrhage may require treatment interruptions and transfusions. Use SPRYCEL with caution in patients requiring medications that inhibit platelet function or anticoagulants. (5.2, 6.1)
- Fluid Retention: SPRYCEL is associated with fluid retention, sometimes severe, including ascites, edema, and pleural and pericardial effusions. Manage with appropriate supportive care measures. (5.3, 6.1)
- QT Prolongation: Use SPRYCEL with caution in patients who have or may develop prolongation of the QT interval. (5.4)
- Congestive Heart Failure, Left Ventricular Dysfunction and Myocardial Infarction: Monitor patients for signs and symptoms consistent with cardiac dysfunction and treat appropriately. (5.5, 6.1)
- Pulmonary Arterial Hypertension (PAH): SPRYCEL may increase the risk of developing PAH which may be reversible on discontinuation. Evaluate patients for signs and symptoms of underlying cardiopulmonary disease prior to initiating SPRYCEL and during treatment. (5.6)
- Use in Pregnancy: Fetal harm may occur when administered to a pregnant woman. Women should be advised of the potential hazard to the fetus and to avoid becoming pregnant. (5.7, 8.1)
ADVERSE REACTIONS
Most common adverse reactions (≥10%) in patients with newly diagnosed chronic phase CML included myelosuppression, fluid retention, diarrhea, headache, musculoskeletal pain, and rash. Most common adverse reactions (≥20%) in patients with resistance or intolerance to prior imatinib therapy included myelosuppression, fluid retention events, diarrhea, headache, dyspnea, skin rash, fatigue, nausea, and hemorrhage. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- CYP3A4 Inhibitors: May increase dasatinib drug levels and should be avoided. If coadministration cannot be avoided, monitor closely and consider reducing SPRYCEL dose. (2.1, 7.1)
- CYP3A4 Inducers: May decrease dasatinib drug levels. If coadministration cannot be avoided, consider increasing SPRYCEL dose. (2.1, 7.2)
- Antacids: May decrease dasatinib drug levels. Avoid simultaneous administration. If needed, administer the antacid at least 2 hours prior to or 2 hours after the dose of SPRYCEL. (7.2)
- H2 Antagonists/Proton Pump Inhibitors: May decrease dasatinib drug levels. Consider antacids in place of H2 antagonists or proton pump inhibitors. (7.2)
USE IN SPECIFIC POPULATIONS
- Hepatic Impairment: Use SPRYCEL with caution in patients with hepatic impairment. (8.6)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2012
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose Modification
2.2 Dose Escalation
2.3 Dose Adjustment for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Bleeding Related Events
5.3 Fluid Retention
5.4 QT Prolongation
5.5 Congestive Heart Failure, Left Ventricular Dysfunction, and Myocardial Infarction
5.6 Pulmonary Arterial Hypertension
5.7 Use in Pregnancy
6 ADVERSE REACTIONS
6.1 Chronic Myeloid Leukemia (CML)
6.2 Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia (Ph+ ALL)
6.3 Additional Data From Clinical Trials
6.4 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs That May Increase Dasatinib Plasma Concentrations
7.2 Drugs That May Decrease Dasatinib Plasma Concentrations
7.3 Drugs That May Have Their Plasma Concentration Altered By Dasatinib
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly Diagnosed Chronic Phase CML
14.2 Imatinib Resistant or Intolerant CML or Ph+ ALL
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
16.3 Handling and Disposal
17 PATIENT COUNSELING INFORMATION
17.1 Bleeding
17.2 Myelosuppression
17.3 Fluid Retention
17.4 Pregnancy
17.5 Gastrointestinal Complaints
17.6 Pain
17.7 Fatigue
17.8 Rash
17.9 Lactose
17.10 Missed Dose
FDA-Approved Patient Labeling
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
SPRYCEL® (dasatinib) is indicated for the treatment of adults with
- newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase. The effectiveness of SPRYCEL is based on cytogenetic response and major molecular response rates [see Clinical Studies (14.1)]. The trial is ongoing and further data will be required to determine long-term outcome.
- chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib.
- Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with resistance or intolerance to prior therapy.
-
2 DOSAGE AND ADMINISTRATION
The recommended starting dosage of SPRYCEL for chronic phase CML is 100 mg administered orally once daily. The recommended starting dosage of SPRYCEL for accelerated phase CML, myeloid or lymphoid blast phase CML, or Ph+ ALL is 140 mg administered orally once daily. Tablets should not be crushed or cut; they should be swallowed whole. SPRYCEL can be taken with or without a meal, either in the morning or in the evening.
In clinical studies, treatment with SPRYCEL was continued until disease progression or until no longer tolerated by the patient. The effect of stopping treatment after the achievement of a complete cytogenetic response (CCyR) has not been investigated.
2.1 Dose Modification
Concomitant Strong CYP3A4 inducers: The use of concomitant strong CYP3A4 inducers may decrease dasatinib plasma concentrations and should be avoided (eg, dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, phenobarbital). St. John’s Wort may decrease dasatinib plasma concentrations unpredictably and should be avoided. If patients must be coadministered a strong CYP3A4 inducer, based on pharmacokinetic studies, a SPRYCEL dose increase should be considered. If the dose of SPRYCEL is increased, the patient should be monitored carefully for toxicity [see Drug Interactions (7.2)].
Concomitant Strong CYP3A4 inhibitors: CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole) may increase dasatinib plasma concentrations. Grapefruit juice may also increase plasma concentrations of dasatinib and should be avoided.
Selection of an alternate concomitant medication with no or minimal enzyme inhibition potential, if possible, is recommended. If SPRYCEL must be administered with a strong CYP3A4 inhibitor, a dose decrease should be considered. Based on pharmacokinetic studies, a dose decrease to 20 mg daily should be considered for patients taking SPRYCEL 100 mg daily. For patients taking SPRYCEL 140 mg daily, a dose decrease to 40 mg daily should be considered. These reduced doses of SPRYCEL are predicted to adjust the area under the curve (AUC) to the range observed without CYP3A4 inhibitors. However, there are no clinical data with these dose adjustments in patients receiving strong CYP3A4 inhibitors. If SPRYCEL is not tolerated after dose reduction, either the strong CYP3A4 inhibitor must be discontinued, or SPRYCEL should be stopped until treatment with the inhibitor has ceased. When the strong inhibitor is discontinued, a washout period of approximately 1 week should be allowed before the SPRYCEL dose is increased. [See Drug Interactions (7.1).]
2.2 Dose Escalation
In clinical studies of adult CML and Ph+ ALL patients, dose escalation to 140 mg once daily (chronic phase CML) or 180 mg once daily (advanced phase CML and Ph+ ALL) was allowed in patients who did not achieve a hematologic or cytogenetic response at the recommended starting dosage.
2.3 Dose Adjustment for Adverse Reactions
Myelosuppression
In clinical studies, myelosuppression was managed by dose interruption, dose reduction, or discontinuation of study therapy. Hematopoietic growth factor has been used in patients with resistant myelosuppression. Guidelines for dose modifications are summarized in Table 1.
