forteo (Teriparatide) injection, solution
[Eli Lilly and Company]
WARNING
In male and female rats, teriparatide caused an increase in the incidence of osteosarcoma (a malignant bone tumor) that was dependent on dose and treatment duration. The effect was observed at systemic exposures to teriparatide ranging from 3 to 60 times the exposure in humans given a 20–mcg dose. Because of the uncertain relevance of the rat osteosarcoma finding to humans, teriparatide should be prescribed only to patients for whom the potential benefits are considered to outweigh the potential risk. Teriparatide should not be prescribed for patients who are at increased baseline risk for osteosarcoma (including those with Paget’s disease of bone or unexplained elevations of alkaline phosphatase, open epiphyses, or prior external beam or implant radiation therapy involving the skeleton) (see WARNINGS and PRECAUTIONS, Carcinogenesis).
DESCRIPTION
FORTEO® [teriparatide (rDNA origin) injection] contains recombinant human parathyroid hormone (1–34), [rhPTH(1–34)], which has an identical sequence to the 34 N–terminal amino acids (the biologically active region) of the 84–amino acid human parathyroid hormone.
Teriparatide has a molecular weight of 4117.8 daltons and its amino acid sequence is shown below:
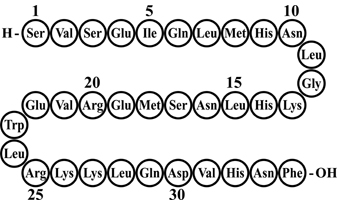
Teriparatide (rDNA origin) is manufactured by Eli Lilly and Company using a strain of Escherichia coli modified by recombinant DNA technology. FORTEO is supplied as a sterile, colorless, clear, isotonic solution in a glass cartridge which is pre–assembled into a disposable pen device for subcutaneous injection. Each mL contains 250 mcg teriparatide (corrected for acetate, chloride, and water content), 0.41 mg glacial acetic acid, 0.1 mg sodium acetate (anhydrous), 45.4 mg mannitol, 3 mg Metacresol, and Water for Injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4.
Each cartridge pre–assembled into a pen device delivers 20 mcg of teriparatide per dose each day for up to 28 days.
See accompanying User Manual: Instructions for Use.
CLINICAL PHARMACOLOGY
Mechanism of Action
Endogenous 84–amino–acid parathyroid hormone (PTH) is the primary regulator of calcium and phosphate metabolism in bone and kidney. Physiological actions of PTH include regulation of bone metabolism, renal tubular reabsorption of calcium and phosphate, and intestinal calcium absorption. The biological actions of PTH and teriparatide are mediated through binding to specific high-affinity cell–surface receptors. Teriparatide and the 34 N–terminal amino acids of PTH bind to these receptors with the same affinity and have the same physiological actions on bone and kidney. Teriparatide is not expected to accumulate in bone or other tissues.
The skeletal effects of teriparatide depend upon the pattern of systemic exposure. Once–daily administration of teriparatide stimulates new bone formation on trabecular and cortical (periosteal and/or endosteal) bone surfaces by preferential stimulation of osteoblastic activity over osteoclastic activity. In monkey studies, teriparatide improved trabecular microarchitecture and increased bone mass and strength by stimulating new bone formation in both cancellous and cortical bone. In humans, the anabolic effects of teriparatide manifest as an increase in skeletal mass, an increase in markers of bone formation and resorption, and an increase in bone strength. By contrast, continuous excess of endogenous PTH, as occurs in hyperparathyroidism, may be detrimental to the skeleton because bone resorption may be stimulated more than bone formation.
Human Pharmacokinetics
Teriparatide is extensively absorbed after subcutaneous injection; the absolute bioavailability is approximately 95% based on pooled data from 20-, 40-, and 80–mcg doses. The rates of absorption and elimination are rapid. The peptide reaches peak serum concentrations about 30 minutes after subcutaneous injection of a 20–mcg dose and declines to non–quantifiable concentrations within 3 hours.
Systemic clearance of teriparatide (approximately 62 L/hr in women and 94 L/hr in men) exceeds the rate of normal liver plasma flow, consistent with both hepatic and extra–hepatic clearance. Volume of distribution, following intravenous injection, is approximately 0.12 L/kg. Intersubject variability in systemic clearance and volume of distribution is 25% to 50%. The half–life of teriparatide in serum is 5 minutes when administered by intravenous injection and approximately 1 hour when administered by subcutaneous injection. The longer half–life following subcutaneous administration reflects the time required for absorption from the injection site.
No metabolism or excretion studies have been performed with teriparatide. However, the mechanisms of metabolism and elimination of PTH(1–34) and intact PTH have been extensively described in published literature. Peripheral metabolism of PTH is believed to occur by non–specific enzymatic mechanisms in the liver followed by excretion via the kidneys.
Special Populations
Pediatric
Pharmacokinetic data in pediatric patients are not available (see WARNINGS).
Geriatric
No age–related differences in teriparatide pharmacokinetics were detected (range 31 to 85 years).
Gender
Although systemic exposure to teriparatide was approximately 20% to 30% lower in men than women, the recommended dose for both genders is 20 mcg/day.
Race
The populations included in the pharmacokinetic analyses were 98.5% Caucasian. The influence of race has not been determined.
Renal insufficiency
No pharmacokinetic differences were identified in 11 patients with mild or moderate renal insufficiency [creatinine clearance (CrCl) 30 to 72 mL/min] administered a single dose of teriparatide. In 5 patients with severe renal insufficiency (CrCl<30 mL/min), the AUC and T1/2 of teriparatide were increased by 73% and 77%, respectively. Maximum serum concentration of teriparatide was not increased. No studies have been performed in patients undergoing dialysis for chronic renal failure (see PRECAUTIONS).
Heart failure
No clinically relevant pharmacokinetic, blood pressure, or pulse rate differences were identified in 13 patients with stable New York Heart Association Class I to III heart failure after the administration of two 20–mcg doses of teriparatide.
Hepatic insufficiency
Non–specific proteolytic enzymes in the liver (possibly Kupffer cells) cleave PTH(1–34) and PTH(1–84) into fragments that are cleared from the circulation mainly by the kidney. No studies have been performed in patients with hepatic impairment.
Drug Interactions
Hydrochlorothiazide — In a study of 20 healthy people, the coadministration of hydrochlorothiazide 25 mg with teriparatide did not affect the serum calcium response to teriparatide 40 mcg. The 24–hour urine excretion of calcium was reduced by a clinically unimportant amount (15%). The effect of coadministration of a higher dose of hydrochlorothiazide with teriparatide on serum calcium levels has not been studied.
