bexxar (tositumomab)
bexxar (tositumomab I-131) injection
[GlaxoSmithKline]
WARNINGS
Hypersensitivity Reactions, including Anaphylaxis
Serious hypersensitivity reactions, including some with fatal outcome, have been reported with the BEXXAR therapeutic regimen. Medications for the treatment of severe hypersensitivity reactions should be available for immediate use. Patients who develop severe hypersensitivity reactions should have infusions of the BEXXAR therapeutic regimen discontinued and receive medical attention (see WARNINGS).
Prolonged and Severe Cytopenias
The majority of patients who received the BEXXAR therapeutic regimen experienced severe thrombocytopenia and neutropenia. The BEXXAR therapeutic regimen should not be administered to patients with >25% lymphoma marrow involvement and/or impaired bone marrow reserve (see WARNINGS and ADVERSE REACTIONS).
Pregnancy Category X
The BEXXAR therapeutic regimen can cause fetal harm when administered to a pregnant woman.
Special requirements
The BEXXAR therapeutic regimen (Tositumomab and Iodine I 131 Tositumomab) contains a radioactive component and should be administered only by physicians and other health care professionals qualified by training in the safe use and handling of therapeutic radionuclides. The BEXXAR therapeutic regimen should be administered only by physicians who are in the process of being or have been certified by GlaxoSmithKline in dose calculation and administration of the BEXXAR therapeutic regimen.
DESCRIPTION
The BEXXAR therapeutic regimen (Tositumomab and Iodine I 131 Tositumomab) is an anti-neoplastic radioimmunotherapeutic monoclonal antibody-based regimen composed of the monoclonal antibody, Tositumomab, and the radiolabeled monoclonal antibody, Iodine I 131 Tositumomab.
Tositumomab
Tositumomab is a murine IgG2a lambda monoclonal antibody directed against the CD20 antigen, which is found on the surface of normal and malignant B lymphocytes. Tositumomab is produced in an antibiotic-free culture of mammaliancells and is composed of two murine gamma 2a heavy chains of 451 amino acids each and two lambda light chains of 220 amino acids each. The approximate molecular weight of Tositumomab is 150 kD.
Tositumomab is supplied as a sterile, pyrogen-free, clear to opalescent, colorless to slightly yellow, preservative-free liquid concentrate. It is supplied at a nominal concentration of 14 mg/mL Tositumomab in 35 mg and 225 mg single-use vials. The formulation contains 10% (w/v) maltose, 145 mM sodium chloride, 10 mM phosphate, and Water for Injection, USP. The pH is approximately 7.2.
Iodine I 131 Tositumomab
Iodine I 131 Tositumomab is a radio-iodinated derivative of Tositumomab that has been covalently linked to Iodine-131. Unbound radio-iodine and other reactants have been removed by chromatographic purification steps. Iodine I 131 Tositumomab is supplied as a sterile, clear, preservative-free liquid for IV administration. The dosimetric dosage form is supplied at nominal protein and activity concentrations of 0.1 mg/mL and 0.61 mCi/mL (at date of calibration), respectively. The therapeutic dosage form is supplied at nominal protein and activity concentrations of 1.1 mg/mL and 5.6 mCi/mL (at date of calibration), respectively. The formulation for the dosimetric and the therapeutic dosage forms contains 4.4%−6.6% (w/v) povidone, 1−2 mg/mL maltose (dosimetric dose) or 9−15 mg/mL maltose (therapeutic dose), 8.5−9.5 mg/mL sodium chloride, and 0.9−1.3 mg/mL ascorbic acid. The pH is approximately 7.0.
BEXXAR Therapeutic Regimen
The BEXXAR therapeutic regimen is administered in two discrete steps: the dosimetric and therapeutic steps. Each step consists of a sequential infusion of Tositumomab followed by Iodine I 131 Tositumomab. The therapeutic step is administered 7-14 days after the dosimetric step. The BEXXAR therapeutic regimen is supplied in two distinct package configurations as follows:
BEXXAR Dosimetric Packaging
- A carton containing two single-use 225 mg vials and one single-use 35 mg vial of Tositumomab supplied by McKesson BioServices and
- A package containing a single-use vial of Iodine I 131 Tositumomab (0.61 mCi/mL at calibration), supplied by MDS Nordion.
BEXXAR Therapeutic Packaging
- A carton containing two single-use 225 mg vials and one single-use 35 mg vial of Tositumomab, supplied by McKesson BioServices and
- A package containing one or two single-use vials of Iodine I 131 Tositumomab (5.6 mCi/mL at calibration), supplied by MDS Nordion.
Physical/Radiochemical Characteristics of Iodine-131
Iodine-131 decays with beta and gamma emissions with a physical half-life of 8.04 days. The principal beta emission has a mean energy of 191.6 keV and the principal gamma emission has an energy of 364.5 keV (Ref 1).
External Radiation
The specific gamma ray constant for Iodine-131 is 2.2 R/millicurie hour at 1 cm. The first half-value layer is 0.24 cm lead (Pb) shielding. A range of values is shown in Table 1 for the relative attenuation of the radiation emitted by this radionuclide that results from interposition of various thicknesses of Pb. To facilitate control of the radiation exposure from this radionuclide, the use of a 2.55 cm thickness of Pb will attenuate the radiation emitted by a factor of about 1,000.
|
Shield Thickness (Pb) cm |
Attenuation Factor |
|
0.24 |
0.5 |
|
0.89 |
10-1 |
|
1.60 |
10-2 |
|
2.55 |
10-3 |
|
3.7 |
10-4 |
The fraction of Iodine-131 radioactivity that remains in the vial after the date of calibration is calculated as follows:
Fraction of remaining radioactivity of Iodine-131 after x days = 2-(x/8.04).
Physical decay is presented in Table 2.
|
Days |
Fraction Remaining |
|
0* |
1.000 |
|
1 |
0.917 |
|
2 |
0.842 |
|
3 |
0.772 |
|
4 |
0.708 |
|
5 |
0.650 |
|
6 |
0.596 |
|
7 |
0.547 |
|
8 |
0.502 |
|
9 |
0.460 |
|
10 |
0.422 |
|
11 |
0.387 |
|
12 |
0.355 |
|
13 |
0.326 |
|
14 |
0.299 |
*(Calibration day)
CLINICAL PHARMACOLOGY
General Pharmacology
Tositumomab binds specifically to the CD20 (human B-lymphocyte−restricted differentiation antigen, Bp 35 or B1) antigen. This antigen is a transmembrane phosphoprotein expressed on pre-B lymphocytes and at higher density on mature B lymphocytes (Ref. 2). The antigen is also expressed on >90% of B-cell non-Hodgkin’s lymphomas (NHL) (Ref. 3). The recognition epitope for Tositumomab is found within the extracellular domain of the CD20 antigen. CD20 does not shed from the cell surface and does not internalize following antibody binding (Ref. 4).
Mechanism of Action
Possible mechanisms of action of the BEXXAR therapeutic regimen include induction of apoptosis (Ref. 5), complement-dependent cytotoxicity (CDC) (Ref. 6), and antibody-dependent cellular cytotoxicity (ADCC) (Ref. 5) mediated by the antibody. Additionally, cell death is associated with ionizing radiation from the radioisotope.
Pharmacokinetics/Pharmacodynamics
The phase 1 study of Iodine I 131 Tositumomab determined that a 475 mg predose of unlabeled antibody decreased splenic targeting and increased the terminal half-life of the radiolabeled antibody. The median blood clearance following administration of 485 mg of Tositumomab in 110 patients with NHL was 68.2 mg/hr (range: 30.2−260.8 mg/hr). Patients with high tumor burden, splenomegaly, or bone marrow involvement were noted to have a faster clearance, shorter terminal half-life, and larger volume of distribution. The total body clearance, as measured by total body gamma camera counts, was dependent on the same factors noted for blood clearance. Patient-specific dosing, based on total body clearance, provided a consistent radiation dose, despite variable pharmacokinetics, by allowing each patient’s administered activity to be adjusted for individual patient variables. The median total body effective half-life, as measured by total body gamma camera counts, in 980 patients with NHL was 67 hours (range: 28-115 hours).
Elimination of Iodine-131 occurs by decay (see Table 2) and excretion in the urine. Urine was collected for 49 dosimetric doses. After 5 days, the whole body clearance was 67% of the injected dose. Ninety-eight percent of the clearance was accounted for in the urine.
Administration of the BEXXAR therapeutic regimen results in sustained depletion of circulating CD20 positive cells. The impact of administration of the BEXXAR therapeutic regimen on circulating CD20 positive cells was assessed in two clinical studies, one conducted in chemotherapy naïve patients and one in heavily pretreated patients. The assessment of circulating lymphocytes did not distinguish normal from malignant cells. Consequently, assessment of recovery of normal B cell function was not directly assessed. At seven weeks, the median number of circulating CD20 positive cells was zero (range: 0-490 cells/mm3). Lymphocyte recovery began at approximately 12 weeks following treatment. Among patients who had CD20 positive cell counts recorded at baseline and at 6 months, 8 of 58 (14%) chemotherapy naïve patients had CD20 positive cell counts below normal limits at six months and 6 of 19 (32%) heavily pretreated patients had CD20 positive cell counts below normal limits at six months. There was no consistent effect of the BEXXAR therapeutic regimen on post-treatment serum IgG, IgA, or IgM levels.