Table 1: Dose Adjustments for Neutropenia and Thrombocytopenia * ANC: absolute neutrophil count Chronic Phase CML
(starting dose 100 mg once daily)ANC* <0.5 × 109/L
or
Platelets <50 × 109/L1. Stop SPRYCEL until ANC ≥1.0 × 109/L and platelets ≥50 × 109/L. 2. Resume treatment with SPRYCEL at the original starting dose if recovery occurs in ≤7 days. 3. If platelets <25 × 109/L or recurrence of ANC <0.5 × 109/L for >7 days, repeat Step 1 and resume SPRYCEL at a reduced dose of 80 mg once daily for second episode. For third episode, further reduce dose to 50 mg once daily (for newly diagnosed patients) or discontinue SPRYCEL (for patients resistant or intolerant to prior therapy including imatinib). Accelerated Phase CML,
Blast Phase CML and
Ph+ ALL
(starting dose 140 mg once daily)ANC* <0.5 × 109/L
or
Platelets <10 × 109/L1. Check if cytopenia is related to leukemia (marrow aspirate or biopsy). 2. If cytopenia is unrelated to leukemia, stop SPRYCEL until ANC ≥1.0 × 109/L and platelets ≥20 × 109/L and resume at the original starting dose. 3. If recurrence of cytopenia, repeat Step 1 and resume SPRYCEL at a reduced dose of 100 mg once daily (second episode) or 80 mg once daily (third episode). 4. If cytopenia is related to leukemia, consider dose escalation to 180 mg once daily. -
3 DOSAGE FORMS AND STRENGTHS
SPRYCEL (dasatinib) Tablets are available as 20-mg, 50-mg, 70-mg, 80-mg, 100-mg, and 140-mg white to off-white, biconvex, film-coated tablets. [See How Supplied (16.1).]
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Treatment with SPRYCEL is associated with severe (NCI CTC Grade 3 or 4) thrombocytopenia, neutropenia, and anemia. Their occurrence is more frequent in patients with advanced phase CML or Ph+ ALL than in chronic phase CML. In a dose-optimization study in patients with resistance or intolerance to prior imatinib therapy and chronic phase CML, Grade 3 or 4 myelosuppression was reported less frequently in patients treated with 100 mg once daily than in patients treated with other dosing regimens.
Perform complete blood counts weekly for the first 2 months and then monthly thereafter, or as clinically indicated. Myelosuppression was generally reversible and usually managed by withholding SPRYCEL temporarily or dose reduction [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.2 Bleeding Related Events
In addition to causing thrombocytopenia in human subjects, dasatinib caused platelet dysfunction in vitro. In all clinical studies, severe central nervous system (CNS) hemorrhages, including fatalities, occurred in 1% of patients receiving SPRYCEL. Severe gastrointestinal hemorrhage, including fatalities, occurred in 4% of patients and generally required treatment interruptions and transfusions. Other cases of severe hemorrhage occurred in 2% of patients. Most bleeding events were associated with severe thrombocytopenia.
Patients were excluded from participation in initial SPRYCEL clinical studies if they took medications that inhibit platelet function or anticoagulants. In subsequent trials, the use of anticoagulants, aspirin, and non-steroidal anti-inflammatory drugs (NSAIDs) was allowed concurrently with SPRYCEL if the platelet count was >50,000–75,000 per microliter. Exercise caution if patients are required to take medications that inhibit platelet function or anticoagulants.
5.3 Fluid Retention
SPRYCEL is associated with fluid retention. In clinical trials, severe fluid retention was reported in up to 10% of patients. Ascites and generalized edema were each reported in <1% of patients. Severe pulmonary edema was reported in 1% of patients. Patients who develop symptoms suggestive of pleural effusion, such as dyspnea or dry cough, should be evaluated by chest X-ray. Severe pleural effusion may require thoracentesis and oxygen therapy. Fluid retention events were typically managed by supportive care measures that include diuretics or short courses of steroids. In dose-optimization studies, fluid retention events were reported less frequently with once daily dosing than with other dosing regimens.
5.4 QT Prolongation
In vitro data suggest that dasatinib has the potential to prolong cardiac ventricular repolarization (QT interval). Of the 2440 patients with CML treated with SPRYCEL in clinical studies, 15 patients (<1%) had QTc prolongation reported as an adverse reaction. Twenty-two patients (1%) experienced a QTcF >500 ms. In 865 patients with leukemia treated with SPRYCEL in five Phase 2 single-arm studies, the maximum mean changes in QTcF (90% upper bound CI) from baseline ranged from 7.0 ms to 13.4 ms.
Administer SPRYCEL with caution to patients who have or may develop prolongation of QTc. These include patients with hypokalemia or hypomagnesemia, patients with congenital long QT syndrome, patients taking anti-arrhythmic medicines or other medicinal products that lead to QT prolongation, and cumulative high-dose anthracycline therapy. Correct hypokalemia or hypomagnesemia prior to SPRYCEL administration.
5.5 Congestive Heart Failure, Left Ventricular Dysfunction, and Myocardial Infarction
Cardiac adverse reactions were reported in 5.8% of 258 patients taking SPRYCEL, including 1.6% of patients with cardiomyopathy, heart failure congestive, diastolic dysfunction, fatal myocardial infarction, and left ventricular dysfunction. Monitor patients for signs or symptoms consistent with cardiac dysfunction and treat appropriately.
5.6 Pulmonary Arterial Hypertension
SPRYCEL may increase the risk of developing pulmonary arterial hypertension (PAH) which may occur anytime after initiation, including after more than one year of treatment. Manifestations include dyspnea, fatigue, hypoxia, and fluid retention. PAH may be reversible on discontinuation of SPRYCEL. Evaluate patients for signs and symptoms of underlying cardiopulmonary disease prior to initiating SPRYCEL and during treatment. If PAH is confirmed, SPRYCEL should be permanently discontinued.
5.7 Use in Pregnancy
SPRYCEL may cause fetal harm when administered to a pregnant woman. In nonclinical studies, at plasma concentrations below those observed in humans receiving therapeutic doses of dasatinib, embryo-fetal toxicities, including skeletal malformations, were observed in rats and rabbits. There are no adequate and well-controlled studies of SPRYCEL in pregnant women. Women of childbearing potential should be advised to avoid becoming pregnant while receiving treatment with SPRYCEL [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Myelosuppression [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
- Bleeding related events [see Warnings and Precautions (5.2)].
- Fluid retention [see Warnings and Precautions (5.3)].
- QT prolongation [see Warnings and Precautions (5.4)].
- Congestive heart failure, left ventricular dysfunction, and myocardial infarction [see Warnings and Precautions (5.5)].
- Pulmonary Arterial Hypertension [see Warnings and Precautions (5.6)].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to SPRYCEL in clinical studies including 258 patients with newly diagnosed chronic phase CML and in 2182 patients with imatinib resistant or intolerant CML or Ph+ ALL.
In the newly diagnosed chronic phase CML trial, the median duration of therapy was 18 months; the median average daily dose was 99 mg.
In the imatinib resistant or intolerant CML or Ph+ ALL clinical trials, patients had a minimum of 2 years follow-up (starting dosage 100 mg once daily, 140 mg once daily, 50 mg twice daily, or 70 mg twice daily). Among patients with chronic phase CML and resistance or intolerance to prior imatinib therapy, the median duration of treatment with SPRYCEL 100 mg once daily was 24 months (range 1–33 months). The median duration of treatment with SPRYCEL 140 mg once daily was 15 months (range 0.03–36 months) for accelerated phase CML, 3 months (range 0.03–29 months) for myeloid blast phase CML, and 3 months (range 0.1–10 months) for lymphoid blast CML.
The majority of SPRYCEL-treated patients experienced adverse reactions at some time. In the newly diagnosed chronic phase CML trial, drug was discontinued for adverse reactions in 6% of SPRYCEL-treated patients. Among patients with resistance or intolerance to prior imatinib therapy, the rates of discontinuation for adverse reaction were 15% in chronic phase CML, 16% in accelerated phase CML, 15% in myeloid blast phase CML, 8% in lymphoid blast phase CML, and 8% in Ph+ ALL. In a dose-optimization study in patients with resistance or intolerance to prior imatinib therapy and chronic phase CML, the rate of discontinuation for adverse reaction was lower in patients treated with 100 mg once daily than in patients treated with other dosing regimens (10% and 16%, respectively).
The most frequently reported adverse reactions reported in ≥10% of patients in newly diagnosed chronic phase CML included myelosuppression, fluid retention events (pleural effusion, superficial localized edema, generalized edema), diarrhea, headache, musculoskeletal pain, and rash. Pleural effusions were reported in 31 patients (see Table 2).