Furosemide — In a study of 9 healthy people and 17 patients with mild, moderate, or severe renal insufficiency (CrCl 13 to 72 mL/min), coadministration of intravenous furosemide (20 to 100 mg) with teriparatide 40 mcg resulted in small increases in the serum calcium (2%) and 24–hour urine calcium (37%) responses to teriparatide that did not appear to be clinically important.
Human Pharmacodynamics
Effects on mineral metabolism
Teriparatide affects calcium and phosphorus metabolism in a pattern consistent with the known actions of endogenous PTH (eg, increases serum calcium and decreases serum phosphorus).
Serum calcium concentrations
When teriparatide 20 mcg is administered once daily, the serum calcium concentration increases transiently, beginning approximately 2 hours after dosing and reaching a maximum concentration between 4 and 6 hours (median increase, 0.4 mg/dL). The serum calcium concentration begins to decline approximately 6 hours after dosing and returns to baseline by 16 to 24 hours after each dose.
In a clinical study of postmenopausal women with osteoporosis, the median peak serum calcium concentration measured 4 to 6 hours after dosing with FORTEO (teriparatide 20 mcg) was 2.42 mmol/L (9.68 mg/dL) at 12 months. The peak serum calcium remained below 2.76 mmol/L (11.0 mg/dL) in >99% of women at each visit. Sustained hypercalcemia was not observed.
In this study, 11.1% of women treated with FORTEO had at least 1 serum calcium value above the upper limit of normal [2.64 mmol/L (10.6 mg/dL)] compared with 1.5% of women treated with placebo. The percentage of women treated with FORTEO whose serum calcium was above the upper limit of normal on consecutive 4- to 6–hour post–dose measurements was 3.0% compared with 0.2% of women treated with placebo. In these women, calcium supplements and/or FORTEO doses were reduced. The timing of these dose reductions was at the discretion of the investigator. FORTEO dose adjustments were made at varying intervals after the first observation of increased serum calcium (median 21 weeks). During these intervals, there was no evidence of progressive increases in serum calcium.
In a clinical study of men with either primary or hypogonadal osteoporosis, the effects on serum calcium were similar to those observed in postmenopausal women. The median peak serum calcium concentration measured 4 to 6 hours after dosing with FORTEO was 2.35 mmol/L (9.44 mg/dL) at 12 months. The peak serum calcium remained below 2.76 mmol/L (11.0 mg/dL) in 98% of men at each visit. Sustained hypercalcemia was not observed.
In this study, 6.0% of men treated with FORTEO daily had at least 1 serum calcium value above the upper limit of normal [2.64 mmol/L (10.6 mg/dL)] compared with none of the men treated with placebo. The percentage of men treated with FORTEO whose serum calcium was above the upper limit of normal on consecutive measurements was 1.3% (2 men) compared with none of the men treated with placebo. Although calcium supplements and/or FORTEO doses could have been reduced in these men, only calcium supplementation was reduced (see PRECAUTIONS and ADVERSE EVENTS).
In a clinical study of women previously treated for 18 to 39 months with raloxifene (n=26) or alendronate (n=33), mean serum calcium >12 hours after FORTEO injection was increased by 0.09 to 0.14 mmol/L (0.36 to 0.56 mg/dL), after 1 to 6 months of FORTEO treatment compared with baseline. Of the women pretreated with raloxifene, 3 (11.5%) had a serum calcium >2.76 mmol/L (11.0 mg/dL), and of those pretreated with alendronate, 3 (9.1%) had a serum calcium >2.76 mmol/L (11.0 mg/dL). The highest serum calcium reported was 3.12 mmol/L (12.5 mg/dL). None of the women had symptoms of hypercalcemia. There were no placebo controls in this study.
Urinary calcium excretion
In a clinical study of postmenopausal women with osteoporosis who received 1000 mg of supplemental calcium and at least 400 IU of vitamin D, daily FORTEO increased urinary calcium excretion. The median urinary excretion of calcium was 4.8 mmol/day (190 mg/day) at 6 months and 4.2 mmol/day (170 mg/day) at 12 months. These levels were 0.76 mmol/day (30 mg/day) and 0.30 mmol/day (12 mg/day) higher, respectively, than in women treated with placebo. The incidence of hypercalciuria (>7.5 mmol Ca/day or 300 mg/day) was similar in the women treated with FORTEO or placebo.
In a clinical study of men with either primary or hypogonadal osteoporosis who received 1000 mg of supplemental calcium and at least 400 IU of vitamin D, daily FORTEO had inconsistent effects on urinary calcium excretion. The median urinary excretion of calcium was 5.6 mmol/day (220 mg/day) at 1 month and 5.3 mmol/day (210 mg/day) at 6 months. These levels were 0.50 mmol/day (20 mg/day) higher and 0.20 mmol/day (8.0 mg/day) lower, respectively, than in men treated with placebo. The incidence of hypercalciuria (>7.5 mmol Ca/day or 300 mg/day) was similar in the men treated with FORTEO or placebo.
Phosphorus and vitamin D
In single–dose studies, teriparatide produced transient phosphaturia and mild transient reductions in serum phosphorus concentration. However, hypophosphatemia (<0.74 mmol/L or 2.4 mg/dL) was not observed in clinical trials with FORTEO.
In clinical trials of daily FORTEO, the median serum concentration of 1,25–dihydroxyvitamin D was increased at 12 months by 19% in women and 14% in men, compared with baseline. In the placebo group, this concentration decreased by 2% in women and increased by 5% in men. The median serum 25–hydroxyvitamin D concentration at 12 months was decreased by 19% in women and 10% in men compared with baseline. In the placebo group, this concentration was unchanged in women and increased by 1% in men.
Effects on markers of bone turnover
Daily administration of FORTEO to men and postmenopausal women with osteoporosis in clinical studies stimulated bone formation, as shown by increases in the formation markers serum bone–specific alkaline phosphatase (BSAP) and procollagen I carboxy–terminal propeptide (PICP). Data on biochemical markers of bone turnover were available for the first 12 months of treatment. Peak concentrations of PICP at 1 month of treatment were approximately 41% above baseline, followed by a decline to near–baseline values by 12 months. BSAP concentrations increased by 1 month of treatment and continued to rise more slowly from 6 through 12 months. The maximum increases of BSAP were 45% above baseline in women and 23% in men. After discontinuation of therapy, BSAP concentrations returned toward baseline. The increases in formation markers were accompanied by secondary increases in the markers of bone resorption: urinary N-telopeptide (NTX) and urinary deoxypyridinoline (DPD), consistent with the physiological coupling of bone formation and resorption in skeletal remodeling. Changes in BSAP, NTX, and DPD were lower in men than in women, possibly because of lower systemic exposure to teriparatide in men.
