INCIVEK
-
telaprevir tablet, film coated
Vertex Pharmaceuticals Incorporated
----------
|
|||||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Chronic Hepatitis C
INCIVEK™ (telaprevir), in combination with peginterferon alfa and ribavirin, is indicated for the treatment of genotype 1 chronic hepatitis C in adult patients with compensated liver disease, including cirrhosis, who are treatment-naïve or who have previously been treated with interferon-based treatment, including prior null responders, partial responders, and relapsers [see Clinical Studies (14.2 and 14.3), including definitions of these terms].
The following points should be considered when initiating treatment with INCIVEK:
- INCIVEK must not be administered as monotherapy and must only be prescribed with both peginterferon alfa and ribavirin [see Warnings and Precautions (5.7)].
- A high proportion of previous null responders (particularly those with cirrhosis) did not achieve a Sustained Virologic Response (SVR) and had telaprevir resistance-associated substitutions emerge on treatment with INCIVEK combination treatment [see Clinical Pharmacology (12.4) and Clinical Studies (14.3)].
- INCIVEK efficacy has not been established for patients who have previously failed therapy with a treatment regimen that includes INCIVEK or other HCV NS3/4A protease inhibitors [see Clinical Pharmacology (12.4)].
2 DOSAGE AND ADMINISTRATION
2.1 INCIVEK/Peginterferon Alfa/Ribavirin Combination Treatment
The recommended dose of INCIVEK tablets is 750 mg (two 375-mg tablets) taken orally 3 times a day (7-9 hours apart) with food (not low fat) [see Clinical Pharmacology (12.3) and Patient Counseling Information (17.4)].
For specific dosage instructions for peginterferon alfa and ribavirin, refer to their respective prescribing information.
Duration of Treatment
The recommended duration of treatment with INCIVEK is 12 weeks in combination with peginterferon alfa and ribavirin. HCV-RNA levels should be monitored at weeks 4 and 12 to determine combination treatment duration and assess for treatment futility (Tables 1 and 2).
| Treatment-Naïve and Prior Relapse Patients | |||
|---|---|---|---|
| HCV RNA* | Triple Therapy INCIVEK, peginterferon alfa and ribavirin | Dual Therapy peginterferon alfa and ribavirin | Total Treatment Duration |
|
|||
| Undetectable (Target Not Detected) at Weeks 4 and 12 | First 12 weeks | Additional 12 weeks | 24 weeks |
| Detectable (1000 IU/mL or less) at Weeks 4 and/or 12 | First 12 weeks | Additional 36 weeks | 48 weeks |
| Prior Partial and Null Responder Patients | |||
| Triple Therapy
INCIVEK, peginterferon alfa and ribavirin | Dual Therapy
peginterferon alfa and ribavirin | Total Treatment Duration | |
| All Patients | First 12 weeks | Additional 36 weeks | 48 weeks |
For the purpose of assessing response-guided therapy eligibility at weeks 4 and 12 (see Table 1), an "undetectable" HCV-RNA (Target Not Detected) result is required; a confirmed "detectable but below limit of quantification" HCV-RNA result should not be considered equivalent to an "undetectable" HCV-RNA (Target Not Detected) result [see Laboratory Tests (5.6)].
Treatment-naïve patients with cirrhosis who have undetectable HCV RNA (Target Not Detected) at weeks 4 and 12 of INCIVEK combination treatment may benefit from an additional 36 weeks of peginterferon alfa and ribavirin (48 weeks total) [see Clinical Studies (14.2)].
2.2 Dose Reduction
To prevent treatment failure, the dose of INCIVEK must not be reduced or interrupted. Refer to the respective prescribing information for dose modification of peginterferon alfa and ribavirin [see Warnings and Precautions (5.7)].
2.3 Discontinuation of Dosing
Patients with inadequate viral response are unlikely to achieve SVR, and may develop treatment-emergent resistance substitutions [see Microbiology (12.4)]. Discontinuation of therapy is recommended in all patients with (1) HCV-RNA levels of greater than 1000 IU/mL at Treatment Week 4 or 12; or (2) confirmed detectable HCV-RNA levels at Treatment Week 24 (see Table 2).
| HCV RNA | Action |
|---|---|
| Week 4 or Week 12: Greater than 1000 IU/mL | Discontinue INCIVEK and peginterferon alfa and ribavirin (INCIVEK treatment complete at 12 weeks) |
| Week 24: Detectable | Discontinue peginterferon alfa and ribavirin |
If peginterferon alfa or ribavirin is discontinued for any reason, INCIVEK must also be discontinued.
3 DOSAGE FORMS AND STRENGTHS
Each tablet contains 375 mg of telaprevir. Tablets are available as purple, film-coated, capsule-shaped tablets debossed with the characters "V 375" on one side.
4 CONTRAINDICATIONS
Contraindications to peginterferon alfa and ribavirin also apply to INCIVEK combination treatment.
INCIVEK combination treatment is contraindicated in:
- women who are or may become pregnant. Ribavirin may cause fetal harm when administered to a pregnant woman. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug treatment, the patient should be apprised of the potential hazard to a fetus [see Warnings and Precautions (5.1), Use in Specific Populations (8.1), and Patient Counseling Information (17.1)].
- men whose female partners are pregnant.