The most frequently reported adverse reactions reported in ≥20% of patients with resistance or intolerance to prior imatinib therapy included myelosuppression, fluid retention events, diarrhea, headache, dyspnea, skin rash, fatigue, nausea, and hemorrhage.
The most frequently reported serious adverse reactions in patients with newly diagnosed chronic phase CML included pleural effusion (2%), hemorrhage (2%), congestive heart failure (1%), and pyrexia (1%). The most frequently reported serious adverse reactions in patients with resistance or intolerance to prior imatinib therapy included pleural effusion (11%), gastrointestinal bleeding (4%), febrile neutropenia (4%), dyspnea (3%), pneumonia (3%), pyrexia (3%), diarrhea (3%), infection (2%), congestive heart failure/cardiac dysfunction (2%), pericardial effusion (1%), and CNS hemorrhage (1%).
6.1 Chronic Myeloid Leukemia (CML)
Adverse reactions (excluding laboratory abnormalities) that were reported in at least 10% of patients are shown in Table 2 for newly diagnosed patients with chronic phase CML and Table 3 for CML patients with resistance or intolerance to prior imatinib therapy.
Table 2: Adverse Reactions Reported in ≥10% of Patients with Newly Diagnosed Chronic Phase CML All Grades Grade 3/4 SPRYCEL
(n=258)Imatinib
(n=258)SPRYCEL
(n=258)Imatinib
(n=258)Preferred Term Percent (%) of Patients a Includes cardiac failure acute, cardiac failure congestive, cardiomyopathy, diastolic dysfunction, ejection fraction decreased, and left ventricular dysfunction. b Includes erythema, erythema multiforme, rash, rash generalized, rash macular, rash papular, rash pustular, skin exfoliation, and rash vesicular. c Adverse reaction of special interest with <10% frequency. d Includes conjunctival hemorrhage, ear hemorrhage, ecchymosis, epistaxis, eye hemorrhage, gingival bleeding, hematoma, hematuria, hemoptysis, intra-abdominal hematoma, petechiae, scleral hemorrhage, uterine hemorrhage, and vaginal hemorrhage. Fluid retention 23 43 1 1 Pleural effusion 12 0 <1 0 Superficial localized edema 10 36 0 <1 Generalized edema 3 7 0 0 Congestive heart failure/
cardiac dysfunctiona2 1 <1 <1 Pericardial effusion 2 <1 <1 0 Pulmonary hypertension 1 0 0 0 Pulmonary edema <1 0 0 0 Diarrhea 18 19 <1 1 Headache 12 10 0 0 Musculoskeletal pain 12 16 0 <1 Rashb 11 17 0 1 Nausea 9 21 0 0 Fatigue 8 11 <1 0 Myalgia 6 12 0 0 Hemorrhagec 6 5 1 1 Gastrointestinal bleeding 2 <1 1 0 Other bleedingd 5 5 0 1 CNS bleeding 0 <1 0 <1 Vomiting 5 10 0 0 Muscle inflammation 4 19 0 <1
Table 3: Adverse Reactions Reported in ≥10% of Patients with CML Resistant or Intolerant to Prior Imatinib Therapy 100 mg Once Daily 140 mg Once Daily Chronic
(n=165)Accelerated
(n=157)Myeloid Blast
(n=74)Lymphoid Blast
(n=33)Preferred Term All Grades Grade 3/4 All Grades Grade 3/4 All Grades Grade 3/4 All Grades Grade 3/4 Percent (%) of Patients a Includes ventricular dysfunction, cardiac failure, cardiac failure congestive, cardiomyopathy, congestive cardiomyopathy, diastolic dysfunction, ejection fraction decreased, and ventricular failure. b Includes drug eruption, erythema, erythema multiforme, erythrosis, exfoliative rash, generalized erythema, genital rash, heat rash, milia, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculopapular, rash papular, rash pruritic, rash pustular, skin exfoliation, skin irritation, urticaria vesiculosa, and rash vesicular. Fluid Retention 34 4 35 8 34 7 21 6 Superficial localized edema 18 0 18 1 14 0 3 0 Pleural effusion 18 2 21 7 20 7 21 6 Generalized edema 3 0 1 0 3 0 0 0 Pericardial effusion 2 1 3 1 0 0 0 0 Congestive heart failure/
cardiac dysfunctiona0 0 0 0 4 0 0 0 Pulmonary edema 0 0 1 0 4 3 0 0 Headache 33 1 27 1 18 1 15 3 Diarrhea 27 2 31 3 20 5 18 0 Fatigue 24 2 19 2 20 1 9 3 Dyspnea 20 2 20 3 15 3 3 3 Musculoskeletal pain 19 2 11 0 8 1 0 0 Nausea 18 1 19 1 23 1 21 3 Skin rashb 17 2 15 0 16 1 21 0 Myalgia 13 0 7 1 7 1 3 0 Arthralgia 12 1 10 0 5 1 0 0 Infection (including bacterial,
viral, fungal, and
non-specified)12 1 10 6 14 7 9 0 Abdominal pain 12 1 6 0 8 3 3 0 Hemorrhage 11 1 26 8 19 9 24 9 Gastrointestinal bleeding 2 1 8 6 9 7 9 3 CNS bleeding 0 0 1 1 0 0 3 3 Vomiting 7 1 11 1 12 0 15 0 Pyrexia 5 1 11 2 18 3 6 0 Febrile neutropenia 1 1 4 4 12 12 12 12 Laboratory Abnormalities
Myelosuppression was commonly reported in all patient populations. The frequency of Grade 3 or 4 neutropenia, thrombocytopenia, and anemia was higher in patients with advanced phase CML than in chronic phase CML (Tables 4 and 5). Myelosuppression was reported in patients with normal baseline laboratory values as well as in patients with pre-existing laboratory abnormalities.
In patients who experienced severe myelosuppression, recovery generally occurred following dose interruption or reduction; permanent discontinuation of treatment occurred in 2% of patients with newly diagnosed chronic phase CML and 5% of patients with resistance or intolerance to prior imatinib therapy [see Warnings and Precautions (5.1)].
Grade 3 or 4 elevations of transaminase or bilirubin and Grade 3 or 4 hypocalcemia, hypokalemia, and hypophosphatemia were reported in patients with all phases of CML but were reported with an increased frequency in patients with myeloid or lymphoid blast phase CML. Elevations in transaminase or bilirubin were usually managed with dose reduction or interruption. Patients developing Grade 3 or 4 hypocalcemia during the course of SPRYCEL therapy often had recovery with oral calcium supplementation.
Laboratory abnormalities reported in patients with newly diagnosed chronic phase CML are shown in Table 4. There were no discontinuations of SPRYCEL therapy in this patient population due to biochemical laboratory parameters.
Table 4: CTC Grade 3/4 Laboratory Abnormalities in Patients with Newly Diagnosed Chronic Phase CML SPRYCEL
(n=258)Imatinib
(n=258)Percent (%) of Patients CTC grades: neutropenia (Grade 3 ≥0.5–<1.0 × 109/L, Grade 4 <0.5 × 109/L); thrombocytopenia (Grade 3 ≥25–<50 × 109/L, Grade 4 <25 × 109/L); anemia (hemoglobin Grade 3 ≥65–<80 g/L, Grade 4 <65 g/L); elevated creatinine (Grade 3 >3–6 × upper limit of normal range (ULN), Grade 4 >6 × ULN); elevated bilirubin (Grade 3 >3–10 × ULN, Grade 4 >10 × ULN); elevated SGOT or SGPT (Grade 3 >5–20 × ULN, Grade 4 >20 × ULN); hypocalcemia (Grade 3 <7.0–6.0 mg/dL, Grade 4 <6.0 mg/dL); hypophosphatemia (Grade 3 <2.0–1.0 mg/dL, Grade 4 <1.0 mg/dL); hypokalemia (Grade 3 <3.0–2.5 mmol/L, Grade 4 <2.5 mmol/L). Hematology Parameters Neutropenia 22 20 Thrombocytopenia 19 10 Anemia 11 7 Biochemistry Parameters Hypophosphatemia 5 24 Hypokalemia 0 2 Hypocalcemia 3 2 Elevated SGPT (ALT) <1 1 Elevated SGOT (AST) <1 1 Elevated Bilirubin 1 0 Elevated Creatinine <1 1 Laboratory abnormalities reported in patients with CML resistant or intolerant to imatinib who received the recommended starting doses of SPRYCEL are shown by disease phase in Table 5.