CLINICAL STUDIES
Treatment of Osteoporosis in Postmenopausal Women
The safety and efficacy of once–daily FORTEO, median exposure of 19 months, were examined in a double–blind, placebo–controlled clinical study of 1637 postmenopausal women with osteoporosis (FORTEO 20 mcg, n=541).
This multicenter study was performed in the US and 16 other countries. All women received 1000 mg of calcium per day and at least 400 IU of vitamin D per day. Baseline and endpoint spinal radiographs were evaluated using the semiquantitative scoring method of Genant et al [J Bone Miner Res 1993;8(9):1137–48]. Ninety percent of the women in the study had 1 or more radiographically diagnosed vertebral fractures at baseline. The primary efficacy endpoint was the occurrence of new radiographically diagnosed vertebral fractures defined as changes in the height of previously undeformed vertebrae. Such fractures are not necessarily symptomatic.
Effect on fracture incidence
New vertebral fractures — FORTEO, when taken with calcium and vitamin D and compared with calcium and vitamin D alone, reduced the risk of 1 or more new vertebral fractures from 14.3% of women in the placebo group to 5.0% in the FORTEO group. This difference was statistically significant (p<0.001); the absolute reduction in risk was 9.3% and the relative reduction was 65%. FORTEO was effective in reducing the risk for vertebral fractures regardless of age, baseline rate of bone turnover, or baseline BMD.
|
||||
|
Percent of Women with Fracture |
||||
|
FORTEO
|
Placebo
|
|
|
|
|
New fracture (≥1) |
5.0* |
14.3 |
9.3 (5.5-13.1) |
65 (45-78) |
|
1 fracture |
3.8 |
9.4 | ||
|
2 fractures |
0.9 |
2.9 | ||
|
≥3 fractures |
0.2 |
2.0 | ||
New nonvertebral osteoporotic fractures — Table 2 shows the effect of FORTEO on the risk of nonvertebral fractures. FORTEO significantly reduced the risk of any nonvertebral fracture from 5.5% in the placebo group to 2.6% in the FORTEO group (p<0.05). The absolute reduction in risk was 2.9% and the relative reduction was 53%.
|
FORTEO*
|
Placebo*
|
|
|
Skeletal site | ||
|
Wrist |
2 (0.4%) |
7 (1.3%) |
|
Ribs |
3 (0.6%) |
5 (0.9%) |
|
Hip |
1 (0.2%) |
4 (0.7%) |
|
Ankle/Foot |
1 (0.2%) |
4 (0.7%) |
|
Humerus |
2 (0.4%) |
2 (0.4%) |
|
Pelvis |
0 |
3 (0.6%) |
|
Other |
6 (1.1%) |
8 (1.5%) |
|
Total |
14 (2.6%)† |
30 (5.5%) |
The cumulative percentage of postmenopausal women with osteoporosis who sustained new nonvertebral fractures was lower in women treated with FORTEO than in women treated with placebo (see Figure 1).
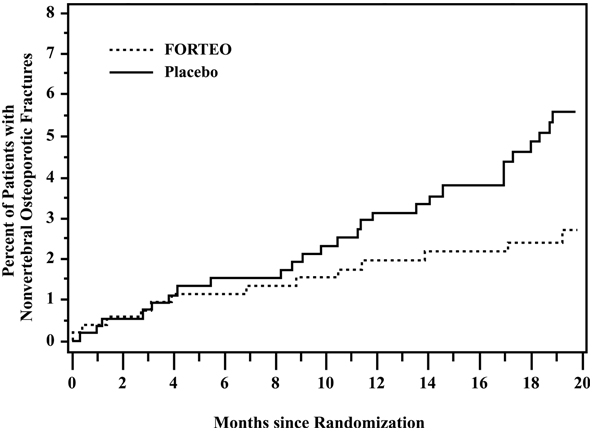
Figure 1. Cumulative percentage of postmenopausal women with osteoporosis sustaining new nonvertebral osteoporotic fractures.1
Effect on bone mineral density (BMD)
FORTEO increased lumbar spine BMD in postmenopausal women with osteoporosis. Statistically significant increases were seen at 3 months and continued throughout the treatment period, as shown in Figure 2.
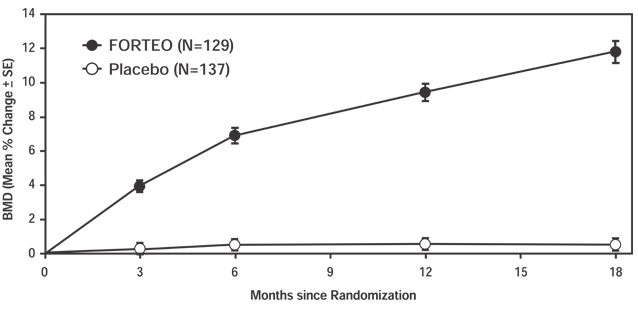
Figure 2. Time course of change in lumbar spine BMD in postmenopausal women with osteoporosis treated with FORTEO vs placebo2 (women with data available at all time points).
Postmenopausal women with osteoporosis who were treated with FORTEO also had statistically significant increases in BMD at the femoral neck, total hip, and total body (see Table 3).
|
FORTEO
|
Placebo
|
|
|
Lumbar spine BMD |
9.7† |
1.1 |
|
Femoral neck BMD |
2.8‡ |
-0.7 |
|
Total hip BMD |
2.6‡ |
-1.0 |
|
Trochanter BMD |
3.5‡ |
-0.2 |
|
Intertrochanter BMD |
2.6‡ |
-1.3 |
|
Ward’s triangle BMD |
4.2‡ |
-0.8 |
|
Total body BMD |
0.6‡ |
-0.5 |
|
Distal 1/3 radius BMD |
-2.1 |
-1.3 |
|
Ultradistal radius BMD |
-0.1 |
-1.6 |
Figure 3 shows the cumulative distribution of the percentage change from baseline of lumbar spine BMD for the FORTEO and placebo groups. FORTEO treatment increased lumbar spine BMD from baseline in 96% of postmenopausal women treated (see Figure 3). Seventy–two percent of patients treated with FORTEO achieved at least a 5% increase in spine BMD, and 44% gained 10% or more.