INCIVEK is contraindicated when combined with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). INCIVEK is contraindicated when combined with drugs that strongly induce CYP3A and thus may lead to lower exposure and loss of efficacy of INCIVEK. Contraindicated drugs are listed below in Table 3 [also see Drug Interactions (7), Table 5 and Clinical Pharmacology (12.3), Tables 6 and 7].
| Drug Class | Drugs within Class that are Contraindicated with INCIVEK | Clinical Comments |
|---|---|---|
|
||
| Alpha 1-adrenoreceptor antagonist | Alfuzosin | Potential for hypotension or cardiac arrhythmia |
| Antimycobacterials | Rifampin | Rifampin significantly reduces telaprevir plasma concentrations. |
| Ergot derivatives | Dihydroergotamine, ergonovine, ergotamine, methylergonovine | Potential for acute ergot toxicity characterized by peripheral vasospasm or ischemia |
| GI motility agent | Cisapride | Potential for cardiac arrhythmias |
| Herbal products | St. John's wort (Hypericum perforatum) | Plasma concentrations of telaprevir can be reduced by concomitant use of the herbal preparation St. John's wort. |
| HMG CoA reductase inhibitors | Lovastatin, simvastatin | Potential for myopathy including rhabdomyolysis |
| Neuroleptic | Pimozide | Potential for serious and/or life-threatening adverse reactions such as cardiac arrhythmias |
| PDE5 inhibitor | Sildenafil (Revatio®) or tadalafil (Adcirca®) [for treatment of pulmonary arterial hypertension]* | Potential for PDE5 inhibitor-associated adverse events, including visual abnormalities, hypotension, prolonged erection, and syncope |
| Sedatives/hypnotics | Orally administered midazolam†, triazolam | Prolonged or increased sedation or respiratory depression |
5 WARNINGS AND PRECAUTIONS
5.1 Pregnancy: Use with Ribavirin and Peginterferon Alfa
Ribavirin may cause birth defects and/or death of the exposed fetus. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients. Ribavirin therapy should not be started unless a report of a negative pregnancy test has been obtained immediately prior to initiation of therapy.
Because INCIVEK must be used in combination with peginterferon alfa and ribavirin, the contraindications and warnings applicable to those drugs are applicable to combination therapy. Female patients of childbearing potential and their male partners as well as male patients and their female partners must use 2 effective contraceptive methods during treatment and for 6 months after all treatment has ended. Female patients should have monthly pregnancy tests during treatment and during the 6-month period after stopping treatment. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients as significant teratogenic and/or embryocidal effects have been demonstrated in all animal species exposed to ribavirin [see Contraindications (4), Use in Specific Populations (8.1), and Patient Counseling Information (17.1)]. Refer also to the prescribing information for ribavirin.
Female Patients
Hormonal contraceptives may be continued but may not be reliable during INCIVEK dosing and for up to two weeks following cessation of INCIVEK [see Drug Interactions (7)]. During this time, female patients of childbearing potential should use two effective non-hormonal methods of contraception. Examples may include barrier methods or intrauterine devices (IUDs) [see also Use in Specific Populations: Pregnancy (8.1) and Patient Counseling Information (17.1)]. Two weeks after completion of INCIVEK treatment, hormonal contraceptives are again appropriate as one of the two required effective methods of birth control; however, specific prescribing information recommendations should be followed for the contraceptives.
5.2 Serious Skin Reactions
Serious skin reactions, including Drug Rash with Eosinophilia and Systemic Symptoms (DRESS) and Stevens-Johnson Syndrome (SJS) were reported in less than 1% of subjects who received INCIVEK combination treatment compared to none who received peginterferon alfa and ribavirin alone. These serious skin reactions required hospitalization, and all patients recovered. The presenting signs of DRESS may include rash, fever, facial edema, and evidence of internal organ involvement (e.g., hepatitis, nephritis). Eosinophilia may or may not be present. The presenting signs of SJS may include fever, target lesions, and mucosal erosions or ulcerations (e.g., conjunctivae, lips).
If a serious skin reaction occurs, all components of INCIVEK combination treatment must be discontinued immediately and the patient should be promptly referred for urgent medical care.
5.3 Rash
Rash developed in 56% of subjects who received INCIVEK combination treatment [see Adverse Reactions (6.1)]. Severe rash (e.g., a generalized rash or rash with vesicles or bullae or ulcerations other than SJS) was reported in 4% of subjects who received INCIVEK combination treatment compared to less than 1% who received peginterferon alfa and ribavirin alone. The severe rash may have a prominent eczematous component.
Patients with mild to moderate rashes should be followed for progression of rash or development of systemic symptoms. If rash progresses and becomes severe or if systemic symptoms develop, INCIVEK should be discontinued. Peginterferon alfa and ribavirin may be continued. If improvement is not observed within 7 days of INCIVEK discontinuation, sequential or simultaneous interruption or discontinuation of ribavirin and/or peginterferon alfa should be considered. If medically indicated, earlier interruption or discontinuation of ribavirin and peginterferon alfa should be considered. Patients should be monitored until the rash has resolved. INCIVEK must not be reduced or restarted if discontinued due to rash. Treatment of rash with oral antihistamines and/or topical corticosteroids may provide symptomatic relief but effectiveness of these measures has not been established. Treatment of rash with systemic corticosteroids is not recommended [see Drug Interactions (7)].
5.4 Anemia
Anemia has been reported with peginterferon alfa and ribavirin therapy. The addition of INCIVEK to peginterferon alfa and ribavirin is associated with an additional decrease in hemoglobin concentrations. Hemoglobin values less than or equal to 10 g per dL were observed in 36% of subjects who received INCIVEK combination treatment compared to 17% of subjects who received peginterferon alfa and ribavirin. Hemoglobin values less than 8.5 g per dL were observed in 14% of subjects who received INCIVEK combination treatment compared to 5% of subjects receiving peginterferon alfa and ribavirin.
In subjects receiving INCIVEK combination treatment, 4% discontinued INCIVEK, 1% discontinued INCIVEK combination treatment, and 32% underwent a ribavirin dose modification (reduction, interruption or discontinuation) due to anemia. In subjects treated with peginterferon alfa and ribavirin alone, there were two discontinuations and 12% underwent ribavirin dose modification due to anemia.