Table 5: CTC Grade 3/4 Laboratory Abnormalities in Clinical Studies of CML: Resistance or Intolerance to Prior Imatinib Therapy Advanced Phase CML Chronic Phase CML 140 mg Once Daily 100 mg Once Daily
(n=165)Accelerated Phase
(n=157)Myeloid Blast Phase
(n=74)Lymphoid Blast Phase
(n=33)Percent (%) of Patients CTC grades: neutropenia (Grade 3 ≥0.5–<1.0 × 109/L, Grade 4 <0.5 × 109/L); thrombocytopenia (Grade 3 ≥25–<50 × 109/L, Grade 4 <25 × 109/L); anemia (hemoglobin Grade 3 ≥65–<80 g/L, Grade 4 <65 g/L); elevated creatinine (Grade 3 >3–6 × upper limit of normal range (ULN), Grade 4 >6 × ULN); elevated bilirubin (Grade 3 >3–10 × ULN, Grade 4 >10 × ULN); elevated SGOT or SGPT (Grade 3 >5–20 × ULN, Grade 4 >20 × ULN); hypocalcemia (Grade 3 <7.0–6.0 mg/dL, Grade 4 <6.0 mg/dL); hypophosphatemia (Grade 3 <2.0–1.0 mg/dL, Grade 4 <1.0 mg/dL); hypokalemia (Grade 3 <3.0–2.5 mmol/L, Grade 4 <2.5 mmol/L). Hematology Parameters Neutropenia 36 58 77 79 Thrombocytopenia 23 63 78 85 Anemia 13 47 74 52 Biochemistry Parameters Hypophosphatemia 10 13 12 18 Hypokalemia 2 7 11 15 Hypocalcemia <1 4 9 12 Elevated SGPT (ALT) 0 2 5 3 Elevated SGOT (AST) <1 0 4 3 Elevated Bilirubin <1 1 3 6 Elevated Creatinine 0 2 8 0 6.2 Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia (Ph+ ALL)
A total of 135 patients with Ph+ ALL were treated with SPRYCEL in clinical studies. The median duration of treatment was 3 months (range 0.03–31 months). The safety profile of patients with Ph+ ALL was similar to those with lymphoid blast phase CML. The most frequently reported adverse reactions included fluid retention events, such as pleural effusion (24%) and superficial edema (19%), and gastrointestinal disorders, such as diarrhea (31%), nausea (24%), and vomiting (16%). Hemorrhage (19%), pyrexia (17%), rash (16%), and dyspnea (16%) were also frequently reported. The most frequently reported serious adverse reactions included pleural effusion (11%), gastrointestinal bleeding (7%), febrile neutropenia (6%), infection (5%), pyrexia (4%), pneumonia (3%), diarrhea (3%), nausea (2%), vomiting (2%), and colitis (2%).
6.3 Additional Data From Clinical Trials
The following adverse reactions were reported in patients in the SPRYCEL clinical studies at a frequency of 1%–<10%, 0.1%–<1%, or <0.1%. These events are included on the basis of clinical relevance.
Gastrointestinal Disorders: 1%–<10% – mucosal inflammation (including mucositis/stomatitis), dyspepsia, abdominal distension, constipation, gastritis, colitis (including neutropenic colitis), oral soft tissue disorder; 0.1%–<1% – ascites, dysphagia, anal fissure, upper gastrointestinal ulcer, esophagitis, pancreatitis; <0.1% – protein losing gastroenteropathy.
General Disorders and Administration Site Conditions: 1%–<10% – asthenia, pain, chest pain, chills; 0.1%–<1% – malaise, temperature intolerance.
Skin and Subcutaneous Tissue Disorders: 1%–<10% – pruritus, alopecia, acne, dry skin, hyperhidrosis, urticaria, dermatitis (including eczema); 0.1%–<1% – pigmentation disorder, skin ulcer, bullous conditions, photosensitivity, nail disorder, acute febrile neutrophilic dermatosis, panniculitis, palmar-plantar erythrodysesthesia syndrome.
Respiratory, Thoracic, and Mediastinal Disorders: 1%–<10% – cough, lung infiltration, pneumonitis, pulmonary hypertension; 0.1%–<1% – asthma, bronchospasm; <0.1% – acute respiratory distress syndrome.
Nervous System Disorders: 1%–<10% – neuropathy (including peripheral neuropathy), dizziness, dysgeusia, somnolence; 0.1%–<1% – amnesia, tremor, syncope; <0.1% – convulsion, cerebrovascular accident, transient ischemic attack, optic neuritis.
Blood and Lymphatic System Disorders: 1%–<10% – pancytopenia; <0.1% – aplasia pure red cell.
Musculoskeletal and Connective Tissue Disorders: 1%–<10% – muscular weakness; 0.1%–<1% – musculoskeletal stiffness, rhabdomyolysis; <0.1% – tendonitis.
Investigations: 1%–<10% – weight increased, weight decreased; 0.1%–<1% – blood creatine phosphokinase increased.
Infections and Infestations: 1%–<10% – pneumonia (including bacterial, viral, and fungal), upper respiratory tract infection/inflammation, herpes virus infection, enterocolitis infection; 0.1%–<1% – sepsis (including fatal outcomes).
Metabolism and Nutrition Disorders: 1%–<10% – anorexia, appetite disturbances; 0.1%–<1% – hyperuricemia, hypoalbuminemia.
Cardiac Disorders: 1%–<10% – arrhythmia (including tachycardia), palpitations; 0.1%–<1% – angina pectoris, cardiomegaly, pericarditis, ventricular arrhythmia (including ventricular tachycardia); <0.1% – cor pulmonale, myocarditis, acute coronary syndrome.
Eye Disorders: 1%–<10% – visual disorder (including visual disturbance, vision blurred, and visual acuity reduced), dry eye; 0.1% –<1% – conjunctivitis.
Vascular Disorders: 1%–<10% – flushing, hypertension; 0.1%–<1% – hypotension, thrombophlebitis; <0.1% – livedo reticularis.
Psychiatric Disorders: 1%–<10% – insomnia, depression; 0.1%–<1% – anxiety, affect lability, confusional state, libido decreased.
Reproductive System and Breast Disorders: 0.1%–<1% – gynecomastia, menstruation irregular.
Injury, Poisoning, and Procedural Complications: 1%–<10% – contusion.
Ear and Labyrinth Disorders: 1%–<10% – tinnitus; 0.1%–<1% – vertigo.
Hepatobiliary Disorders: 0.1%–<1% – cholestasis, cholecystitis, hepatitis.
Renal and Urinary Disorders: 0.1%–<1% – urinary frequency, renal failure, proteinuria.
Neoplasms Benign, Malignant, and Unspecified: 0.1%–<1% – tumor lysis syndrome.
Immune System Disorders: 0.1%–<1% – hypersensitivity (including erythema nodosum).
6.4 Postmarketing Experience
The following additional adverse reactions have been identified during post approval use of SPRYCEL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders: atrial fibrillation/atrial flutter
Vascular disorders: thrombosis/embolism (including pulmonary embolism, deep vein thrombosis)
Respiratory, thoracic, and mediastinal disorders: interstitial lung disease, pulmonary arterial hypertension
-
7 DRUG INTERACTIONS
7.1 Drugs That May Increase Dasatinib Plasma Concentrations
CYP3A4 Inhibitors: Dasatinib is a CYP3A4 substrate. In a study of 18 patients with solid tumors, 20-mg SPRYCEL once daily coadministered with 200 mg of ketoconazole twice daily increased the dasatinib Cmax and AUC by four- and five-fold, respectively. Concomitant use of SPRYCEL and drugs that inhibit CYP3A4 may increase exposure to dasatinib and should be avoided. In patients receiving treatment with SPRYCEL, close monitoring for toxicity and a SPRYCEL dose reduction should be considered if systemic administration of a potent CYP3A4 inhibitor cannot be avoided [see Dosage and Administration (2.1)].