Radiation Dosimetry
Estimations of radiation-absorbed doses for Iodine I 131 Tositumomab were performed using sequential whole body images and the MIRDOSE 3 software program. Patients with apparent thyroid, stomach, or intestinal imaging were selected for organ dosimetry analyses. The estimated radiation-absorbed doses to organs and marrow from a course of the BEXXAR therapeutic regimen are presented in Table 3.
|
BEXXAR |
BEXXAR |
||
|
mGy/MBq |
mGy/MBq |
||
|
Median |
Range |
||
|
From Organ ROIs | |||
|
Thyroid |
2.71 |
1.4 - 6.2 |
|
|
Kidneys |
1.96 |
1.5 - 2.5 |
|
|
ULI Wall |
1.34 |
0.8 - 1.7 |
|
|
LLI Wall |
1.30 |
0.8 - 1.6 |
|
|
Heart Wall |
1.25 |
0.5 - 1.8 |
|
|
Spleen |
1.14 |
0.7 - 5.4 |
|
|
Testes |
0.83 |
0.3 - 1.3 |
|
|
Liver |
0.82 |
0.6 - 1.3 |
|
|
Lungs |
0.79 |
0.5 - 1.1 |
|
|
Red Marrow |
0.65 |
0.5 - 1.1 |
|
|
Stomach Wall |
0.40 |
0.2 - 0.8 |
|
|
From Whole Body ROIs | |||
|
Urine Bladder Wall |
0.64 |
0.6 - 0.9 |
|
|
Bone Surfaces |
0.41 |
0.4 - 0.6 |
|
|
Pancreas |
0.31 |
0.2 - 0.4 |
|
|
Gall Bladder Wall |
0.29 |
0.2 - 0.3 |
|
|
Adrenals |
0.28 |
0.2 - 0.3 |
|
|
Ovaries |
0.25 |
0.2 - 0.3 |
|
|
Small Intestine |
0.23 |
0.2 - 0.3 |
|
|
Thymus |
0.22 |
0.1 - 0.3 |
|
|
Uterus |
0.20 |
0.2 - 0.2 |
|
|
Muscle |
0.18 |
0.1 - 0.2 |
|
|
Breasts |
0.16 |
0.1 - 0.2 |
|
|
Skin |
0.13 |
0.1 - 0.2 |
|
|
Brain |
0.13 |
0.1 - 0.2 |
|
|
Total Body |
0.24 |
0.2 - 0.3 |
CLINICAL STUDIES
The efficacy of the BEXXAR therapeutic regimen was evaluated in 2 studies conducted in patients with low-grade, transformed low-grade, or follicular large-cell lymphoma. Determination of clinical benefit of the BEXXAR therapeutic regimen was based on evidence of durable responses without evidence of an effect on survival. All patients had received prior treatment without an objective response or had progression of disease following treatment. Patients were required to have a granulocyte count >1500 cells/mm3, a platelet count ≥100,000/mm3, an average of ≤25% of the intratrabecular marrow space involved by lymphoma, and no evidence of progressive disease arising in a field irradiated with >3500 cGy within 1 year of completion of irradiation.
Study 1 was a multicenter, single-arm study of 40 patients whose disease had not responded to or had progressed after at least four doses of Rituximab therapy. The median age was 57 (range: 35−78); the median time from diagnosis to protocol entry was 50 months (range: 12−170); and the median number of prior chemotherapy regimens was 4 (range: 1−11). The efficacy outcome data from this study, as determined by an independent panel that reviewed patient records and radiologic studies, are summarized in Table 4.
Among the forty patients in the study, twenty-four patients had disease that did not respond to their last treatment with Rituximab, 11 patients had disease that responded to Rituximab for less than 6 months, and five patients had disease that responded to Rituximab, with a duration of response of 6 months or greater. Overall, 35 of the 40 patients met the definition of “Rituximab refractory”, defined as no response or a response of less than 6 months duration. In this subset of patients the overall objective response was 63% (95% confidence interval 45%, 79%) with a median duration of 25 months (range of 4 - 38+ months). The complete response in this subset of patients was 29% (95% CI of 15%, 46%) with a median duration of response not yet reached (range of 4 - 38+ months).
Study 2 was a multicenter, single arm, open-label study of 60 chemotherapy refractory patients. The median age was 60 (range 38-82), the median time from diagnosis to protocol entry was 53 months (range: 9-334), and the median number of prior chemotherapy regimens was 4 (range 2-13). Fifty-three patients had not responded to prior therapy and 7 patients had responded with a duration of response of<6 months. The efficacy outcome data from this study, as determined by an independent panel that reviewed patient records and radiologic studies are also summarized in Table 4. Investigators continued to follow eight patients with complete response after the last independent review panel assessment. The updated duration of ongoing response as per investigators was reported to range from 42 to 85 months.
|
Study 1 (n = 40) |
Study 2 (n = 60) |
|
|
Overall Response | ||
|
Rate 95% CI a |
68% (51%, 81%) |
47% (34%, 60%) |
|
Response Duration (mos) | ||
|
Median 95% CI a Range |
16 (10, NRb) 1+ to 38+ |
12 (7, 47) 2 to 47 |
|
Complete Response c | ||
|
Rate 95% CI a |
33% (19%, 49%) |
20% (11%, 32%) |
|
Complete response c duration (mos) | ||
|
Median 95% CI a Range |
NR b (15, NR) 4 to 38+ |
47 (47, NR) 9 to 47 |
aCI = Confidence Interval
bNR = Not reached, Median duration of follow up: Study 1 = 26 months; Study 2 = 30 months
cComplete response rate = Pathologic and clinical complete responses
The results of these studies were supported by demonstration of durable objective responses in three single-arm studies. In these studies, 130 patients with Rituximab-naïve follicular non-Hodgkin’s lymphoma with or without transformation were evaluated for efficacy. All patients had relapsed following, or were refractory to, chemotherapy. The overall response rates ranged from 49% to 64% and the median durations of response ranged from 13 to 16 months. Due to small sample sizes in the supportive studies, as in studies 1 and 2, the 95% confidence intervals for the median durations of response are wide.
INDICATIONS AND USAGE
The BEXXAR therapeutic regimen (Tositumomab and Iodine I 131 Tositumomab) is indicated for the treatment of patients with CD20 antigen-expressing relapsed or refractory, low grade, follicular, or transformed non-Hodgkin's lymphoma, including patients with Rituximab-refractory non-Hodgkin’s lymphoma. Determination of the effectiveness of the BEXXAR therapeutic regimen is based on overall response rates in patients whose disease is refractory to chemotherapy alone or to chemotherapy and Rituximab. The effects of the BEXXAR therapeutic regimen on survival are not known.
The BEXXAR therapeutic regimen is not indicated for the initial treatment of patients with CD20 positive non-Hodgkin’s lymphoma. (See ADVERSE REACTIONS, Immunogenicity.)
The BEXXAR therapeutic regimen is intended as a single course of treatment. The safety of multiple courses of the BEXXAR therapeutic regimen, or combination of this regimen with other forms of irradiation or chemotherapy, has not been evaluated.
CONTRAINDICATIONS
The BEXXAR therapeutic regimen is contraindicated in patients with known hypersensitivity to murine proteins or any other component of the BEXXAR therapeutic regimen.
PREGNANCY CATEGORY X
Iodine I 131 Tositumomab (a component of the BEXXAR therapeutic regimen) is contraindicated for use in women who are pregnant. Iodine-131 may cause harm to the fetal thyroid gland when administered to pregnant women. Review of the literature has shown that transplacental passage of radioiodide may cause severe, and possibly irreversible, hypothyroidism in neonates. While there are no adequate and well-controlled studies of the BEXXAR therapeutic regimen in pregnant animals or humans, use of the BEXXAR therapeutic regimen in women of childbearing age should be deferred until the possibility of pregnancy has been ruled out. If the patient becomes pregnant while being treated with the BEXXAR therapeutic regimen, the patient should be apprised of the potential hazard to the fetus (see BOXED WARNING, Pregnancy Category X).
WARNINGS
Prolonged and Severe Cytopenias
(see BOXED WARNINGS; ADVERSE REACTIONS, Hematologic Events): The most common adverse reactions associated with the BEXXAR therapeutic regimen were severe or life-threatening cytopenias (NCI CTC grade 3 or 4) with 71% of the 230 patients enrolled in clinical studies experiencing grade 3 or 4 cytopenias. These consisted primarily of grade 3 or 4 thrombocytopenia (53%) and grade 3 or 4 neutropenia (63%). The time to nadir was 4 to 7 weeks and the duration of cytopenias was approximately 30 days. Thrombocytopenia, neutropenia, and anemia persisted for more than 90 days following administration of the BEXXAR therapeutic regimen in 16 (7%), 15 (7%), and 12 (5%) patients respectively (this includes patients with transient recovery followed by recurrent cytopenia). Due to the variable nature in the onset of cytopenias, complete blood counts should be obtained weekly for 10-12 weeks. The sequelae of severe cytopenias were commonly observed in the clinical studies and included infections (45% of patients), hemorrhage (12%), a requirement for growth factors (12% G- or GM-CSF; 7% Epoetin alfa)and blood product support (15% platelet transfusions; 16% red blood cell transfusions). Prolonged cytopenias may also influence subsequent treatment decisions.
The safety of the BEXXAR therapeutic regimen has not been established in patients with >25% lymphoma marrow involvement, platelet count<100,000 cells/mm3 or neutrophil count <1,500 cells/mm3.
Hypersensitivity Reactions Including Anaphylaxis
(see BOXED WARNINGS; ADVERSE REACTIONS, Hypersensitivity Reactions and Immunogenicity): Serious hypersensitivity reactions, including some with fatal outcome, were reported during and following administration of the BEXXAR therapeutic regimen. Emergency supplies including medications for the treatment of hypersensitivity reactions, e.g., epinephrine, antihistamines and corticosteroids, should be available for immediate use in the event of an allergic reaction during administration of the BEXXAR therapeutic regimen. Patients who have received murine proteins should be screened for human anti-mouse antibodies (HAMA). Patients who are positive for HAMA may be at increased risk of anaphylaxis and serious hypersensitivity reactions during administration of the BEXXAR therapeutic regimen.
