DILANTIN
-
phenytoin sodium capsule, extended release
Parke-Davis
----------
Dilantin®
(Phenytoin Sodium) 100 mg Extended Oral Capsule
DESCRIPTION
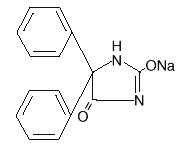
Phenytoin sodium is an antiepileptic drug. Phenytoin sodium is related to the barbiturates in chemical structure, but has a five-membered ring. The chemical name is sodium 5,5-diphenyl-2, 4-imidazolidinedione, having the following structural formula:
Each Dilantin— 100 mg Extended Oral Capsule—contains 100 mg phenytoin sodium. Also contains lactose monohydrate, NF; confectioner's sugar, NF; talc, USP; and magnesium stearate, NF. The capsule body contains titanium dioxide, USP and gelatin, NF. The capsule cap contains FD&C red No. 28; FD&C yellow No. 6; and gelatin NF. Product in vivo performance is characterized by a slow and extended rate of absorption with peak blood concentrations expected in 4 to 12 hours as contrasted to Prompt Phenytoin Sodium Capsules, USP with a rapid rate of absorption with peak blood concentration expected in 1½ to 3 hours.
CLINICAL PHARMACOLOGY
Phenytoin is an antiepileptic drug which can be used in the treatment of epilepsy. The primary site of action appears to be the motor cortex where spread of seizure activity is inhibited. Possibly by promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of posttetanic potentiation at synapses. Loss of posttetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas. Phenytoin reduces the maximal activity of brain stem centers responsible for the tonic phase of tonic-clonic (grand mal) seizures.
The plasma half-life in man after oral administration of phenytoin averages 22 hours, with a range of 7 to 42 hours. Steady-state therapeutic levels are achieved at least 7 to 10 days (5–7 half-lives) after initiation of therapy with recommended doses of 300 mg/day.
When serum level determinations are necessary, they should be obtained at least 5–7 half-lives after treatment initiation, dosage change, or addition or subtraction of another drug to the regimen so that equilibrium or steady-state will have been achieved. Trough levels provide information about clinically effective serum level range and confirm patient compliance and are obtained just prior to the patient's next scheduled dose. Peak levels indicate an individual's threshold for emergence of dose-related side effects and are obtained at the time of expected peak concentration. For Dilantin capsules, peak serum levels occur 4 to 12 hours after administration.
Optimum control without clinical signs of toxicity occurs more often with serum levels between 10 and 20 mcg/mL, although some mild cases of tonic-clonic (grand mal) epilepsy may be controlled with lower serum levels of phenytoin.
In most patients maintained at a steady dosage, stable phenytoin serum levels are achieved. There may be wide interpatient variability in phenytoin serum levels with equivalent dosages. Patients with unusually low levels may be noncompliant or hypermetabolizers of phenytoin. Unusually high levels result from liver disease, congenital enzyme deficiency, or drug interactions which result in metabolic interference. The patient with large variations in phenytoin plasma levels, despite standard doses, presents a difficult clinical problem. Serum level determinations in such patients may be particularly helpful. As phenytoin is highly protein bound, free phenytoin levels may be altered in patients whose protein binding characteristics differ from normal.
Most of the drug is excreted in the bile as inactive metabolites which are then reabsorbed from the intestinal tract and excreted in the urine. Urinary excretion of phenytoin and its metabolites occurs partly with glomerular filtration but more importantly by tubular secretion. Because phenytoin is hydroxylated in the liver by an enzyme system which is saturable at high plasma levels, small incremental doses may increase the half-life and produce very substantial increases in serum levels, when these are in the upper range. The steady-state level may be disproportionately increased, with resultant intoxication, from an increase in dosage of 10% or more.
INDICATIONS AND USAGE
Dilantin is indicated for the control of generalized tonic-clonic (grand mal) and complex partial (psychomotor, temporal lobe) seizures and prevention and treatment of seizures occurring during or following neurosurgery.