7.2 Drugs That May Decrease Dasatinib Plasma Concentrations
CYP3A4 Inducers: When a single morning dose of SPRYCEL was administered following 8 days of continuous evening administration of 600 mg of rifampin, a potent CYP3A4 inducer, the mean Cmax and AUC of dasatinib were decreased by 81% and 82%, respectively. Alternative agents with less enzyme induction potential should be considered. If SPRYCEL must be administered with a CYP3A4 inducer, a dose increase in SPRYCEL should be considered [see Dosage and Administration (2.1)].
Antacids: Nonclinical data demonstrate that the solubility of dasatinib is pH dependent. In a study of 24 healthy subjects, administration of 30 mL of aluminum hydroxide/magnesium hydroxide 2 hours prior to a single 50-mg dose of SPRYCEL was associated with no relevant change in dasatinib AUC; however, the dasatinib Cmax increased 26%. When 30 mL of aluminum hydroxide/magnesium hydroxide was administered to the same subjects concomitantly with a 50-mg dose of SPRYCEL, a 55% reduction in dasatinib AUC and a 58% reduction in Cmax were observed. Simultaneous administration of SPRYCEL with antacids should be avoided. If antacid therapy is needed, the antacid dose should be administered at least 2 hours prior to or 2 hours after the dose of SPRYCEL.
H2 Antagonists/Proton Pump Inhibitors: Long-term suppression of gastric acid secretion by H2 antagonists or proton pump inhibitors (eg, famotidine and omeprazole) is likely to reduce dasatinib exposure. In a study of 24 healthy subjects, administration of a single 50-mg dose of SPRYCEL 10 hours following famotidine reduced the AUC and Cmax of dasatinib by 61% and 63%, respectively. In a study of 14 healthy subjects, administration of a single 100-mg dose of SPRYCEL 22 hours following a 40-mg omeprazole dose at steady state reduced the AUC and Cmax of dasatinib by 43% and 42%, respectively. The concomitant use of H2 antagonists or proton pump inhibitors with SPRYCEL is not recommended. The use of antacids (at least 2 hours prior to or 2 hours after the dose of SPRYCEL) should be considered in place of H2 antagonists or proton pump inhibitors in patients receiving SPRYCEL therapy.
7.3 Drugs That May Have Their Plasma Concentration Altered By Dasatinib
CYP3A4 Substrates: Single-dose data from a study of 54 healthy subjects indicate that the mean Cmax and AUC of simvastatin, a CYP3A4 substrate, were increased by 37% and 20%, respectively, when simvastatin was administered in combination with a single 100-mg dose of SPRYCEL. Therefore, CYP3A4 substrates known to have a narrow therapeutic index such as alfentanil, astemizole, terfenadine, cisapride, cyclosporine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus, or ergot alkaloids (ergotamine, dihydroergotamine) should be administered with caution in patients receiving SPRYCEL.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D
SPRYCEL may cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of SPRYCEL in pregnant women. Women of childbearing potential should be advised of the potential hazard to the fetus and to avoid becoming pregnant. If SPRYCEL is used during pregnancy, or if the patient becomes pregnant while taking SPRYCEL, the patient should be apprised of the potential hazard to the fetus.
In nonclinical studies, at plasma concentrations below those observed in humans receiving therapeutic doses of dasatinib, embryo-fetal toxicities were observed in rats and rabbits. Fetal death was observed in rats. In both rats and rabbits, the lowest doses of dasatinib tested (rat: 2.5 mg/kg/day [15 mg/m2/day] and rabbit: 0.5 mg/kg/day [6 mg/m2/day]) resulted in embryo-fetal toxicities. These doses produced maternal AUCs of 105 ng•hr/mL (0.3-fold the human AUC in females at a dose of 70 mg twice daily) and 44 ng•hr/mL (0.1-fold the human AUC) in rats and rabbits, respectively. Embryo-fetal toxicities included skeletal malformations at multiple sites (scapula, humerus, femur, radius, ribs, and clavicle), reduced ossification (sternum; thoracic, lumbar, and sacral vertebrae; forepaw phalanges; pelvis; and hyoid body), edema, and microhepatia.
8.3 Nursing Mothers
It is unknown whether SPRYCEL is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from SPRYCEL, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and efficacy of SPRYCEL in patients less than 18 years of age have not been established.
8.5 Geriatric Use
In the newly diagnosed chronic phase CML study, 25 patients (10%) were 65 years of age and over and 7 patients (3%) were 75 years of age and over. Of the 2182 patients in clinical studies of SPRYCEL with resistance or intolerance to imatinib therapy, 547 (25%) were 65 years of age and over and 105 (5%) were 75 years of age and over. No differences in efficacy were observed between older and younger patients. Compared to patients under age 65 years, patients aged 65 years and older are more likely to experience toxicity.
8.6 Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of dasatinib was evaluated in healthy volunteers with normal liver function and patients with moderate (Child-Pugh class B) and severe (Child-Pugh class C) hepatic impairment. Compared to the healthy volunteers with normal hepatic function, the dose normalized pharmacokinetic parameters were decreased in the patients with hepatic impairment.
No dosage adjustment is necessary in patients with hepatic impairment [see Clinical Pharmacology (12.3)]. Caution is recommended when administering SPRYCEL to patients with hepatic impairment.
-
10 OVERDOSAGE
Experience with overdose of SPRYCEL in clinical studies is limited to isolated cases. Overdosage of 280 mg per day for 1 week was reported in two patients and both developed severe myelosuppression and bleeding. Since SPRYCEL is associated with severe myelosuppression [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)], patients who ingested more than the recommended dosage should be closely monitored for myelosuppression and given appropriate supportive treatment.
Acute overdose in animals was associated with cardiotoxicity. Evidence of cardiotoxicity included ventricular necrosis and valvular/ventricular/atrial hemorrhage at single doses ≥100 mg/kg (600 mg/m2) in rodents. There was a tendency for increased systolic and diastolic blood pressure in monkeys at single doses ≥10 mg/kg (120 mg/m2).
-
11 DESCRIPTION
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
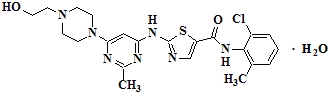
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol. SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dasatinib, at nanomolar concentrations, inhibits the following kinases: BCR-ABL, SRC family (SRC, LCK, YES, FYN), c-KIT, EPHA2, and PDGFRβ. Based on modeling studies, dasatinib is predicted to bind to multiple conformations of the ABL kinase.
In vitro, dasatinib was active in leukemic cell lines representing variants of imatinib mesylate sensitive and resistant disease. Dasatinib inhibited the growth of chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) cell lines overexpressing BCR-ABL. Under the conditions of the assays, dasatinib was able to overcome imatinib resistance resulting from BCR-ABL kinase domain mutations, activation of alternate signaling pathways involving the SRC family kinases (LYN, HCK), and multi-drug resistance gene overexpression.
12.3 Pharmacokinetics
Absorption
Maximum plasma concentrations (Cmax) of dasatinib are observed between 0.5 and 6 hours (Tmax) following oral administration. Dasatinib exhibits dose proportional increases in AUC and linear elimination characteristics over the dose range of 15 mg to 240 mg/day. The overall mean terminal half-life of dasatinib is 3–5 hours.
Data from a study of 54 healthy subjects administered a single, 100-mg dose of dasatinib 30 minutes following consumption of a high-fat meal resulted in a 14% increase in the mean AUC of dasatinib. The observed food effects were not clinically relevant.