Secondary Malignancies
Myelodysplastic syndrome (MDS) and/or acute leukemia were reported in 10% (24/230) of patients enrolled in the clinical studies and 3% (20/765) of patients included in expanded access programs, with median follow-up of 39 and 27 months, respectively. Among the 44 reported cases, the median time to development of MDS/leukemia was 31 months following treatment; however, the cumulative rate continues to increase.
Additional non-hematological malignancies were also reported in 54 of the 995 patients enrolled in clinical studies or included in the expanded access program. Approximately half of these were non-melanomatous skin cancers. The remainder, which occurred in 2 or more patients, included colorectal cancer (7), head and neck cancer (6), breast cancer (5), lung cancer (4), bladder cancer (4), melanoma (3), and gastric cancer (2). The relative risk of developing secondary malignancies in patients receiving the BEXXAR therapeutic regimen over the background rate in this population cannot be determined, due to the absence of controlled studies (see ADVERSE REACTIONS).
Pregnancy Category X
(see BOXED WARNINGS; CONTRAINDICATIONS).
Hypothyroidism
Administration of the BEXXAR therapeutic regimen may result in hypothyroidism (see ADVERSE REACTIONS, Hypothyroidism). Thyroid-blocking medications should be initiated at least 24 hours before receiving the dosimetric dose and continued until 14 days after the therapeutic dose (see DOSAGE and ADMINISTRATION). All patients must receive thyroid-blocking agents; any patient who is unable to tolerate thyroid-blocking agents should not receive the BEXXAR therapeutic regimen. Patients should be evaluated for signs and symptoms of hypothyroidism and screened for biochemical evidence of hypothyroidism annually.
PRECAUTIONS
Radionuclide Precautions
Iodine I 131 Tositumomab is radioactive. Care should be taken, consistent with the institutional radiation safety practices and applicable federal guidelines, to minimize exposure of medical personnel and other patients.
Renal Function
Iodine I 131 Tositumomab and Iodine-131 are excreted primarily by the kidneys. Impaired renal function may decrease the rate of excretion of the radiolabeled iodine and increase patient exposure to the radioactive component of the BEXXAR therapeutic regimen. There are no data regarding the safety of administration of the BEXXAR therapeutic regimen in patients with impaired renal function.
Immunization
The safety of immunization with live viral vaccines following administration of the BEXXAR therapeutic regimen has not been studied. The ability of patients who have received the BEXXAR therapeutic regimen to generate a primary or anamnestic humoral response to any vaccine has not been studied.
Information for Patients
Prior to administration of the BEXXAR therapeutic regimen, patients should be advised that they will have a radioactive material in their body for several days upon their release from the hospital or clinic. After discharge, patients should be provided with both oral and written instructions for minimizing exposure of family members, friends and the general public. Patients should be given a copy of the written instructions for use as a reference for the recommended precautionary actions.
The pregnancy status of women of childbearing potential should be assessed and these women should be advised of the potential risks to the fetus (see CONTRAINDICATIONS). Women who are breastfeeding should be instructed to discontinue breastfeeding and should be apprised of the resultant potential harmful effects to the infant if these instructions are not followed.
Patients should be advised of the potential risk of toxic effects on the male and female gonads following the BEXXAR therapeutic regimen, and be instructed to use effective contraceptive methods during treatment and for 12 months following the administration of the BEXXAR therapeutic regimen.
Patients should be informed of the risks of hypothyroidism and be advised of the importance of compliance with thyroid blocking agents and need for life-long monitoring.
Patients should be informed of the possibility of developing a HAMA immune response and that HAMA may affect the results of in vitro and in vivo diagnostic tests as well as results of therapies that rely on murine antibody technology.
Patients should be informed of the risks of cytopenias and symptoms associated with cytopenia, the need for frequent monitoring for up to 12 weeks after treatment, and the potential for persistent cytopenias beyond 12 weeks.
Patients should be informed that MDS, secondary leukemia, and solid tumors have also been observed in patients receiving the BEXXAR therapeutic regimen.
Due to lack of controlled clinical studies, and high background incidence in the heavily pretreated patient population, the relative risk of development of myelodysplastic syndrome/acute leukemia and solid tumors due to the BEXXAR therapeutic regimen cannot be determined.
Laboratory Monitoring
A complete blood count (CBC) with differential and platelet count should be obtained prior to, and at least weekly following administration of the BEXXAR therapeutic regimen. Weekly monitoring of blood counts should continue for a minimum of 10 weeks or, if persistent, until severe cytopenias have completely resolved. More frequent monitoring is indicated in patients with evidence of moderate or more severe cytopenias (see BOXED WARNINGS and WARNINGS). Thyroid stimulating hormone (TSH) level should be monitored before treatment and annually thereafter. Serum creatinine levels should be measured immediately prior to administration of the BEXXAR therapeutic regimen.
Drug Interactions
No formal drug interaction studies have been performed. Due to the frequent occurrence of severe and prolonged thrombocytopenia, the potential benefits of medications that interfere with platelet function and/or anticoagulation should be weighed against the potential increased risk of bleeding and hemorrhage.
Drug/Laboratory Test Interactions
Administration of the BEXXAR therapeutic regimen may result in the development of HAMA. The presence of HAMA may affect the accuracy of the results of in vitro and in vivo diagnostic tests and may affect the toxicity profile and efficacy of therapeutic agents that rely on murine antibody technology. Patients who are HAMA positive may be at increased risk for serious allergic reactions and other side effects if they undergo in vivo diagnostic testing or treatment with murine monoclonal antibodies.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies have been performed to establish the carcinogenic or mutagenic potential of the BEXXAR therapeutic regimen or to determine its effects on fertility in males or females. However, radiation is a potential carcinogen and mutagen. Administration of the BEXXAR therapeutic regimen results in delivery of a significant radiation dose to the testes. The radiation dose to the ovaries has not been established. There have been no studies to evaluate whether administration of the BEXXAR therapeutic regimen causes hypogonadism, premature menopause, azoospermia and/or mutagenic alterations to germ cells. There is a potential risk that the BEXXAR therapeutic regimen may cause toxic effects on the male and female gonads. Effective contraceptive methods should be used during treatment and for 12 months following administration of the BEXXAR therapeutic regimen.
Pregnancy Category X
(See CONTRAINDICATIONS; WARNINGS.)
Nursing Mothers
Radioiodine is excreted in breast milk and may reach concentrations equal to or greater than maternal plasma concentrations. Immunoglobulins are also known to be excreted in breast milk. The absorption potential and potential for adverse effects of the monoclonal antibody component (Tositumomab) in the infant are not known. Therefore, formula feedings should be substituted for breast feedings before starting treatment. Women should be advised to discontinue nursing.
Pediatric Use
The safety and effectiveness of the BEXXAR therapeutic regimen in children have not been established.
Geriatric Use
Clinical studies of the BEXXAR therapeutic regimen did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. In clinical studies, 230 patients received the BEXXAR therapeutic regimen at the recommended dose. Of these, 27% (61 patients) were age 65 or older and 4% (10 patients) were age 75 or older. Across all studies, the overall response rate was lower in patients age 65 and over (41% vs. 61%) and the duration of responses was shorter (10 months vs. 16 months); however, these findings are primarily derived from 2 of the 5 studies. While the incidence of severe hematologic toxicity was lower, the duration of severe hematologic toxicity was longer in those age 65 or older as compared to patients less than 65 years of age. Due to the limited experience greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS
The most serious adverse reactions observed in the clinical trials were severe and prolonged cytopenias and the sequelae of cytopenias which included infections (sepsis) and hemorrhage in thrombocytopenic patients, allergic reactions (bronchospasm and angioedema), secondary leukemia and myelodysplasia (see BOXED WARNINGS and WARNINGS).
The most common adverse reactions occurring in the clinical trials included neutropenia, thromobocytopenia, and anemia that are both prolonged and severe. Less common but severe adverse reactions included pneumonia, pleural effusion and dehydration.
Data regarding adverse events were primarily obtained in 230 patients with non-Hodgkin’s lymphoma enrolled in five clinical trials using the recommended dose and schedule. Patients had a median follow-up of 39 months and 79% of the patients were followed at least 12 months for survival and selected adverse events. Patients had a median of 3 prior chemotherapy regimens, a median age of 55 years, 60% male, 27% had transformation to a higher grade histology, 29% were intermediate grade and 2% high grade histology (IWF) and 68% had Ann Arbor stage IV disease. Patients enrolled in these studies were not permitted to have prior hematopoietic stem cell transplantation or irradiation to more than 25% of the red marrow. In the expanded access program, which included 765 patients, data regarding clinical serious adverse events and HAMA and TSH levels were used to supplement the characterization of delayed adverse events (see ADVERSE REACTIONS, Hypothyroidism, Secondary Leukemia and Myelodysplastic Syndrome, Immunogenicity).
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Hematologic Events
Hematologic toxicity was the most frequently observed adverse event in clinical trials with the BEXXAR therapeutic regimen (Table 6). Sixty-three (27%) of 230 patients received one or more hematologic supportive care measures following the therapeutic dose: 12% received G-CSF; 7% received Epoetin alfa; 15% received platelet transfusions; and 16% received packed red blood cell transfusions. Twenty-eight (12%) patients experienced hemorrhagic events; the majority were mild to moderate.