Phenytoin serum level determinations may be necessary for optimal dosage adjustments (see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY sections).
CONTRAINDICATIONS
Phenytoin is contraindicated in those patients who are hypersensitive to phenytoin or other hydantoins.
WARNINGS
Abrupt withdrawal of phenytoin in epileptic patients may precipitate status epilepticus. When, in the judgment of the clinician, the need for dosage reduction, discontinuation, or substitution of alternative antiepileptic medication arises, this should be done gradually. However, in the event of an allergic or hypersensitivity reaction, rapid substitution of alternative therapy may be necessary. In this case, alternative therapy should be an antiepileptic drug not belonging to the hydantoin chemical class.
There have been a number of reports suggesting a relationship between phenytoin and the development of lymphadenopathy (local or generalized) including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin's disease. Although a cause and effect relationship has not been established, the occurrence of lymphadenopathy indicates the need to differentiate such a condition from other types of lymph node pathology. Lymph node involvement may occur with or without symptoms and signs resembling serum sickness, e.g., fever, rash, and liver involvement.
In all cases of lymphadenopathy, follow-up observation for an extended period is indicated and every effort should be made to achieve seizure control using alternative antiepileptic drugs.
Acute alcoholic intake may increase phenytoin serum levels, while chronic alcohol use may decrease serum levels.
In view of isolated reports associating phenytoin with exacerbation of porphyria, caution should be exercised in using this medication in patients suffering from this disease.
Usage In Pregnancy
Clinical
- A.
-
Risks to Mother. An increase in seizure frequency may occur during pregnancy because of altered phenytoin pharmacokinetics. Periodic measurement of plasma phenytoin concentrations may be valuable in the management of pregnant women as a guide to appropriate adjustment of dosage (see PRECAUTIONS, Laboratory Tests). However, postpartum restoration of the original dosage will probably be indicated.
- B.
-
Risks to the Fetus. If this drug is used during pregnancy, or if the patient becomes pregnant while taking the drug, the patient should be apprised of the potential harm to the fetus.
Prenatal exposure to phenytoin may increase the risks for congenital malformations and other adverse developmental outcomes. Increased frequencies of major malformations (such as orofacial clefts and cardiac defects), minor anomalies (dysmorphic facial features, nail and digit hypoplasia), growth abnormalities (including microcephaly), and mental deficiency have been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy. There have also been several reported cases of malignancies, including neuroblastoma, in children whose mothers received phenytoin during pregnancy. The overall incidence of malformations for children of epileptic women treated with antiepileptic drugs (phenytoin and/or others) during pregnancy is about 10%, or two- to three-fold that in the general population. However, the relative contributions of antiepileptic drugs and other factors associated with epilepsy to this increased risk are uncertain and in most cases it has not been possible to attribute specific developmental abnormalities to particular antiepileptic drugs.
Patients should consult with their physicians to weigh the risks and benefits of phenytoin during pregnancy.
- C.
- Postpartum Period. A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.
Preclinical
Increased resorption and malformation rates have been reported following administration of phenytoin doses of 75 mg/kg or higher (approximately 120% of the maximum human loading dose or higher on a mg/m2 basis) to pregnant rabbits.
PRECAUTIONS
General
The liver is the chief site of biotransformation of phenytoin; patients with impaired liver function, elderly patients, or those who are gravely ill may show early signs of toxicity.
A small percentage of individuals who have been treated with phenytoin have been shown to metabolize the drug slowly. Slow metabolism may be due to limited enzyme availability and lack of induction; it appears to be genetically determined.
Phenytoin should be discontinued if a skin rash appears (see WARNINGS section regarding drug discontinuation). If the rash is exfoliative, purpuric, or bullous or if lupus erythematosus, Stevens-Johnson syndrome, or toxic epidermal necrolysis is suspected, use of this drug should not be resumed and alternative therapy should be considered. (See ADVERSE REACTIONS section.) If the rash is of a milder type (measles-like or scarlatiniform), therapy may be resumed after the rash has completely disappeared. If the rash recurs upon reinstitution of therapy, further phenytoin medication is contraindicated.
Phenytoin and other hydantoins are contraindicated in patients who have experienced phenytoin hypersensitivity (see CONTRAINDICATIONS). Additionally, caution should be exercised if using structurally similar compounds (e.g., barbiturates, succinimides, oxazolidinediones, and other related compounds) in these same patients.
Hyperglycemia, resulting from the drug's inhibitory effects on insulin release, has been reported. Phenytoin may also raise the serum glucose level in diabetic patients.
Osteomalacia has been associated with phenytoin therapy and is considered to be due to phenytoin's interference with vitamin D metabolism.
Phenytoin is not indicated for seizures due to hypoglycemic or other metabolic causes. Appropriate diagnostic procedures should be performed as indicated.
Phenytoin is not effective for absence (petit mal) seizures. If tonic-clonic (grand mal) and absence (petit mal) seizures are present, combined drug therapy is needed.
Serum levels of phenytoin sustained above the optimal range may produce confusional states referred to as "delirium," "psychosis," or "encephalopathy," or rarely irreversible cerebellar dysfunction. Accordingly, at the first sign of acute toxicity, plasma levels are recommended. Dose reduction of phenytoin therapy is indicated if plasma levels are excessive; if symptoms persist, termination is recommended. (See WARNINGS section.)
Information for Patients
Patients taking phenytoin should be advised of the importance of adhering strictly to the prescribed dosage regimen, and of informing the physician of any clinical condition in which it is not possible to take the drug orally as prescribed, e.g., surgery, etc.
Patients should also be cautioned on the use of other drugs or alcoholic beverages without first seeking the physician's advice.
Patients should be instructed to call their physician if skin rash develops.
The importance of good dental hygiene should be stressed in order to minimize the development of gingival hyperplasia and its complications.
Do not use capsules which are discolored.
Laboratory Tests
Phenytoin serum level determinations may be necessary to achieve optimal dosage adjustments.
Drug Interactions
There are many drugs which may increase or decrease phenytoin levels or which phenytoin may affect. Serum level determinations for phenytoin are especially helpful when possible drug interactions are suspected. The most commonly occurring drug interactions are listed below:
- Drugs which may increase phenytoin serum levels include: acute alcohol intake, amiodarone, chloramphenicol, chlordiazepoxide, cimetidine, diazepam, dicumarol, disulfiram, estrogens, ethosuximide, fluoxetine, H2-antagonists, halothane, isoniazid, methylphenidate, phenothiazines, phenylbutazone, salicylates, succinimides, sulfonamides, ticlopidine, tolbutamide, trazodone.
- Drugs which may decrease phenytoin levels include: carbamazepine, chronic alcohol abuse, reserpine, and sucralfate. Moban® brand of molindone hydrochloride contains calcium ions which interfere with the absorption of phenytoin. Ingestion times of phenytoin and antacid preparations containing calcium should be staggered in patients with low serum phenytoin levels to prevent absorption problems.
- Drugs which may either increase or decrease phenytoin serum levels include: phenobarbital, sodium valproate, and valproic acid. Similarly, the effect of phenytoin on phenobarbital, valproic acid, and sodium valproate serum levels is unpredictable.
- Although not a true drug interaction, tricyclic antidepressants may precipitate seizures in susceptible patients and phenytoin dosage may need to be adjusted.
- Drugs whose efficacy is impaired by phenytoin include: corticosteroids, coumarin anticoagulants, digitoxin, doxycycline, estrogens, furosemide, oral contraceptives, paroxetine, quinidine, rifampin, theophylline, vitamin D.
Drug Enteral Feeding/Nutritional Preparations Interaction
Literature reports suggest that patients who have received enteral feeding preparations and/or related nutritional supplements have lower than expected phenytoin plasma levels. It is therefore suggested that phenytoin not be administered concomitantly with an enteral feeding preparation. More frequent serum phenytoin level monitoring may be necessary in these patients.
Drug/Laboratory Test Interactions
Phenytoin may decrease serum concentrations of T4. It may also produce lower than normal values for dexamethasone or metyrapone tests. Phenytoin may cause increased serum levels of glucose, alkaline phosphatase, and gamma glutamyl transpeptidase (GGT).
Care should be taken when using immunoanalytical methods to measure plasma phenytoin concentrations.
Carcinogenesis
See WARNINGS section for information on carcinogenesis.
Pregnancy
Pregnancy Category D
See WARNINGS section.
Nursing Mothers
Infant breast-feeding is not recommended for women taking this drug because phenytoin appears to be secreted in low concentrations in human milk.
Pediatric Use
See DOSAGE AND ADMINISTRATION section.
ADVERSE REACTIONS
Central Nervous System
The most common manifestations encountered with phenytoin therapy are referable to this system and are usually dose-related. These include nystagmus, ataxia, slurred speech, decreased coordination, and mental confusion. Dizziness, insomnia, transient nervousness, motor twitchings, and headaches have also been observed. There have also been rare reports of phenytoin-induced dyskinesias, including chorea, dystonia, tremor, and asterixis, similar to those induced by phenothiazine and other neuroleptic drugs.
A predominantly sensory peripheral polyneuropathy has been observed in patients receiving long-term phenytoin therapy.
Gastrointestinal System
Nausea, vomiting, constipation, toxic hepatitis, and liver damage.
Integumentary System
Dermatological manifestations sometimes accompanied by fever have included scarlatiniform or morbilliform rashes. A morbilliform rash (measles-like) is the most common; other types of dermatitis are seen more rarely. Other more serious forms which may be fatal have included bullous, exfoliative or purpuric dermatitis, lupus erythematosus, Stevens-Johnson syndrome, and toxic epidermal necrolysis (see PRECAUTIONS section).
Hemopoietic System
Hemopoietic complications, some fatal, have occasionally been reported in association with administration of phenytoin. These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression. While macrocytosis and megaloblastic anemia have occurred, these conditions usually respond to folic acid therapy. Lymphadenopathy including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin's disease have been reported (see WARNINGS section).
Connective Tissue System
Coarsening of the facial features, enlargement of the lips, gingival hyperplasia, hypertrichosis, and Peyronie's disease.
Immunologic
Hypersensitivity syndrome (which may include, but is not limited to, symptoms such as arthralgias, eosinophilia, fever, liver dysfunction, lymphadenopathy, or rash), systemic lupus erythematosus, periarteritis nodosa and immunoglobulin abnormalities.
OVERDOSAGE
The lethal dose in pediatric patients is not known. The lethal dose in adults is estimated to be 2 to 5 grams. The initial symptoms are nystagmus, ataxia, and dysarthria. Other signs are tremor, hyperreflexia, lethargy, slurred speech, nausea, vomiting. The patient may become comatose and hypotensive. Death is due to respiratory and circulatory depression.
There are marked variations among individuals with respect to phenytoin plasma levels where toxicity may occur. Nystagmus, on lateral gaze, usually appears at 20 mcg/mL, ataxia at 30 mcg/mL; dysarthria and lethargy appear when the plasma concentration is over 40 mcg/mL, but as high a concentration as 50 mcg/mL has been reported without evidence of toxicity. As much as 25 times the therapeutic dose has been taken to result in a serum concentration over 100 mcg/mL with complete recovery.
Treatment
Treatment is nonspecific since there is no known antidote.
The adequacy of the respiratory and circulatory systems should be carefully observed and appropriate supportive measures employed. Hemodialysis can be considered since phenytoin is not completely bound to plasma proteins. Total exchange transfusion has been used in the treatment of severe intoxication in pediatric patients.
In acute overdosage, the possibility of other CNS depressants, including alcohol, should be borne in mind.
DOSAGE AND ADMINISTRATION
Serum concentrations should be monitored in changing from Phenytoin Sodium Extended Oral Capsules (Dilantin) to Prompt Phenytoin Sodium Capsules, USP, and from the sodium salt to the free acid form.
Dilantin® capsules are formulated with the sodium salt of phenytoin. The free acid form of phenytoin is used in Dilantin-125 Suspension and Dilantin Infatabs. Because there is approximately an 8% increase in drug content with the free acid form over that of the sodium salt, dosage adjustments and serum level monitoring may be necessary when switching from a product formulated with the free acid to a product formulated with the sodium salt and vice versa.
General
Dosage should be individualized to provide maximum benefit. In some cases, serum blood level determinations may be necessary for optimal dosage adjustments—the clinically effective serum level is usually 10–20 mcg/mL. With recommended dosage, a period of seven to ten days may be required to achieve steady-state blood levels with phenytoin and changes in dosage (increase or decrease) should not be carried out at intervals shorter than seven to ten days.
Adult Dosage
Divided daily dosage
Patients who have received no previous treatment may be started on one 100-mg Dilantin (Phenytoin sodium) Extended Oral Capsule three times daily and the dosage then adjusted to suit individual requirements. For most adults, the satisfactory maintenance dosage will be one capsule three to four times a day. An increase up to two capsules three times a day may be made, if necessary.
Once-a-day dosage
In adults, if seizure control is established with divided doses of three 100-mg Dilantin capsules daily, once-a-day dosage with 300 mg of extended phenytoin sodium capsules may be considered. Studies comparing divided doses of 300 mg with a single daily dose of this quantity indicated absorption, peak plasma levels, biologic half-life, difference between peak and minimum values, and urinary recovery were equivalent. Once-a-day dosage offers a convenience to the individual patient or to nursing personnel for institutionalized patients and is intended to be used only for patients requiring this amount of drug daily. A major problem in motivating noncompliant patients may also be lessened when the patient can take this drug once a day. However, patients should be cautioned not to miss a dose, inadvertently.
Only extended phenytoin sodium capsules are recommended for once-a-day dosing. Inherent differences in dissolution characteristics and resultant absorption rates of phenytoin due to different manufacturing procedures and/or dosage forms preclude such recommendation for other phenytoin products. When a change in the dosage form or brand is prescribed, careful monitoring of phenytoin serum levels should be carried out.
Loading dose
Some authorities have advocated use of an oral loading dose of phenytoin in adults who require rapid steady-state serum levels and where intravenous administration is not desirable. This dosing regimen should be reserved for patients in a clinic or hospital setting where phenytoin serum levels can be closely monitored. Patients with a history of renal or liver disease should not receive the oral loading regimen.
Initially, one gram of phenytoin capsules is divided into three doses (400 mg, 300 mg, 300 mg) and administered at two-hour intervals. Normal maintenance dosage is then instituted 24 hours after the loading dose, with frequent serum level determinations.
Pediatric Dosage
Initially, 5 mg/kg/day in two or three equally divided doses, with subsequent dosage individualized to a maximum of 300 mg daily. A recommended daily maintenance dosage is usually 4 to 8 mg/kg. Children over 6 years old and adolescents may require the minimum adult dose (300 mg/day).
HOW SUPPLIED
Hard, filled No. 3 capsules containing a white powder. The medium orange cap having "PD" printed in black ink and the white, opaque body having "DILANTIN" over "100 mg" printed in black ink.
100's (NDC 0071-0369-24)
1,000's (NDC 0071-0369-32)
Unit Dose 100's (NDC 0071-0369-40)
Store at 20–25°C (68–77°F) [See USP Controlled Room Temperature]. Preserve in tight, light-resistant containers. Protect from moisture.
Rx only

LAB-0334-5.0
September 2007
| DILANTIN
phenytoin sodium capsule, extended release |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
Revised: 09/2008Parke-Davis