Distribution
In patients, dasatinib has an apparent volume of distribution of 2505 L, suggesting that the drug is extensively distributed in the extravascular space. Binding of dasatinib and its active metabolite to human plasma proteins in vitro was approximately 96% and 93%, respectively, with no concentration dependence over the range of 100–500 ng/mL.
Metabolism
Dasatinib is extensively metabolized in humans, primarily by the cytochrome P450 enzyme 3A4. CYP3A4 was the primary enzyme responsible for the formation of the active metabolite. Flavin-containing monooxygenase 3 (FMO-3) and uridine diphosphate-glucuronosyltransferase (UGT) enzymes are also involved in the formation of dasatinib metabolites.
The exposure of the active metabolite, which is equipotent to dasatinib, represents approximately 5% of the dasatinib AUC. This indicates that the active metabolite of dasatinib is unlikely to play a major role in the observed pharmacology of the drug. Dasatinib also had several other inactive oxidative metabolites.
Dasatinib is a weak time-dependent inhibitor of CYP3A4. At clinically relevant concentrations, dasatinib does not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, or 2E1. Dasatinib is not an inducer of human CYP enzymes.
Elimination
Elimination is primarily via the feces. Following a single oral dose of [14C]-labeled dasatinib, approximately 4% and 85% of the administered radioactivity was recovered in the urine and feces, respectively, within 10 days. Unchanged dasatinib accounted for 0.1% and 19% of the administered dose in urine and feces, respectively, with the remainder of the dose being metabolites.
Effects of Age and Gender
Pharmacokinetic analyses of demographic data indicate that there are no clinically relevant effects of age and gender on the pharmacokinetics of dasatinib.
Hepatic Impairment
Dasatinib doses of 50 mg and 20 mg were evaluated in eight patients with moderate (Child-Pugh class B) and seven patients with severe (Child-Pugh class C) hepatic impairment, respectively. Matched controls with normal hepatic function (n=15) were also evaluated and received a dasatinib dose of 70 mg. Compared to subjects with normal liver function, patients with moderate hepatic impairment had decreases in dose normalized Cmax and AUC by 47% and 8%, respectively. Patients with severe hepatic impairment had dose normalized Cmax decreased by 43% and AUC decreased by 28% compared to the normal controls.
These differences in Cmax and AUC are not clinically relevant. Dose adjustment is not necessary in patients with hepatic impairment.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a two-year carcinogenicity study, rats were administered oral doses of dasatinib at 0.3, 1, and 3 mg/kg/day. The highest dose resulted in a plasma drug exposure (AUC) level equivalent to human exposure at 70 mg twice daily. Dasatinib induced a statistically significant increase in the combined incidence of squamous cell carcinomas and papillomas in the uterus and cervix of high-dose females and prostate adenoma in low-dose males.
Dasatinib was clastogenic when tested in vitro in Chinese hamster ovary cells, with and without metabolic activation. Dasatinib was not mutagenic when tested in an in vitro bacterial cell assay (Ames test) and was not genotoxic in an in vivo rat micronucleus study.
The effects of dasatinib on male and female fertility have not been studied. However, results of repeat-dose toxicity studies in multiple species indicate the potential for dasatinib to impair reproductive function and fertility. Effects evident in male animals included reduced size and secretion of seminal vesicles, and immature prostate, seminal vesicle, and testis. The administration of dasatinib resulted in uterine inflammation and mineralization in monkeys, and cystic ovaries and ovarian hypertrophy in rodents.
-
14 CLINICAL STUDIES
14.1 Newly Diagnosed Chronic Phase CML
An open-label, multicenter, international, randomized trial was conducted in adult patients with newly diagnosed chronic phase CML. A total of 519 patients were randomized to receive either SPRYCEL 100 mg once daily or imatinib 400 mg once daily. The primary endpoint was the rate of confirmed complete cytogenetic response (CCyR) within 12 months. Confirmed CCyR was defined as a CCyR noted on two consecutive occasions (at least 28 days apart).
Median age was 46 years in the SPRYCEL group and 49 years in the imatinib groups, with 10% and 11% of patients ≥65 years of age. There were slightly more male than female patients in both groups (59% vs 41%). Fifty-three percent of all patients were Caucasian, and 39% were Asian. At baseline, the distribution of Hasford Scores was similar in the SPRYCEL and imatinib treatment groups (low risk: 33% and 34%; intermediate risk: 48% and 47%; high risk: 19% and 19%, respectively).
The median duration of treatment was 14 months for SPRYCEL and 14 months for imatinib. With a minimum of 12 months follow-up, 85% of patients randomized to SPRYCEL and 81% of patients randomized to imatinib were still on study.
Efficacy results are summarized in Table 6.
Table 6: Efficacy Results in Newly Diagnosed Patients with Chronic Phase CML SPRYCEL
(n=259)Imatinib
(n=260)p-value Response rate (95% CI) a Confirmed CCyR is defined as a CCyR noted on two consecutive occasions at least 28 days apart. b Major molecular response (at any time) was defined as BCR-ABL ratios ≤0.1% by RQ-PCR in peripheral blood samples standardized on the International scale. *Adjusted for Hasford Score and indicated statistical significance at a pre-defined nominal level of significance. CI = confidence interval. Confirmed CCyR within 12 monthsa 76.8% (71.2–81.8) 66.2% (60.1–71.9) p=0.007* Major Molecular Responseb 52.1% (45.9–58.3) 33.8% (28.1–39.9) p<0.0001* Median time to confirmed CCyR was 3.1 months in 199 SPRYCEL responders and 5.6 months in 177 imatinib responders. Median time to MMR was 6.3 months in 135 SPRYCEL responders and 9.2 months in 88 imatinib responders.
Five patients on the dasatinib arm progressed to either accelerated phase or blast crisis while nine patients on the imatinib arm progressed to either accelerated phase or blast crisis.
14.2 Imatinib Resistant or Intolerant CML or Ph+ ALL
The efficacy and safety of SPRYCEL were investigated in adult patients with CML or Ph+ ALL whose disease was resistant to or who were intolerant to imatinib: 1158 patients had chronic phase CML, 858 patients had accelerated phase, myeloid blast phase, or lymphoid blast phase CML, and 130 patients had Ph+ ALL. In a clinical study in chronic phase CML, resistance to imatinib was defined as failure to achieve a complete hematologic response (CHR; after 3 months), major cytogenetic response (MCyR; after 6 months), or complete cytogenetic response (CCyR; after 12 months); or loss of a previous molecular response (with concurrent ≥10% increase in Ph+ metaphases), cytogenetic response, or hematologic response. Imatinib intolerance was defined as inability to tolerate 400 mg or more of imatinib per day or discontinuation of imatinib because of toxicity.
Results described below are based on a minimum of 2 years follow-up after the start of SPRYCEL therapy in patients with a median time from initial diagnosis of approximately 5 years. Across all studies, 48% of patients were women, 81% were white, 15% were black or Asian, 25% were 65 years of age or older, and 5% were 75 years of age or older. Most patients had long disease histories with extensive prior treatment, including imatinib, cytotoxic chemotherapy, interferon, and stem cell transplant. Overall, 80% of patients had imatinib-resistant disease and 20% of patients were intolerant to imatinib. The maximum imatinib dose had been 400–600 mg/day in about 60% of the patients and >600 mg/day in 40% of the patients.
The primary efficacy endpoint in chronic phase CML was MCyR, defined as elimination (CCyR) or substantial diminution (by at least 65%, partial cytogenetic response) of Ph+ hematopoietic cells. The primary efficacy endpoint in accelerated phase, myeloid blast phase, lymphoid blast phase CML, and Ph+ ALL was major hematologic response (MaHR), defined as either a CHR or no evidence of leukemia (NEL).