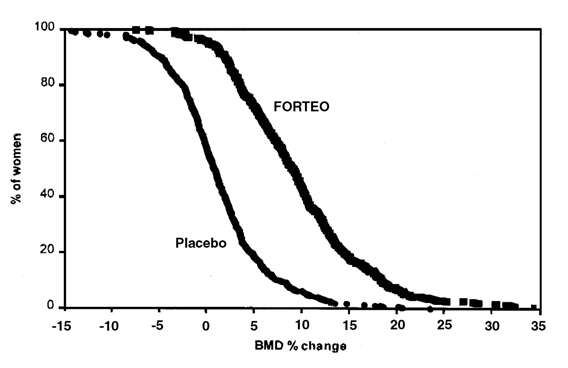
Figure 3. Percent of postmenopausal women with osteoporosis attaining a lumbar spine BMD percent change from baseline at least as great as the value on the x-axis (median duration of treatment 19 months).
Both treatment groups lost height during the trial. The mean decreases were 3.61 and 2.81 mm in the placebo and FORTEO groups, respectively.
Bone histology — The effects of teriparatide on bone histology were evaluated in iliac crest biopsies of 35 postmenopausal women treated for 12 to 24 months with calcium and vitamin D and teriparatide 20 or 40 mcg/day. Normal mineralization was observed with no evidence of cellular toxicity. The new bone formed with teriparatide was of normal quality (as evidenced by the absence of woven bone and marrow fibrosis).
Treatment to increase bone mass in men with primary or hypogonadal osteoporosis — The safety and efficacy of once–daily FORTEO, median exposure of 10 months, were examined in a double–blind, placebo–controlled clinical study of 437 men with either primary (idiopathic) or hypogonadal osteoporosis (FORTEO 20 mcg, n=151). This multicenter efficacy study was performed in the US and 10 other countries. All men received 1000 mg of calcium per day and at least 400 IU of vitamin D per day. The primary efficacy endpoint was change in lumbar spine BMD.
FORTEO increased lumbar spine BMD in men with primary or hypogonadal osteoporosis. Statistically significant increases were seen at 3 months and continued throughout the treatment period. FORTEO was effective in increasing lumbar spine BMD regardless of age, baseline rate of bone turnover, and baseline BMD. The effects of FORTEO at additional skeletal sites are shown in Table 4.
|
FORTEO
|
Placebo
|
|
|
Lumbar spine BMD |
5.9† |
0.5 |
|
Femoral neck BMD |
1.5‡ |
0.3 |
|
Total hip BMD |
1.2 |
0.5 |
|
Trochanter BMD |
1.3 |
1.1 |
|
Intertrochanter BMD |
1.2 |
0.6 |
|
Ward’s triangle BMD |
2.8 |
1.1 |
|
Total body BMD |
0.4 |
-0.4 |
|
Distal 1/3 radius BMD |
-0.5 |
-0.2 |
|
Ultradistal radius BMD |
-0.5 |
-0.3 |
Figure 4 shows the cumulative distribution of the percentage change from baseline of lumbar spine BMD for the FORTEO and placebo groups. FORTEO treatment for a median of 10 months increased lumbar spine BMD from baseline in 94% of men treated. Fifty–three percent of patients treated with FORTEO achieved at least a 5% increase in spine BMD, and 14% gained 10% or more.
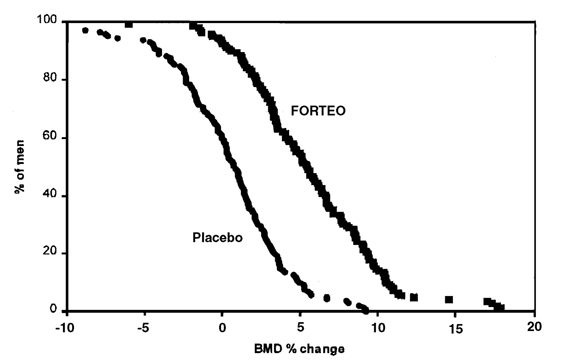
Figure 4. Percent of men with primary or hypogonadal osteoporosis attaining a lumbar spine BMD percent change from baseline at least as great as the value on the x-axis (median duration of treatment 10 months).
- 1
- This graph includes all fractures listed above in Table 2.
- 2
- p<0.001 for FORTEO compared with placebo at each post-baseline time point
INDICATIONS AND USAGE
FORTEO is indicated for the treatment of postmenopausal women with osteoporosis who are at high risk for fracture. These include women with a history of osteoporotic fracture, or who have multiple risk factors for fracture, or who have failed or are intolerant of previous osteoporosis therapy, based upon physician assessment (see BLACK BOX WARNING). In postmenopausal women with osteoporosis, FORTEO increases BMD and reduces the risk of vertebral and nonvertebral fractures.
FORTEO is indicated to increase bone mass in men with primary or hypogonadal osteoporosis who are at high risk for fracture. These include men with a history of osteoporotic fracture, or who have multiple risk factors for fracture, or who have failed or are intolerant to previous osteoporosis therapy, based upon physician assessment (see BLACK BOX WARNING). In men with primary or hypogonadal osteoporosis, FORTEO increases BMD. The effects of FORTEO on risk for fracture in men have not been studied.
-
FORTEO reduces the risk of vertebral fractures in postmenopausal women with osteoporosis.
-
FORTEO reduces the risk of nonvertebral fractures in postmenopausal women with osteoporosis.
-
FORTEO increases vertebral and femoral neck BMD in postmenopausal women with osteoporosis and in men with primary or hypogonadal osteoporosis.
-
The effects of FORTEO on fracture risk have not been studied in men.
CONTRAINDICATIONS
FORTEO should not be given to patients with hypersensitivity to teriparatide or to any of its excipients.
WARNINGS
In male and female rats, teriparatide caused an increase in the incidence of osteosarcoma (a malignant bone tumor) that was dependent on dose and treatment duration (see BLACK BOX WARNING and PRECAUTIONS; Carcinogenesis).
The following categories of patients have increased baseline risk of osteosarcoma and therefore should not be treated with FORTEO:
-
Paget’s disease of bone. FORTEO should not be given to patients with Paget’s disease of bone. Unexplained elevations of alkaline phosphatase may indicate Paget’s disease of bone.