Infectious Events
One hundred and four of the 230 (45%) patients experienced one or more adverse events possibly related to infection. The majority were viral (e.g., rhinitis, pharyngitis, flu symptoms, or herpes) or other minor infections. Twenty of 230 (9%) patients experienced infections that were considered serious because the patient was hospitalized to manage the infection. Documented infections included pneumonia, bacteremia, septicemia, bronchitis, and skin infections.
Hypersensitivity Reactions
Fourteen patients (6%) experienced one or more of the following adverse events: allergic reaction, face edema, injection site hypersensitivity, anaphylactic reaction, laryngismus, and serum sickness. In the post-marketing setting, severe hypersensitivity reactions, including fatal anaphylaxis have been reported.
Gastrointestinal Toxicity
Eighty-seven patients (38%) experienced one or more gastrointestinal adverse events, including nausea, emesis, abdominal pain, and diarrhea. These events were temporally related to the infusion of the antibody. Nausea, vomiting, and abdominal pain were often reported within days of infusion, whereas diarrhea was generally reported days to weeks after infusion.
Infusional Toxicity
A constellation of symptoms, including fever, rigors or chills, sweating, hypotension, dyspnea, bronchospasm, and nausea, have been reported during or within 48 hours of infusion. Sixty-seven patients (29%) reported fever, rigors/chills, or sweating within 14 days following the dosimetric dose. Although all patients in the clinical studies received pretreatment with acetaminophen and an antihistamine, the value of premedication in preventing infusion-related toxicity was not evaluated in any of the clinical studies. Infusional toxicities were managed by slowing and/or temporarily interrupting the infusion. Symptomatic management was required in more severe cases. Adjustment of the rate of infusion to control adverse reactions occurred in 16 patients (7%); seven patients required adjustments for only the dosimetric infusion, two required adjustments for only the therapeutic infusion, and seven required adjustments for both the dosimetric and the therapeutic infusions. Adjustments included reduction in the rate of infusion by 50%, temporary interruption of the infusion, and in 2 patients, infusion was permanently discontinued.
Table 5 lists clinical adverse events that occurred in ≥5% of patients. Table 6 provides a detailed description of the hematologic toxicity.
|
Body System Preferred Term |
All Grades |
Grade 3/4 |
|
Total |
(96%) |
(48%) |
|
Non-Hematologic AEs |
||
|
Body as a Whole |
81% |
12% |
|
Asthenia |
46% |
2% |
|
Fever |
37% |
2% |
|
Infectionb |
21% |
<1% |
|
Pain |
19% |
1% |
|
Chills |
18% |
1% |
|
Headache |
16% |
0% |
|
Abdominal pain |
15% |
3% |
|
Back pain |
8% |
1% |
|
Chest pain |
7% |
0% |
|
Neck pain |
6% |
1% |
|
Cardiovascular System |
26% |
3% |
|
Hypotension |
7% |
1% |
|
Vasodilatation |
5% |
0% |
|
Digestive System |
56% |
9% |
|
Nausea |
36% |
3% |
|
Vomiting |
15% |
1% |
|
Anorexia |
14% |
0% |
|
Diarrhea |
12% |
0% |
|
Constipation |
6% |
1% |
|
Dyspepsia |
6% |
<1% |
|
Endocrine System |
7% |
0% |
|
Hypothyroidism |
7% |
0% |
|
Metabolic and Nutritional Disorders |
21% |
3% |
|
Peripheral edema |
9% |
0% |
|
Weight loss |
6% |
<1% |
|
Musculoskeletal System |
23% |
3% |
|
Myalgia |
13% |
<1% |
|
Arthralgia |
10% |
1% |
|
Nervous System |
26% |
3% |
|
Dizziness |
5% |
0% |
|
Somnolence |
5% |
0% |
|
Respiratory System |
44% |
8% |
|
Cough increased |
21% |
1% |
|
Pharyngitis |
12% |
0% |
|
Dyspnea |
11% |
3% |
|
Rhinitis |
10% |
0% |
|
Pneumonia |
6% |
0% |
|
Skin and Appendages |
44% |
5% |
|
Rash |
17% |
<1% |
|
Pruritus |
10% |
0% |
|
Sweating |
8% |
<1% |
aExcludes laboratory derived hematologic adverse events (see Table 6).
bThe COSTART term for infection includes a subset of infections (e.g., upper respiratory infection). Other terms are mapped to preferred terms (e.g., pneumonia and sepsis). For a more inclusive summary see ADVERSE REACTIONS, Infectious Events.
|
Endpoint |
Values |
|
Platelets | |
|
Median nadir (cells/mm3) |
43,000 |
|
Per patient incidencea platelets<50,000/mm3 |
53% (n = 123) |
|
Medianb duration of platelets<50,000/mm3 (days) |
32 |
|
Grade 3/4 without recovery to Grade 2, N (%) |
16 (7%) |
|
Per patient incidencec platelets<25,000/mm3 |
21% (n = 47) |
|
ANC | |
|
Median nadir (cells/mm3) |
690 |
|
Per patient incidencea ANC<1,000 cells/mm3 |
63% (n = 145) |
|
Medianb duration of ANC<1,000 cells/mm3 (days) |
31 |
|
Grade 3/4 without recovery to Grade 2, N (%) |
15 (7%) |
|
Per patient incidencec ANC<500 cells/mm3 |
25% (n = 57) |
|
Hemoglobin | |
|
Median nadir (gm/dL) |
10 |
|
Per patient incidencea<8 gm/dL |
29% (n = 66) |
|
Medianb duration of hemoglobin<8.0 gm/dL (days) |
23 |
|
Grade 3/4 without recovery to Grade 2, N (%) |
12 (5%) |
|
Per patient incidencec hemoglobin<6.5 gm/dL |
5% (n = 11) |
aGrade 3/4 toxicity was assumed if patient was missing 2 or more weeks of hematology data between Week 5 and Week 9.
bDuration of Grade 3/4 of 1,000+ days (censored) was assumed for those patients with undocumented Grade 3/4 and no hematologic data on or after Week 9.
cGrade 4 toxicity was assumed if patient had documented Grade 3 toxicity and was missing 2 or more weeks of hematology data between Week 5 and Week 9.
Delayed Adverse Reactions
Delayed adverse reactions, including hypothyroidism, HAMA, and myelodysplasia/leukemia, were assessed in 230 patients included in clinical studies and 765 patients included in expanded access programs. The entry characteristics of patients included from the expanded access programs were similar to the characteristics of patients enrolled in the clinical studies, except that the median number of prior chemotherapy regimens was fewer (2 vs. 3) and the proportion with low-grade histology was higher (77% vs. 70%) in patients from the expanded access programs.
Secondary Leukemia and Myelodysplastic Syndrome (MDS)
There were 44 cases of MDS/secondary leukemia reported among 995 (4.0%) patients included in clinical studies and expanded access programs, with a median follow-up of 29 months. The overall incidence of MDS/secondary leukemia among the 230 patients included in the clinical studies was 10% (24/230), with a median follow-up of 39 months and a median time to development of MDS of 34 months. The cumulative incidence of MDS/secondary leukemia in this patient population was 4.7% at 2 years and 15% at 5 years. The incidence of MDS/secondary leukemia among the 765 patients in the expanded access programs was 3% (20/765), with a median follow-up of 27 months and a median time to development of MDS of 31 months. The cumulative incidence of MDS/secondary leukemia in this patient population was 1.6% at 2 years and 6% at 5 years.
Secondary Malignancies
Of the 995 patients in clinical studies and the expanded access programs, there were 65 reports of second malignancies in 54 patients, excluding secondary leukemias. The most common included non-melanomatous skin cancers (26), colorectal cancer (7), head and neck cancer (6), breast cancer (5), lung cancer (4), bladder cancer (4), melanoma (3), and gastric cancer (2). Some of these events included recurrence of an earlier diagnosis of cancer.
Hypothyroidism
Of the 230 patients in the clinical studies, 203 patients did not have elevated TSH upon study entry. Of these, 137 patients had at least one post-treatment TSH value available and were not taking thyroid hormonal treatment upon study entry. With a median follow up period of 46 months, the incidence of hypothyroidism based on elevated TSH or initiation of thyroid replacement therapy in these patients was 18% with a median time to development of hypothyroidism of 16 months. The cumulative incidences of hypothyroidism at 2 and 5 years in these 137 patients were 11% and 19% respectively. New events have been observed up to 90 months post-treatment.
Of the 765 patients in the expanded access programs, 670 patients did not have elevated TSH upon study entry. Of these, 455 patients had at least one post-treatment TSH value available and were not taking thyroid hormonal treatment upon study entry. With a median follow up period of 33 months, the incidence of hypothyroidism based on elevated TSH or initiation of thyroid replacement therapy in these 455 patients was 13% with a median time to development of hypothyroidism of 15 months. The cumulative incidences of hypothyroidism at 2 and 5 years in these patients were 9% and 17%, respectively.
Immunogenicity
One percent (11/989) of the chemotherapy-relapsed or refractory patients included in the clinical studies or the expanded access program had a positive serology for HAMA prior to treatment and six patients had no baseline assessment for HAMA. Of the 230 patients in the clinical studies, 220 patients were seronegative for HAMA prior to treatment, and 219 had at least one post-treatment HAMA value obtained. With a median observation period of 6 months, a total of 23 patients (11%) became seropositive for HAMA post-treatment. The median time of HAMA development was 6 months. The cumulative incidences of HAMA seropositivity at 6 months, 12 months, and 18 months were 6%, 17% and 21% respectively.