Hemoglobin should be monitored prior to and at least at weeks 2, 4, 8 and 12 during INCIVEK combination treatment and as clinically appropriate. For the management of anemia, ribavirin dose reductions should be used (refer to the prescribing information for ribavirin for its dose reduction guidelines). If ribavirin dose reductions are inadequate, discontinuation of INCIVEK should be considered. If ribavirin is permanently discontinued for the management of anemia, INCIVEK must also be permanently discontinued. Ribavirin may be restarted per the dosing modification guidelines for ribavirin. The dose of INCIVEK must not be reduced and INCIVEK must not be restarted if discontinued.
5.5 Drug Interactions
See Table 3 for a listing of drugs that are contraindicated for use with INCIVEK due to potentially life-threatening adverse events or potential loss of therapeutic effect to INCIVEK [see Contraindications (4)]. Refer to Table 5 for established and other potentially significant drug-drug interactions [see Drug Interactions (7)].
5.6 Laboratory Tests
HCV-RNA levels should be monitored at weeks 4 and 12 and as clinically indicated. Use of a sensitive real-time RT-PCR assay for monitoring HCV-RNA levels during treatment is recommended. The assay should have a lower limit of HCV-RNA quantification equal to or less than 25 IU per mL and a limit of HCV-RNA detection of approximately 10-15 IU per mL. For the purpose of assessing response-guided therapy eligibility, an "undetectable" HCV-RNA (Target Not Detected) result is required; a confirmed "detectable but below limit of quantification" HCV-RNA result should not be considered equivalent to an "undetectable" HCV-RNA result (reported as "Target Not Detected" or "HCV RNA Not Detected").
Hematology evaluations (including white cell differential count) are recommended prior to and at weeks 2, 4, 8 and 12 and as clinically appropriate.
Chemistry evaluations (electrolytes, serum creatinine, uric acid, hepatic enzymes, bilirubin, and TSH) are recommended as frequently as the hematology evaluations or as clinically indicated [see Adverse Reactions (6)].
Refer to the prescribing information for peginterferon alfa and ribavirin, including pregnancy testing requirements.
5.7 General
INCIVEK must not be administered as monotherapy and must only be prescribed with both peginterferon alfa and ribavirin. Therefore, the prescribing information for peginterferon alfa and ribavirin must be consulted before starting treatment with INCIVEK.
There are no clinical data on re-treating patients who have failed an HCV NS3/4A protease inhibitor-based treatment, nor are there data on repeated courses of INCIVEK [see Microbiology (12.4)].
5.8 Hepatic Impairment
INCIVEK is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh B or C, score greater than or equal to 7) or patients with decompensated liver disease. Refer to prescribing information for peginterferon alfa and ribavirin which must be co-administered with INCIVEK [see Use in Specific Populations: Hepatic Impairment (8.6)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Pregnancy: Use with Ribavirin and Peginterferon alfa [see Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.1), and Patient Counseling Information (17.1)]
- Serious Skin Reactions/Rash [see Warnings and Precautions (5.2 and 5.3)]
- Anemia [see Warnings and Precautions (5.4)]
INCIVEK must be administered with peginterferon alfa and ribavirin. Refer to their respective prescribing information for their associated adverse reactions.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety assessment is based on data from pooled adequate and well-controlled clinical trials including 1797 subjects who received INCIVEK combination treatment and 493 who received peginterferon alfa and ribavirin.
Serious adverse drug reactions occurred in 3% of subjects who received INCIVEK combination treatment compared to none of the subjects treated with peginterferon alfa and ribavirin. The most frequent serious adverse events in subjects treated with INCIVEK combination treatment were skin disorders (rash and/or pruritus) and anemia [see Warnings and Precautions (5.2, 5.3, and 5.4)]. Fourteen percent of subjects discontinued INCIVEK due to adverse drug reactions. Rash, anemia, fatigue, pruritus, nausea, and vomiting were the most frequent adverse drug reactions leading to discontinuation of INCIVEK.
INCIVEK was administered in combination with peginterferon alfa and ribavirin. The following table lists adverse drug reactions that occurred in INCIVEK-treated subjects with an incidence at least 5% greater than in subjects receiving peginterferon alfa and ribavirin alone (Table 4).
| INCIVEK, peginterferon alfa, and ribavirin Combination Treatment N=1797 | Peginterferon alfa and ribavirin N=493 |
|
|---|---|---|
|
||
| Rash* | 56% | 34% |
| Fatigue | 56% | 50% |
| Pruritus | 47% | 28% |
| Nausea | 39% | 28% |
| Anemia* | 36% | 17% |
| Diarrhea | 26% | 17% |
| Vomiting | 13% | 8% |
| Hemorrhoids | 12% | 3% |
| Anorectal discomfort | 11% | 3% |
| Dysgeusia | 10% | 3% |
| Anal pruritus | 6% | 1% |
Description of Selected Adverse Drug Reactions
Rash
In controlled clinical trials, rash events (all grades) were reported in 56% of subjects who received INCIVEK combination treatment and in 34% of subjects who received peginterferon alfa and ribavirin. Rash most frequently began during the first 4 weeks, but could occur at any time during INCIVEK combination treatment. Improvement of rash occurs after INCIVEK dosing completion or discontinuation; however, rashes may take weeks for complete resolution.
Rash events led to discontinuation of INCIVEK alone in 6% of subjects and discontinuation of INCIVEK combination treatment in 1% of subjects.
For serious skin reactions and severe rash, see Warnings and Precautions (5.2 and 5.3).