-
Pediatric populations. FORTEO has not been studied in pediatric populations. FORTEO should not be used in pediatric patients or young adults with open epiphyses.
-
Prior external beam or implant radiation therapy involving the skeleton. FORTEO should not be given to such patients.
Patients with bone metastases or a history of skeletal malignancies should be excluded from treatment with FORTEO.
Patients with metabolic bone diseases other than osteoporosis should be excluded from treatment with FORTEO.
FORTEO has not been studied in patients with pre–existing hypercalcemia. These patients should be excluded from treatment with FORTEO because of the possibility of exacerbating hypercalcemia.
PRECAUTIONS
General
The safety and efficacy of FORTEO have not been evaluated beyond 2 years of treatment. Consequently, use of the drug for more than 2 years is not recommended.
In clinical trials, the frequency of urolithiasis was similar in patients treated with FORTEO and placebo. However, FORTEO has not been studied in patients with active urolithiasis. If active urolithiasis or pre–existing hypercalciuria are suspected, measurement of urinary calcium excretion should be considered. FORTEO should be used with caution in patients with active or recent urolithiasis because of the potential to exacerbate this condition.
Hypotension
In short–term clinical pharmacology studies with teriparatide, transient episodes of symptomatic orthostatic hypotension were observed infrequently. Typically, an event began within 4 hours of dosing and spontaneously resolved within a few minutes to a few hours. When transient orthostatic hypotension occurred, it happened within the first several doses, it was relieved by placing the person in a reclining position, and it did not preclude continued treatment.
Concomitant treatment with digitalis
In a study of 15 healthy people administered digoxin daily to steady state, a single FORTEO dose did not alter the effect of digoxin on the systolic time interval (from electrocardiographic Q–wave onset to aortic valve closure, a measure of digoxin’s calcium–mediated cardiac effect). However, sporadic case reports have suggested that hypercalcemia may predispose patients to digitalis toxicity. Because FORTEO transiently increases serum calcium, FORTEO should be used with caution in patients taking digitalis.
Hepatic, renal, and cardiac
Limited information is available to evaluate safety in patients with hepatic, renal, and cardiac disease.
Information for Patients
For safe and effective use of FORTEO, the physician should inform patients about the following:
General
Patients should read the Medication Guide and User Manual provided with the delivery device (pen) before starting therapy with FORTEO and re–read them each time the prescription is renewed.
Osteosarcomas in rats
Patients should be made aware that FORTEO caused osteosarcomas in rats and that the clinical relevance of these findings is unknown.
Orthostatic hypotension
FORTEO should be administered initially under circumstances where the patient can immediately sit or lie down if symptoms occur. Patients should be instructed that if they feel lightheaded or have palpitations after the injection, they should sit or lie down until the symptoms resolve. If symptoms persist or worsen, patients should be instructed to consult a physician before continuing treatment (see PRECAUTIONS, General).
Hypercalcemia
Although symptomatic hypercalcemia was not observed in clinical trials, physicians should instruct patients to contact a health care provider if they develop persistent symptoms of hypercalcemia (ie, nausea, vomiting, constipation, lethargy, muscle weakness).
Use of the the delivery device (pen)
Patients should be instructed on how to properly use the delivery device (refer to User Manual), properly dispose of needles, and be advised not to share their pens with other patients.
Other osteoporosis treatments
Patients should be informed regarding the roles of supplemental calcium and/or vitamin D, weight–bearing exercise, and modification of certain behavioral factors such as cigarette smoking and/or alcohol consumption.
Laboratory Tests
Serum calcium
FORTEO transiently increases serum calcium, with the maximal effect observed at approximately 4 to 6 hours post–dose. By 16 hours post–dose, serum calcium generally has returned to or near baseline. These effects should be kept in mind because serum calcium concentrations observed within 16 hours after a dose may reflect the pharmacologic effect of teriparatide. Persistent hypercalcemia was not observed in clinical trials with FORTEO. If persistent hypercalcemia is detected, treatment with FORTEO should be discontinued pending further evaluation of the cause of hypercalcemia.
Patients known to have an underlying hypercalcemic disorder, such as primary hyperparathyroidism, should not be treated with FORTEO (see WARNINGS).
Urinary calcium
FORTEO increases urinary calcium excretion, but the frequency of hypercalciuria in clinical trials was similar for patients treated with FORTEO and placebo (see CLINICAL PHARMACOLOGY, Human Pharmacodynamics).
Renal function
No clinically important adverse renal effects were observed in clinical studies. Assessments included creatinine clearance; measurements of blood urea nitrogen (BUN), creatinine, and electrolytes in serum; urine specific gravity and pH; and examination of urine sediment. Long–term evaluation of patients with severe renal insufficiency, patients undergoing acute or chronic dialysis, or patients who have functioning renal transplants has not been performed.
Serum uric acid
FORTEO increases serum uric acid concentrations. In clinical trials, 2.8% of FORTEO patients had serum uric acid concentrations above the upper limit of normal compared with 0.7% of placebo patients. However, the hyperuricemia did not result in an increase in gout, arthralgia, or urolithiasis.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two carcinogenicity bioassays were conducted in Fischer 344 rats. In the first study, male and female rats were given daily subcutaneous teriparatide injections of 5, 30, or 75 mcg/kg/day for 24 months from 2 months of age. These doses resulted in systemic exposures that were, respectively, 3, 20, and 60 times higher than the systemic exposure observed in humans following a subcutaneous dose of 20 mcg (based on AUC comparison). Teriparatide treatment resulted in a marked dose–related increase in the incidence of osteosarcoma, a rare malignant bone tumor, in both male and female rats. Osteosarcomas were observed at all doses and the incidence reached 40% to 50% in the high–dose groups. Teriparatide also caused a dose–related increase in osteoblastoma and osteoma in both sexes. No osteosarcomas, osteoblastomas or osteomas were observed in untreated control rats. The bone tumors in rats occurred in association with a large increase in bone mass and focal osteoblast hyperplasia.
The second 2–year study was carried out in order to determine the effect of treatment duration and animal age on the development of bone tumors. Female rats were treated for different periods between 2 and 26 months of age with subcutaneous doses of 5 and 30 mcg/kg (equivalent to 3 and 20 times the human exposure at the 20–mcg dose, based on AUC comparison). The study showed that the occurrence of osteosarcoma, osteoblastoma and osteoma was dependent upon dose and duration of exposure. Bone tumors were observed when immature 2–month old rats were treated with 30 mcg/kg/day for 24 months or with 5 or 30 mcg/kg/day for 6 months. Bone tumors were also observed when mature 6–month old rats were treated with 30 mcg/kg/day for 6 or 20 months. Tumors were not detected when mature 6–month old rats were treated with 5 mcg/kg/day for 6 or 20 months. The results did not demonstrate a difference in susceptibility to bone tumor formation, associated with teriparatide treatment, between mature and immature rats.