Of the 765 patients in the expanded access programs, 758 patients were seronegative for HAMA prior to treatment, and 569 patients had at least one post-treatment HAMA value obtained. With a median observation period of 7 months, a total of 57 patients (10%) became seropositive for HAMA post-treatment. The median time of HAMA development was 5 months. The cumulative incidences of HAMA seropositivity at 6 months, 12 months, and 18 months were 7%, 12% and 13%, respectively.
In a study of 76 previously untreated patients with low-grade non-Hodgkin’s lymphoma who received the BEXXAR therapeutic regimen, the incidence of conversion to HAMA seropositivity was 70%, with a median time to development of HAMA of 27 days.
The data reflect the percentage of patients whose test results were consideredpositive for HAMA in an ELISA assay that detects antibodies to the Fc portion of IgG1 murine immunoglobulin and are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of HAMA in patients treated with the BEXXAR therapeutic regimen with the incidence of HAMA in patients treated with other products may be misleading.
OVERDOSAGE
The maximum dose of the BEXXAR therapeutic regimen that was administered in clinical trials was 88 cGy. Three patients were treated with a total body dose of 85 cGy of Iodine I 131 Tositumomab in a dose escalation study. Two of the 3 patients developed Grade 4 toxicity of 5 weeks duration with subsequent recovery. In addition, accidental overdose of the BEXXAR therapeutic regimen occurred in one patient at a total body dose of 88 cGy. The patient developed Grade 3 hematologic toxicity of 18 days duration. Patients who receive an accidental overdose of Iodine I 131 Tositumomab should be monitored closely for cytopenias and radiation-related toxicity. The effectiveness of hematopoietic stem cell transplantation as a supportive care measure for marrow injury has not been studied; however, the timing of such support should take into account the pharmacokinetics of the BEXXAR therapeutic regimen and decay rate of the Iodine-131 in order to minimize the possibility of irradiation of infused hematopoietic stem cells.
DOSAGE AND ADMINISTRATION
Recommended Dose
The BEXXAR therapeutic regimen consists of four components administered in two discrete steps: the dosimetric step, followed 7-14 days later by a therapeutic step.
Note: The safety of the BEXXAR therapeutic regimen was established only in the setting of patients receiving thyroid blocking agents and premedication to ameliorate/prevent infusion reactions (see Concomitant Medications).
Dosimetric step
- Tositumomab 450 mg intravenously in 50 ml 0.9% Sodium Chloride over 60 minutes. Reduce the rate of infusion by 50% for mild to moderate infusional toxicity; interrupt infusion for severe infusional toxicity. After complete resolution of severe infusional toxicity, infusion may be resumed with a 50% reduction in the rate of infusion.
- Iodine I 131 Tositumomab (containing 5.0 mCi Iodine-131 and 35 mg Tositumomab) intravenously in 30 ml 0.9% Sodium Chloride over 20 minutes. Reduce the rate of infusion by 50% for mild to moderate infusional toxicity; interrupt infusion for severe infusional toxicity. After complete resolution of severe infusional toxicity, infusion may be resumed with a 50% reduction in the rate of infusion.
Therapeutic step
Note: Do not administer the therapeutic step if biodistribution is altered (see Assessment of Biodistribution of Iodine I 131 Tositumomab).
- Tositumomab 450 mg intravenously in 50 ml 0.9% Sodium Chloride over 60 minutes. Reduce the rate of infusion by 50% for mild to moderate infusional toxicity; interrupt infusion for severe infusional toxicity. After complete resolution of severe infusional toxicity, infusion may be resumed with a 50% reduction in the rate of infusion.
- Iodine I 131 Tositumomab (see CALCULATION
OF IODINE-131 ACTIVITY FOR THE THERAPEUTIC DOSE). Reduce
the rate of infusion by 50% for mild to moderate infusional toxicity;
interrupt infusion for severe infusional toxicity. After complete
resolution of severe infusional toxicity, infusion may be resumed
with a 50% reduction in the rate of infusion.
- Patients with ≥150,000 platelets/mm3: The recommended dose is the activity of Iodine-131 calculated to deliver 75 cGy total body irradiation and 35 mg Tositumomab, administered intravenously over 20 minutes.
- Patients with NCI Grade 1 thrombocytopenia (platelet counts ≥100,000 but <150,000 platelets/mm3): The recommended dose is the activity of Iodine-131 calculated to deliver 65 cGy total body irradiation and 35 mg Tositumomab, administered intravenously over 20 minutes.
Concomitant Medications
The safety of the BEXXAR therapeutic regimen was established in studies in which all patients received the following concurrent medications:
- Thyroid protective agents: Saturated solution of potassium iodide (SSKI) 4 drops orally t.i.d.; Lugol’s solution 20 drops orally t.i.d.; or potassium iodide tablets 130 mg orally q.d. Thyroid protective agents should be initiated at least 24 hours prior to administration of the Iodine I 131 Tositumomab dosimetric dose and continued until 2 weeks after administration of the Iodine I 131 Tositumomab therapeutic dose.
Patients should not receive the dosimetric dose of Iodine I 131 Tositumomab if they have not yet received at least three doses of SSKI, three doses of Lugol’s solution, or one dose of 130 mg potassium iodide tablet (at least 24 hours prior to the dosimetric dose).
- Acetaminophen 650 mg orally and diphenhydramine 50 mg orally 30 minutes prior to administration of Tositumomab in the dosimetric and therapeutic steps.
The BEXXAR therapeutic regimen is administered via an IV tubing set with an in-line 0.22 micron filter. THE SAME IV TUBING SET AND FILTER MUST BE USED THROUGHOUT THE ENTIRE DOSIMETRIC OR THERAPEUTIC STEP. A CHANGE IN FILTER CAN RESULT IN LOSS OF DRUG.
Figure1 shows an overview of the dosing schedule.
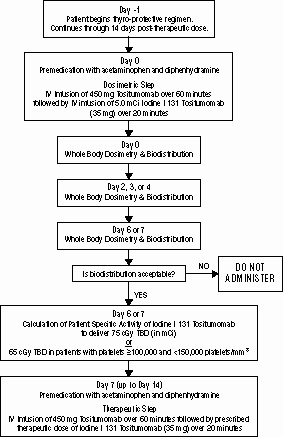
PREPARATION OF THE BEXXAR THERAPEUTIC REGIMEN
GENERAL
Read all directions thoroughly and assemble all materials before preparing the dose for administration.
The Iodine I 131 Tositumomab dosimetric and therapeutic doses should be measured by a suitable radioactivity calibration system immediately prior to administration. The dose calibrator must be operated in accordance with the manufacturer’s specifications and quality control for the measurement of Iodine-131.
All supplies for preparation and administration of the BEXXAR therapeutic regimen should be sterile. Use appropriate aseptic technique and radiation precautions for the preparation of the components of the BEXXAR therapeutic regimen.
Waterproof gloves should be utilized in the preparation and administration of the product. Iodine I 131 Tositumomab doses should be prepared, assayed, and administered by personnel who are licensed to handle and/or administer radionuclides. Appropriate shielding should be used during preparation and administration of the product.
Restrictions on patient contact with others and release from the hospital must follow all applicable federal, state, and institutional regulations.
Preparation for the Dosimetric Step
Tositumomab Dose
Required materials not supplied:
A. One 50 mL syringe with attached 18-gauge needle (to withdraw 450 mg of Tositumomab from two vials each containing 225 mg Tositumomab)
B. One 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
C. One 50 mL syringe for drawing up 32 mL of saline for disposal from the 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
Method:
- Withdraw and dispose of 32 mL of saline from a 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP.
- Withdraw the entire contents from each of the two 225 mg vials (a total of 450 mg Tositumomab in 32 mL) and transfer to the infusion bag containing 18 mL of 0.9% Sodium Chloride for Injection, USP to yield a final volume of 50 mL.
- Gently mix the solution by inverting/rotating the bag. DO NOT SHAKE.
- The diluted Tositumomab may be stored for up to 24 hours when stored refrigerated at 2°C−8°C (36°F−46°F) and for up to 8 hours at room temperature.
Note: Tositumomab solution may contain particulates that are generally white in nature. The product should appear clear to opalescent, colorless to slightly yellow.
Preparation of Iodine I 131 Tositumomab Dosimetric Dose
Required materials not supplied:
A. Lead shielding for preparation vial and syringe pump
B. One 30 mL syringe with 18-gauge needle to withdraw the calculated volume of Iodine I 131 Tositumomab from the Iodine I 131 Tositumomab vial. One 60 mL syringe with 18-gauge needle to withdraw the volume from the preparation vial for administration
C. One 20 mL syringe with attached needle, filled with 0.9% Sodium Chloride for Injection, USP
D. One 3 mL syringe with attached needle to withdraw Tositumomab from 35 mg vial
E. One sterile, 30 or 50 mL preparation vial
F. Two lead pots, both kept at room temperature. One pot is used to thaw the labeled antibody and the second pot is used to hold the preparation vial
Method:
- Allow approximately 60 minutes for thawing (at ambient temperature) of the Iodine I 131 Tositumomab dosimetric vial with appropriate lead shielding.
- Based on the activity concentration of the vial (see actual product specification sheet for the vial supplied in the dosimetric package), calculate the volume required for an Iodine I 131 Tositumomab activity of 5.0 mCi.
- Withdraw the calculated volume from the Iodine I 131 Tositumomab vial.
- Transfer this volume to the shielded preparation vial.