Chronic Phase CML
Dose-Optimization Study: A randomized, open-label study was conducted in patients with chronic phase CML to evaluate the efficacy and safety of SPRYCEL administered once daily compared with SPRYCEL administered twice daily. Patients with significant cardiac diseases, including myocardial infarction within 6 months, congestive heart failure within 3 months, significant arrhythmias, or QTc prolongation were excluded from the study. The primary efficacy endpoint was MCyR in patients with imatinib-resistant CML. A total of 670 patients, of whom 497 had imatinib-resistant disease, were randomized to the SPRYCEL 100 mg once daily, 140 mg once daily, 50 mg twice daily, or 70 mg twice daily group. Median duration of treatment was 22 months.
Efficacy was achieved across all SPRYCEL treatment groups with the once daily schedule demonstrating comparable efficacy (non-inferiority) to the twice daily schedule on the primary efficacy endpoint (difference in MCyR 1.9%; 95% CI [-6.8%–10.6%]).
Efficacy results are presented in Table 7 for patients with chronic phase CML who received the recommended starting dose of 100 mg once daily. Additional efficacy results in this patient population are described after the table. Results for all patients with chronic phase CML, regardless of dosage (a starting dosage of 100 mg once daily, 140 mg once daily, 50 mg twice daily, or 70 mg twice daily), were consistent with those for patients treated with 100 mg once daily.
Table 7: Efficacy of SPRYCEL in Imatinib Resistant or Intolerant Chronic Phase CML 100 mg Once Daily
(n=167)a CHR (response confirmed after 4 weeks): WBC ≤ institutional ULN, platelets <450,000/mm3, no blasts or promyelocytes in peripheral blood, <5% myelocytes plus metamyelocytes in peripheral blood, basophils in peripheral blood <20%, and no extramedullary involvement. b MCyR combines both complete (0% Ph+ metaphases) and partial (>0%–35%) responses. CHRa% (95% CI) 92% (86–95) MCyRb% (95% CI) 63% (56–71) CCyR% (95% CI) 50% (42–58) In the SPRYCEL 100 mg once daily group, median time to MCyR was 2.9 months (95% CI: [2.8–3.0]). Based on the Kaplan-Meier estimates, 93% (95% CI: [88%–98%]) of patients who had achieved an MCyR maintained that response for 18 months. The estimated rate of progression-free survival and overall survival in all patients treated with 100 mg once daily was 80% (95% CI: [73%–87%]) and 91% (95% CI: [86%–96%]), respectively, at 2 years.
Advanced Phase CML and Ph+ ALL
Dose-Optimization Study: One randomized open-label study was conducted in patients with advanced phase CML (accelerated phase CML, myeloid blast phase CML, or lymphoid blast phase CML) to evaluate the efficacy and safety of SPRYCEL administered once daily compared with SPRYCEL administered twice daily. The primary efficacy endpoint was MaHR. A total of 611 patients were randomized to either the SPRYCEL 140 mg once daily or 70 mg twice daily group. Median duration of treatment was approximately 6 months for both treatment groups. The once daily schedule demonstrated comparable efficacy (non-inferiority) to the twice daily schedule on the primary efficacy endpoint.
The efficacy and safety of SPRYCEL were also investigated in patients with Ph+ ALL in one randomized study (starting dosage 140 mg once daily or 70 mg twice daily) and one single-arm study (starting dosage 70 mg twice daily). The primary efficacy endpoint was MaHR. A total of 130 patients were enrolled in these studies. The median duration of therapy was 3 months.
Response rates are presented in Table 8.
Table 8: Efficacy of SPRYCEL in Imatinib Resistant or Intolerant Advanced Phase CML and Ph+ ALL 140 mg Once Daily Accelerated
(n=158)Myeloid Blast
(n=75)Lymphoid Blast
(n=33)Ph+ ALL
(n=40)a Hematologic response criteria (all responses confirmed after 4 weeks): Major hematologic response: (MaHR) = complete hematologic response (CHR) + no evidence of leukemia (NEL).
CHR: WBC ≤ institutional ULN, ANC ≥1000/mm3, platelets ≥100,000/mm3, no blasts or promyelocytes in peripheral blood, bone marrow blasts ≤5%, <5% myelocytes plus metamyelocytes in peripheral blood, basophils in peripheral blood <20%, and no extramedullary involvement.
NEL: same criteria as for CHR but ANC ≥500/mm3 and <1000/mm3, or platelets ≥20,000/mm3 and ≤100,000/mm3.b MCyR combines both complete (0% Ph+ metaphases) and partial (>0%–35%) responses. CI = confidence interval ULN = upper limit of normal range. MaHRa 66% 28% 42% 38% (95% CI) (59–74) (18–40) (26–61) (23–54) CHRa 47% 17% 21% 33% (95% CI) (40–56) (10–28) (9–39) (19–49) NELa 19% 11% 21% 5% (95% CI) (13–26) (5–20) (9–39) (1–17) MCyRb 39% 28% 52% 70% (95% CI) (31–47) (18–40) (34–69) (54–83) CCyR 32% 17% 39% 50% (95% CI) (25–40) (10–28) (23–58) (34–66) In the SPRYCEL 140 mg once daily group, the median time to MaHR was 1.9 months for patients with accelerated phase CML, 1.9 months for patients with myeloid blast phase CML, and 1.8 months for patients with lymphoid blast phase CML.
In patients with myeloid blast phase CML, the median duration of MaHR was 8 months and 9 months for the 140 mg once daily group and the 70 mg twice daily group, respectively. In patients with lymphoid blast phase CML, the median duration of MaHR was 5 months and 8 months for the 140 mg once daily group and the 70 mg twice daily group, respectively. In patients with Ph+ ALL who were treated with SPRYCEL 140 mg once daily, the median duration of MaHR was 4.6 months. The medians of progression-free survival for patients with Ph+ ALL treated with SPRYCEL 140 mg once daily and 70 mg twice daily were 4.0 months and 3.5 months, respectively.
-
15 REFERENCES
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004–165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999, http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html.
- American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. (2006) 63:1172–1193.
- Polovich M, White JM, Kelleher LO (eds). 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd ed). Pittsburgh, PA: Oncology Nursing Society.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SPRYCEL® (dasatinib) tablets are available as described in Table 9.
Table 9: SPRYCEL Trade Presentations NDC Number Strength Description Tablets per Bottle 54868-5759-0 70 mg white to off-white, biconvex, round, film-coated tablet with “BMS” debossed on one side and “524” on the other side 60 16.2 Storage
SPRYCEL® tablets should be stored at 20° to 25°C (68° to 77°F); excursions permitted between 15°–30°C (59°–86°F) [see USP Controlled Room Temperature].
16.3 Handling and Disposal
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published [see References (15)].
SPRYCEL (dasatinib) tablets consist of a core tablet (containing the active drug substance), surrounded by a film coating to prevent exposure of pharmacy and clinical personnel to the active drug substance. However, if tablets are inadvertently crushed or broken, pharmacy and clinical personnel should wear disposable chemotherapy gloves. Personnel who are pregnant should avoid exposure to crushed or broken tablets.
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling.
17.1 Bleeding
Patients should be informed of the possibility of serious bleeding and to report immediately any signs or symptoms suggestive of hemorrhage (unusual bleeding or easy bruising).
17.2 Myelosuppression
Patients should be informed of the possibility of developing low blood cell counts; they should be instructed to report immediately should fever develop, particularly in association with any suggestion of infection.
17.3 Fluid Retention
Patients should be informed of the possibility of developing fluid retention (swelling, weight gain, or shortness of breath) and to seek medical attention if those symptoms arise.
17.4 Pregnancy
Patients should be informed that dasatinib may cause fetal harm when administered to a pregnant woman. Women should be advised of the potential hazard to the fetus and to avoid becoming pregnant. If SPRYCEL is used during pregnancy, or if the patient becomes pregnant while taking SPRYCEL, the patient should be apprised of the potential hazard to the fetus [see Warnings and Precautions (5.7)].
17.5 Gastrointestinal Complaints
Patients should be informed that they may experience nausea, vomiting, or diarrhea with SPRYCEL. If these symptoms are significant, they should seek medical attention.