Anemia
In controlled clinical trials, the overall incidence and severity of anemia increased with INCIVEK combination treatment compared to peginterferon alfa and ribavirin alone. The incidence of anemia adverse events was 36% with INCIVEK combination treatment compared to 17% with peginterferon alfa and ribavirin alone. A decrease in hemoglobin levels occurred during the first 4 weeks of treatment, with lowest values reached at the end of INCIVEK dosing. Hemoglobin values gradually returned to levels observed with peginterferon alfa and ribavirin after INCIVEK dosing was completed [see Warnings and Precautions (5.4)].
Anorectal Signs and Symptoms
In the controlled clinical trials, 29% of subjects treated with INCIVEK combination treatment experienced anorectal adverse events, compared to 7% of those treated with peginterferon alfa and ribavirin alone. The majority of these events (e.g., hemorrhoids, anorectal discomfort, anal pruritus, and rectal burning) were mild to moderate in severity; less than 1% led to treatment discontinuation and all resolved during or after completion of INCIVEK dosing.
Laboratory abnormalities
White Blood Cells: Treatment with peginterferon alfa is associated with decreases in mean values for total white blood cell, absolute neutrophil, and absolute lymphocyte count. More INCIVEK-treated subjects had decreases in lymphocyte counts to 499/mm3 or less (15% compared to 5%). Decreases in total white cell counts to 1,499/mm3 or less were comparable (8% compared to 5%). The incidence of decreases in absolute neutrophil counts to 749/mm3 or less was 15% in subjects treated with peginterferon alfa and ribavirin alone compared to 12% among those treated with INCIVEK combination treatment.
Platelets: Treatment with peginterferon alfa is associated with decreases in mean platelet counts. More patients treated with INCIVEK combination treatment had decreases in mean platelet values of all grades: 47% compared to 36% treated with peginterferon alfa and ribavirin alone. Three percent of INCIVEK combination treatment subjects had decreases to 49,999/mm3 or less compared to 1% of those treated with peginterferon alfa and ribavirin-treated alone.
Bilirubin: Forty one percent of INCIVEK-treated subjects compared to 28% of peginterferon alfa and ribavirin-treated subjects had all grade elevations in bilirubin levels; 4% and 2% of subjects, respectively, had greater than or equal to 2.6 x ULN elevations. Bilirubin levels increased most steeply during the first 1 to 2 weeks of INCIVEK dosing, stabilized and between Weeks 12 and 16 were at baseline levels.
Uric Acid: During the INCIVEK combination treatment period, 73% of subjects had elevated uric acid levels compared to 29% for those treated with peginterferon alfa and ribavirin alone. Shifts to greater than or equal to 12.1 mg per dL from baseline in uric acid levels were also more frequent among subjects treated with INCIVEK (7%) compared to peginterferon alfa and ribavirin (1%). Less than 1% of subjects had clinical events of gout/gouty arthritis; none were serious and none resulted in treatment discontinuation.
7 DRUG INTERACTIONS
7.1 Potential for INCIVEK to Affect Other Drugs
INCIVEK is an inhibitor of CYP3A. Co-administration of INCIVEK with drugs that are primarily metabolized by CYP3A may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and adverse reactions (see Table 5). INCIVEK is also an inhibitor of P-gp. Co-administration of INCIVEK with drugs that are substrates for P-gp transport may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and adverse reactions (see Table 5). If dose adjustments of concomitant medications are made during INCIVEK treatment, they should be re-adjusted after administration of INCIVEK is completed.
7.2 Potential for Other Drugs to Affect INCIVEK
INCIVEK is a substrate of CYP3A and P-gp; therefore, drugs that induce CYP3A and/or P-gp may decrease INCIVEK plasma concentrations and reduce the therapeutic effect of INCIVEK. Co-administration of INCIVEK with drugs that inhibit CYP3A and/or P-gp may increase INCIVEK plasma concentrations.
7.3 Established and Other Potentially Significant Drug Interactions
Table 5 provides effect of concentration of INCIVEK or concomitant drug with INCIVEK. These recommendations are based on either drug interaction trials (indicated with *) or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of efficacy.