The relevance of these rat findings to humans is uncertain.
Mutagenesis
Teriparatide was not genotoxic in any of the following test systems: the Ames test for bacterial mutagenesis; the mouse lymphoma assay for mammalian cell mutation; the chromosomal aberration assay in Chinese hamster ovary cells, with and without metabolic activation; and the in vivo micronucleus test in mice.
Impairment of fertility
No effects on fertility were observed in male and female rats given subcutaneous teriparatide doses of 30, 100, or 300 mcg/kg/day prior to mating and in females continuing through gestation Day 6 (16 to 160 times the human dose of 20 mcg based on surface area, mcg/m2).
Pregnancy
Pregnancy Category C — In pregnant rats given subcutaneous teriparatide doses up to 1000 mcg/kg/day, there were no findings. In pregnant mice given subcutaneous doses of 225 or 1000 mcg/kg/day (≥60 times the human dose based on surface area, mcg/m2) from gestation Day 6 through 15, the fetuses showed an increased incidence of skeletal deviations or variations (interrupted rib, extra vertebra or rib).
Developmental effects in a perinatal/postnatal study in pregnant rats given subcutaneous doses of teriparatide from gestation Day 6 through postpartum Day 20 included mild growth retardation in female offspring at doses ≥225 mcg/kg/day (≥120 times the human dose based on surface area, mcg/m2), and in male offspring at 1000 mcg/kg/day (540 times the human dose based on surface area, mcg/m2). There was also reduced motor activity in both male and female offspring at 1000 mcg/kg/day. There were no developmental or reproductive effects in mice or rats at a dose of 30 mcg/kg (8 or 16 times the human dose based on surface area, mcg/m2). The effect of teriparatide treatment on human fetal development has not been studied. FORTEO is not indicated for use in pregnancy.
Nursing Mothers
Because FORTEO is indicated for the treatment of osteoporosis in postmenopausal women, it should not be administered to women who are nursing their children. There have been no clinical studies to determine if teriparatide is secreted into breast milk.
Pediatric Use
The safety and efficacy of FORTEO have not been established in pediatric populations. FORTEO is not indicated for use in pediatric patients (see WARNINGS).
Geriatric Use
Of the patients receiving FORTEO in the osteoporosis trial of 1637 postmenopausal women, 75% were 65 years of age and over and 23% were 75 years of age and over. Of the patients receiving FORTEO in the osteoporosis trial of 437 men, 39% were 65 years of age and over and 13% were 75 years of age and over. No significant differences in bone response or adverse reactions were seen in geriatric patients receiving FORTEO as compared with younger patients. Nonetheless, as with many medications, elderly patients may have greater sensitivity to the adverse effects of FORTEO.
ADVERSE EVENTS
The safety of teriparatide has been evaluated in 24 clinical trials that enrolled over 2800 women and men. Four long–term Phase 3 clinical trials included 1 large placebo–controlled, double–blind, multinational trial with 1637 postmenopausal women; 1 placebo–controlled, double–blind, multinational trial with 437 men; and 2 active–controlled trials including 393 postmenopausal women. Teriparatide doses ranged from 5 to 100 mcg/day in short–term trials and 20 to 40 mcg/day in the other trials. A total of 1943 of the patients studied received teriparatide, including 815 patients at 20 mcg/day and 1107 patients at 40 mcg/day. In the clinical trials, a total of 1432 patients were treated with teriparatide for 3 months to 2 years, of whom 1137 were treated for greater than 1 year (500 at 20 mcg/day and 637 at 40 mcg/day). The maximum duration of treatment was 2 years. Adverse events associated with FORTEO usually were mild and generally did not require discontinuation of therapy.
In the two Phase 3 placebo–controlled clinical trials in men and postmenopausal women, early discontinuation due to adverse events occurred in 5.6% of patients assigned to placebo and 7.1% of patients assigned to FORTEO. Reported adverse events that appeared to be increased by FORTEO treatment were dizziness and leg cramps.
Table 5 lists adverse events that occurred in the two Phase 3 placebo–controlled clinical trials in men and postmenopausal women at a frequency ≥2.0% in the FORTEO groups and in more FORTEO–treated patients than in placebo–treated patients, without attribution of causality.
|
FORTEO
|
Placebo
|
|
|
Event Classification |
(%) |
(%) |
|
Body as a Whole | ||
|
Pain |
21.3 |
20.5 |
|
Headache |
7.5 |
7.4 |
|
Asthenia |
8.7 |
6.8 |
|
Neck pain |
3.0 |
2.7 |
|
Cardiovascular | ||
|
Hypertension |
7.1 |
6.8 |
|
Angina pectoris |
2.5 |
1.6 |
|
Syncope |
2.6 |
1.4 |
|
Digestive System | ||
|
Nausea |
8.5 |
6.7 |
|
Constipation |
5.4 |
4.5 |
|
Diarrhea |
5.1 |
4.6 |
|
Dyspepsia |
5.2 |
4.1 |
|
Vomiting |
3.0 |
2.3 |
|
Gastrointestinal disorder |
2.3 |
2.0 |
|
Tooth disorder |
2.0 |
1.3 |
|
Musculoskeletal | ||
|
Arthralgia |
10.1 |
8.4 |
|
Leg cramps |
2.6 |
1.3 |
|
Nervous System | ||
|
Dizziness |
8.0 |
5.4 |
|
Depression |
4.1 |
2.7 |
|
Insomnia |
4.3 |
3.6 |
|
Vertigo |
3.8 |
2.7 |
|
Respiratory System | ||
|
Rhinitis |
9.6 |
8.8 |
|
Cough increased |
6.4 |
5.5 |
|
Pharyngitis |
5.5 |
4.8 |
|
Dyspnea |
3.6 |
2.6 |
|
Pneumonia |
3.9 |
3.3 |
|
Skin and Appendages | ||
|
Rash |
4.9 |
4.5 |
|
Sweating |
2.2 |
1.7 |
Serum calcium — FORTEO transiently increases serum calcium, with the maximal effect observed at approximately 4 to 6 hours post–dose. Serum calcium measured at least 16 hours post–dose was not different from pretreatment levels. In clinical trials, the frequency of at least 1 episode of transient hypercalcemia in the 4 to 6 hours after FORTEO administration was increased from 1.5% of women and none of the men treated with placebo to 11.1% of women and 6.0% of men treated with FORTEO. The number of patients treated with FORTEO whose transient hypercalcemia was verified on consecutive measurements was 3.0% of women and 1.3% of men.