- Assay the dose to ensure that the appropriate activity (mCi)
has been prepared.
a.If the assayed dose is 5.0 mCi (±10%) proceed with step 6.
b.If the assayed dose does not contain 5.0 mCi (±10%) recalculate the activity concentration of the Iodine I 131 Tositumomab at this time, based on the volume and the activity in the preparation vial. Recalculate the volume required for an Iodine I 131 Tositumomab activity of 5.0 mCi. Using the same 30 mL syringe, add or subtract the appropriate volume from the Iodine I 131 Tositumomab vial so that the preparation vial contains the volume required for an Iodine I 131 Tositumomab activity of 5.0 mCi (±10%). Re-assay the preparation vial and proceed with step 6.
- Calculate the amount of Tositumomab contained in the solution of Iodine I 131 Tositumomab in the shielded preparation vial, based on the volume and protein concentration (see actual product specification sheet supplied in the dosimetric package).
- If the shielded preparation vial contains less than 35 mg, calculate the amount of additional Tositumomab needed to yield a total of 35 mg protein. Calculate the volume needed from the 35 mg vial of Tositumomab, based on the protein concentration. Withdraw the calculated volume of Tositumomab from the 35 mg vial of Tositumomab, and transfer this volume to the shielded preparation vial. The preparation vial should now contain a total of 35 mg of Tositumomab.
- Using the 20 mL syringe containing 0.9% Sodium Chloride for Injection, USP, add a sufficient quantity to the shielded preparation vial to yield a final volume of 30 mL. Gently mix the solutions.
- Withdraw the entire contents from the preparation vial into a 60 mL syringe using a large bore needle (18-gauge).
- Assay and record the activity.
Administration of the Dosimetric Step
Required materials not supplied: For questions about required materials call the BEXXAR Service Center at 1-877-423-9927.
A. IV Filter set (0.22 micron filter), 15 inch with injection site (port) and luer lock
B. One Primary IV infusion set
C. One 100 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
D. Two Secondary IV infusion sets
E. One IV Extension set, 30 inch luer lock
F. One 3-way stopcock
G. One 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
H. One Infusion pump for Tositumomab infusion
I. One Syringe Pump for Iodine I 131 Tositumomab infusion
J. Lead shielding for use in the administration of the dosimetric dose
Tositumomab Infusion:
(See Figure 1 in the “Workbook for Dosimetry Methodology and Administration Set-Up” for diagrammatic illustration of the configuration of the infusion set components.)
- Attach a primary IV infusion set (Item B) to the 0.22 micron in-line filter set (Item A) and the 100 mL bag of sterile 0.9% Sodium Chloride for Injection, USP (Item C).
- After priming the primary IV infusion set (Item B) and IV filter set (Item A), connect the infusion bag containing 450 mg Tositumomab (50 mL) via a secondary IV infusion set (Item D) to the primary IV infusion set (Item B) at a port distal to the 0.22 micron in-line filter. Infuse Tositumomab over 60 minutes.
- After completion of the Tositumomab infusion, disconnect the secondary IV infusion set (Item D) and flush the primary IV infusion set (Item B) and the in-line IV filter set (Item A) with sterile 0.9% Sodium Chloride for Injection, USP. Discard the Tositumomab bag and secondary IV infusion set.
Iodine I 131 Tositumomab Dosimetric Infusion:
(See Figure 2 in the “Workbook for Dosimetry Methodology and Administration Set-Up” for diagrammatic illustration of the configuration of the infusion set components.)
- Appropriate shielding should be used in the administration of the dosimetric dose.
- The dosimetric dose is delivered in a 60 mL syringe.
- Connect the extension set (Item E) to the 3-way stopcock (Item F).
- Connect the 50 mL bag of sterile 0.9% Sodium Chloride for Injection,
USP (Item G) to a secondary IV infusion set (Item D) and connect the
infusion set to the 3-way stopcock (Item F). Prime the secondary IV
infusion set (Item D) and the extension set (Item E). Connect the
extension set (Item E) to a port in the primary IV infusion set (Item
B), distal to the filter.
(Note: You must use the same primary infusion set (Item B) and IV filter set (Item A) with pre-wetted filter that was used for the Tositumomab infusion. A change in filter can result in loss of up to 7% of the Iodine I 131 Tositumomab dose.)
- Attach the syringe filled with the Iodine I 131 Tositumomab to the 3-way stopcock (Item F)
- Set syringe pump to deliver the entire 5.0 mCi (35 mg) dose of Iodine I 131 Tositumomab over 20 minutes.
- After completion of the infusion of Iodine I 131 Tositumomab, close the stopcock (Item F) to the syringe. Flush the extension set (Item E) and the secondary IV infusion set (Item D) with 0.9% Sodium Chloride for Injection, USP from the 50 mL bag (Item G).
- After the flush, disconnect the extension set (Item E), 3-way stopcock (Item F) and syringe. Disconnect the primary IV infusion set (Item B) and in-line filter set (Item A). Determine the combined residual activity of the syringe and infusion set components (stopcock, extension set, primary infusion set and in-line filter set) by assaying these items in a suitable radioactivity calibration system immediately following completion of administration of all components of the dosimetric step. Calculate and record the dose delivered to the patient by subtracting the residual activity in the syringe and the infusion set components from the activity of Iodine I 131 Tositumomab in the syringe prior to infusion.
- Discard all materials used to deliver the Iodine I 131 Tositumomab (e.g., syringes, vials, in-line filter set, extension set and infusion sets) in accordance with local, state, and federal regulations governing radioactive and biohazardous waste.
Determination of Dose for the Therapeutic Step (see Calculation of Iodine-131 Activity for Therapeutic Dose): The method for determining and calculating the patient-specific dose of Iodine-131 activity (mCi) to be administered in the therapeutic step is described below. The derived values obtained in steps 3 and 4 and calculation of the therapeutic dose as described in step 6 may be determined manually [see “Workbook for Dosimetry Methodology and Administration Set-Up”] or calculated automatically using the GlaxoSmithKline proprietary software program [BEXXAR Patient Management Templates]. To receive training and to obtain the “BEXXAR Patient Management Templates” call the BEXXAR Service Center at 1-877-423-9927. For assistance with either manual or automated calculations call the BEXXAR Service Center at 1-877-423-9927.
- Following infusion of the Iodine I 131 Tositumomab dosimetric
dose, obtain total body gamma camera counts and whole body images
at the following timepoints:
a.Within one hour of infusion and prior to urination
b.2-4 days after infusion of the dosimetric dose, following urination
c.6-7 days after infusion of the dosimetric dose, following urination
- Assess biodistribution. If biodistribution is altered, the therapeutic step should not be administered.
- Determine total body residence time (see Graph 1, “Determination of Residence Time”, in the “Workbook for Dosimetry Methodology and Administration Set-Up”).
- Determine activity hours (see Table 2, “Determination of Activity Hours”, in the “Workbook for Dosimetry Methodology and Administration Set-Up”), according to gender. Use actual patient mass (in kg) or maximum effective mass (in kg) whichever is lower (see Table 1, “Determination of Maximum Effective Mass”, in the “Workbook for Dosimetry Methodology and Administration Set-Up”).
- Determine whether the desired total body dose should be reduced (to 65 cGy) due to a platelet count of 100,000 to <150,000 cells/mm3.
- Based on the total body residence time and activity hours, calculate the Iodine-131 activity (mCi) to be administered to deliver the therapeutic dose of 65 or 75 cGy.
The following equation is used to calculate the activity of Iodine-131 required for delivery of the desired total body dose of radiation.
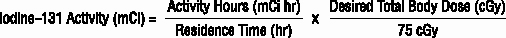
Preparation for the Therapeutic Step
Tositumomab Dose
Required materials not supplied:
A. One 50 mL syringe with attached 18-gauge needle (to withdraw 450 mg of Tositumomab from two vials each containing 225 mg Tositumomab)
B. One 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
C. One 50 mL syringe for drawing up 32 mL of saline for disposal from the 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
Method:
- Withdraw and dispose of 32 mL of saline from a 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP.
- Withdraw the entire contents from each of the two 225 mg vials (a total of 450 mg Tositumomab in 32 mL) and transfer to the infusion bag containing 18 mL of 0.9% Sodium Chloride for Injection, USP to yield a final volume of 50 mL.
- Gently mix the solutions by inverting/rotating the bag. DO NOT SHAKE.
- The diluted Tositumomab may be stored for up to 24 hours when stored refrigerated at 2°C−8°C (36°F−46°F)and for up to 8 hours at room temperature.
Note: Tositumomab solution may contain particulates that are generally white in nature. The product should appear clear to opalescent, colorless to slightly yellow.
Preparation of Iodine I 131 Tositumomab Therapeutic Dose
Required materials not supplied:
A.Lead shielding for preparation vial and syringe pump
B.One or two 30 mL syringes with 18-gauge needles to withdraw the calculated volume of Iodine I 131 Tositumomab from the Iodine I 131 Tositumomab vial(s). One or two 60 mL syringes with 18-gauge needles to withdraw the volume from the preparation vial for administration
C. One 20 mL syringe with attached needle filled with 0.9% Sodium Chloride for Injection, USP
D. One 3 mL sterile syringe with attached needle to draw up Tositumomab from the 35 mg vial
E. One sterile, 30 or 50 mL preparation vial
F.Two lead pots both kept at room temperature. One pot is used to thaw the labeled antibody, and the second pot is used to hold the preparation vial
Method:
- Allow approximately 60 minutes for thawing (at ambient temperature) of the Iodine I 131 Tositumomab therapeutic vial with appropriate lead shielding.
- Calculate the dose of Iodine I 131 Tositumomab required (see CALCULATION OF IODINE-131 ACTIVITY FOR THERAPEUTIC DOSE).