| Concomitant Drug Class: Drug Name | Effect on concentration of INCIVEK or Concomitant Drug | Clinical Comment |
|---|---|---|
| The direction of the arrow (↑ = increase, ↓ = decrease, ↔ = no change) indicates the direction of the change in PK. | ||
|
||
| ANTIARRHYTHMICS | ||
| lidocaine (systemic), amiodarone, bepridil, flecainide, propafenone, quinidine | ↑ antiarrhythmics | Co-administration with telaprevir has the potential to produce serious and/or life-threatening adverse events and has not been studied. Caution is warranted and clinical monitoring is recommended when co-administered with telaprevir. |
| digoxin* | ↑ digoxin | Concentrations of digoxin were increased when co-administered with telaprevir. The lowest dose of digoxin should be initially prescribed. The serum digoxin concentrations should be monitored and used for titration of digoxin dose to obtain the desired clinical effect. |
| ANTIBACTERIALS | ||
| clarithromycin erythromycin telithromycin | ↑ telaprevir ↑ antibacterials | Concentrations of both telaprevir and the antibacterial may be increased during co-administration. Caution is warranted and clinical monitoring is recommended when co-administered with telaprevir. QT interval prolongation and Torsade de Pointes have been reported with clarithromycin and erythromycin. QT interval prolongation has been reported with telithromycin. |
| ANTICOAGULANT | ||
| warfarin | ↑ or ↓ warfarin | Concentrations of warfarin may be altered when co-administered with telaprevir. The international normalized ratio (INR) should be monitored when warfarin is co-administered with telaprevir. |
| ANTICONVULSANTS | ||
| carbamazepine phenobarbital phenytoin | ↓ telaprevir ↑ carbamazepine ↑ or ↓ phenytoin ↑ or ↓ phenobarbital | Concentrations of the anticonvulsant may be altered and concentrations of telaprevir may be decreased. Caution should be used when prescribing carbamazepine, phenobarbital, and phenytoin. Telaprevir may be less effective in patients taking these agents concomitantly. Clinical or laboratory monitoring of carbamazepine, phenobarbital, and phenytoin concentrations and dose titration are recommended to achieve the desired clinical response. |
| ANTIDEPRESSANTS | ||
| escitalopram* | ↔ telaprevir ↓ escitalopram | Concentrations of escitalopram were decreased when co-administered with telaprevir. Selective serotonin reuptake inhibitors such as escitalopram have a wide therapeutic index, but doses may need to be adjusted when combined with telaprevir. |
| trazodone | ↑ trazodone | Concomitant use of trazodone and telaprevir may increase plasma concentrations of trazodone which may lead to adverse events such as nausea, dizziness, hypotension and syncope. If trazodone is used with telaprevir, the combination should be used with caution and a lower dose of trazodone should be considered. |
| ANTIFUNGALS | ||
| ketoconazole*
itraconazole posaconazole voriconazole | ↑ ketoconazole ↑ telaprevir ↑ itraconazole ↑ posaconazole ↑ or ↓ voriconazole | Ketoconazole increases the plasma concentrations of telaprevir. Concomitant systemic use of itraconazole or posaconazole with telaprevir may increase plasma concentration of telaprevir. Plasma concentrations of itraconazole, ketoconazole, or posaconazole may be increased in the presence of telaprevir. When co-administration is required, high doses of itraconazole or ketoconazole (greater than 200 mg/day) are not recommended. Caution is warranted and clinical monitoring is recommended for itraconazole, posaconazole and voriconazole. QT interval prolongation and Torsade de Pointes have been reported with voriconazole and posaconazole. QT interval prolongation has been reported with ketoconazole. Due to multiple enzymes involved with voriconazole metabolism, it is difficult to predict the interaction with telaprevir. Voriconazole should not be administered to patients receiving telaprevir unless an assessment of the benefit/risk ratio justifies its use. |
| ANTI GOUT | ||
| colchicine | ↑ colchicine | Patients with renal or hepatic impairment should not be given colchicine with telaprevir, due to the risk of colchicine toxicity. A reduction in colchicine dosage or an interruption of colchicine treatment is recommended in patients with normal renal or hepatic function. Treatment of gout flares: co-administration of colchicine in patients on telaprevir: 0.6 mg (1 tablet) for 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Not to be repeated before 3 days. If used for prophylaxis of gout flares: co-administration of colchicine in patients on telaprevir: If the original regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day. If the original regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day. Treatment of familial Mediterranean fever (FMF): co-administration of colchicine in patients on telaprevir: Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day). |
| ANTIMYCOBACTERIAL | ||
| rifabutin | ↓ telaprevir ↑ rifabutin | Concentrations of telaprevir may be decreased, while rifabutin concentrations may be increased during co-administration. Telaprevir may be less effective due to decreased concentrations. The concomitant use of rifabutin and telaprevir is not recommended. |
| BENZODIAZEPINES | ||
| alprazolam*
| ↑ alprazolam | Concomitant use of alprazolam and telaprevir increases exposure to alprazolam. Clinical monitoring is warranted. |
| parenterally administered midazolam* | ↑ midazolam | Concomitant use of parenterally administered midazolam with telaprevir increased exposure to midazolam. Co-administration should be done in a setting which ensures clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dose reduction for midazolam should be considered, especially if more than a single dose of midazolam is administered. Co-administration of oral midazolam with telaprevir is contraindicated. |
| zolpidem (non-benzodiazepine sedative)* | ↓ zolpidem | Exposure to zolpidem was decreased when co-administered with telaprevir. Clinical monitoring and dose titration of zolpidem is recommended to achieve the desired clinical response. |
| CALCIUM CHANNEL BLOCKERS | ||
| amlodipine* | ↑amlodipine | Exposure to amlodipine was increased when co-administered with telaprevir. Caution should be used and dose reduction for amlodipine should be considered. Clinical monitoring is recommended. |
| diltiazem felodipine nicardipine nifedipine nisoldipine verapamil | ↑calcium channel blockers | Concentrations of other calcium channel blockers may be increased when telaprevir is co-administered. Caution is warranted and clinical monitoring of patients is recommended. |
| CORTICOSTEROIDS | ||
| Systemic