Immunogenicity — In a large clinical trial, antibodies that cross–reacted with teriparatide were detected in 2.8% of women receiving FORTEO. Generally, antibodies were first detected following 12 months of treatment and diminished after withdrawal of therapy. There was no evidence of hypersensitivity reactions, allergic reactions, effects on serum calcium, or effects on BMD response.
Postmarketing Reports
Since market introduction, adverse events reported have included:
-
Possible allergic events soon after injection: acute dyspnea, oro/facial edema, generalized urticaria, chest pain (less than 1 in 1000 patients treated).
-
Hypercalcemia greater than 2.76 mmol/L (11 mg/dL) (less than 1 in 100 patients treated); hypercalcemia greater than 3.25 mmol/L (13 mg/dL) (less than 1 in 1000 patients treated).
-
Injection site and injection technique events including pain, swelling, erythema, localized bruising, pruritus and minor bleeding at the injection site (less than 1 in 30 patients treated). These usually have been mild and transient.
-
Muscle spasms, such as of the leg or back, are reported commonly (between 1 and 10 patients per 100 patients treated), sometimes shortly after the first dose. Serious back spasms have been reported very rarely (less than 1 in 10,000 patients treated).
OVERDOSAGE
Incidents of overdose in humans have not been reported in clinical trials. Teriparatide has been administered in single doses of up to 100 mcg and in repeated doses of up to 60 mcg/day for 6 weeks. The effects of overdose that might be expected include a delayed hypercalcemic effect and risk of orthostatic hypotension. Nausea, vomiting, dizziness, and headache might also occur.
In postmarketing spontaneous reports, there have been cases of medication error in which the entire contents (up to 800 mcg) of the FORTEO delivery device have been administered as a single dose. Transient events reported have included nausea, weakness/lethargy and hypotension. In some cases, no adverse events occurred as a result of the overdose. No fatalities associated with overdose have been reported.
In single–dose rodent studies using subcutaneous injection of teriparatide, no mortality was seen in rats given doses of 1000 mcg/kg (540 times the human dose based on surface area, mcg/m2) or in mice given 10,000 mcg/kg (2700 times the human dose based on surface area, mcg/m2).
Overdose management — There is no specific antidote for teriparatide. Treatment of suspected overdose should include discontinuation of FORTEO, monitoring of serum calcium and phosphorus, and implementation of appropriate supportive measures, such as hydration.
DOSAGE AND ADMINISTRATION
FORTEO should be administered as a subcutaneous injection into the thigh or abdominal wall. The recommended dosage is 20 mcg once a day.
FORTEO should be administered initially under circumstances in which the patient can sit or lie down if symptoms of orthostatic hypotension occur (see PRECAUTIONS, Information for the Patient).
FORTEO is a clear and colorless liquid. Do not use if solid particles appear or if the solution is cloudy or colored. The delivery device (pen) used to administer FORTEO should not be used past the stated expiration date.
No data are available on the safety or efficacy of intravenous or intramuscular injection of FORTEO.
The safety and efficacy of FORTEO have not been evaluated beyond 2 years of treatment. Consequently, use of the drug for more than 2 years is not recommended.
INSTRUCTIONS FOR PEN USE
Patients and caregivers who administer FORTEO should receive appropriate training and instruction on the proper use of the FORTEO delivery device from a qualified health professional. It is important to read, understand, and follow the instructions in the FORTEO delivery device User Manual. Failure to do so may result in inaccurate dosing. Each FORTEO delivery device can be used for up to 28 days, including the first injection from the pen. After the 28–day use period, discard the FORTEO delivery device, even if it still contains some unused solution. Never share a FORTEO delivery device.
STORAGE
The FORTEO delivery device should be stored under refrigeration at 2° to 8°C (36° to 46°F) at all times. Recap the pen when not in use to protect the cartridge from physical damage and light. During the use period, time out of the refrigerator should be minimized; the dose may be delivered immediately following removal from the refrigerator.
Do not freeze. Do not use FORTEO if it has been frozen.
HOW SUPPLIED
The FORTEO delivery device is available in the following:
- 3 mL prefilled pen delivery device NDC 0002-8971-01 (MS8971)
- 2.4 mL prefilled pen delivery device NDC 0002-8400-01 (MS8400).
Literature revised June 25, 2008
Manufactured by Lilly France - F-67640 Fegersheim, France
for Eli Lilly and Company - Indianapolis, IN 46285, USA
www.forteo.com
Copyright © 2002, 2008, Eli Lilly and Company. All rights reserved.
Medication Guide
FORTEO®(for-TAY-o)
teriparatide (rDNA origin) injection
Read this Medication Guide carefully before you start taking FORTEO and each time you get a refill. The information may have changed. Also, read the User Manual that comes with the FORTEO delivery device (pen) for information on how to use the device to inject your medicine the right way. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or your treatment. Ask your healthcare provider if there is something you do not understand or if you want to learn more about the benefits and risks of FORTEO.
What is the most important information I should know about FORTEO?
As part of drug testing, teriparatide, the active ingredient in FORTEO, was given to rats for a significant part of their lifetime. In these studies, teriparatide caused some rats to develop osteosarcoma, a bone cancer. Osteosarcoma in humans is a serious but very rare cancer. Osteosarcoma occurs in about 4 out of every million older adults each year. It is not known if humans treated with FORTEO also have a higher chance of getting osteosarcoma.
What is FORTEO?
FORTEO is a prescription medicine that contains teriparatide, a man-made medicine that is like the natural hormone called parathyroid hormone or PTH. PTH is produced by the body. FORTEO forms new bone, increases bone mineral density and bone strength. This lowers the chance of getting a fracture. In postmenopausal (after the “change of life”) women with osteoporosis, FORTEO can lessen the number of fractures of the spine and other bones. The effect on fractures has not been studied in men.
FORTEO is used in both men and postmenopausal women with osteoporosis who are at high risk for having fractures. FORTEO can be used by people who have had a fracture related to osteoporosis, or who have multiple risk factors for fracture, or who cannot use other osteoporosis treatments.