- Based on the activity concentration of the vial (see actual product specification sheet for each vial supplied in the therapeutic package), calculate the volume required for the Iodine I 131 Tositumomab activity required for the therapeutic dose.
- Using one or more 30 mL syringes with an 18-gauge needle, withdraw the calculated volume from the Iodine I 131 Tositumomab vial.
- Transfer this volume to the shielded preparation vial.
- Assay the dose to ensure that the appropriate activity (mCi)
has been prepared.
a.If the assayed dose is the calculated dose (±10%) needed for the therapeutic step, proceed with step 7.
b. If the assayed dose does not contain the desired dose (±10%), re-calculate the activity concentration of the Iodine I 131 Tositumomab at this time, based on the volume and the activity in the preparation vial. Re-calculate the volume required for an Iodine I 131 Tositumomab activity for the therapeutic dose. Using the same 30 mL syringe, add or subtract the appropriate volume from the Iodine I 131 Tositumomab vial so that the preparation vial contains the volume required for the Iodine I 131 Tositumomab activity required for the therapeutic dose. Re-assay the preparation vial. Proceed to step 7.
- Calculate the amount of Tositumomab protein contained in the solution of Iodine I 131 Tositumomab in the shielded preparation vial, based on the volume and protein concentration (see product specification sheet).
- If the shielded preparation vial contains less than 35 mg, calculate
the amount of additional Tositumomab needed to yield a total of 35
mg protein. Calculate the volume needed from the 35 mg vial of Tositumomab,
based on the protein concentration. Withdraw the calculated volume
of Tositumomab from the 35 mg vial of Tositumomab, and transfer this
volume to the shielded preparation vial. The preparation vial should
now contain a total of 35 mg of Tositumomab.
Note: If the dose of Iodine I 131 Tositumomab requires the use of 2 vials of Iodine I 131 Tositumomab or the entire contents of a single vial of Iodine I 131 Tositumomab, there may be no need to add protein from the 35 mg vial of Tositumomab.
- Using the 20 mL syringe containing 0.9% Sodium Chloride for Injection, USP, add a sufficient volume (if needed) to the shielded preparation vial to yield a final volume of 30 mL. Gently mix the solution.
- Withdraw the entire volume from the preparation vial into a one or more sterile 60 mL syringes using a large bore needle (18-gauge).
- Assay and record the activity.
Administration of the Therapeutic Step
Note: Restrictions on patient contact with others and release from the hospital must follow all applicable federal, state, and institutional regulations.
Required materials not supplied: For questions about required materials call the BEXXAR Service Center at 1-877-423-9927.
A. One IV Filter set (0.22 micron, filter), 15 inch with injection site (port) and luer lock
B. One Primary IV infusion set
C. One 100 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
D. Two Secondary IV infusion sets
E. One IV extension set, 30 inch luer lock
F. One 3-way stopcock
G. One 50 mL bag of sterile 0.9% Sodium Chloride for Injection, USP
H. One Infusion pump for Tositumomab infusion
I. One Syringe Pump for Iodine I 131 Tositumomab infusion
J. Lead shielding for use in the administration of the therapeutic dose
Tositumomab Infusion:
(See Figure 1 in the “Workbook for Dosimetry Methodology and Administration Set-Up” for diagrammatic illustration of the configuration of the infusion set components.)
- Attach a primary IV infusion set (Item B) to the 0.22 micron in-line filter set (Item A) and a 100 mL bag of sterile 0.9% Sodium Chloride for Injection, USP (Item C).
- After priming the primary IV infusion set (Item B) and filter set (Item A), connect the infusion bag containing 450 mg Tositumomab (50 mL) via a secondary IV infusion set (Item D) to the primary IV infusion set (Item B) at a port distal to the 0.22 micron in-line filter. Infuse Tositumomab over 60 minutes.
- After completion of the Tositumomab infusion, disconnect the secondary IV infusion set (Item D) and flush the primary IV infusion set (Item B) and the IV filter set (Item A) with sterile 0.9% Sodium Chloride for Injection, USP. Discard the Tositumomab bag and secondary IV infusion set.
Iodine I 131 Tositumomab Therapeutic Infusion:
(See Figure 2 in the “Workbook for Dosimetry Methodology and Administration Set-Up” for diagrammatic illustration of the configuration of the infusion set components.)
- Appropriate shielding should be used in the administration of the therapeutic dose.
- The therapeutic dose is delivered in one or more 60 mL syringes.
- Connect the extension set (Item E) to the 3-way stopcock (Item F).
- Connect the 50 mL bag of sterile 0.9% Sodium Chloride for Injection,
USP (Item G) to a secondary IV infusion set (Item D) and connect the
infusion set to the 3-way stopcock (Item F). Prime the secondary IV
infusion set (Item D) and the extension set (Item E). Connect the
extension set (Item E) to a port in the primary IV infusion set (Item
B), distal to the filter.
(Note: You must use the same primary infusion set (Item B) and IV filter set (Item A) with pre-wetted filter that was used for the Tositumomab infusion. A change in filter can result in loss of up to 7% of the Iodine I 131 Tositumomab dose.)
- Attach the syringe filled with the Iodine I 131 Tositumomab to the 3-way stopcock (Item F).
- Set syringe pump to deliver the entire therapeutic dose of Iodine I 131 Tositumomab over 20 minutes. (Note: If more than one syringe is required, remove the syringe and repeat steps 5 and 6.)
- After completion of the infusion of Iodine I 131 Tositumomab, close the stopcock (Item F) to the syringe. Flush the secondary IV infusion set (Item D) and the extension set (Item E) with 0.9% Sodium Chloride from the 50 mL bag of sterile, 0.9% Sodium Chloride for Injection, USP (Item G).
- After the flush, disconnect the extension set (Item E), 3-way stopcock (Item F) and syringe. Disconnect the primary IV infusion set (Item B) and in-line filter set (Item A). Determine the combined residual activity of the syringe(s) and infusion set components (stopcock, extension set, primary infusion set and in-line filter set) by assaying these items in a suitable radioactivity calibration system immediately following completion of administration of all components of the therapeutic step. Calculate and record the dose delivered to the patient by subtracting the residual activity in the syringe and infusion set components from the activity of Iodine I 131 Tositumomab in the syringe prior to infusion.
- Discard all materials used to deliver the Iodine I 131 Tositumomab (e.g., syringes, vials, in-line filter set, extension set and infusion sets) in accordance with local, state, and federal regulations governing radioactive and biohazardous waste.
DOSIMETRY
The following sections describe the procedures for image acquisition for collection of dosimetry data, interpretation of biodistribution images, calculation of residence time, and calculation of activity hours. Please read all sections carefully.
IMAGE ACQUISITION AND INTERPRETATION
Gamma Camera and Dose Calibrator Procedures
Manufacturer-specific quality control procedures should be followed for the gamma camera/computer system, the collimator, and the dose calibrator. Less than 20% variance between maximum and minimum pixel count values in the useful field of view is acceptable on Iodine-131 intrinsic flood fields and variability<10% is preferable. Iodine-131-specific camera uniformity corrections are strongly recommended, rather than applying lower energy correction to the Iodine-131 window. Camera extrinsic uniformity should be assessed at least monthly using 99mTc or 57Co as a source with imaging at the appropriate window.
Additional (non-routine) quality control procedures are required. To assure the accuracy and precision of the patient total body counts, the gamma camera must undergo validation and daily quality control on each day it is used to collect patient images.
Use the same setup and region of interest (ROI) for calibration, determination of background, and whole body patient studies.
Gamma Camera Set-Up
The same camera, collimator, scanning speed, energy window, and setup must be used for all studies. The gamma camera must be capable of whole body imaging and have a large or extra large field of view with a digital interface. It must be equipped with a parallel-hole collimator rated to at least364 keV by the manufacturer with a septal penetration for Iodine-131 of <7%.
The camera and computer must be set up for scanning as follows:
- Parallel hole collimator rated to at least 364 keV with a septal penetration for Iodine-131 of <7%
- Symmetric window (20-25%) centered on the 364 keV photo peak of Iodine-131 (314-414 keV)
- Matrix: appropriate whole body matrix
- Scanning speed: 10-30 cm/minute
Counts from Calibrated Source for Quality Control
Camera sensitivity for Iodine-131 must be determined each day. Determination of the gamma camera’s sensitivity is obtained by scanning a calibrated activity of Iodine-131 (e.g., 200−250 μCi in at least 20 mL of saline within a sealed pharmaceutical vial). The radioactivity of the Iodine-131 source is first determined using a NIST-traceable-calibrated clinical dose calibrator at the Iodine-131 setting.
Background Counts
The background count is obtained from a scan with no radioactive source. This should be obtained following the count of the calibrated source and just prior to obtaining the patient count.
If abnormally high background counts are measured, the source should be identified and, if possible, removed. If abnormally low background counts are measured, the camera energy window setting and collimator should be verified before repeating the background counts.
The counts per μCi are obtained by dividing the background-corrected source count by the calibrated activity for that day. For a specific camera and collimator, the counts per μCi should be relatively constant. When values vary more than 10% from the established ratio, the reason for the discrepancy should be ascertained and corrected and the source countrepeated.
Patient Total Body Counts
The source and background counts are obtained first and the camera sensitivity (i.e., constant counting efficiency) is established prior to obtaining the patient count. The same rectangular region of interest (ROI) must be used for the whole body counts, the quality control counts of the radioactive source, and the background counts.
Acquire anterior and posterior whole body images for gamma camera counts. For any particular patient, the same gamma camera must be used for all scans. To obtain proper counts, extremities must be included in the images, and arms should not cross over the body. The scans should be centered on the midline of the patient. Record the time of the start of the radiolabeled dosimetric infusion and the time of the start of each count acquisition.