prednisone methylprednisolone | ↑ prednisone ↑ methylprednisolone | Systemic corticosteroids such as prednisone and methylprednisolone are CYP3A substrates. Since telaprevir is a potent CYP3A inhibitor, plasma concentrations of these corticosteroids can be increased significantly. Co-administration of systemic corticosteroids and telaprevir is not recommended [see Warnings and Precautions (5.3)]. |
| Systemic
dexamethasone | ↓ telaprevir | Systemic dexamethasone induces CYP3A and can thereby decrease telaprevir plasma concentrations. This may result in loss of therapeutic effect of telaprevir. Therefore this combination should be used with caution or alternatives should be considered. |
| Inhaled/Nasal
fluticasone budesonide | ↑ fluticasone ↑ budesonide | Concomitant use of inhaled fluticasone or budesonide and telaprevir may increase plasma concentrations of fluticasone or budesonide resulting in significantly reduced serum cortisol concentrations. Co-administration of fluticasone or budesonide and telaprevir is not recommended unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effects. |
| ENDOTHELIN RECEPTOR ANTAGONIST | ||
| bosentan | ↑ bosentan | Concentrations of bosentan may be increased when co-administered with telaprevir. Caution is warranted and clinical monitoring is recommended. |
| HIV-ANTIVIRAL AGENTS: HIV-PROTEASE INHIBITORS (PIs)
|
||
| atazanavir/ritonavir* | ↓ telaprevir ↑ atazanavir | Concomitant administration of telaprevir and atazanavir/ritonavir resulted in reduced steady-state telaprevir exposure, while steady-state atazanavir exposure was increased. |
| darunavir/ritonavir* | ↓ telaprevir ↓ darunavir | Concomitant administration of telaprevir and darunavir/ritonavir resulted in reduced steady-state exposures to telaprevir and darunavir. It is not recommended to co-administer darunavir/ritonavir and telaprevir. |
| fosamprenavir/ritonavir* | ↓ telaprevir ↓ fosamprenavir | Concomitant administration of telaprevir and fosamprenavir/ritonavir resulted in reduced steady-state exposures to telaprevir and amprenavir. It is not recommended to co-administer fosamprenavir/ritonavir and telaprevir. |
| lopinavir/ritonavir* | ↓ telaprevir ↔ lopinavir | Concomitant administration of telaprevir and lopinavir/ritonavir resulted in reduced steady-state telaprevir exposure, while the steady-state exposure to lopinavir was not affected. It is not recommended to co-administer lopinavir/ritonavir and telaprevir. |
| HIV-ANTIVIRAL AGENTS: REVERSE TRANSCRIPTASE INHIBITORS | ||
| efavirenz* | ↓ telaprevir ↓ efavirenz | Concomitant administration of telaprevir and efavirenz resulted in reduced steady-state exposures to telaprevir and efavirenz. |
| tenofovir disoproxil fumarate* | ↔ telaprevir ↑ tenofovir | Concomitant administration of telaprevir and tenofovir disoproxil fumarate resulted in increased tenofovir exposure. Increased clinical and laboratory monitoring are warranted. Tenofovir disoproxil fumarate should be discontinued in patients who develop tenofovir-associated toxicities. |
| HMG-CoA REDUCTASE INHIBITORS | ||
| atorvastatin | ↑ atorvastatin | Plasma concentrations of atorvastatin are markedly increased when co-administered with telaprevir. Avoid concomitant administration of telaprevir and atorvastatin. |
| HORMONAL CONTRACEPTIVES/ESTROGEN | ||
| ethinyl estradiol*
norethindrone | ↓ ethinyl estradiol ↔ norethindrone | Exposure to ethinyl estradiol was decreased when co-administered with telaprevir. Two effective non-hormonal methods of contraception should be used during treatment with telaprevir. Patients using estrogens as hormone replacement therapy should be clinically monitored for signs of estrogen deficiency. Refer also to Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.1), and Patient Counseling Information (17.1). |
| IMMUNOSUPPRESSANTS | ||
| cyclosporine*
sirolimus tacrolimus* | ↑ cyclosporine ↑ sirolimus ↑ tacrolimus | Plasma concentrations of cyclosporine and tacrolimus are markedly increased when co-administered with telaprevir. Plasma concentration of sirolimus may be increased when co-administered with telaprevir, though this has not been studied. Significant dose reductions and prolongation of the dosing interval of the immunosuppressant to achieve the desired blood levels should be anticipated. Close monitoring of the immunosuppressant blood levels, and frequent assessments of renal function and immunosuppressant-related side effects are recommended when co-administered with telaprevir. Tacrolimus may prolong the QT interval. The use of telaprevir in organ transplant patients has not been studied. |
| INHALED BETA AGONIST | ||
| salmeterol | ↑ salmeterol | Concentrations of salmeterol may be increased when co-administered with telaprevir. Concurrent administration of salmeterol and telaprevir is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia. |
| NARCOTIC ANALGESIC | ||
| methadone* | ↓ R-methadone | Concentrations of methadone were reduced when co-administered with telaprevir. No adjustment of methadone dose is required when initiating co-administration of telaprevir. However, clinical monitoring is recommended as the dose of methadone during maintenance therapy may need to be adjusted in some patients. |
| PDE5 INHIBITORS | ||
| sildenafil tadalafil vardenafil | ↑ PDE5 inhibitors | Concentrations of PDE5 inhibitors may be increased when co-administered with telaprevir. For the treatment of erectile dysfunction, sildenafil at a single dose not exceeding 25 mg in 48 hours, vardenafil at a single dose not exceeding 2.5 mg dose in 72 hours, or tadalafil at a single dose not exceeding 10 mg dose in 72 hours can be used with increased monitoring for PDE5 inhibitor-associated adverse events. QT interval prolongation has been reported with vardenafil. Caution is warranted and clinical monitoring is recommended. Co-administration of sildenafil or tadalafil and telaprevir in the treatment of pulmonary arterial hypertension is contraindicated [see Contraindications (4)]. |
In addition to the drugs included in Table 5, the interaction between INCIVEK and the following drug was evaluated in clinical trials and no dose adjustment is needed for either drug [see Clinical Pharmacology (12.3)]: esomeprazole, raltegravir, or buprenorphine.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Because INCIVEK must be used in combination with ribavirin and peginterferon alfa, the contraindications and warnings applicable to those drugs are applicable to combination treatment. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients.
INCIVEK/Peginterferon Alfa/Ribavirin Combination Treatment
Pregnancy Category X: Animal studies have shown that ribavirin causes birth defects and/or fetal deaths while peginterferon alfa is abortifacient [see Contraindications (4) and Warnings and Precautions (5.1)]. See the prescribing information for ribavirin.