FORTEO has not been studied in children.
Who should not use FORTEO?
Do not use FORTEO if you:
- are allergic to any of the ingredients in FORTEO. See the end of this Medication Guide for a complete list of the ingredients in FORTEO.
- have Paget's disease of bone.
- have unexplained high levels of alkaline phosphatase in your blood, which means you might have Paget's disease of bone. If you are not sure, ask your doctor.
- are a child or growing adult.
- have ever been diagnosed with bone cancer or other cancers that have spread (metastasized) to your bones.
- have had radiation therapy involving your bones.
- have certain bone diseases. If you have a bone disease, tell your doctor.
- have too much calcium in your blood (hypercalcemia).
FORTEO should not be used to prevent osteoporosis. FORTEO should be used to treat patients who are considered to be at high risk for fracture.
What should I tell my healthcare provider before taking FORTEO?
Tell your healthcare provider about all of your medical conditions, including if you:
- have one of the conditions listed in the section “Who should not use FORTEO?”
- have trouble injecting yourself and do not have someone who can help you.
- have or have had kidney stones.
- are pregnant or thinking about becoming pregnant. It is not known if FORTEO will harm your unborn baby.
- are breast-feeding or thinking about breast-feeding. It is not known if FORTEO passes into breast milk. You should not breast-feed while taking FORTEO.
Tell your healthcare provider about all the medicines you take including prescription and non-prescription medicines, vitamins, and herbal supplements. Your healthcare provider needs this information to help keep you from taking FORTEO with other medicines that may harm you.
- Especially tell your doctor if you take medicines that contain digoxin (for example, Digoxin, Lanoxicaps, Lanoxin).
How should I use FORTEO?
- Use FORTEO one time each day. Your healthcare provider should teach you how to use the FORTEO delivery device (see the User Manual).
- The use of FORTEO for more than 2 years is not recommended.
- The FORTEO delivery device has enough medicine for 28 days. It is set to give a 20 microgram dose of medicine each day (see the User Manual). Do not inject all the medicine in the FORTEO delivery device at any one time.
- Do not transfer the contents of the FORTEO delivery device to a syringe. This can result in taking the wrong dose of FORTEO. If you do not have pen needles available to use with your FORTEO delivery device, talk with your healthcare provider.
- Inject FORTEO one time each day in your thigh or abdomen (lower stomach area). Talk to your healthcare provider about how to rotate injection sites.
- FORTEO should look clear and colorless. Do not use FORTEO if it has particles in it, or if it is cloudy or colored.
- Inject FORTEO right away after you take the delivery device out of the refrigerator.
- After each use, safely remove the needle, recap the delivery device, and put it back in the refrigerator right away (see the User Manual).
- You can take FORTEO with or without food or drink.
- You can take FORTEO at any time of the day. To help you remember to take FORTEO, take it at about the same time each day.
- If you forget or are unable to take FORTEO at your usual time, take it as soon as you can on that day. Do not take more than one injection in the same day.
Follow your healthcare provider's instructions about other ways you can help your osteoporosis, such as exercise, diet, and reducing or stopping your use of tobacco and alcohol. If your healthcare provider recommends calcium and vitamin D supplements, you can take them at the same time you take FORTEO.
What are the possible side effects of FORTEO?
Most side effects are mild and include:
- nausea.
- dizziness or fast heartbeat. Some people get dizzy or get a fast heartbeat right after the first few doses. This usually happens within 4 hours of taking FORTEO and goes away within a few hours. For the first few doses, take your injections of FORTEO in a place where you can sit or lie down right away if you get these symptoms. If your symptoms get worse or do not go away, stop taking FORTEO and call your healthcare provider.
- leg cramps.
- joint aches.
- increased calcium in your blood. Tell your healthcare provider if you have continuing nausea, vomiting, constipation, low energy, or muscle weakness. These may be signs there is too much calcium in your blood.
- injection site reactions including redness, swelling, pain, itching, a few drops of blood, and bruising.
Your healthcare provider may take samples of blood and urine during treatment to check your response to FORTEO. Also, your healthcare provider may ask you to have follow-up tests of bone mineral density. Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of FORTEO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store FORTEO?
- Keep your FORTEO delivery device in the refrigerator at 36° to 46°F (2° to 8°C).
- Do not freeze the FORTEO delivery device. Do not use FORTEO if it has been frozen.
- Do not use FORTEO after the expiration date printed on the delivery device and packaging.
- Throw away the FORTEO delivery device after 28 days even if it has medicine in it (see the User Manual).
General information about FORTEO
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FORTEO for a condition for which it was not prescribed. Do not give FORTEO to other people, even if they have the same condition you have.
This Medication Guide summarizes the most important information about FORTEO. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about FORTEO that is written for healthcare professionals. For more information, go to www.FORTEO.com or call Lilly toll free at 1-866-4FORTEO (1-866-436-7836).
What are the ingredients in FORTEO?
Active ingredient: teriparatide
Inactive ingredients: glacial acetic acid, sodium acetate (anhydrous), mannitol, metacresol, and water for injection. In addition, hydrochloric acid solution 10% and/or sodium hydroxide solution 10% may have been added to adjust the product to pH 4.
What is Osteoporosis?
Osteoporosis is a disease in which the bones become thin and weak, increasing the chance of having a broken bone. Osteoporosis usually causes no symptoms until a fracture happens. The most common fractures are in the spine (backbone). They can shorten height, even without causing pain. Over time, the spine can become curved or deformed and the body bent over. Fractures from osteoporosis can also happen in almost any bone in the body, for example, the wrist, rib, or hip. Once you have had a fracture, the chance for more fractures greatly increases.
The following risk factors increase your chance of getting fractures from osteoporosis:
- past broken bones from osteoporosis.
- very low bone mineral density (BMD).
- frequent falls.
- limited movement, such as using a wheelchair.
- medical conditions likely to cause bone loss, such as some kinds of arthritis.
- medicines that may cause bone loss, for example: seizure medicines (such as phenytoin), blood thinners (such as heparin), steroids, high doses of vitamin A.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised June 25, 2008
Manufactured by Lilly France - F-67640 Fegersheim, France
for Eli Lilly and Company - Indianapolis, IN 46285, USA
Copyright © 2002, 2008, Eli Lilly and Company. All rights reserved.
| Forteo (Teriparatide) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
| Forteo (Teriparatide) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
Revised: 07/2008Eli Lilly and Company