Gamma camera counts will be obtained at the three imaging timepoints:
- Count 1: Within an hour of end of the infusion of the Iodine I 131 Tositumomab dosimetric dose prior to patient voiding.
- Count 2: Two to 4 days after administration of the Iodine I 131 Tositumomab dosimetric dose and immediately following patient voiding.
- Count 3: Six to 7 days after the administration of the Iodine I 131 Tositumomab dosimetric dose and immediately following patient voiding.
Assessment of Biodistribution of Iodine I 131 Tositumomab
The biodistribution of Iodine I 131 Tositumomab should be assessed by determination of total body residence time and by visual examination of whole body camera images from the first image taken at the time of Count 1 (within an hour of the end of the infusion) and from the second image taken at the time of Count 2 (at 2 to 4 days after administration). To resolve ambiguities, an evaluation of the third image at the time of Count 3 (6 to 7 days after administration) may be necessary. If either of these methods indicates that the biodistribution is altered, the Iodine I 131 Tositumomab therapeutic dose should not be administered.
Expected Biodistribution
- On the first imaging timepoint: Most of the activity is in the blood pool (heart and major blood vessels) and the uptake in normal liver and spleen is less than in the heart.
- On the second and third imaging timepoints: The activity in the blood pool decreases significantly and there is decreased accumulation of activity in normal liver and spleen. Images may show uptake by thyroid, kidney, and urinary bladder and minimal uptake in the lungs. Tumor uptake in soft tissues and in normal organs is seen as areas of increased intensity.
Results Indicating Altered Biodistribution
- On the first imaging timepoint: If the blood pool is not visualized or if there is diffuse, intense tracer uptake in the liver and/or spleen or uptake suggestive of urinary obstruction the biodistribution is altered. Diffuse lung uptake greater than that of blood pool on the first day represents altered biodistribution.
- On the second and third imaging timepoints: Uptake suggestive of urinary obstruction and diffuse lung uptake greater than that of the blood pool represent altered biodistribution.
- Total body residence times of less than 50 hours and more than 150 hours.
CALCULATION OF IODINE-131 ACTIVITY FOR THE THERAPEUTIC DOSE
There are two options for calculation of the Iodine-131 activity for the therapeutic dose. The derived values and calculation of the therapeutic dose may be determined manually [see “Workbook for Dosimetry Methodology and Administration Set-Up”] or calculated automatically using the GlaxoSmithKline proprietary software program [BEXXAR Patient Management Templates]. The following describes in greater detail the stepwise method for manual determination of the Iodine-131 activity for the therapeutic dose.
Residence Time (hr)
For each timepoint, calculate the background corrected total body count at each timepoint (defined as the geometric mean). The following equation is used:
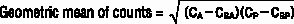
In this equation, CA = the anterior counts, CBA = the anterior background counts, CP = the posterior counts, and CBP = the posterior background counts.
Once the geometric mean of the counts has been calculated for each of the 3 timepoints, the % injected activity remaining for each timepoint is calculated by dividing the geometric mean of the counts from that timepoint by the geometric mean of the counts from Day 0 and multiplying by 100.
The residence time (h) is then determined by plotting the time from the start of
infusion and the % injected activity values for the 3 imaging timepoints on
Graph 1 (see Worksheet “Determination of Residence Time” in the “Workbook for Dosimetry Methodology and Administration Set-Up” supplied with Dosimetric Dose Packaging). A best-fit line is then drawn from 100% (the pre-plotted Day 0 value) through the other 2 plotted points (if the line does not intersect the two points, one point must lie above the best-fit line and one point must lie below the best-fit line). The residence time (h) is read from the x-axis of the graph at the point where the fitted line intersects with the horizontal 37% injected activity line.
Activity Hours (mCi hr)
In order to determine the activity hours (mCi hr), look up the patient’s maximum effective mass derived from the patient’s sex and height (see Worksheet “Determination of Maximum Effective Mass” in the “Workbook for Dosimetry Methodology and Administration Set-Up” supplied with Dosimetric Dose Packaging). If the patient’s actual weight is less than the maximum effective mass, the actual weight should be used in the activity hours table (see Worksheet “Determination of Activity Hours” in the “Workbook for Dosimetry Methodology and Administration Set-Up” supplied with Dosimetric Dose Packaging). If the patient’s actual weight is greater than the maximum effective mass, the mass from the worksheet for “Determination of Maximum Effective Mass” should be used.
Calculation of Iodine-131 Activity for the Therapeutic Dose
The following equation is used to calculate the activity of Iodine-131 required for delivery of the desired total body dose of radiation.
HOW SUPPLIED
TOSITUMOMAB DOSIMETRIC PACKAGING
The components of the dosimetric step will be shipped ONLY to individuals who are participating in the certification program or have been certified in the preparation and administration of the BEXXAR therapeutic regimen. The components are shipped from separate sites; when ordering, ensure that the components are scheduled to arrive on the same day. The components of the Tositumomab Dosimetric Step include:
- Tositumomab: Two single-use 225 mg vials (16.1 mL) and one single-use
35 mg vial (2.5 mL) of Tositumomab at a protein concentration of 14
mg/mL supplied by McKesson BioServices.
NDC 0007-3260-31
- Iodine I 131 Tositumomab: A single-use vial of Iodine I 131
Tositumomab within a lead pot, supplied by MDS Nordion. Each single-use
vial contains not less than 20 mL of Iodine I 131 Tositumomab at nominal
protein and activity concentrations of 0.1 mg/mL and 0.61 mCi/mL
(at calibration), respectively. (Refer to the product specification
sheet for the lot-specific protein concentration, activity concentration,
total activity and expiration date.)
NDC 0007-3261-01
TOSITUMOMAB THERAPEUTIC PACKAGING
The components of the therapeutic step will be shipped ONLY to individuals who are participating in the certification program or have been certified in the preparation and administration of the BEXXAR therapeutic regimen for an individual patient who has completed the Dosimetric Step. The components of the therapeutic step are shipped from separate sites; when ordering, ensure that the components are scheduled to arrive on the same day. The components of the Tositumomab Therapeutic Step include:
- Tositumomab: Two single-use 225 mg vials (16.1 mL) and one single-use
35 mg vial (2.5 mL) of Tositumomab at a protein concentration of 14
mg/mL supplied by McKesson BioServices.
NDC 0007-3260-36
- Iodine I 131 Tositumomab: One or two single-use vials of Iodine
I 131 Tositumomab within a lead pot(s), supplied by MDS Nordion. Each
single-use vial contains not less than 20 mL of Iodine I 131 Tositumomab
at nominal protein and activity concentrations of 1.1 mg/mL and 5.6 mCi/mL
(at calibration), respectively. Refer to the product specification
sheet for the lot-specific protein concentration, activity concentration,
total activity and expiration date.
NDC 0007-3262-01
STABILITY AND STORAGE
TOSITUMOMAB
Vials of Tositumomab (35 mg and 225 mg) should be stored refrigerated at 2oC-8oC (36oF-46oF) prior to dilution. Do not use beyond expiration date. Protect from strong light. DO NOT SHAKE. Do not freeze. Discard any unused portions left in the vial.
Solutions of diluted Tositumomab are stable for up to 24 hours when stored refrigerated at 2°C-8°C (36°F-46°F) and for up to 8 hours at room temperature. However, it is recommended that the diluted solution be stored refrigerated at 2°C-8°C (36°F-46°F) prior to administration because it does not contain preservatives. Any unused portion must be discarded. Do not freeze solutions of diluted Tositumomab.
IODINE I 131 TOSITUMOMAB
Store frozen in the original lead pots. The lead pot containing the product must be stored in a freezer at a temperature of -20oC or below until it is removed for thawing prior to administration to the patient. Do not use beyond the expiration date on the label of the lead pot.
Thawed dosimetric and therapeutic doses of Iodine I 131 Tositumomab are stable for up to 8 hours at 2°C-8°C (36°F-46°F) or at room temperature. Solutions of Iodine I 131 Tositumomab diluted for infusion contain no preservatives and should be stored refrigerated at 2°C-8°C (36°F-46°F) prior to administration (do not freeze). Any unused portion must be discarded according to federal and state laws.
REFERENCES
- Weber DA, Eckman KF, Dillman LT, Ryman JC. In: MIRD: radionuclide data and decay schemes. New York: Society of Nuclear Medicine Inc. 1989:229.
- Tedder T, Boyd A, Freedman A, Nadler L, Schlossman S. The B cell surface molecule is functionally linked with B cell activation and differentiation. J Immunol 1985;135(2):973-979.
- Anderson, KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood 1984;63(6):1424-1433.
- Press OW, Howell-Clark J, Anderson S, Bernstein I. Retention of B-cell-specific monoclonal antibodies by human lymphoma cells. Blood 1994;83:1390−7.
- Cardarelli PM, Quinn M, Buckman D, Fang Y, Colcher D, King DJ, Bebbington C, et al. Binding to CD20 by anti-B1 antibody or F(ab')(2) is sufficient for induction of apoptosis in B-cell lines.Cancer Immunol Immunother 2002 Mar;51(1):15-24.
- Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol 1980;125:1678−85.
U.S. Lic. 1727
GlaxoSmithKline
Research Triangle Park, NC 27709
BEXXAR is a registered trademark of GlaxoSmithKline.
©2005, GlaxoSmithKline. All rights reserved.
October 2005RL-2245
| BEXXAR (tositumomab) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| BEXXAR (tositumomab I-131) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| BEXXAR (tositumomab) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| BEXXAR (tositumomab I-131) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Revised: 01/2008GlaxoSmithKline