Significant teratogenic and/or embryocidal effects have been demonstrated in all animal species exposed to ribavirin; and therefore ribavirin is contraindicated in women who are pregnant and in the male partners of women who are pregnant [see Contraindications (4), Warnings and Precautions (5.1) and ribavirin prescribing information]. Interferons have abortifacient effects in animals and should be assumed to have abortifacient potential in humans (see peginterferon alfa prescribing information).
Extreme caution must be taken to avoid pregnancy in female patients and female partners of male patients while taking this combination. Women of childbearing potential and their male partners should not receive ribavirin unless they are using effective contraception (two reliable forms) during treatment with ribavirin and for 6 months after treatment. Systemic hormonal contraceptives may not be as effective in women while taking INCIVEK. Therefore, two alternative effective methods of contraception, including intrauterine devices and barrier methods, should be used in women during treatment with INCIVEK and concomitant ribavirin [see Warnings and Precautions (5.1)].
A Ribavirin Pregnancy Registry has been established to monitor maternal-fetal outcomes of pregnancies in female patients and female partners of male patients exposed to ribavirin during treatment and for 6 months following cessation of treatment. Health care providers and patients are encouraged to report such cases by calling 1-800-593-2214.
INCIVEK (telaprevir) Tablets
Pregnancy Category B: Telaprevir treatment alone in mice and rats did not result in harm to the fetus. The highest doses tested produced exposures equal to 1.84- and 0.60-fold the exposures in humans at the recommended clinical dose, respectively. Telaprevir treatment alone had effects on fertility parameters in rats. The no observed adverse effect level (NOAEL) for testicular toxicity was established at exposures 0.17-fold the human exposures at the recommended clinical dose. Potential effects on sperm (e.g., decreased % motile sperm and increased non-motile sperm count) were observed in a rat fertility study at exposures 0.30-fold the human exposures at the recommended clinical dose. Additional effects on fertility include minor increases in percent preimplantation loss, in percent of dams with nonviable embryos and percent of nonviable conceptuses per litter. These effects are likely associated with testicular toxicity in male but contributions of the female cannot be ruled out. There are, however, no adequate and well-controlled trials in pregnant women.
Significant teratogenic and/or embryocidal effects have been demonstrated in all animal species exposed to ribavirin. Extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients—both during treatment and for 6 months after the completion of all treatment. INCIVEK combination treatment should not be started unless a female patient has a negative pregnancy test immediately prior to initiation of treatment. Pregnancy testing should occur monthly during INCIVEK combination treatment and for 6 months after all treatment has ended [see Contraindications (4) and Patient Counseling Information (17.1)]. Pregnancy testing in non-pregnant female partners is recommended before INCIVEK combination therapy, every month during INCIVEK combination therapy, and for 6 months after ribavirin therapy has ended.
Hormonal contraceptives may be continued but may not be reliable during INCIVEK dosing and for up to two weeks following cessation of INCIVEK [see Drug Interactions (7)]. During this time, female patients of childbearing potential should use 2 effective non-hormonal methods of contraception. Examples may include barrier methods or IUDs [see also Warnings and Precautions (5.1) and Patient Counseling Information (17.1)]. Refer also to the prescribing information for ribavirin.
Two weeks after completion of INCIVEK treatment, hormonal contraceptives are again appropriate as one of the 2 required effective methods of birth control; however, specific prescribing information recommendations should be followed for the contraceptives. Refer also to the prescribing information for ribavirin.
8.3 Nursing Mothers
It is not known whether telaprevir is excreted in human breast milk. When administered to lactating rats, levels of telaprevir were higher in milk compared to those observed in plasma. Rat offspring exposed to telaprevir in utero showed no effects on body weight at birth. However, when fed via milk from telaprevir-treated dams, body weight gain of pups was lower than pups fed milk from control dams. After weaning, rat pup body weight gain was similar in offspring from telaprevir-treated and control dams. Because of the potential for adverse reactions in nursing infants, nursing must be discontinued prior to initiation of treatment. See also the prescribing information for ribavirin.
8.4 Pediatric Use
The safety, efficacy and pharmacokinetic profile of INCIVEK in pediatric patients have not been established.
8.5 Geriatric Use
Clinical trials of INCIVEK did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration and monitoring of INCIVEK in geriatric patients reflecting the greater frequency of decreased hepatic function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
INCIVEK is not recommended for use in patients with moderate or severe hepatic impairment (Child-Pugh B or C, score greater than or equal to 7) because no pharmacokinetic or safety data are available regarding the use of INCIVEK in HCV-infected patients with moderate or severe hepatic impairment, and appropriate doses have not been established [see Warnings and Precautions (5.8) and Clinical Pharmacology (12.3)]. No dose adjustment of INCIVEK is necessary for patients with mild hepatic impairment (Child-Pugh A, score 5-6). Refer also to the prescribing information for peginterferon alfa and ribavirin which must be co-administered with INCIVEK.
8.7 Renal Impairment
No dose adjustment is necessary for INCIVEK in HCV-infected patients with mild, moderate or severe renal impairment. INCIVEK has not been studied in HCV-infected patients with CrCl less than or equal to 50 mL per min.
The pharmacokinetics of telaprevir were assessed after administration of a single dose of 750 mg to HCV-negative subjects with severe renal impairment (CrCl less than 30 mL per min). INCIVEK has not been studied in subjects with end-stage renal disease (ESRD) or on hemodialysis [see Clinical Pharmacology (12.3)]. Refer also to the prescribing information for peginterferon alfa and ribavirin which must be co-administered with INCIVEK.
8.8 Co-infection
The safety and efficacy of INCIVEK have not been established in patients co-infected with HCV/HIV or HCV/HBV [see Drug Interactions (7)].
8.9 Solid Organ Transplantation
The safety and efficacy of INCIVEK have not been established in solid organ transplant patients [see Drug Interactions (7)].