17.6 Pain
Patients should be informed that they may experience headache or musculoskeletal pain with SPRYCEL. If these symptoms are significant, they should seek medical attention.
17.7 Fatigue
Patients should be informed that they may experience fatigue with SPRYCEL. If this symptom is significant, they should seek medical attention.
17.8 Rash
Patients should be informed that they may experience skin rash with SPRYCEL. If this symptom is significant, they should seek medical attention.
-
PATIENT PACKAGE INSERT
FDA-Approved Patient Labeling
PATIENT INFORMATION
SPRYCEL® (Spry-sell)
(dasatinib)
Tablets
Read the Patient Information that comes with SPRYCEL before you start taking it and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is SPRYCEL?
SPRYCEL® is a prescription medicine used to treat adults who have:
- newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.
- Ph+ CML who no longer benefit from, or did not tolerate, other treatment, including Gleevec® (imatinib mesylate).
- Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) who no longer benefit from, or did not tolerate, other treatment.
It is not known if SPRYCEL is safe and effective in children younger than 18 years old.
What should I tell my healthcare provider before taking SPRYCEL?
Before you take SPRYCEL, tell your healthcare provider if you:
- have problems with your immune system
- have liver problems
- have heart problems
- are lactose intolerant
- have any other medical conditions
- are pregnant or planning to become pregnant. SPRYCEL may harm your unborn baby. Women should not become pregnant while taking SPRYCEL. Talk to your healthcare provider right away if you are pregnant or plan to become pregnant.
- are breastfeeding or plan to breastfeed. It is not known if SPRYCEL passes into your breast milk or if it can harm your baby. You and your healthcare provider should decide if you will take SPRYCEL or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, antacids, and herbal supplements.
Especially tell your healthcare provider if you take:
- medicines that increase the amount of SPRYCEL in your bloodstream, such as:
Nizoral® (ketoconazole), Nefazodone (serzone, nefadar), Sporanox® (itraconazole), Invirase® (saquinavir), Norvir® (ritonavir), Ketek® (telithromycin), Reyataz® (atazanavir sulfate), E-mycin® (erythromycin), Crixivan® (indinavir), Biaxin® (clarithromycin). Viracept® (nelfinavir), - medicines that decrease the amount of SPRYCEL in your bloodstream, such as:
Decadron® (dexamethasone), Rimactane® (rifampin), Dilantin® (phenytoin), Luminal® (phenobarbital). Tegretol® (carbamazepine), - medicines whose blood levels might change by taking SPRYCEL, such as:
Sandimmune® (cyclosporine), Rapamune® (sirolimus), Alfenta® (alfentanil), Prograf® (tacrolimus), Fentanyl® (fentanyl), Ergomar® (ergotamine). Orap® (pimozide), SPRYCEL is best absorbed from your stomach into your bloodstream in the presence of stomach acid. You should avoid taking medicines that reduce stomach acid, such as:
Tagamet® (cimetidine), Protonix® (pantoprazole sodium), Pepcid® (famotidine), Nexium® (esomeprazole), Zantac® (ranitidine), AcipHex® (rabeprazole), Prilosec® (omeprazole), Prevacid® (lansoprazole). Medicines that neutralize stomach acid, such as Maalox® (aluminum hydroxide/magnesium hydroxide), Tums® (calcium carbonate), or Rolaids® (calcium carbonate and magnesia), may be taken up to 2 hours before or 2 hours after SPRYCEL.
Since SPRYCEL therapy may cause bleeding, tell your healthcare provider if you are using blood thinner medicine, such as Coumadin® (warfarin sodium) or aspirin.
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take SPRYCEL?
Take SPRYCEL exactly as prescribed by your healthcare provider.
- Take SPRYCEL with or without food. Try to take SPRYCEL at the same time each day.
- Swallow SPRYCEL tablets whole. Do not break, cut, or crush the tablets.
- You should not drink grapefruit juice while taking SPRYCEL.
-
Your healthcare provider may:
- change your dose of SPRYCEL or
- tell you to temporarily stop taking SPRYCEL.
- Do not change your dose or stop taking SPRYCEL without first talking with your healthcare provider.
- If you miss a dose of SPRYCEL, take your next scheduled dose at its regular time. Do not take two doses at the same time. Call your healthcare provider or pharmacist if you are not sure what to do.
- If you take too much SPRYCEL, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of SPRYCEL?
SPRYCEL may cause serious side effects, including:
- Low Blood Cell Counts: SPRYCEL may cause low red blood cell counts (anemia), low white blood cell counts (neutropenia), and low platelet counts (thrombocytopenia). Your healthcare provider will do blood tests to check your blood cell counts regularly during your treatment with SPRYCEL. Call your healthcare provider right away if you have a fever or any signs of an infection while taking SPRYCEL.
-
Bleeding: SPRYCEL may cause severe bleeding that can lead to death. Call your healthcare provider right away if you have:
- unusual bleeding or bruising of your skin
- bright red or dark tar-like stools
- a decrease in your level of consciousness, headache, or change in speech.
-
Your body may hold too much fluid (fluid retention): In severe cases, fluid may build up in the lining of your lungs, the sac around your heart, or your stomach cavity. Call your healthcare provider right away if you get any of these symptoms during treatment with SPRYCEL:
- swelling all over your body
- weight gain
- shortness of breath and cough.
- Heart problems. SPRYCEL may cause an abnormal heart rate, heart problems or a heart attack. It may also cause changes in the blood vessels supplying the lungs. Your healthcare provider will monitor the potassium and magnesium levels in your blood, and your heart function.
- Pulmonary Arterial Hypertension (PAH). SPRYCEL may cause high blood pressure in the vessels of your lungs. PAH may happen at anytime during your treatment with SPRYCEL. Your healthcare provider should check your heart and lungs before and during your treatment with SPRYCEL. Call your healthcare provider right away if you have shortness of breath, tiredness, or swelling all over your body (fluid retention).
Other common side effects of SPRYCEL therapy include:
• diarrhea • tiredness • headache • vomiting • cough • muscle pain • skin rash • weakness • fever • infections • nausea Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of SPRYCEL. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SPRYCEL?
- Store SPRYCEL at room temperature, between 68°F to 77°F (20°C to 25°C).
- Ask your healthcare provider or pharmacist about the right way to throw away outdated or unused SPRYCEL.
- Women who are pregnant should not handle crushed or broken SPRYCEL tablets.
- Keep SPRYCEL and all medicines out of the reach of children and pets.
General information about SPRYCEL
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information leaflet. Do not use SPRYCEL for a condition for which it is not prescribed. Do not give SPRYCEL to other people even if they have the same symptoms you have. It may harm them.
This Patient Information leaflet summarizes the most important information about SPRYCEL. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about SPRYCEL that is written for healthcare professionals.
For more information, go to www.sprycel.com or call 1-800-332-2056.
What are the ingredients in SPRYCEL?
Active ingredient: dasatinib
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
This Patient Package Insert has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USARevised: October 2011
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA1284903A0
Rev October 2011Additional barcode labeling by:
Physicians Total Care, Inc.
Tulsa, Oklahoma 74146
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SPRYCEL
dasatinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5759(NDC:0003-0524) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength dasatinib (UNII: RBZ1571X5H) (dasatinib - UNII:RBZ1571X5H) dasatinib 70 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) hydroxypropyl cellulose (UNII: RFW2ET671P) hypromelloses (UNII: 3NXW29V3WO) polyethylene glycols (UNII: 3WJQ0SDW1A) lactose monohydrate (UNII: EWQ57Q8I5X) cellulose, microcrystalline (UNII: OP1R32D61U) hydroxypropyl cellulose (UNII: RFW2ET671P) magnesium stearate (UNII: 70097M6I30) titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color WHITE Score no score Shape ROUND (biconvex) Size 9mm Flavor Imprint Code BMS;524 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5759-0 1 in 1 CARTON 1 60 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021986 02/12/2007 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel
