|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
To reduce the development of drug-resistant bacteria and maintain the effectiveness of LEVAQUIN® and other antibacterial drugs, LEVAQUIN® should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
LEVAQUIN® Tablets/Injection and Oral Solution are indicated for the treatment of adults (≥18 years of age) with mild, moderate, and severe infections caused by susceptible strains of the designated microorganisms in the conditions listed in this section. LEVAQUIN® Injection is indicated when intravenous administration offers a route of administration advantageous to the patient (e.g., patient cannot tolerate an oral dosage form).
Culture and susceptibility testing
Appropriate culture and susceptibility tests should be performed before treatment in order to isolate and identify organisms causing the infection and to determine their susceptibility to levofloxacin [see Clinical Pharmacology (12.4)]. Therapy with LEVAQUIN® may be initiated before results of these tests are known; once results become available, appropriate therapy should be selected.
As with other drugs in this class, some strains of Pseudomonas aeruginosa may develop resistance fairly rapidly during treatment with LEVAQUIN®. Culture and susceptibility testing performed periodically during therapy will provide information about the continued susceptibility of the pathogens to the antimicrobial agent and also the possible emergence of bacterial resistance.
1.1 Nosocomial Pneumonia
LEVAQUIN® is indicated for the treatment of nosocomial pneumonia due to methicillin-susceptible Staphylococcus aureus, Pseudomonas aeruginosa, Serratia marcescens, Escherichia coli, Klebsiella pneumoniae, Haemophilus influenzae, or Streptococcus pneumoniae. Adjunctive therapy should be used as clinically indicated. Where Pseudomonas aeruginosa is a documented or presumptive pathogen, combination therapy with an anti-pseudomonal β-lactam is recommended [see Clinical Studies (14.1)].
1.2 Community-Acquired Pneumonia: 7–14 day Treatment Regimen
LEVAQUIN® is indicated for the treatment of community-acquired pneumonia due to methicillin-susceptible Staphylococcus aureus, Streptococcus pneumoniae (including multi-drug-resistant Streptococcus pneumoniae [MDRSP]), Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella pneumoniae, Moraxella catarrhalis, Chlamydophila pneumoniae, Legionella pneumophila, or Mycoplasma pneumoniae [see Dosage and Administration (2.1) and Clinical Studies (14.2)].
MDRSP isolates are strains resistant to two or more of the following antibacterials: penicillin (MIC ≥2 mcg/mL), 2nd generation cephalosporins, e.g., cefuroxime, macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
1.3 Community-Acquired Pneumonia: 5-day Treatment Regimen
LEVAQUIN® is indicated for the treatment of community-acquired pneumonia due to Streptococcus pneumoniae (excluding multi-drug-resistant strains [MDRSP]), Haemophilus influenzae, Haemophilus parainfluenzae, Mycoplasma pneumoniae, or Chlamydophila pneumoniae [see Dosage and Administration (2.1) and Clinical Studies (14.3)].
1.4 Acute Bacterial Sinusitis: 5-day and 10–14 day Treatment Regimens
LEVAQUIN® is indicated for the treatment of acute bacterial sinusitis due to Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis [see Clinical Studies (14.4)].
1.5 Acute Bacterial Exacerbation of Chronic Bronchitis
LEVAQUIN® is indicated for the treatment of acute bacterial exacerbation of chronic bronchitis due to methicillin-susceptible Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, Haemophilus parainfluenzae, or Moraxella catarrhalis.
1.6 Complicated Skin and Skin Structure Infections
LEVAQUIN® is indicated for the treatment of complicated skin and skin structure infections due to methicillin-susceptible Staphylococcus aureus, Enterococcus faecalis, Streptococcus pyogenes, or Proteus mirabilis [see Clinical Studies (14.5)].
1.7 Uncomplicated Skin and Skin Structure Infections
LEVAQUIN® is indicated for the treatment of uncomplicated skin and skin structure infections (mild to moderate) including abscesses, cellulitis, furuncles, impetigo, pyoderma, wound infections, due to methicillin-susceptible Staphylococcus aureus , or Streptococcus pyogenes.
1.8 Chronic Bacterial Prostatitis
LEVAQUIN® is indicated for the treatment of chronic bacterial prostatitis due to Escherichia coli, Enterococcus faecalis, or methicillin-susceptible Staphylococcus epidermidis [see Clinical Studies (14.6)].
1.9 Complicated Urinary Tract Infections: 5-day Treatment Regimen
LEVAQUIN® is indicated for the treatment of complicated urinary tract infections due to Escherichia coli, Klebsiella pneumoniae, or Proteus mirabilis [see Clinical Studies (14.7)].
1.10 Complicated Urinary Tract Infections: 10-day Treatment Regimen
LEVAQUIN® is indicated for the treatment of complicated urinary tract infections (mild to moderate) due to Enterococcus faecalis, Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, or Pseudomonas aeruginosa [see Clinical Studies (14.8)].
1.11 Acute Pyelonephritis: 5 or 10-day Treatment Regimen
LEVAQUIN® is indicated for the treatment of acute pyelonephritis caused by Escherichia coli, including cases with concurrent bacteremia [see Clinical Studies (14.7, 14.8)] .
1.12 Uncomplicated Urinary Tract Infections
LEVAQUIN® is indicated for the treatment of uncomplicated urinary tract infections (mild to moderate) due to Escherichia coli, Klebsiella pneumoniae, or Staphylococcus saprophyticus.
1.13 Inhalational Anthrax (Post-Exposure)
LEVAQUIN® is indicated for inhalational anthrax (post-exposure) to reduce the incidenceor progression of disease following exposure to aerosolized Bacillus anthracis. The effectiveness of LEVAQUIN® is based on plasma concentrations achieved in humans, a surrogate endpoint reasonably likely to predict clinical benefit. LEVAQUIN® has not been tested in humans for the post-exposure prevention of inhalation anthrax. The safety of LEVAQUIN® in adults for durations of therapy beyond 28 days or in pediatric patients for durations of therapy beyond 14 days has not been studied. Prolonged LEVAQUIN® therapy should only be used when the benefit outweighs the risk [see Dosage and Administration (2.1, 2.2) and Clinical Studies (14.9)].
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adult Patients with Normal Renal Function
The usual dose of LEVAQUIN® Tablets or Oral Solution is 250 mg, 500 mg, or 750 mg administered orally every 24 hours, as indicated by infection and described in Table 1. The usual dose of LEVAQUIN® Injection is 250 mg or 500 mg administered by slow infusion over 60 minutes every 24 hours or 750 mg administered by slow infusion over 90 minutes every 24 hours, as indicated by infection and described in Table 1.
These recommendations apply to patients with creatinine clearance ≥ 50 mL/min. For patients with creatinine clearance <50 mL/min, adjustments to the dosing regimen are required [see Dosage and Administration (2.3)].
| Type of Infection* | Dosed Every 24 hours | Duration (days)† |
|---|---|---|
|
||
| Nosocomial Pneumonia | 750 mg | 7–14 |
| Community Acquired Pneumonia‡ | 500 mg | 7–14 |
| Community Acquired Pneumonia§ | 750 mg | 5 |
| Acute Bacterial Sinusitis | 750 mg | 5 |
| 500 mg | 10–14 | |
| Acute Bacterial Exacerbation of Chronic Bronchitis | 500 mg | 7 |
| Complicated Skin and Skin Structure Infections (SSSI) | 750 mg | 7–14 |
| Uncomplicated SSSI | 500 mg | 7–10 |
| Chronic Bacterial Prostatitis | 500 mg | 28 |
| Complicated Urinary
Tract Infection (cUTI) or Acute Pyelonephritis (AP)¶ | 750 mg | 5 |
| Complicated Urinary Tract Infection (cUTI) or Acute Pyelonephritis (AP)# | 250 mg | 10 |
| Uncomplicated Urinary Tract Infection | 250 mg | 3 |
| Inhalational Anthrax
(Post-Exposure), adult and pediatric patients > 50 kg and ≥ 6 months
of age Þ,ß
Pediatric patients < 50 kg and ≥ 6 months of age Þ,ß | 500 mg see Table 3 below (2.2) | 60ß
60ß |
2.2 Dosage in Pediatric Patients
The dosage in pediatric patients ≥ 6 months of age is described below in Table 2.
|
|||
| Type of infection * | Dose | Freq. Once every | Duration† |
| Inhalational Anthrax (post-exposure)ठ| |||
| Pediatric patients > 50 kg and ≥ 6 months of age | 500 mg | 24 hr | 60 days§ |
| Pediatric patients < 50 kg and ≥ 6 months of age | 8 mg/kg (not to exceed 250 mg per dose) | 12 hr | 60 days§ |
2.3 Dosage Adjustment in Adults with Renal Impairment
Administer LEVAQUIN® with caution in the presence of renal insufficiency. Careful clinical observation and appropriate laboratory studies should be performed prior to and during therapy since elimination of levofloxacin may be reduced.
No adjustment is necessary for patients with a creatinine clearance ≥ 50 mL/min.
In patients with impaired renal function (creatinine clearance<50 mL/min), adjustment of the dosage regimen is necessary to avoid the accumulation of levofloxacin due to decreased clearance [see Use in Specific Populations (8.6)].
Table 3 shows how to adjust dose based on creatinine clearance.
| Dosage in Normal Renal Function Every 24 hours | Creatinine Clearance 20 to 49 mL/min | Creatinine Clearance 10 to 19 mL/min | Hemodialysis or Chronic Ambulatory Peritoneal Dialysis (CAPD) |
|---|---|---|---|
| 750 mg | 750 mg every 48 hours | 750 mg initial dose, then 500 mg every 48 hours | 750 mg initial dose, then 500 mg every 48 hours |
| 500 mg | 500 mg initial dose, then 250 mg every 24 hours | 500 mg initial dose, then 250 mg every 48 hours | 500 mg initial dose, then 250 mg every 48 hours |
| 250 mg | No dosage adjustment required | 250 mg every 48 hours. If treating uncomplicated UTI, then no dosage adjustment is required | No information on dosing adjustment is available |
2.4 Drug Interaction With Chelation Agents: Antacids, Sucralfate, Metal Cations, Multivitamins
LEVAQUIN ® Tablets and Oral Solution
LEVAQUIN® Tablets and Oral Solution should be administered at least two hours before or two hours after antacids containing magnesium, aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc or didanosine chewable/buffered tablets or the pediatric powder for oral solution [see Drug Interactions (7.1) and Patient Counseling Information (17.2)].
LEVAQUIN ® Injection
LEVAQUIN® Injection should not be co-administered with any solution containing multivalent cations, e.g., magnesium, through the same intravenous line [see Dosage and Administration (2.6)].
2.5 Administration Instructions
Food and LEVAQUIN® Tablets and Oral Solution
LEVAQUIN® Tablets can be administered without regard to food. It is recommended that LEVAQUIN® Oral Solution be taken 1 hour before or 2 hours after eating.
LEVAQUIN ® Injection
Caution: Rapid or bolus intravenous infusion of LEVAQUIN® has been associated with hypotension and must be avoided. LEVAQUIN® Injection should be infused intravenously slowly over a period of not less than 60 or 90 minutes, depending on the dosage. LEVAQUIN® Injection should be administered only by intravenous infusion. It is not for intramuscular, intrathecal, intraperitoneal, or subcutaneous administration.
Hydration for Patients Receiving LEVAQUIN® Tablets, Oral Solution, and Injection
Adequate hydration of patients receiving oral or intravenous LEVAQUIN® should be maintained to prevent the formation of highly concentrated urine. Crystalluria and cylindruria have been reported with quinolones [see Adverse Reactions (6.1) and Patient Counseling Information (17.2)].
2.6 Preparation of Intravenous Product
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Because only limited data are available on the compatibility of LEVAQUIN® Injection with other intravenous substances, additives or other medications should not be added to LEVAQUIN® Injection Premix in Single-Use Flexible Containers and LEVAQUIN® Injection in Single-Use Vials, or infused simultaneously through the same intravenous line. If the same intravenous line is used for sequential infusion of several different drugs, the line should be flushed before and after infusion of LEVAQUIN® Injection with an infusion solution compatible with LEVAQUIN® Injection and with any other drug(s) administered via this common line.
LEVAQUIN ® Injection in Single-Use Vials
Single-use vials require dilution prior to administration.
LEVAQUIN® Injection is supplied in single-use vials containing a concentrated levofloxacin solution with the equivalent of 500 mg (20 mL vial) and 750 mg (30 mL vial) of levofloxacin in Water for Injection, USP. The 20 mL and 30 mL vials each contain 25 mg of levofloxacin/mL. These LEVAQUIN® Injection single-use vials must be further diluted with an appropriate solution prior to intravenous administration [see Table 4]. The concentration of the resulting diluted solution should be 5 mg/mL prior to administration.
Compatible Intravenous Solutions: Any of the following intravenous solutions may be used to prepare a 5 mg/mL levofloxacin solution with the approximate pH values:
| Intravenous Fluids | Final pH of LEVAQUIN® Solution |
|---|---|
| 0.9% Sodium Chloride Injection, USP | 4.71 |
| 5% Dextrose Injection, USP | 4.58 |
| 5% Dextrose/0.9% NaCl Injection | 4.62 |
| 5% Dextrose in Lactated Ringers | 4.92 |
| Plasma‑Lyte® 56/5% Dextrose Injection | 5.03 |
| 5% Dextrose, 0.45% Sodium Chloride, and 0.15% Potassium Chloride Injection | 4.61 |
| Sodium Lactate Injection (M/6) | 5.54 |
Since no preservative or bacteriostatic agent is present in this product, aseptic technique must be used in preparation of the final intravenous solution. Since the vials are for single-use only, any unused portion remaining in the vial should be discarded. When used to prepare two 250 mg doses from the 20 mL vial containing 500 mg of levofloxacin, the full content of the vial should be withdrawn at once using a single-entry procedure, and a second dose should be prepared and stored for subsequent use [see Stability of LEVAQUIN® Injection Following Dilution] .
Prepare the desired dosage of levofloxacin according to Table 5:
| Desired Dosage Strength | From Appropriate Vial, Withdraw Volume | Volume of Diluent | Infusion Time |
|---|---|---|---|
| 250 mg | 10 mL (20 mL Vial) | 40 mL | 60 min |
| 500 mg | 20 mL (20 mL Vial) | 80 mL | 60 min |
| 750 mg | 30 mL (30 mL Vial) | 120 mL | 90 min |
For example, to prepare a 500 mg dose using the 20 mL vial (25 mg/mL), withdraw 20 mL and dilute with a compatible intravenous solution to a total volume of 100 mL.
This intravenous drug product should be inspected visually for particulate matter prior to administration. Samples containing visible particles should be discarded.
Stability of LEVAQUIN® Injection Following Dilution: LEVAQUIN® Injection, when diluted in a compatible intravenous fluid to a concentration of 5 mg/mL, is stable for 72 hours when stored at or below 25°C (77°F) and for 14 days when stored under refrigeration at 5°C (41°F) in plastic intravenous containers. Solutions that are diluted in a compatible intravenous solution and frozen in glass bottles or plastic intravenous containers are stable for 6 months when stored at - 20°C (- 4°F). Thaw frozen solutions at room temperature 25°C (77°F) or in a refrigerator 8°C (46°F). Do not force thaw by microwave irradiation or water bath immersion. Do not refreeze after initial thawing.
LEVAQUIN ® Injection Premix in Single-Use Flexible Containers (5 mg/mL)
LEVAQUIN® Injection is also supplied in flexible containers within a foil overwrap. These contain a premixed, ready to use levofloxacin solution in 5% dextrose (D5W) for single-use. The 100 mL premixed flexible containers contain either 250 mg/50 mL or 500 mg/100 mL of levofloxacin solution. The 150 mL flexible container contains 750 mg/150 mL of levofloxacin solution. The concentration of each container is 5 mg/mL. No further dilution of these preparations is necessary.Because the premix flexible containers are for single-use only, any unused portion should be discarded.
Instructions for the Use of LEVAQUIN® Injection Premix in Flexible Containers:
- Tear outer wrap at the notch and remove solution container.
- Check the container for minute leaks by squeezing the inner bag firmly. If leaks are found, or if the seal is not intact, discard the solution, as the sterility may be compromised.
- Do not use if the solution is cloudy or a precipitate is present.
- Use sterile equipment.
- WARNING: Do not use flexible containers in series connections. Such use could result in air embolism due to residual air being drawn from the primary container before administration of the fluid from the secondary container is complete.
Preparation for Administration:
- Close flow control clamp of administration set.
- Remove cover from port at bottom of container.
- Insert piercing pin of administration set into port with a twisting motion until the pin is firmly seated. NOTE: See full directions on administration set carton.
- Suspend container from hanger.
- Squeeze and release drip chamber to establish proper fluid level in chamber during infusion of LEVAQUIN® Injection Premix in Flexible Containers.
- Open flow control clamp to expel air from set. Close clamp.
- Regulate rate of administration with flow control clamp.
3 DOSAGE FORMS AND STRENGTHS
TABLETS, Film-coated, capsule-shaped
- 250 mg terra cotta pink tablets, imprinted with "250" on one side and "LEVAQUIN" on the other
- 500 mg peach tablets, imprinted with "500" on one side and "LEVAQUIN" on the other
- 750 mg white tablets, imprinted with "750" on one side and "LEVAQUIN" on the other
ORAL SOLUTION, 25mg/mL, clear yellow to clear greenish-yellow color
INJECTION, Single-Use Vials of concentrated solution for dilution for intravenous infusion, clear yellow to clear greenish-yellow in appearance
- 20 mL vial of 25 mg/mL levofloxacin solution, equivalent to 500 mg of levofloxacin
- 30 mL vial of 25 mg/mL levofloxacin solution, equivalent to 750 mg of levofloxacin
INJECTION (5 mg/mL in 5% Dextrose) Premix in Single-Use Flexible Containers, for intravenous infusion
- 100 mL container, fill volume 50 mL (equivalent to 250 mg levofloxacin)
- 100 mL container, fill volume 100 mL (equivalent to 500 mg levofloxacin)
- 150 mL container, fill volume 150 mL (equivalent to 750 mg levofloxacin)
4 CONTRAINDICATIONS
LEVAQUIN® is contraindicated in persons with known hypersensitivity to levofloxacin, or other quinolone antibacterials [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity and/or anaphylactic reactions have been reported in patients receiving therapy with quinolones, including LEVAQUIN®. These reactions often occur following the first dose. Some reactions have been accompanied by cardiovascular collapse, hypotension/shock, seizure, loss of consciousness, tingling, angioedema (including tongue, laryngeal, throat, or facial edema/swelling), airway obstruction (including bronchospasm, shortness of breath, and acute respiratory distress), dyspnea, urticaria, itching, and other serious skin reactions. LEVAQUIN® should be discontinued immediately at the first appearance of a skin rash or any other sign of hypersensitivity. Serious acute hypersensitivity reactions may require treatment with epinephrine and other resuscitative measures, including oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines, and airway management, as clinically indicated [see Adverse Reactions (6); Patient Counseling Information (17.3)].
5.2 Other Serious and Sometimes Fatal Reactions
Other serious and sometimes fatal events, some due to hypersensitivity, and some due to uncertain etiology, have been reported rarely in patients receiving therapy with quinolones, including LEVAQUIN®. These events may be severe and generally occur following the administration of multiple doses. Clinical manifestations may include one or more of the following:
- fever, rash, or severe dermatologic reactions (e.g., toxic epidermal necrolysis, Stevens-Johnson Syndrome);
- vasculitis; arthralgia; myalgia; serum sickness;
- allergic pneumonitis;
- interstitial nephritis; acute renal insufficiency or failure;
- hepatitis; jaundice; acute hepatic necrosis or failure;
- anemia, including hemolytic and aplastic; thrombocytopenia, including thrombotic thrombocytopenic purpura; leukopenia; agranulocytosis; pancytopenia; and/or other hematologic abnormalities.
The drug should be discontinued immediately at the first appearance of skin rash, jaundice, or any other sign of hypersensitivity and supportive measures instituted [see Adverse Reactions (6); Patient Counseling Information (17.3)].
5.3 Hepatotoxicity
Post-marketing reports of severe hepatotoxicity (including acute hepatitis and fatal events) have been received for patients treated with LEVAQUIN®. No evidence of serious drug-associated hepatotoxicity was detected in clinical trials of over 7,000 patients. Severe hepatotoxicity generally occurred within 14 days of initiation of therapy and most cases occurred within 6 days. Most cases of severe hepatotoxicity were not associated with hypersensitivity [see Warnings and Precautions (5.2)]. The majority of fatal hepatotoxicity reports occurred in patients 65 years of age or older and most were not associated with hypersensitivity. LEVAQUIN ® should be discontinued immediately if the patient develops signs and symptoms of hepatitis [see Adverse Reactions (6); Patient Counseling Information (17.3)].
5.4 Tendon Effects
Ruptures of the shoulder, hand, Achilles tendon, or other tendons that required surgical repair or resulted in prolonged disability have been reported in patients receiving quinolones, including LEVAQUIN®. Postmarketing surveillance reports indicate that this risk is increased in patients receiving concomitant corticosteroids and in patients over 65 years of age. LEVAQUIN® should be discontinued if the patient experiences pain, inflammation, or rupture of a tendon. Patients should rest and refrain from exercise until the diagnosis of tendonitis or tendon rupture has been confidently excluded. Tendon rupture can occur during or after therapy withquinolones, including LEVAQUIN®[see Adverse Reactions (6); Patient Counseling Information (17.3)].
5.5 Central Nervous System Effects
Convulsions and toxic psychoses have been reported in patients receiving quinolones, including LEVAQUIN®. Quinolones may also cause increased intracranial pressure and central nervous system stimulation which may lead to tremors, restlessness, anxiety, lightheadedness, confusion, hallucinations, paranoia, depression, nightmares, insomnia, and, rarely, suicidal thoughts or acts. These reactions may occur following the first dose. If these reactions occur in patients receiving LEVAQUIN®, the drug should be discontinued and appropriate measures instituted. As with other quinolones, LEVAQUIN® should be used with caution in patients with a known or suspected central nervous system (CNS) disorder that may predispose them to seizures or lower the seizure threshold (e.g., severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose them to seizures or lower the seizure threshold (e.g., certain drug therapy, renal dysfunction.) [see Adverse Reactions (6); Drug Interactions (7.4, 7.5); Patient Counseling Information (17.3)].
5.6 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including LEVAQUIN ®, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated [see Adverse Reactions (6.2), Patient Counseling Information (17.3)].
5.7 Peripheral Neuropathy
Rare cases of sensory or sensorimotor axonal polyneuropathy affecting small and/or large axons resulting in paresthesias, hypoesthesias, dysesthesias and weakness have been reported in patients receiving quinolones, including LEVAQUIN®. LEVAQUIN® should be discontinued if the patient experiences symptoms of neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation in order to prevent the development of an irreversible condition [see Adverse Reactions (6), Patient Counseling Information (17.3)].
5.8 Prolongation of the QT Interval
Some quinolones, including LEVAQUIN®, have been associated with prolongation of the QT interval on the electrocardiogram and infrequent cases of arrhythmia. Rare cases of torsade de pointes have been spontaneously reported during postmarketing surveillance in patients receiving quinolones, including LEVAQUIN®. LEVAQUIN® should be avoided in patients with known prolongation of the QT interval, patients with uncorrected hypokalemia, and patients receiving class IA (quinidine, procainamide), or class III (amiodarone, sotalol) antiarrhythmic agents. Elderly patients may be more susceptible to drug-associated effects on the QT interval [see Adverse Reactions (6.3), Use in Specific Populations (8.5), and Patient Counseling Information (17.3)].
5.9 Musculoskeletal Disorders in Pediatric Patients and Arthropathic Effects in Animals
LEVAQUIN® is indicated in pediatric patients (≥ 6 months of age) only for the prevention of inhalational anthrax (post-exposure) [see Indications and Usage (1.13)]. An increased incidence of musculoskeletal disorders (arthralgia, arthritis, tendonopathy, and gait abnormality) compared to controls has been observed in pediatric patients receiving LEVAQUIN®[see Use in Specific Populations (8.4)].
In immature rats and dogs, the oral and intravenous administration of levofloxacin resulted in increased osteochondrosis. Histopathological examination of the weight-bearing joints of immature dogs dosed with levofloxacin revealed persistent lesions of the cartilage. Other fluoroquinolones also produce similar erosions in the weight-bearing joints and other signs of arthropathy in immature animals of various species [see Animal Toxicology and/or Pharmacology (13.2)].
5.10 Blood Glucose Disturbances
As with other quinolones, disturbances of blood glucose, including symptomatic hyper- and hypoglycemia, have been reported with LEVAQUIN®, usually in diabetic patients receiving concomitant treatment with an oral hypoglycemic agent (e.g., glyburide) or with insulin. In these patients, careful monitoring of blood glucose is recommended. If a hypoglycemic reaction occurs in a patient being treated with LEVAQUIN®, LEVAQUIN® should be discontinued and appropriate therapy should be initiated immediately [see Adverse Reactions (6.2); Drug Interactions (7.3); Patient Counseling Information (17.4)].
5.11 Photosensitivity/Phototoxicity
Moderate to severe photosensitivity/phototoxicity reactions, the latter of which may manifest as exaggerated sunburn reactions (e.g., burning, erythema, exudation, vesicles, blistering, edema) involving areas exposed to light (typically the face, “V” area of the neck, extensor surfaces of the forearms, dorsa of the hands), can be associated with the use of quinolones after sun or UV light exposure. Therefore, excessive exposure to these sources of light should be avoided. Drug therapy should be discontinued if photosensitivity/phototoxicity occurs [see Adverse Reactions (6.3); Patient Counseling Information (17.3)].
5.12 Development of Drug Resistant Bacteria
Prescribing LEVAQUIN® in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria [see Patient Counseling Information (17.1)].
6 ADVERSE REACTIONS
6.1 Serious and Otherwise Important Adverse Reactions
The following serious and otherwise important adverse drug reactions are discussed in greater detail in other sections of labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Other Serious and Sometimes Fatal Reactions [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Tendon Effects [see Warnings and Precautions (5.4)]
- Central Nervous System Effects [see Warnings and Precautions (5.5)]
- Clostridium difficile-Associated Diarrhea [see Warnings and Precautions (5.6)]
- Peripheral Neuropathy [see Warnings and Precautions (5.7)]
- Prolongation of the QT Interval [see Warnings and Precautions (5.8)]
- Musculoskeletal Disorders in Pediatric Patients [see Warnings and Precautions (5.9)]
- Blood Glucose Disturbances [see Warnings and Precautions (5.10)]
- Photosensitivity/Phototoxicity [see Warnings and Precautions (5.11)]
- Development of Drug Resistant Bacteria [see Warnings and Precautions (5.12)]
Hypotension has been associated with rapid or bolus intravenous infusion of LEVAQUIN®. LEVAQUIN® should be infused slowly over 60 to 90 minutes, depending on dosage [see Dosage and Administration (2.5)].
Crystalluria and cylindruria have been reported with quinolones, including LEVAQUIN®. Therefore, adequate hydration of patients receiving LEVAQUIN® should be maintained to prevent the formation of a highly concentrated urine [see Dosage and Administration (2.5)].
6.2 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to LEVAQUIN® in7537 patients in 29 pooled Phase 3 clinical trials. The population studied had a mean age of 50 years (approximately 74% of the population was < 65 years of age), 50% were male, 71% were Caucasian, 19% were Black. Patients were treated with LEVAQUIN® for a wide variety of infectious diseases [see Indications and Usage (1)]. Patients received LEVAQUIN® doses of 750 mg once daily, 250 mg once daily, or 500 mg once or twice daily. Treatment duration was usually 3–14 days, and the mean number of days on therapy was 10 days.
The overall incidence, type and distribution of adverse reactions was similar in patients receiving LEVAQUIN® doses of 750 mg once daily, 250 mg once daily, and 500 mg once or twice daily. Discontinuation of LEVAQUIN® due to adverse drug reactions occurred in 4.3% of patients overall, 3.8% of patients treated with the 250 mg and 500 mg doses and 5.4% of patients treated with the 750 mg dose. The most common adverse drug reactions leading to discontinuation with the 250 and 500 mg doses were gastrointestinal (1.4%), primarily nausea (0.6%); vomiting (0.4%); dizziness (0.3%); and headache (0.2%). The most common adverse drug reactions leading to discontinuation with the 750 mg dose were gastrointestinal (1.2%), primarily nausea (0.6%), vomiting (0.5%); dizziness (0.3%); and headache (0.3%).
Adverse reactions occurring in ≥1% of LEVAQUIN®-treated patients and less common adverse reactions, occurring in 0.1 to <1% of LEVAQUIN®-treated patients, are shown in Table 6 and Table 7, respectively. The most common adverse drug reactions (≥3%) are nausea, headache, diarrhea, insomnia, constipation, and dizziness.
| System/Organ Class | Adverse Reaction | % (N=7537) |
|---|---|---|
| Infections and Infestations | moniliasis | 1 |
| Psychiatric Disorders | insomnia* [see Warnings and Precautions
(5.5)]
| 4 |
| Nervous System Disorders | headache dizziness [see Warnings and Precautions (5.5)] | 6 3 |
| Respiratory, Thoracic and Mediastinal Disorders | dyspnea [see Warnings and Precautions (5.1)] | 1 |
| Gastrointestinal Disorders | nausea diarrhea constipation abdominal pain vomiting dyspepsia | 7 5 3 2 2 2 |
| Skin and Subcutaneous Tissue Disorders | rash [see Warnings
and Precautions (5.1)]
pruritus | 2 1 |
| Reproductive System and Breast Disorders | vaginitis | 1† |
| General Disorders and Administration Site Conditions | edema injection site reaction chest pain | 1 1 1 |
| System/Organ Class | Adverse Reaction |
|---|---|
|
|
| Infections and Infestations | genital moniliasis |
| Blood and
Lymphatic System Disorders
| anemia thrombocytopenia granulocytopenia [see Warnings and Precautions (5.2)] |
| Immune
System Disorders
| allergic reaction [See Warnings and Precautions (5.1, 5.2)] |
| Metabolism and Nutrition Disorders | hyperglycemia hypoglycemia [see Warnings and Precautions (5.10)] hyperkalemia |
| Psychiatric Disorders | anxiety agitation confusion depression hallucination nightmare* [see Warnings and Precautions (5.5)] sleep disorder* anorexia abnormal dreaming* |
| Nervous System Disorders | tremor convulsions [see Warnings and Precautions (5.5)] paresthesia [see Warnings and Precautions (5.7)] vertigo hypertonia hyperkinesias abnormal gait somnolence* syncope |
| Respiratory, Thoracic and Mediastinal Disorders | epistaxis |
| Cardiac Disorders | cardiac arrest palpitation ventricular tachycardia ventricular arrhythmia |
| Vascular Disorders | phlebitis |
| Gastrointestinal Disorders | gastritis stomatitis pancreatitis esophagitis gastroenteritis glossitis pseudomembraneous/ C. difficile colitis [see Warnings and Precautions (5.6)] |
| Hepatobiliary Disorders | abnormal hepatic function increased hepatic enzymes increased alkaline phosphatase |
| Skin and Subcutaneous Tissue Disorders | urticaria [see Warnings and Precautions (5.1)] |
| Musculoskeletal and Connective Tissue Disorders | arthralgia tendonitis [see Warnings and Precautions (5.4)] myalgia skeletal pain |
| Renal and Urinary Disorders | abnormal renal function acute renal failure [see Warnings and Precautions (5.2)] |
In clinical trials using multiple-dose therapy, ophthalmologic abnormalities, including cataracts and multiple punctate lenticular opacities, have been noted in patients undergoing treatment with quinolones, including LEVAQUIN®. The relationship of the drugs to these events is not presently established.
6.3 Postmarketing Experience
Table 8 lists adverse reactions that have been identified during post-approval use of LEVAQUIN®. Because these reactions are reported voluntarily from a population of uncertain size, reliably estimating their frequency or establishing a causal relationship to drug exposure is not always possible.
| System/Organ Class | Adverse Reaction |
|---|---|
| Blood and Lymphatic System Disorders | pancytopenia aplastic anemia leukopenia hemolytic anemia [see Warnings and Precautions (5.2)] eosinophilia |
| Immune System Disorders | hypersensitivity reactions, sometimes fatal including: anaphylactic/anaphylactoid reactions anaphylactic shock angioneurotic edema serum sickness [see Warnings and Precautions (5.1, 5.2)] |
| Psychiatric Disorders | psychosis paranoia isolated reports of suicide attempt and suicidal ideation [see Warnings and Precautions (5.5)] |
| Nervous
System Disorders
| anosmia ageusia parosmia dysgeusia peripheral neuropathy [see Warnings and Precautions (5.7)] isolated reports of encephalopathy abnormal electroencephalogram (EEG) dysphonia |
| Eye Disorders | vision disturbance, including diplopia visual acuity reduced vision blurred scotoma |
| Ear and Labyrinth Disorders | hypoacusis tinnitus |
| Cardiac Disorders | isolated reports of torsade de pointes electrocardiogram QT prolonged [see Warnings and Precautions (5.8) ] tachycardia |
| Vascular Disorders | vasodilatation |
| Respiratory, Thoracic and Mediastinal Disorders | isolated reports of allergic pneumonitis [see
Warnings and Precautions (5.2)]
|
| Hepatobiliary Disorders | hepatic failure (including fatal cases) hepatitis jaundice [see Warnings and Precautions 5.2, 5.3)] |
| Skin and Subcutaneous Tissue Disorders | bullous eruptions to include: Stevens-Johnson Syndrome toxic epidermal necrolysis erythema multiforme [see Warnings and Precautions (5.2)] photosensitivity/photoxicity reaction [see Warnings and Precautions (5.11)] leukocytoclastic vasculitis |
| Musculoskeletal and Connective Tissue Disorders | tendon rupture [see Warnings and Precautions (5.4)]
muscle injury, including rupture rhabdomyolysis |
| Renal and Urinary Disorders | interstitial nephritis [see Warnings and Precautions (5.2)] |
| General Disorders and Administration Site Conditions | multi-organ failure pyrexia |
| Investigations | prothrombin time prolonged international normalized ratio prolonged muscle enzymes increased |
7 DRUG INTERACTIONS
7.1 Chelation Agents: Antacids, Sucralfate, Metal Cations, Multivitamins
LEVAQUIN ® Tablets and Oral Solution
While the chelation by divalent cations is less marked than with other quinolones, concurrent administration of LEVAQUIN® Tablets and Oral Solution with antacids containing magnesium, or aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc may interfere with the gastrointestinal absorption of levofloxacin, resulting in systemic levels considerably lower than desired. Tablets with antacids containing magnesium, aluminum, as well as sucralfate, metal cations such as iron, and multivitamins preparations with zinc or didanosine may substantially interfere with the gastrointestinal absorption of levofloxacin, resulting in systemic levels considerably lower than desired. These agents should be taken at least two hours before or two hours after oral LEVAQUIN® administration.
LEVAQUIN ® Injection
There are no data concerning an interaction of intravenous quinolones with oral antacids, sucralfate, multivitamins, didanosine, or metal cations. However, no quinolone should be co-administered with any solution containing multivalent cations, e.g., magnesium, through the same intravenous line [see Dosage and Administration (2.5)].
7.2 Warfarin
No significant effect of LEVAQUIN® on the peak plasma concentrations, AUC, and other disposition parameters for R- and S- warfarin was detected in a clinical study involving healthy volunteers. Similarly, no apparent effect of warfarin on levofloxacin absorption and disposition was observed. However, there have been reports during the postmarketing experience in patients that LEVAQUIN® enhances the effects of warfarin. Elevations of the prothrombin time in the setting of concurrent warfarin and LEVAQUIN® use have been associated with episodes of bleeding. Prothrombin time, International Normalized Ratio (INR), or other suitable anticoagulation tests should be closely monitored if LEVAQUIN® is administered concomitantly with warfarin. Patients should also be monitored for evidence of bleeding [see Adverse Reactions (6.3); Patient Counseling Information (17.4)].
7.3 Antidiabetic Agents
Disturbances of blood glucose, including hyperglycemia and hypoglycemia, have been reported in patients treated concomitantly with quinolones and an antidiabetic agent. Therefore, careful monitoring of blood glucose is recommended when these agents are co-administered [see Warnings and Precautions (5.10); Adverse Reactions (6.2), Patient Counseling Information (17.4)].
7.4 Non-Steroidal Anti-Inflammatory Drugs
The concomitant administration of a non-steroidal anti-inflammatory drug with a quinolone, including LEVAQUIN®, may increase the risk of CNS stimulation and convulsive seizures [see Warnings and Precautions (5.5)].
7.5 Theophylline
No significant effect of LEVAQUIN® on the plasma concentrations, AUC, and other disposition parameters for theophylline was detected in a clinical study involving healthy volunteers. Similarly, no apparent effect of theophylline on levofloxacin absorption and disposition was observed. However, concomitant administration of other quinolones with theophylline has resulted in prolonged elimination half-life, elevated serum theophylline levels, and a subsequent increase in the risk of theophylline-related adverse reactions in the patient population. Therefore, theophylline levels should be closely monitored and appropriate dosage adjustments made when LEVAQUIN® is co-administered. Adverse reactions, including seizures, may occur with or without an elevation in serum theophylline levels [see Warnings and Precautions (5.5)].
7.6 Cyclosporine
No significant effect of LEVAQUIN® on the peak plasma concentrations, AUC, and other disposition parameters for cyclosporine was detected in a clinical study involving healthy volunteers. However, elevated serum levels of cyclosporine have been reported in the patient population when co-administered with some other quinolones. Levofloxacin Cmax and ke were slightly lower while Tmax and t½ were slightly longer in the presence of cyclosporine than those observed in other studies without concomitant medication. The differences, however, are not considered to be clinically significant. Therefore, no dosage adjustment is required for LEVAQUIN® or cyclosporine when administered concomitantly.
7.7 Digoxin
No significant effect of LEVAQUIN® on the peak plasma concentrations, AUC, and other disposition parameters for digoxin was detected in a clinical study involving healthy volunteers. Levofloxacin absorption and disposition kinetics were similar in the presence or absence of digoxin. Therefore, no dosage adjustment for LEVAQUIN® or digoxin is required when administered concomitantly.
7.8 Probenecid and Cimetidine
No significant effect of probenecid or cimetidine on the Cmax of levofloxacin was observed in a clinical study involving healthy volunteers. The AUC and t½ of levofloxacin were higher while CL/F and CLR were lower during concomitant treatment of LEVAQUIN® with probenecid or cimetidine compared to LEVAQUIN® alone. However, these changes do not warrant dosage adjustment for LEVAQUIN® when probenecid or cimetidine is co-administered.
7.9 Interactions with Laboratory or Diagnostic Testing
Some quinolones, including LEVAQUIN®, may produce false-positive urine screening results for opiates using commercially available immunoassay kits. Confirmation of positive opiate screens by more specific methods may be necessary.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Levofloxacin was not teratogenic in rats at oral doses as high as 810 mg/kg/day which corresponds to 9.4 times the highest recommended human dose based upon relative body surface area, or at intravenous doses as high as 160 mg/kg/day corresponding to 1.9 times the highest recommended human dose based upon relative body surface area. The oral dose of 810 mg/kg/day to rats caused decreased fetal body weight and increased fetal mortality. No teratogenicity was observed when rabbits were dosed orally as high as 50 mg/kg/day which corresponds to 1.1 times the highest recommended human dose based upon relative body surface area, or when dosed intravenously as high as 25 mg/kg/day, corresponding to 0.5 times the highest recommended human dose based upon relative body surface area.
There are, however, no adequate and well-controlled studies in pregnant women. LEVAQUIN® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
8.3 Nursing Mothers
Based on data on other quinolones and very limited data on LEVAQUIN®, it can be presumed that levofloxacin will be excreted in human milk. Because of the potential for serious adverse reactions from LEVAQUIN® in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Quinolones, including levofloxacin, cause arthropathy and osteochondrosis in juvenile animals of several species. [see Warnings and Precautions [5.9] and Animal Toxicology and/or Pharmacology (13.2)].
Inhalational Anthrax (Post-Exposure)
Levofloxacin is indicated in pediatric patients for inhalational anthrax (post-exposure). The risk-benefit assessment indicates that administration of levofloxacin to pediatric patients is appropriate. The safety of levofloxacin in pediatric patients treated for more than 14 days has not been studied. The pharmacokinetics of levofloxacin following a single intravenous dose were investigated in pediatric patients ranging in age from six months to 16 years. Pediatric patients cleared levofloxacin faster than adult patients resulting in lower plasma exposures than adults for a given mg/kg dose [see Indications and Usage (1.13), Dosage and Administration (2.2), Clinical Pharmacology (12.3) and Clinical Studies (14.9)].
Adverse Events
In clinical trials, 1534 children (6 months to 16 years of age) were treated with oral and intravenous LEVAQUIN®. Children 6 months to 5 years of age received LEVAQUIN® 10 mg/kg twice a day and children greater than 5 years of age received 10 mg/kg once a day (maximum 500 mg per day) for approximately 10 days.
A subset of children in the clinical trials (1340 LEVAQUIN®-treated and 893 non-fluoroquinolone-treated) enrolled in a prospective, long-term surveillance study to assess the incidence of protocol-defined musculoskeletal disorders (arthralgia, arthritis, tendonopathy, gait abnormality) during 60 days and 1 year following the first dose of study drug. Children treated with LEVAQUIN® had a significantly higher incidence of musculoskeletal disorders when compared to the non-fluoroquinolone-treated children as illustrated in Table 9.
| Follow-up Period | LEVAQUIN®
N = 1340 | Non-Fluoroquinolone*
N = 893 | p-value† |
|---|---|---|---|
|
|||
| 60 days | 28 (2.1%) | 8 (0.9%) | p = 0.038 |
| 1 year ‡ | 46 (3.4%) | 16 (1.8%) | p = 0.025 |
Arthralgia was the most frequently occurring musculoskeletal disorder in both treatment groups. Most of the musculoskeletal disorders in both groups involved multiple weight-bearing joints. Disorders were moderate in 8/46 (17%) children and mild in 35/46 (76%) LEVAQUIN®-treated children and most were treated with analgesics. The median time to resolution was 7 days for LEVAQUIN®-treated children and 9 for non-fluoroquinolone-treated children (approximately 80% resolved within 2 months in both groups). No child had a severe or serious disorder and all musculoskeletal disorders resolved without sequelae.
Vomiting and diarrhea were the most frequently reported adverse events, occurring in similar frequency in the LEVAQUIN®-treated and non-fluoroquinolone-treated children.
In addition to the events reported in pediatric patients in clinical trials, events reported in adults during clinical trials or post-marketing experience [see Adverse Reactions (6)] may also be expected to occur in pediatric patients.
8.5 Geriatric Use
In phase 3 clinical trials, 1,945 LEVAQUIN®-treated patients (26%) were ≥ 65 years of age. Of these, 1,081 patients (14%) were between the ages of 65 and 74 and 864 patients (12%) were 75 years or older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
Severe, and sometimes fatal, cases of hepatotoxicity have been reported post-marketing in association with LEVAQUIN®. The majority of fatal hepatotoxicity reports occurred in patients 65 years of age or older and most were not associated with hypersensitivity. LEVAQUIN® should be discontinued immediately if the patient develops signs and symptoms of hepatitis [see Warnings and Precautions (5.3)].
Patients over 65 are at increased risk for developing severe tendon disorders including tendon rupture when being treated with a fluoroquinolone such as LEVAQUIN®. This risk is further increased with concomitant steroid therapy. Tendon rupture usually involves the Achilles, hand or shoulder tendons and can occur during therapy or up to a few months post completion of therapy. Caution should be used when prescribing LEVAQUIN® to elderly patients especially those on corticosteroids. Patients should be informed of this potential side effect and advised to discontinue therapy and inform their physicians if any tendon symptoms occur [see Warnings and Precautions (5.4)].
Elderly patients may be more susceptible to drug-associated effects on the QT interval. Therefore, precaution should be taken when using LEVAQUIN® with concomitant drugs that can result in prolongation of the QT interval (e.g., class IA or class III antiarrhythmics) or in patients with risk factors for torsade de pointes (e.g., known QT prolongation, uncorrected hypokalemia) [see Warnings and Precautions (5.8)].
The pharmacokinetic properties of levofloxacin in younger adults and elderly adults do not differ significantly when creatinine clearance is taken into consideration. However, since the drug is known to be substantially excreted by the kidney, the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Clearance of levofloxacin is substantially reduced and plasma elimination half-life is substantially prolonged in patients with impaired renal function (creatinine clearance < 50 mL/min), requiring dosage adjustment in such patients to avoid accumulation. Neither hemodialysis nor continuous ambulatory peritoneal dialysis (CAPD) is effective in removal of levofloxacin from the body, indicating that supplemental doses of LEVAQUIN® are not required following hemodialysis or CAPD [see Dosage and Administration (2.3)].
8.7 Hepatic Impairment
Pharmacokinetic studies in hepatically impaired patients have not been conducted. Due to the limited extent of levofloxacin metabolism, the pharmacokinetics of levofloxacin are not expected to be affected by hepatic impairment.
10 OVERDOSAGE
In the event of an acute overdosage, the stomach should be emptied. The patient should be observed and appropriate hydration maintained. Levofloxacin is not efficiently removed by hemodialysis or peritoneal dialysis.
LEVAQUIN® exhibits a low potential for acute toxicity. Mice, rats, dogs and monkeys exhibited the following clinical signs after receiving a single high dose of LEVAQUIN®: ataxia, ptosis, decreased locomotor activity, dyspnea, prostration, tremors, and convulsions. Doses in excess of 1500 mg/kg orally and 250 mg/kg IV produced significant mortality in rodents.
11 DESCRIPTION
LEVAQUIN® is a synthetic broad-spectrum antibacterial
agent for oral and intravenous administration. Chemically, levofloxacin, a
chiral fluorinated carboxyquinolone, is the pure (-)-(S)-enantiomer of the
racemic drug substance ofloxacin. The chemical name is (-)-(S)-9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic
acid hemihydrate.
Figure 1: The Chemical Structure
of Levofloxacin 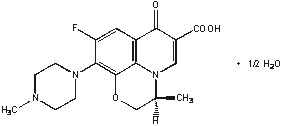
The empirical formula is C18H20FN3O4 •½ H2O and the molecular weight is 370.38. Levofloxacin is a light yellowish-white to yellow-white crystal or crystalline powder. The molecule exists as a zwitterion at the pH conditions in the small intestine.
The data demonstrate that from pH 0.6 to 5.8, the solubility of levofloxacin is essentially constant (approximately 100 mg/mL). Levofloxacin is considered soluble to freely soluble in this pH range, as defined by USP nomenclature. Above pH 5.8, the solubility increases rapidly to its maximum at pH 6.7 (272 mg/mL) and is considered freely soluble in this range. Above pH 6.7, the solubility decreases and reaches a minimum value (about 50 mg/mL) at a pH of approximately 6.9.
Levofloxacin has the potential to form stable coordination compounds with many metal ions. This in vitro chelation potential has the following formation order: Al+3>Cu+2>Zn+2>Mg+2>Ca+2.
Excipients and Description of Dosage Forms
LEVAQUIN® Tablets
LEVAQUIN® Tablets are available as film-coated tablets and contain the following inactive ingredients:
- 250 mg (as expressed in the anhydrous form): hypromellose, crospovidone, microcrystalline cellulose, magnesium stearate, polyethylene glycol, titanium dioxide, polysorbate 80 and synthetic red iron oxide.
- 500 mg (as expressed in the anhydrous form): hypromellose, crospovidone, microcrystalline cellulose, magnesium stearate, polyethylene glycol, titanium dioxide, polysorbate 80 and synthetic red and yellow iron oxides.
- 750 mg (as expressed in the anhydrous form): hypromellose, crospovidone, microcrystalline cellulose, magnesium stearate, polyethylene glycol, titanium dioxide, polysorbate 80.
LEVAQUIN® Oral Solution
LEVAQUIN® Oral Solution, 25 mg/mL, is a multi-use self-preserving aqueous solution of levofloxacin with pH ranging from 5.0 – 6.0. The appearance of LEVAQUIN® Oral Solution may range from clear yellow to clear greenish-yellow. This does not adversely affect product potency.
LEVAQUIN® Oral Solution contains the following inactive ingredients: sucrose, glycerin, sucralose, hydrochloric acid, purified water, propylene glycol, artificial and natural flavors, benzyl alcohol, ascorbic acid, and caramel color. It may also contain a solution of sodium hydroxide for pH adjustment.
LEVAQUIN® Injection
The appearance of LEVAQUIN® Injection may range from a clear yellow to a clear greenish-yellow solution. This does not adversely affect product potency.
LEVAQUIN ®Injection in Single-Use Vials is a sterile, preservative-free aqueous solution of levofloxacin in Water for Injection, with pH ranging from 3.8 to 5.8.
LEVAQUIN®Injection Premix in Single-Use Flexible Containers is a sterile, preservative-free aqueous solution of levofloxacin with pH ranging from 3.8 to 5.8. This is a dilute, non-pyrogenic, nearly isotonic premixed solution that contains levofloxacin in 5% Dextrose (D5W). Solutions of hydrochloric acid and sodium hydroxide may have been added to adjust the pH.
The flexible container is fabricated from a specially formulated non-plasticized, thermoplastic copolyester (CR3). The amount of water that can permeate from the container into the overwrap is insufficient to affect the solution significantly. Solutions in contact with the flexible container can leach out certain of the container's chemical components in very small amounts within the expiration period. The suitability of the container material has been confirmed by tests in animals according to USP biological tests for plastic containers.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Levofloxacin is a member of the fluoroquinolone class of antibacterial agents [see Clinical Pharmacology (12.4)].
12.3 Pharmacokinetics
The mean ±SD pharmacokinetic parameters of levofloxacin determined under single and steady-state conditions following oral tablet, oral solution, or intravenous (IV) doses of LEVAQUIN® are summarized in Table 10.
| Regimen | Cmax | Tmax | AUC | CL/F1 | Vd/F2 | t1/2 | CLR |
|---|---|---|---|---|---|---|---|
| (mcg/mL) | (h) | (mcg∙h/mL) | (mL/min) | (L) | (h) | (mL/min) | |
| 1 clearance/bioavailability | |||||||
| 2 volume of distribution/bioavailability | |||||||
| 3 healthy males 18–53 years of age | |||||||
| 4 60 min infusion for 250 mg and 500 mg doses, 90 min infusion for 750 mg dose | |||||||
| 5 healthy male and female subjects 18–54 years of age | |||||||
| 6 500 mg every 48h for patients with moderate renal impairment (CLCR 20–50 mL/min) and infections of the respiratory tract or skin | |||||||
| 7 dose-normalized values (to 500 mg dose), estimated by population pharmacokinetic modeling | |||||||
| 8 healthy males 22–75 years of age | |||||||
| 9 healthy females 18–80 years of age | |||||||
| 10 young healthy male and female subjects 18–36 years of age | |||||||
| 11 healthy elderly male and female subjects 66–80 years of age | |||||||
| 12 healthy males and females 19–55 years of age. | |||||||
| *Absolute bioavailability; F=0.99 ± 0.08 from a 500 mg tablet and F=0.99 ± 0.06 from a 750 mg tablet; | |||||||
| ND=not determined. | |||||||
| Single dose | |||||||
| 250 mg oral tablet3 | 2.8 ± 0.4 | 1.6 ± 1.0 | 27.2 ± 3.9 | 156 ± 20 | ND | 7.3 ± 0.9 | 142 ± 21 |
| 500 mg oral tablet3* | 5.1 ± 0.8 | 1.3 ± 0.6 | 47.9 ± 6.8 | 178 ± 28 | ND | 6.3 ± 0.6 | 103 ± 30 |
| 500 mg oral solution12 | 5.8 ± 1.8 | 0.8 ± 0.7 | 47.8 ± 10.8 | 183 ± 40 | 112 ± 37.2 | 7.0 ± 1.4 | ND |
| 500 mg IV3 | 6.2 ± 1.0 | 1.0 ± 0.1 | 48.3 ± 5.4 | 175 ± 20 | 90 ± 11 | 6.4 ± 0.7 | 112 ± 25 |
| 750 mg oral tablet5* | 9.3 ± 1.6 | 1.6 ± 0.8 | 101 ± 20 | 129 ± 24 | 83 ± 17 | 7.5 ± 0.9 | ND |
| 750 mg IV5 | 11.5 ±4.04 | ND | 110 ±40 | 126 ±39 | 75 ± 13 | 7.5 ± 1.6 | ND |
| Multiple dose | |||||||
| 500 mg every 24h oral tablet3 | 5.7 ± 1.4 | 1.1 ± 0.4 | 47.5 ± 6.7 | 175 ± 25 | 102 ± 22 | 7.6 ± 1.6 | 116 ± 31 |
| 500 mg every 24h IV3 | 6.4 ± 0.8 | ND | 54.6 ± 11.1 | 158 ± 29 | 91 ± 12 | 7.0 ± 0.8 | 99 ± 28 |
| 500 mg or 250 mg every 24h IV, patients with bacterial infection6 | 8.7± 4.07 | ND | 72.5 ± 51.27 | 154 ± 72 | 111 ± 58 | ND | ND |
| 750 mg every 24h oral tablet5 | 8.6 ± 1.9 | 1.4 ± 0.5 | 90.7 ± 17.6 | 143 ± 29 | 100 ± 16 | 8.8 ± 1.5 | 116 ± 28 |
| 750 mg every 24h IV5 | 12.1 ± 4.14 | ND | 108 ± 34 | 126 ± 37 | 80 ± 27 | 7.9 ± 1.9 | ND |
| 500 mg oral tablet single dose, effects of gender and age: | |||||||
| Male8 | 5.5 ± 1.1 | 1.2 ± 0.4 | 54.4 ± 18.9 | 166 ± 44 | 89 ± 13 | 7.5 ± 2.1 | 126 ± 38 |
| Female9 | 7.0 ± 1.6 | 1.7 ± 0.5 | 67.7 ± 24.2 | 136 ± 44 | 62 ± 16 | 6.1 ± 0.8 | 106 ± 40 |
| Young10 | 5.5 ± 1.0 | 1.5 ± 0.6 | 47.5 ± 9.8 | 182 ± 35 | 83 ± 18 | 6.0 ± 0.9 | 140 ± 33 |
| Elderly11 | 7.0 ± 1.6 | 1.4 ± 0.5 | 74.7 ± 23.3 | 121 ± 33 | 67 ± 19 | 7.6 ± 2.0 | 91 ± 29 |
| 500 mg oral single dose tablet, patients with renal insufficiency: | |||||||
| CLCR 50–80 mL/min | 7.5 ± 1.8 | 1.5 ± 0.5 | 95.6 ± 11.8 | 88 ± 10 | ND | 9.1 ± 0.9 | 57 ± 8 |
| CLCR 20–49 mL/min | 7.1 ± 3.1 | 2.1 ± 1.3 | 182.1 ± 62.6 | 51 ± 19 | ND | 27 ± 10 | 26 ± 13 |
| CLCR <20 mL/min | 8.2 ± 2.6 | 1.1 ± 1.0 | 263.5 ± 72.5 | 33 ± 8 | ND | 35 ± 5 | 13 ± 3 |
| Hemodialysis | 5.7 ± 1.0 | 2.8 ± 2.2 | ND | ND | ND | 76 ± 42 | ND |
| CAPD | 6.9 ± 2.3 | 1.4 ± 1.1 | ND | ND | ND | 51 ± 24 | ND |
Absorption
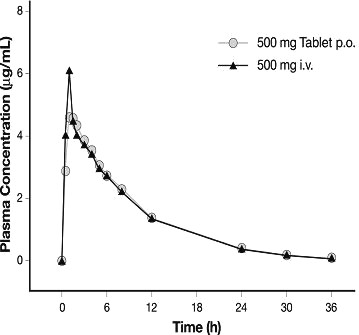
Levofloxacin is rapidly and essentially completely absorbed after oral administration. Peak plasma concentrations are usually attained one to two hours after oral dosing. The absolute bioavailability of levofloxacin from a 500 mg tablet and a 750 mg tablet of LEVAQUIN® are both approximately 99%, demonstrating complete oral absorption of levofloxacin. Following a single intravenous dose of LEVAQUIN® to healthy volunteers, the mean ±SD peak plasma concentration attained was 6.2 ±1.0 mcg/mL after a 500 mg dose infused over 60 minutes and 11.5 ±4.0 mcg/mL after a 750 mg dose infused over 90 minutes. LEVAQUIN® Oral Solution and Tablet formulations are bioequivalent.
Levofloxacin pharmacokinetics are linear and predictable after single and multiple oral or IV dosing regimens. Steady-state conditions are reached within 48 hours following a 500 mg or 750 mg once-daily dosage regimen. The mean ±SD peak and trough plasma concentrations attained following multiple once-daily oral dosage regimens were approximately 5.7 ±1.4 and 0.5 ±0.2 mcg/mL after the 500 mg doses, and 8.6 ±1.9 and 1.1 ±0.4 mcg/mL after the 750 mg doses, respectively. The mean ±SD peak and trough plasma concentrations attained following multiple once-daily IV regimens were approximately 6.4±0.8 and 0.6 ±0.2 mcg/mL after the 500 mg doses, and 12.1 ±4.1 and 1.3 ±0.71 mcg/mL after the 750 mg doses, respectively. Oral administration of a 500 mg dose of LEVAQUIN® with food prolongs the time to peak concentration by approximately 1 hour and decreases the peak concentration by approximately 14% following tablet and approximately 25% following oral solution administration. Therefore, LEVAQUIN® Tablets can be administered without regard to food. It is recommended that LEVAQUIN® Oral Solution be taken 1 hour before, or 2 hours after eating.
The plasma concentration profile of levofloxacin after IV
administration is similar and comparable in extent of exposure (AUC) to that
observed for LEVAQUIN® Tablets when equal doses (mg/mg) are
administered. Therefore, the oral and IV routes of administration can be considered
interchangeable (see Figure
2 and Figure 3).
Figure 2: Mean Levofloxacin Plasma Concentration vs. Time Profile: 750 mg
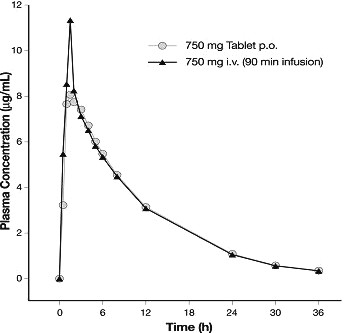
Figure 3: Mean Levofloxacin Plasma Concentration vs. Time Profile: 500 mg
Distribution
The mean volume of distribution of levofloxacin generally ranges from 74 to 112 L after single and multiple 500 mg or 750 mg doses, indicating widespread distribution into body tissues. Levofloxacin reaches its peak levels in skin tissues and in blister fluid of healthy subjects at approximately 3 hours after dosing. The skin tissue biopsy to plasma AUC ratio is approximately 2 and the blister fluid to plasma AUC ratio is approximately 1 following multiple once-daily oral administration of 750 mg and 500 mg doses of LEVAQUIN®, respectively, to healthy subjects. Levofloxacin also penetrates well into lung tissues. Lung tissue concentrations were generally 2- to 5- fold higher than plasma concentrations and ranged from approximately 2.4 to 11.3 mcg/g over a 24-hour period after a single 500 mg oral dose.
In vitro, over a clinically relevant range (1 to 10 mcg/mL) of serum/plasma levofloxacin concentrations, levofloxacin is approximately 24 to 38% bound to serum proteins across all species studied, as determined by the equilibrium dialysis method. Levofloxacin is mainly bound to serum albumin in humans. Levofloxacin binding to serum proteins is independent of the drug concentration.
Metabolism
Levofloxacin is stereochemically stable in plasma and urine and does not invert metabolically to its enantiomer, D-ofloxacin. Levofloxacin undergoes limited metabolism in humans and is primarily excreted as unchanged drug in the urine. Following oral administration, approximately 87% of an administered dose was recovered as unchanged drug in urine within 48 hours, whereas less than 4% of the dose was recovered in feces in 72 hours. Less than 5% of an administered dose was recovered in the urine as the desmethyl and N-oxide metabolites, the only metabolites identified in humans. These metabolites have little relevant pharmacological activity.
Excretion
Levofloxacin is excreted largely as unchanged drug in the urine. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously. The mean apparent total body clearance and renal clearance range from approximately 144 to 226 mL/min and 96 to 142 mL/min, respectively. Renal clearance in excess of the glomerular filtration rate suggests that tubular secretion of levofloxacin occurs in addition to its glomerular filtration. Concomitant administration of either cimetidine or probenecid results in approximately 24% and 35% reduction in the levofloxacin renal clearance, respectively, indicating that secretion of levofloxacin occurs in the renal proximal tubule. No levofloxacin crystals were found in any of the urine samples freshly collected from subjects receiving LEVAQUIN®.
Geriatric
There are no significant differences in levofloxacin pharmacokinetics between young and elderly subjects when the subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of LEVAQUIN® to healthy elderly subjects (66 – 80 years of age), the mean terminal plasma elimination half-life of levofloxacin was about 7.6 hours, as compared to approximately 6 hours in younger adults. The difference was attributable to the variation in renal function status of the subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by age. LEVAQUIN® dose adjustment based on age alone is not necessary [See Use in Specific Populations (8.5)].
Pediatrics
The pharmacokinetics of levofloxacin following a single 7 mg/kg intravenous dose were investigated in pediatric patients ranging in age from 6 months to 16 years. Pediatric patients cleared levofloxacin faster than adult patients, resulting in lower plasma exposures than adults for a given mg/kg dose. Subsequent pharmacokinetic analyses predicted that a dosage regimen of 8 mg/kg every 12 hours (not to exceed 250 mg per dose) for pediatric patients 6 months to 17 years of age would achieve comparable steady state plasma exposures (AUC0-24 and Cmax) to those observed in adult patients administered 500 mg of levofloxacin once every 24 hours.
Gender
There are no significant differences in levofloxacin pharmacokinetics between male and female subjects when subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of LEVAQUIN® to healthy male subjects, the mean terminal plasma elimination half-life of levofloxacin was about 7.5 hours, as compared to approximately 6.1 hours in female subjects. This difference was attributable to the variation in renal function status of the male and female subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by the gender of the subjects. Dose adjustment based on gender alone is not necessary.
Race
The effect of race on levofloxacin pharmacokinetics was examined through a covariate analysis performed on data from 72 subjects: 48 white and 24 non-white. The apparent total body clearance and apparent volume of distribution were not affected by the race of the subjects.
Renal Impairment
Clearance of levofloxacin is substantially reduced and plasma elimination half-life is substantially prolonged in patients with impaired renal function (creatinine clearance < 50 mL/min), requiring dosage adjustment in such patients to avoid accumulation. Neither hemodialysis nor continuous ambulatory peritoneal dialysis (CAPD) is effective in removal of levofloxacin from the body, indicating that supplemental doses of LEVAQUIN® are not required following hemodialysis or CAPD [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Hepatic Impairment
Pharmacokinetic studies in hepatically impaired patients have not been conducted. Due to the limited extent of levofloxacin metabolism, the pharmacokinetics of levofloxacin are not expected to be affected by hepatic impairment [See Use in Specific Populations (8.7)].
Bacterial Infection
The pharmacokinetics of levofloxacin in patients with serious community-acquired bacterial infections are comparable to those observed in healthy subjects.
Drug-Drug Interactions
The potential for pharmacokinetic drug interactions between LEVAQUIN® and antacids warfarin, theophylline, cyclosporine, digoxin, probenecid, and cimetidine has been evaluated [see Drug Interactions (7)].
12.4 Microbiology
Mechanism of Action
Levofloxacin is the L-isomer of the racemate, ofloxacin, a quinolone antimicrobial agent. The antibacterial activity of ofloxacin resides primarily in the L-isomer. The mechanism of action of levofloxacin and other fluoroquinolone antimicrobials involves inhibition of bacterial topoisomerase IV and DNA gyrase (both of which are type II topoisomerases), enzymes required for DNA replication, transcription, repair and recombination.
Drug Resistance
Fluoroquinolone resistance can arise through mutations in defined regions of DNA gyrase or topoisomerase IV, termed the Quinolone-Resistance Determining Regions (QRDRs), or through altered efflux.
Fluoroquinolones, including levofloxacin, differ in chemical structure and mode of action from aminoglycosides, macrolides and β-lactam antibiotics, including penicillins. Fluoroquinolones may, therefore, be active against bacteria resistant to these antimicrobials.
Resistance to levofloxacin due to spontaneous mutation in vitro is a rare occurrence (range: 10-9 to 10-10). Although cross-resistance has been observed between levofloxacin and some other fluoroquinolones, some microorganisms resistant to other fluoroquinolones may be susceptible to levofloxacin.
Activity in vitro and in vivo
Levofloxacin has in vitro activity against a wide range of Gram-negative and Gram-positive microorganisms.
Levofloxacin is often bactericidal at concentrations equal to or slightly greater than inhibitory concentrations.
Levofloxacin has been shown to be active against most strains of the following microorganisms both in vitro and in clinical infections as described in Indications and Usage (1):
-
Aerobic
Gram-Positive Microorganisms
- Enterococcus faecalis (many strains are only moderately susceptible)
- Staphylococcus aureus (methicillin-susceptible strains)
- Staphylococcus epidermidis (methicillin-susceptible strains)
- Staphylococcus saprophyticus
- Streptococcus pneumoniae (including multi-drug resistant strains [MDRSP]1)
- Streptococcus pyogenes
- 1
- MDRSP (Multi-drug resistant Streptococcus pneumoniae) isolates are strains resistant to two or more of the following antibiotics: penicillin (MIC ≥2 mcg/mL), 2nd generation cephalosporins, e.g., cefuroxime; macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
-
Aerobic
Gram-Negative Microorganisms
- Enterobacter cloacae
- Escherichia coli
- Haemophilus influenzae
- Haemophilus parainfluenzae
- Klebsiella pneumoniae
- Legionella pneumophila
- Moraxella catarrhalis
- Proteus mirabilis
- Pseudomonas aeruginosa2
- Serratia marcescens
- 2
- As with other drugs in this class, some strains of Pseudomonas aeruginosa may develop resistance fairly rapidly during treatment with LEVAQUIN®.
-
Other
Microorganisms
- Chlamydophila pneumoniae
- Mycoplasma pneumoniae
Levofloxacin has been shown to be active against Bacillus anthracis both in vitro and by use of plasma levels as a surrogate marker in a rhesus monkey model for anthrax (post-exposure) [see Indications and Usage (1.13), Clinical Studies (14.9)].
The following in vitro data are available, but their clinical significance is unknown: Levofloxacin exhibits in vitro minimum inhibitory concentrations (MIC values) of 2 mcg/mL or less against most (≥90%) strains of the following microorganisms; however, the safety and effectiveness of LEVAQUIN® in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled trials.
-
Aerobic
Gram-Positive Microorganisms
- Staphylococcus haemolyticus
- β-hemolytic Streptococcus (Group C/F)
- β-hemolytic Streptococcus (Group G)
- Streptococcus agalactiae
- Streptococcus milleri
- Viridans group streptococci
-
Aerobic
Gram-Negative Microorganisms
- Acinetobacter baumannii
- Acinetobacter lwoffii
- Bordetella pertussis
- Citrobacter koseri
- Citrobacter freundii
- Enterobacter aerogenes
- Enterobacter sakazakii
- Klebsiella oxytoca
- Morganella morganii
- Pantoea agglomerans
- Proteus vulgaris
- Providencia rettgeri
- Providencia stuartii
- Pseudomonas fluorescens
-
Anaerobic
Gram-Positive Microorganisms
- Clostridium perfringens
Susceptibility Tests
Susceptibility testing for levofloxacin should be performed, as it is the optimal predictor of activity.
-
Dilution techniques:
Quantitative methods are used to determine antimicrobial minimal inhibitory concentrations (MIC values). These MIC values provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MIC values should be determined using a standardized procedure. Standardized procedures are based on a dilution method1 (broth or agar) or equivalent with standardized inoculum concentrations and standardized concentrations of levofloxacin powder. The MIC values should be interpreted according to the criteria outlined in Table 11. -
Diffusion techniques:
Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure2 requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 5 mcg levofloxacin to test the susceptibility of microorganisms to levofloxacin.
Reports from the laboratory providing results of the standard single-disk susceptibility test with a 5 mcg levofloxacin disk should be interpreted according the criteria outlined in Table 11. Interpretation involves correlation of the diameter obtained in the disk test with the MIC for levofloxacin.
A report of Susceptible indicates that the pathogen is likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable. A report of Intermediate indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where a high dosage of drug can be used. This category also provides a buffer zone which prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of Resistant indicates that the pathogen is not likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable; other therapy should be selected.Table 11: Susceptibility Interpretive Criteria for LEVAQUIN® Minimum Inhibitory
Concentrations (mcg/mL)Disk Diffusion
(zone diameter in mm)S = Susceptible, I = Intermediate, R = Resistant - *
- These interpretive standards
are applicable only to broth microdilution susceptibility testing with Haemophilus influenzae and Haemophilus
parainfluenzae using Haemophilus Test Medium.1
- †
- The current absence of data on resistant strains precludes defining any categories other than "Susceptible." Strains yielding MIC /zone diameter results suggestive of a "nonsusceptible" category should be submitted to a reference laboratory for further testing.
- ‡
- These interpretive standards are applicable only to disk diffusion susceptibility testing with Haemophilus influenzae and Haemophilus parainfluenzae using Haemophilus Test Medium.2
- §
- These interpretive standards are applicable only to broth microdilution susceptibility tests using cation-adjusted Mueller-Hinton broth with 2–5% lysed horse blood.
- ¶
- These zone diameter standards for Streptococcus spp. including S. pneumoniae apply only to tests performed using Mueller-Hinton agar supplemented with 5% sheep blood and incubated in 5% CO2.
Pathogen S I R S I R Enterobacteriaceae ≤2 4 ≥8 ≥17 14–16 ≤13 Enterococcus faecalis ≤2 4 ≥8 ≥17 14–16 ≤13 Methicillin-susceptible
Staphylococcus species≤2 4 ≥8 ≥17 14–16 ≤13 Pseudomonas aeruginosa ≤2 4 ≥8 ≥17 14–16 ≤13 Haemophilus influenzae ≤2* --† --† ≥17‡ --† --† Haemophilus parainfluenzae ≤2* --† --† ≥17‡ --† --† Streptococcus pneumoniae ≤2§ 4§ ≥8§ ≥17¶ 14–16¶ ≤13 ¶ Streptococcus pyogenes ≤2 4 ≥8 ≥17 14–16 ≤13 -
Quality Control:
Standardized susceptibility test procedures require the use of laboratory control microorganisms to control the technical aspects of the laboratory procedures. For dilution technique, standard levofloxacin powder should give the MIC values provided in Table 12. For diffusion technique, the 5 mcg levofloxacin disk should provide zone diameters provided in Table 12.
Table 12: Quality Control for Susceptibility Testing Microorganism Microorganism
QC NumberMIC (mcg/mL) Disk Diffusion
(zone diameter in mm)- *
- This quality control range is applicable to only H. influenzae ATCC 49247 tested by a broth microdilution procedure using Haemophilus Test Medium (HTM).1
- †
- This quality control range is applicable to only H. influenzae ATCC 49247 tested by a disk diffusion procedure using Haemophilus Test Medium (HTM).2
- ‡
- This quality control range is applicable to only S. pneumoniae ATCC 49619 tested by a broth microdilution procedure using cation-adjusted Mueller-Hinton broth with 2–5% lysed horse blood.
- §
- This quality control range is applicable to only S. pneumoniae ATCC 49619 tested by a disk diffusion procedure using Mueller-Hinton agar supplemented with 5% sheep blood and incubated in 5% CO2.
Enterococcus faecalis ATCC 29212 0.25 – 2 Not applicable Escherichia coli ATCC 25922 0.008 – 0.06 29 – 37 Escherichia coli ATCC 35218 0.015 – 0.06 Not applicable Haemophilus influenzae ATCC 49247 0.008 – 0.03* 32 – 40† Pseudomonas aeruginosa ATCC 27853 0.5 – 4 19 – 26 Staphylococcus aureus ATCC 29213 0.06 – 0.5 Not applicable Staphylococcus aureus ATCC 25923 Not applicable 25 – 30 Streptococcus pneumoniae ATCC 49619 0.5 – 2‡ 20 – 25§
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a lifetime bioassay in rats, levofloxacin exhibited no carcinogenic potential following daily dietary administration for 2 years; the highest dose (100 mg/kg/day) was 1.4 times the highest recommended human dose (750 mg) based upon relative body surface area. Levofloxacin did not shorten the time to tumor development of UV-induced skin tumors in hairless albino (Skh-1) mice at any levofloxacin dose level and was therefore not photo-carcinogenic under conditions of this study. Dermal levofloxacin concentrations in the hairless mice ranged from 25 to 42 mcg/g at the highest levofloxacin dose level (300 mg/kg/day) used in the photo-carcinogenicity study. By comparison, dermal levofloxacin concentrations in human subjects receiving 750 mg of LEVAQUIN® averaged approximately 11.8 mcg/g at Cmax.
Levofloxacin was not mutagenic in the following assays: Ames bacterial mutation assay (S. typhimurium and E. coli), CHO/HGPRT forward mutation assay, mouse micronucleus test, mouse dominant lethal test, rat unscheduled DNA synthesis assay, and the mouse sister chromatid exchange assay. It was positive in the in vitro chromosomal aberration (CHL cell line) and sister chromatid exchange (CHL/IU cell line) assays.
Levofloxacin caused no impairment of fertility or reproductive performance in rats at oral doses as high as 360 mg/kg/day, corresponding to 4.2 times the highest recommended human dose based upon relative body surface area and intravenous doses as high as 100 mg/kg/day, corresponding to 1.2 times the highest recommended human dose based upon relative body surface area.
13.2 Animal Toxicology and/or Pharmacology
Levofloxacin and other quinolones have been shown to cause arthropathy in immature animals of most species tested [see Warnings and Precautions (5.9)]. In immature dogs (4–5 months old), oral doses of 10 mg/kg/day for 7 days and intravenous doses of 4 mg/kg/day for 14 days of levofloxacin resulted in arthropathic lesions. Administration at oral doses of 300 mg/kg/day for 7 days and intravenous doses of 60 mg/kg/day for 4 weeks produced arthropathy in juvenile rats. Three-month old beagle dogs dosed orally with levofloxacin at 40 mg/kg/day exhibited clinically severe arthrotoxicity resulting in the termination of dosing at Day 8 of a 14-day dosing routine. Slight musculoskeletal clinical effects, in the absence of gross pathological or histopathological effects, resulted from the lowest dose level of 2.5 mg/kg/day (approximately 0.2-fold the pediatric dose based upon AUC comparisons). Synovitis and articular cartilage lesions were observed at the 10 and 40 mg/kg dose levels (approximately 0.7-fold and 2.4-fold the pediatric dose, respectively, based on AUC comparisons). Articular cartilage gross pathology and histopathology persisted to the end of the 18-week recovery period for those dogs from the 10 and 40 mg/kg/day dose levels.
When tested in a mouse ear swelling bioassay, levofloxacin exhibited phototoxicity similar in magnitude to ofloxacin, but less phototoxicity than other quinolones.
While crystalluria has been observed in some intravenous rat studies, urinary crystals are not formed in the bladder, being present only after micturition and are not associated with nephrotoxicity.
In mice, the CNS stimulatory effect of quinolones is enhanced by concomitant administration of non-steroidal anti-inflammatory drugs.
In dogs, levofloxacin administered at 6 mg/kg or higher by rapid intravenous injection produced hypotensive effects. These effects were considered to be related to histamine release.
In vitro and in vivo studies in animals indicate that levofloxacin is neither an enzyme inducer nor inhibitor in the human therapeutic plasma concentration range; therefore, no drug metabolizing enzyme-related interactions with other drugs or agents are anticipated.
14 CLINICAL STUDIES
14.1 Nosocomial Pneumonia
Adult patients with clinically and radiologically documented nosocomial pneumonia were enrolled in a multicenter, randomized, open-label study comparing intravenous LEVAQUIN® (750 mg once daily) followed by oral LEVAQUIN® (750 mg once daily) for a total of 7–15 days to intravenous imipenem/cilastatin (500–1000 mg every 6–8 hours daily) followed by oral ciprofloxacin (750 mg every 12 hours daily) for a total of 7–15 days. LEVAQUIN®-treated patients received an average of 7 days of intravenous therapy (range: 1–16 days); comparator-treated patients received an average of 8 days of intravenous therapy (range: 1–19 days).
Overall, in the clinically and microbiologically evaluable population, adjunctive therapy was empirically initiated at study entry in 56 of 93 (60.2%) patients in the LEVAQUIN® arm and 53 of 94 (56.4%) patients in the comparator arm. The average duration of adjunctive therapy was 7 days in the LEVAQUIN® arm and 7 days in the comparator. In clinically and microbiologically evaluable patients with documented Pseudomonas aeruginosa infection, 15 of 17 (88.2%) received ceftazidime (N=11) or piperacillin/tazobactam (N=4) in the LEVAQUIN® arm and 16 of 17 (94.1%) received an aminoglycoside in the comparator arm. Overall, in clinically and microbiologically evaluable patients, vancomycin was added to the treatment regimen of 37 of 93 (39.8%) patients in the LEVAQUIN® arm and 28 of 94 (29.8%) patients in the comparator arm for suspected methicillin-resistant S. aureus infection.
Clinical success rates in clinically and microbiologically evaluable patients at the posttherapy visit (primary study endpoint assessed on day 3–15 after completing therapy) were 58.1% for LEVAQUIN® and 60.6% for comparator. The 95% CI for the difference of response rates (LEVAQUIN® minus comparator) was [-17.2, 12.0]. The microbiological eradication rates at the posttherapy visit were 66.7% for LEVAQUIN® and 60.6% for comparator. The 95% CI for the difference of eradication rates (LEVAQUIN® minus comparator) was [-8.3, 20.3]. Clinical success and microbiological eradication rates by pathogen are detailed in Table 13.
| Pathogen | N | LEVAQUIN® No. (%) of Patients Microbiologic/ Clinical Outcomes | N | Imipenem/Cilastatin No. (%) of Patients Microbiologic/ Clinical Outcomes |
|---|---|---|---|---|
| MSSA* | 21 | 14 (66.7)/13 (61.9) | 19 | 13 (68.4)/15 (78.9) |
| P. aeruginosa † | 17 | 10 (58.8)/11 (64.7) | 17 | 5 (29.4)/7 (41.2) |
| S. marcescens | 11 | 9 (81.8)/7 (63.6) | 7 | 2 (28.6)/3 (42.9) |
| E. coli | 12 | 10 (83.3)/7 (58.3) | 11 | 7 (63.6 )/8 (72.7) |
| K. pneumoniae ‡ | 11 | 9 (81.8)/5 (45.5) | 7 | 6 (85.7)/3 (42.9) |
| H. influenzae | 16 | 13 (81.3)/10 (62.5) | 15 | 14 (93.3)/11 (73.3) |
| S. pneumoniae | 4 | 3 (75.0)/3 (75.0) | 7 | 5 (71.4)/4 (57.1) |
14.2 Community-Acquired Pneumonia: 7–14 day Treatment Regimen
Adult inpatients and outpatients with a diagnosis of community-acquired bacterial pneumonia were evaluated in 2 pivotal clinical studies. In the first study, 590 patients were enrolled in a prospective, multi-center, unblinded randomized trial comparing LEVAQUIN® 500 mg once daily orally or intravenously for 7 to 14 days to ceftriaxone 1 to 2 grams intravenously once or in equally divided doses twice daily followed by cefuroxime axetil 500 mg orally twice daily for a total of 7 to 14 days. Patients assigned to treatment with the control regimen were allowed to receive erythromycin (or doxycycline if intolerant of erythromycin) if an infection due to atypical pathogens was suspected or proven. Clinical and microbiologic evaluations were performed during treatment, 5 to 7 days posttherapy, and 3 to 4 weeks posttherapy. Clinical success (cure plus improvement) with LEVAQUIN® at 5 to 7 days posttherapy, the primary efficacy variable in this study, was superior (95%) to the control group (83%). The 95% CI for the difference of response rates (LEVAQUIN® minus comparator) was [-6, 19]. In the second study, 264 patients were enrolled in a prospective, multi-center, non-comparative trial of 500 mg LEVAQUIN® administered orally or intravenously once daily for 7 to 14 days. Clinical success for clinically evaluable patients was 93%. For both studies, the clinical success rate in patients with atypical pneumonia due to Chlamydophila pneumoniae, Mycoplasma pneumoniae, and Legionella pneumophila were 96%, 96%, and 70%, respectively. Microbiologic eradication rates across both studies are presented in Table 14.
| Pathogen | No. Pathogens | Microbiologic Eradication Rate (%) |
|---|---|---|
| H. influenzae | 55 | 98 |
| S. pneumoniae | 83 | 95 |
| S. aureus | 17 | 88 |
| M. catarrhalis | 18 | 94 |
| H. parainfluenzae | 19 | 95 |
| K. pneumoniae | 10 | 100.0 |
Community-Acquired Pneumonia Due to Multi-Drug Resistant Streptococcus pneumoniae
LEVAQUIN® was effective for the treatment of community-acquired pneumonia caused by multi-drug resistant Streptococcus pneumoniae (MDRSP). MDRSP isolates are strains resistant to two or more of the following antibacterials: penicillin (MIC ≥2 mcg/mL), 2nd generation cephalosporins (e.g., cefuroxime, macrolides, tetracyclines and trimethoprim/sulfamethoxazole). Of 40 microbiologically evaluable patients with MDRSP isolates, 38 patients (95.0%) achieved clinical and bacteriologic success at post-therapy. The clinical and bacterial success rates are shown in Table 15.
| Screening Susceptibility | Clinical Success | Bacteriological Success* | ||
|---|---|---|---|---|
| n/N† | % | n/N‡ | % | |
|
||||
| Penicillin-resistant | 16/17 | 94.1 | 16/17 | 94.1 |
| 2nd generation Cephalosporin resistant | 31/32 | 96.9 | 31/32 | 96.9 |
| Macrolide-resistant | 28/29 | 96.6 | 28/29 | 96.6 |
| Trimethoprim/ Sulfamethoxazole resistant | 17/19 | 89.5 | 17/19 | 89.5 |
| Tetracycline-resistant | 12/12 | 100 | 12/12 | 100 |
Not all isolates were resistant to all antimicrobial classes tested. Success and eradication rates are summarized in Table 16.
| Type of Resistance | Clinical Success | Bacteriologic Eradication |
|---|---|---|
| Resistant to 2 antibacterials | 17/18 (94.4%) | 17/18 (94.4%) |
| Resistant to 3 antibacterials | 14/15 (93.3%) | 14/15 (93.3%) |
| Resistant to 4 antibacterials | 7/7 (100%) | 7/7 (100%) |
| Resistant to 5 antibacterials | 0 | 0 |
| Bacteremia with MDRSP | 8/9 (89%) | 8/9 (89%) |
14.3 Community-Acquired Pneumonia: 5-Day Treatment Regimen
To evaluate the safety and efficacy of higher dose and shorter course of LEVAQUIN®, 528 outpatient and hospitalized adults with clinically and radiologically determined mild to severe community-acquired pneumonia were evaluated in a double-blind, randomized, prospective, multicenter study comparing LEVAQUIN® 750 mg, IV or orally, every day for five days or LEVAQUIN® 500 mg IV or orally, every day for 10 days.
Clinical success rates (cure plus improvement) in the clinically evaluable population were 90.9% in the LEVAQUIN® 750 mg group and 91.1% in the LEVAQUIN® 500 mg group. The 95% CI for the difference of response rates (LEVAQUIN® 750 minus LEVAQUIN® 500) was [-5.9, 5.4]. In the clinically evaluable population (31–38 days after enrollment) pneumonia was observed in 7 out of 151 patients in the LEVAQUIN® 750 mg group and 2 out of 147 patients in the LEVAQUIN® 500 mg group. Given the small numbers observed, the significance of this finding cannot be determined statistically. The microbiological efficacy of the 5-day regimen was documented for infections listed in Table 17.
| Penicillin susceptible S. pneumoniae | 19/20 |
| Haemophilus influenzae | 12/12 |
| Haemophilus parainfluenzae | 10/10 |
| Mycoplasma pneumoniae | 26/27 |
| Chlamydophila pneumoniae | 13/15 |
14.4 Acute Bacterial Sinusitis: 5-day and 10–14 day Treatment Regimens
LEVAQUIN® is approved for the treatment of acute bacterial sinusitis (ABS) using either 750 mg by mouth × 5 days or 500 mg by mouth once daily × 10–14 days. To evaluate the safety and efficacy of a high dose short course of LEVAQUIN®, 780 outpatient adults with clinically and radiologically determined acute bacterialsinusitis were evaluated in a double-blind, randomized, prospective, multicenter study comparing LEVAQUIN® 750 mg by mouth once daily for five days to LEVAQUIN® 500 mg by mouth once daily for 10 days.
Clinical success rates (defined as complete or partial resolution of the pre-treatment signs and symptoms of ABS to such an extent that no further antibiotic treatment was deemed necessary) in the microbiologically evaluable population were 91.4% (139/152) in the LEVAQUIN® 750 mg group and 88.6% (132/149) in the LEVAQUIN® 500 mg group at the test-of-cure (TOC) visit (95% CI [-4.2, 10.0] for LEVAQUIN® 750 mg minus LEVAQUIN® 500 mg).
Rates of clinical success by pathogen in the microbiologically evaluable population who had specimens obtained by antral tap at study entry showed comparable results for the five- and ten-day regimens at the test-of-cure visit 22 days post treatment.
| Pathogen | LEVAQUIN® 750 mg × 5 days | LEVAQUIN® 500 mg × 10 days |
|---|---|---|
|
||
| Streptococcus pneumoniae* | 25/27 (92.6%) | 26/27 (96.3%) |
| Haemophilus influenzae* | 19/21 (90.5%) | 25/27 (92.6%) |
| Moraxella catarrhalis* | 10/11 (90.9%) | 13/13 (100%) |
14.5 Complicated Skin and Skin Structure Infections
Three hundred ninety-nine patients were enrolled in an open-label, randomized, comparative study for complicated skin and skin structure infections. The patients were randomized to receive either LEVAQUIN® 750 mg once daily (IV followed by oral), or an approved comparator for a median of 10 ± 4.7 days. As is expected in complicated skin and skin structure infections, surgical procedures were performed in the LEVAQUIN® and comparator groups. Surgery (incision and drainage or debridement) was performed on 45% of the LEVAQUIN®-treated patients and 44% of the comparator treated patients, either shortly before or during antibiotic treatment and formed an integral part of therapy for this indication.
Among those who could be evaluated clinically 2–5 days after completion of study drug, overall success rates (improved or cured) were 116/138 (84.1%) for patients treated with LEVAQUIN® and 106/132 (80.3%) for patients treated with the comparator.
Success rates varied with the type of diagnosis ranging from 68% in patients with infected ulcers to 90% in patients with infected wounds and abscesses. These rates were equivalent to those seen with comparator drugs.
14.6 Chronic Bacterial Prostatitis
Adult patients with a clinical diagnosis of prostatitis and microbiological culture results from urine sample collected after prostatic massage (VB3) or expressed prostatic secretion (EPS) specimens obtained via the Meares-Stamey procedure were enrolled in a multicenter, randomized, double-blind study comparing oral LEVAQUIN® 500 mg, once daily for a total of 28 days to oral ciprofloxacin 500 mg, twice daily for a total of 28 days. The primary efficacy endpoint was microbiologic efficacy in microbiologically evaluable patients. A total of 136 and 125 microbiologically evaluable patients were enrolled in the LEVAQUIN® and ciprofloxacin groups, respectively. The microbiologic eradication rate by patient infection at 5–18 days after completion of therapy was 75.0% in the LEVAQUIN® group and 76.8% in the ciprofloxacin group (95% CI [-12.58, 8.98] for LEVAQUIN® minus ciprofloxacin). The overall eradication rates for pathogens of interest are presented in Table 19.
| LEVAQUIN® (N=136) | Ciprofloxacin (N=125) | |||
|---|---|---|---|---|
| Pathogen | N | Eradication | N | Eradication |
|
||||
| E. coli | 15 | 14 (93.3%) | 11 | 9 (81.8%) |
| E. faecalis | 54 | 39 (72.2%) | 44 | 33 (75.0%) |
| S. epidermidis * | 11 | 9 (81.8%) | 14 | 11 (78.6%) |
Eradication rates for S. epidermidis when found with other co-pathogens are consistent with rates seen in pure isolates.
Clinical success (cure + improvement with no need for further antibiotic therapy) rates in microbiologically evaluable population 5–18 days after completion of therapy were 75.0% for LEVAQUIN®-treated patients and 72.8% for ciprofloxacin-treated patients (95% CI [-8.87, 13.27] for LEVAQUIN® minus ciprofloxacin). Clinical long-term success (24–45 days after completion of therapy) rates were 66.7% for the LEVAQUIN®-treated patients and 76.9% for the ciprofloxacin-treated patients (95% CI [-23.40, 2.89] for LEVAQUIN® minus ciprofloxacin).
14.7 Complicated Urinary Tract Infections and Acute Pyelonephritis: 5-day Treatment Regimen
To evaluate the safety and efficacy of the higher dose and shorter course of LEVAQUIN®, 1109 patients with cUTI and AP were enrolled in a randomized, double-blind, multicenter clinical trial conducted in the US from November 2004 to April 2006 comparing LEVAQUIN® 750 mg IV or orally once daily for 5 days (546 patients) with ciprofloxacin 400 mg IV or 500 mg orally twice daily for 10 days (563 patients). Patients with AP complicated by underlying renal diseases or conditions such as complete obstruction, surgery, transplantation, concurrent infection or congenital malformation were excluded. Efficacy was measured by bacteriologic eradication of the baseline organism(s) at the post-therapy visit in patients with a pathogen identified at baseline. The post-therapy (test-of-cure) visit occurred 10 to 14 days after the last active dose of LEVAQUIN® and 5 to 9 days after the last dose of active ciprofloxacin.
The bacteriologic cure rates overall for LEVAQUIN® and control at the test-of-cure (TOC) visit for the group of all patients with a documented pathogen at baseline (modified intent to treat or mITT) and the group of patients in the mITT population who closely followed the protocol (Microbiologically Evaluable) are summarized in Table 20.
| LEVAQUIN® 750 mg orally or IV once daily for 5 days | Ciprofloxacin 400 mg IV/500 mg orally twice daily for 10 days | Overall Difference [95% CI] | |||
|---|---|---|---|---|---|
| n/N | % | n/N | % | LEVAQUIN®-Ciprofloxacin | |
|
|||||
| mITT Population* | |||||
| Overall (cUTI or AP) | 252/333 | 75.7 | 239/318 | 75.2 | 0.5 (-6.1, 7.1) |
| cUTI | 168/230 | 73.0 | 157/213 | 73.7 | |
| AP | 84/103 | 81.6 | 82/105 | 78.1 | |
| Microbiologically Evaluable Population† | |||||
| Overall (cUTI or AP) | 228/265 | 86.0 | 215/241 | 89.2 | -3.2 [-8.9, 2.5] |
| cUTI | 154/185 | 83.2 | 144/165 | 87.3 | |
| AP | 74/80 | 92.5 | 71/76 | 93.4 | |
Microbiologic eradication rates in the Microbiologically Evaluable population at TOC for individual pathogens recovered from patients randomized to LEVAQUIN® treatment are presented in Table 21.
| Pathogen | Microbiologic Eradication Rate (n/N) | % |
|---|---|---|
|
||
| Escherichia coli* | 155/172 | 90 |
| Klebsiella pneumoniae | 20/23 | 87 |
| Proteus mirabilis | 12/12 | 100 |
14.8 Complicated Urinary Tract Infections and Acute Pyelonephritis: 10-day Treatment Regimen
To evaluate the safety and efficacy of the 250 mg dose, 10 day regimen of LEVAQUIN®, 567 patients with uncomplicated UTI, mild-to-moderate cUTI, and mild-to-moderate AP were enrolled in a randomized, double-blind, multicenter clinical trial conducted in the US from June 1993 to January 1995 comparing LEVAQUIN® 250 orally once daily for 10 days (285 patients) with ciprofloxacin 500 mg orally twice daily for 10 days (282 patients). Patients with a resistant pathogen, recurrent UTI, women over age 55 years, and with an indwelling catheter were initially excluded, prior to protocol amendment which took place after 30% of enrollment. Microbiological efficacy was measured by bacteriologic eradication of the baseline organism(s) at 1–12 days post-therapy in patients with a pathogen identified at baseline.
The bacteriologic cure rates overall for LEVAQUIN® and control at the test-of-cure (TOC) visit for the group of all patients with a documented pathogen at baseline (modified intent to treat or mITT) and the group of patients in the mITT population who closely followed the protocol (Microbiologically Evaluable) are summarized in Table 22.
| LEVAQUIN®
250 mg once daily for 10 days | Ciprofloxacin 500 mg twice daily for 10 days |
|||
|---|---|---|---|---|
| n/N | % | n/N | % | |
|
||||
| mITT Population † | 174/209 | 83.3 | 184/219 | 84.0 |
| Microbiologically Evaluable Population‡ | 164/177 | 92.7 | 159/171 | 93.0 |
14.9 Inhalational Anthrax (Post-Exposure)
The effectiveness of LEVAQUIN® for this indication is based on plasma concentrations achieved in humans, a surrogate endpoint reasonably likely to predict clinical benefit. LEVAQUIN® has not been tested in humans for the post-exposure prevention of inhalation anthrax. The mean plasma concentrations of LEVAQUIN® associated with a statistically significant improvement in survival over placebo in the rhesus monkey model of inhalational anthrax are reached or exceeded in adult and pediatric patients receiving the recommended oral and intravenous dosage regimens [see Indications and Usage (1.13); Dosage and Administration (2.1, 2.2)].
Levofloxacin pharmacokinetics have been evaluated in adult and pediatric patients. The mean (± SD) steady state peak plasma concentration in human adults receiving 500 mg orally or intravenously once daily is 5.7 ± 1.4 and 6.4 ± 0.8 mcg/mL, respectively; and the corresponding total plasma exposure (AUC0-24) is 47.5 ± 6.7 and 54.6 ± 11.1 mcg.h/mL, respectively. The predicted steady-state pharmacokinetic parameters in pediatric patients ranging in age from 6 months to 17 years receiving 8 mg/kg orally every 12 hours (not to exceed 250 mg per dose) were calculated to be comparable to those observed in adults receiving 500 mg orally once daily [see Clinical Pharmacology (12.3)].
In adults, the safety of LEVAQUIN® for treatment durations of up to 28 days is well characterized. However, information pertaining to extended use at 500 mg daily up to 60 days is limited. Prolonged LEVAQUIN® therapy in adults should only be used when the benefit outweighs the risk.
In pediatric patients, the safety of levofloxacin for treatment durations of more than 14 days has not been studied. An increased incidence of musculoskeletal adverse events (arthralgia, arthritis, tendonopathy, gait abnormality) compared to controls has been observed in clinical studies with treatment duration of up to 14 days. Long-term safety data, including effects on cartilage, following the administration of levofloxacin to pediatric patients is limited [see Warnings and Precautions (5.9), Use in Specific Populations (8.4)].
A placebo-controlled animal study in rhesus monkeys exposed to an inhaled mean dose of 49 LD50 (~2.7 X 106) spores (range 17 - 118 LD50) of B. anthracis (Ames strain) was conducted. The minimal inhibitory concentration (MIC) of levofloxacin for the anthrax strain used in this study was 0.125 mcg/mL. In the animals studied, mean plasma concentrations of levofloxacin achieved at expected Tmax (1 hour post dose) following oral dosing to steady state ranged from 2.79 to 4.87 mcg/mL. Steady state trough concentrations at 24 hours post-dose ranged from 0.107 to 0.164 mcg/mL. Mean (SD) steady state AUC0-24 was 33.4 ± 3.2 mcg.h/mL (range 30.4 to 36.0 mcg.h/mL). Mortality due to anthrax for animals that received a 30 day regimen of oral LEVAQUIN® beginning 24 hrs post exposure was significantly lower (1/10), compared to the placebo group (9/10) [P=0.0011, 2-sided Fisher’s Exact Test]. The one levofloxacin treated animal that died of anthrax did so following the 30-day drug administration period.
15 REFERENCES
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically Approved Standard – Seventh Edition. Clinical and Laboratory Standards Institute document M7-A7, Vol. 26, No. 2, CLSI, Wayne, PA, January 2006.
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Disk Susceptibility Tests. Approved Standard – Ninth Edition. Clinical and Laboratory Standards Institute document M2-A9, Vol. 26, No. 1, CLSI, Wayne, PA, January 2006.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 LEVAQUIN® Tablets
LEVAQUIN® Tablets are supplied as 250, 500, and 750 mg capsule-shaped, coated tablets. LEVAQUIN® Tablets are packaged in bottles and in unit-dose blister strips in the following configurations:
- 250 mg tablets are terra cotta pink and are imprinted: "LEVAQUIN" on
one side and "250" on the other side.
- –
- bottles of 50 (NDC 0045-1520-50)
- –
- unit-dose/100 tablets (NDC 0045-1520-10)
- 500 mg tablets are peach and are imprinted: "LEVAQUIN" on one side and
"500" on the other side
- –
- bottles of 50 (NDC 0045-1525-50)
- –
- unit-dose/100 tablets (NDC 0045-1525-10)
- 750 mg tablets are white and are imprinted "LEVAQUIN" on one side and
"750" on the other side
- –
- bottles of 20 (NDC 0045-1530-20)
- –
- unit-dose/100 tablets (NDC 0045-1530-10)
- –
- LĒVA-pak 5 tablets (NDC 0045-1530-05)
LEVAQUIN® Tablets should be stored at 15° to 30°C (59° to 86°F) in well-closed containers.
LEVAQUIN® Tablets are manufactured for OMP DIVISION, ORTHO-McNEIL PHARMACEUTICAL, INC. by Janssen Ortho LLC, Gurabo, Puerto Rico 00778.
16.2 LEVAQUIN® Oral Solution
LEVAQUIN® Oral Solution is supplied in a 16 oz. multi-use bottle (NDC 0045-1515-01). Each bottle contains 480 mL of the 25 mg/mL levofloxacin oral solution
LEVAQUIN® Oral Solution should be stored at 25°C (77°F); excursions permitted to 15° – 30°C (59° to 86°F) [refer to USP controlled room temperature].
LEVAQUIN® Oral Solution is manufactured for OMP DIVISION, ORTHO-McNEIL PHARMACEUTICAL, INC. by Janssen Pharmaceutica N.V., Beerse, Belgium.
16.3 LEVAQUIN® Injection, Single-Use Vials
LEVAQUIN® Injection is supplied in single-use vials. Each vial contains a concentrated solution with the equivalent of 500 mg of levofloxacin in 20 mL vials and 750 mg of levofloxacin in 30 mL vials.
- 25 mg/mL, 20 mL vials (NDC 0045-0069-51)
- 25 mg/mL, 30 mL vials (NDC 0045-0065-55)
LEVAQUIN® Injection in Single-Use Vials should be stored at controlled room temperature and protected from light.
LEVAQUIN® Injection in Single-Use Vials is manufactured for OMP DIVISION, ORTHO-McNEIL PHARMACEUTICAL, INC. by Janssen Pharmaceutica N.V., Beerse, Belgium.
16.4 LEVAQUIN® Injection Pre-Mixed Solution, Single-Use in Flexible Container
LEVAQUIN® (levofloxacin in 5% dextrose) Injection is supplied as a single-use, premixed solution in flexible containers. Each bag contains a dilute solution with the equivalent of 250, 500, or 750 mg of levofloxacin, respectively, in 5% Dextrose (D5W).
- 5 mg/mL (250 mg), 100 mL flexible container, 50 mL fill (NDC 0045-0067-01)
- 5 mg/mL (500 mg), 100 mL flexible container, 100 mL fill (NDC 0045-0068-01)
- 5 mg/mL (750 mg), 150 mL flexible container, 150 mL fill (NDC 0045-0066-01)
LEVAQUIN® Injection Premix in Flexible Containers should be stored at or below 25°C (77°F); however, brief exposure up to 40°C (104°F) does not adversely affect the product. Avoid excessive heat and protect from freezing and light.
LEVAQUIN® Injection Premix in Flexible Containers is manufactured for OMP DIVISION, ORTHO-McNEIL PHARMACEUTICAL, INC. by Hospira, Inc., Lake Forest, IL 60045.
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (17.5)
17.1 Antibacterial Resistance
Antibacterial drugs including LEVAQUIN® should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When LEVAQUIN® is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by LEVAQUIN® or other antibacterial drugs in the future.
17.2 Administration with Food, Fluids, and Concomitant Medications
Patients should be informed that LEVAQUIN® Tablets may be taken with or without food. LEVAQUIN® Oral Solution should be taken 1 hour before or 2 hours after eating. The tablet and oral solution should be taken at the same time each day.
Patients should drink fluids liberally while taking LEVAQUIN® to avoid formation of a highly concentrated urine and crystal formation in the urine.
Antacids containing magnesium, or aluminum, as well as sucralfate, metal cations such as iron, and multivitamin preparations with zinc or didanosine should be taken at least two hours before or two hours after oral LEVAQUIN® administration.
17.3 Serious and Potentially Serious Adverse Reactions
Patients should be informed of the following serious adverse reactions that have been associated with LEVAQUIN® or other quinolone use:
- Hypersensitivity Reactions: Patients should be informed that LEVAQUIN® can cause hypersensitivity reactions, even following the first dose. Patients should discontinue the drug at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (e.g., swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
- Hepatotoxicity: Severe hepatotoxicity (including acute hepatitis and fatal events) has been reported in patients taking LEVAQUIN®. Patients should inform their physician and be instructed to discontinue LEVAQUIN® treatment immediately if they experience any signs or symptoms of liver injury including: loss of appetite, nausea, vomiting, fever, weakness, tiredness, right upper quadrant tenderness, itching, yellowing of the skin and eyes, light colored bowel movements or dark colored urine.
- Tendon Disorders: Patients should discontinue LEVAQUIN® treatment and inform their physician if they experience pain, inflammation, or rupture of a tendon, and to rest and refrain from exercise until the diagnosis of tendonitis or tendon rupture has been excluded. The risk of serious tendon disorders is higher in those over 65 years of age, especially those on corticosteroids.
- Convulsions: Convulsions have been reported in patients taking quinolones, including LEVAQUIN®. Patients should notify their physician before taking this drug if they have a history of convulsions.
- Neurologic Adverse Effects (e.g., dizziness, lightheadedness): Patients should know how they react to LEVAQUIN® before they operate an automobile or machinery or engage in other activities requiring mental alertness and coordination.
- Diarrhea: Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
- Peripheral Neuropathies: If symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness develop, patients should discontinue treatment and contact their physician.
- Prolongation of the QT Interval: Patients should inform their physician of any personal or family history of QT prolongation or proarrhythmic conditions such as hypokalemia, bradycardia, or recent myocardial ischemia; if they are taking any class IA (quinidine, procainamide), or class III (amiodarone, sotalol) antiarrhythmic agents. Patients should notify their physicians if they have any symptoms of prolongation of the QT interval, including prolonged heart palpitations or a loss of consciousness.
- Musculoskeletal Disorders in Pediatric Patients: Parents should inform their child's physician if their child has a history of joint-related problems before taking this drug. Parents of pediatric patients should also notify their child's physician of any tendon or joint-related problems that occur during or following LEVAQUIN® therapy [see Warnings and Precautions (5.9) and Use in Specific Populations (8.4)].
- Photosensitivity/Phototoxicity: Patients should be advised that photosensitivity/phototoxicity has been reported in patients receiving quinolone antibiotics. Patients should minimize or avoid exposure to natural or artificial sunlight (tanning beds or UVA/B treatment) while taking quinolones. If patients need to be outdoors when taking quinolones, they should wear loose-fitting clothes that protect skin from sun exposure and discuss other sun protection measures with their physician. If a sunburn like reaction or skin eruption occurs, patients should contact their physician.
17.4 Drug Interactions with Insulin, Oral Hypoglycemic Agents, and Warfarin
Patients should be informed that if they are diabetic and are being treated with insulin or an oral hypoglycemic agent and a hypoglycemic reaction occurs, they should discontinue LEVAQUIN® and consult a physician.
Patients should be informed that concurrent administration of warfarin and LEVAQUIN® has been associated with increases of the International Normalized Ratio (INR) or prothrombin time and clinical episodes of bleeding. Patients should notify their physician if they are taking warfarin, be monitored for evidence of bleeding, and also have their anticoagulation tests closely monitored while taking warfarin concomitantly.
17.5 FDA-Approved Patient Labeling
Patient Information
About:
LEVAQUIN® (levofloxacin) Tablets
250 mg Tablets, 500 mg Tablets, and 750 mg Tablets
And
LEVAQUIN® (levofloxacin) Oral Solution, 25 mg/mL
This leaflet contains important information about LEVAQUIN®, and should be read completely before you begin treatment. This leaflet does not take the place of discussions with your doctor or healthcare professional about your medical condition or your treatment. This leaflet does not list all benefits and risks of LEVAQUIN®. The medicine described here can be prescribed only by a licensed health care professional. If you have any questions about LEVAQUIN® talk to your health care professional. Only your healthcare professional can determine if LEVAQUIN® is right for you.
What is LEVAQUIN®?
LEVAQUIN® is a quinolone antibiotic used to treat lung, sinus, skin, and urinary tract infections caused by certain germs called bacteria. LEVAQUIN® kills many of the types of bacteria that can infect the lungs, sinuses, skin, and urinary tract and has been shown in a large number of clinical trials to be safe and effective for the treatment of bacterial infections.
Sometimes viruses rather than bacteria may infect the lungs and sinuses (for example, the common cold). LEVAQUIN®, like other antibiotics, does not kill viruses.
You should contact your healthcare professional if you think that your condition is not improving while taking LEVAQUIN®. LEVAQUIN® Tablets are terra cotta pink for the 250 mg tablet, peach colored for the 500 mg tablet, or white for the 750 mg tablet. The appearance of LEVAQUIN® Oral Solution may range from clear yellow to clear greenish-yellow.
How and when should I take LEVAQUIN®?
LEVAQUIN® should be taken once a day for 3, 5, 7, 10, 14 or 28 days depending on your prescription. LEVAQUIN® Tablets should be swallowed and may be taken with or without food. LEVAQUIN® Oral Solution should be taken 1 hour before or 2 hours after eating. Try to take the tablet and oral solution at the same time each day and drink fluids liberally.
You may begin to feel better quickly; however, in order to make sure that all bacteria are killed, you should complete the full course of medication. Do not take more than the prescribed dose of LEVAQUIN® even if you missed a dose by mistake. You should not take a double dose.
Who should not take LEVAQUIN®?
You should not take LEVAQUIN® if you have ever had a severe allergic reaction to any of the group of antibiotics known as "quinolones" such as ciprofloxacin. Serious and occasionally fatal allergic reactions have been reported in patients receiving therapy with quinolones, including LEVAQUIN®.
If you are pregnant or are planning to become pregnant while taking LEVAQUIN®, talk to your healthcare professional before taking this medication. LEVAQUIN® is not recommended for use during pregnancy or nursing, as the effects on the unborn child or nursing infant are unknown.
Due to possible side effects, LEVAQUIN® is not recommended for pediatric patients except in the prevention of anthrax after inhalational exposure.
What are possible side effects of LEVAQUIN®?
LEVAQUIN® is generally well tolerated. The most common adverse drug reactions (≥3%) are nausea, headache, diarrhea, insomnia, constipation, and dizziness.
You should be careful about driving or operating machinery until you are sure LEVAQUIN® is not causing dizziness.
Allergic reactions have been reported in patients receiving quinolones including LEVAQUIN®, even after just one dose. If you develop hives, skin rash or other symptoms of an allergic reaction, you should stop taking this medication and call your healthcare professional.
Hepatotoxicity (liver damage) has been reported in patients receiving LEVAQUIN®. Call your doctor right away if you have unexplained symptoms such as: nausea or vomiting, stomach pain, fever, weakness, abdominal pain or tenderness, itching, unusual or unexplained tiredness, loss of appetite, light colored bowel movements, dark colored urine or yellowing of your skin or the whites of your eyes.
Pain, swelling, and tears of Achilles, shoulder, or hand tendons have been reported in patients receiving fluoroquinolones, including LEVAQUIN®. The risk for tendon effects is higher if you are over 65 years old, and especially if you are taking corticosteroids. If you develop pain, swelling, or rupture of a tendon, you should stop taking LEVAQUIN®, avoid exercise and strenuous use of the affected area, and contact your healthcare professional.
Sun sensitivity (photosensitivity), which can appear as skin eruption or severe sunburn, can occur in some patients taking quinolone antibiotics after exposure to sunlight or artificial ultraviolet (UV) light (e.g., tanning beds). LEVAQUIN® has been infrequently associated with photosensitivity. Avoid excessive exposure to sunlight or artificial UV light while taking LEVAQUIN®. Use a sunscreen and wear protective clothing if out in the sun. If photosensitivity develops, contact your physician.
If you have diabetes and you develop a hypoglycemic reaction while on LEVAQUIN®, you should stop taking LEVAQUIN® and call your healthcare professional.
Convulsions have been reported in patients receiving quinolone antibiotics including LEVAQUIN®. If you have experienced convulsions in the past, be sure to let your physician know that you have a history of convulsions.
Quinolones, including LEVAQUIN®, may also cause central nervous system stimulation which may lead to tremors, restlessness, anxiety, lightheadedness, confusion, hallucinations, paranoia, depression, nightmares, insomnia, and rarely, suicidal thoughts or acts.
Diarrhea that usually ends after treatment is a common problem caused by antibiotics. A more serious form of diarrhea can occur during or up to 2 months after the use of antibiotics. This has been reported with all antibiotics including with LEVAQUIN®. If you develop a watery and bloody stool with or without stomach cramps and fever, contact your physician as soon as possible.
In a few people, LEVAQUIN®, like some other antibiotics, may produce a small effect on the heart that is seen on an electrocardiogram test. The rare heart problem is called QT prolongation and can cause an abnormal heartbeat and can be very dangerous. The chances of this event are increased in those with a family history of prolonged QT interval, low potassium (hypokalemia), and who are taking drugs to control heart rhythm, called class IA (quinidine, procainamide) or class III (amiodarone, sotalol) antiarrhythmic agents. You should call your healthcare professional right away if you have any prolonged heart palpitations (a change in the way your heart beats) or a loss of consciousness (fainting spells).
If you notice any side effects not mentioned in this leaflet or you have concerns about the side effects you are experiencing, please inform your healthcare professional.
For more complete information regarding LEVAQUIN®, please refer to the full prescribing information, which may be obtained from your healthcare professional, pharmacist, or the Physicians Desk Reference (PDR).
What about other medicines I am taking?
Taking warfarin and LEVAQUIN® together can further predispose you to the development of bleeding problems. If you take warfarin, be sure to tell your healthcare professional.
Many antacids and multivitamins may interfere with the absorption of LEVAQUIN® and may prevent it from working properly. You should take LEVAQUIN® either 2 hours before or 2 hours after taking these products.
It is important to let your healthcare professional know all of the medicines you are using.
What if I have been prescribed LEVAQUIN® for possible anthrax exposure?
LEVAQUIN® has been approved to reduce the chance of developing anthrax infection following exposure to the anthrax bacteria. With the exception of using LEVAQUIN® to prevent anthrax after possible exposure, LEVAQUIN® is not recommended for children. If you are pregnant, or plan to become pregnant while taking LEVAQUIN®, you and your doctor should discuss if the benefits of taking LEVAQUIN® for anthrax outweigh the risks.
LEVAQUIN® is generally well tolerated. Side effects that may occur during treatment to prevent anthrax might be acceptable due to the seriousness of the disease. You and your doctor should discuss the risks of not taking your medicine against the risks of experiencing side effects.
Other information
Take your dose of LEVAQUIN® once a day.
Complete the course of medication even if you are feeling better.
Keep this medication out of the reach of children.
Some quinolones, including LEVAQUIN®, may produce false-positive urine screening results for opiates using commercially available immunoassay kits. Confirmation of positive opiate screens by more specific methods may be necessary.
This information does not take the place of discussions with your doctor or healthcare professional about your medical condition or your treatment.
OMP
DIVISION
ORTHO-McNEIL PHARMACEUTICAL, INC.
Raritan, New Jersey,
USA 08869
U.S. Patent No. 5,053,407.
May 2008
| Levaquin (levofloxacin) | ||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| Levaquin (levofloxacin) | |||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||
| Levaquin (levofloxacin) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
| Levaquin (levofloxacin) | |||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||
| Levaquin (levofloxacin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Levaquin (levofloxacin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Levaquin (levofloxacin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Levaquin (levofloxacin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Levaquin (levofloxacin) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Revised: 07/2008PriCara Unit of Ortho-McNeil Pharmaceutical, Inc.