10 OVERDOSAGE
The highest documented dose administered is 1875 mg every 8 hours for 4 days in healthy subjects with INCIVEK alone. In that trial, the following common adverse events were reported more frequently with the 1875 mg q8h regimen compared to the 750 mg q8h regimen: nausea, headache, diarrhea, decreased appetite, dysgeusia, and vomiting.
No specific antidote is available for overdose with INCIVEK. Treatment of overdose with INCIVEK consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. In the event of an overdose, it is reasonable to employ the standard supportive measures, such as, removing unabsorbed material from the gastrointestinal tract, employing clinical monitoring (including obtaining an electrocardiogram), and instituting supportive therapy if required.
It is not known whether telaprevir is dialyzable by peritoneal or hemodialysis.
11 DESCRIPTION
INCIVEK (telaprevir) is an inhibitor of the HCV NS3/4A protease.
The IUPAC name for telaprevir is (1S,3aR,6aS)-2-[(2S)-2-({(2S)-2-cyclohexyl-2-[(pyrazin-2-ylcarbonyl)amino]acetyl}amino)-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1,2-dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-1-carboxamide. Its molecular formula is C36H53N7O6 and its molecular weight is 679.85. Telaprevir has the following structural formula:
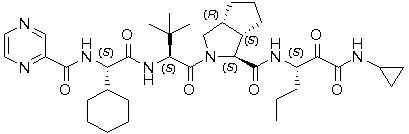
Telaprevir drug substance is a white to off-white powder with a solubility in water of 0.0047 mg/mL.
Telaprevir interconverts to an R-diastereomer, VRT-127394, which is the major metabolite in plasma and is approximately 30-fold less potent than telaprevir.
INCIVEK is available as a purple, capsule-shaped, film-coated tablet for oral administration containing 375 mg of telaprevir. Each tablet contains the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, D&C Red No. 40, dibasic calcium phosphate (anhydrous), FD&C Blue No. 2, hypromellose acetate succinate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, sodium lauryl sulfate, sodium stearyl fumarate, talc, and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Telaprevir is a direct-acting antiviral agent (DAA) against the hepatitis C virus [see Clinical Pharmacology (12.4)].
12.2 Pharmacodynamics
ECG Evaluation
The effect of telaprevir 750 and 1875 mg on QTc interval was evaluated in a double-blind, double-dummy, randomized, placebo-, and active-controlled (moxifloxacin 400 mg) four period crossover thorough QT trial in 44 subjects. In the trial with demonstrated ability to detect small effects, the upper bound of the one-sided 95% confidence interval for the largest placebo adjusted, baseline-corrected QTc based on Fridericia correction method (QTcF) was below 10 ms, the threshold for regulatory concern. The dose of 1875 mg is adequate to represent the high exposure clinical scenario.
12.3 Pharmacokinetics
The pharmacokinetic properties of telaprevir have been evaluated in healthy adult subjects and in subjects with chronic hepatitis C. Following multiple doses of telaprevir (750 mg q8h) in combination with peginterferon alfa and ribavirin in treatment-naïve subjects with genotype 1 chronic hepatitis C, mean (SD) Cmax was 3510 (1280) ng/mL, Cmin was 2030 (930) ng/mL, and AUC8h was 22,300 (8650) ng•hr/mL.
Absorption and Bioavailabilty
Telaprevir is orally available, most likely absorbed in the small intestine, with no evidence for absorption in the colon. Maximum plasma concentrations after a single dose of telaprevir are generally achieved after 4 to 5 hours. In vitro studies performed with human Caco-2 cells indicated that telaprevir is a substrate of P-glycoprotein (P-gp). Exposure to telaprevir is higher during co-administration of peginterferon alfa and ribavirin than after administration of telaprevir alone.
Effects of Food on Oral Absorption
The systemic exposure (AUC) to telaprevir was increased by 237% when telaprevir was administered with a standard fat meal (containing 533 kcal and 21 g fat) compared to when telaprevir was administered under fasting conditions. In addition, the type of meal significantly affects exposure to telaprevir. Relative to fasting, when telaprevir was administered with a low-fat meal (249 kcal, 3.6 g fat) and a high-fat meal (928 kcal, 56 g fat), the systemic exposure (AUC) to telaprevir was increased by approximately 117% and 330%, respectively. Doses of INCIVEK were administered within 30 minutes of completing a meal or snack containing approximately 20 grams of fat in the Phase 3 trials. Therefore, INCIVEK should always be taken with food (not low fat).
Distribution
In vitro, within a concentration range of 0.1 µM (68 ng per mL) to 20 µM (13600 ng per mL), telaprevir is approximately 59% to 76% bound to plasma proteins. Telaprevir binds primarily to alpha 1-acid glycoprotein and albumin and the binding is concentration dependent, decreasing with increasing concentrations of telaprevir. After oral administration, the typical apparent volume of distribution (Vd/F) was estimated to be 252 L, with an inter-individual variability of 72%.
Metabolism
Telaprevir is extensively metabolized in the liver, involving hydrolysis, oxidation, and reduction. Multiple metabolites were detected in feces, plasma, and urine. After repeated-oral administration, the R-diastereomer of telaprevir (30-fold less active), pyrazinoic acid, and a metabolite that underwent reduction at the α-ketoamide bond of telaprevir (not active) were found to be the predominant metabolites of telaprevir. In vitro studies using recombinant human cytochrome P450 (CYP) isoforms indicated that CYP3A4 was the major CYP isoform responsible for telaprevir metabolism. However, non-CYP mediated metabolism likely plays a role after multiple dosing of telaprevir.
Elimination
Following administration of a single oral dose of 750 mg 14C-telaprevir in healthy subjects, 90% of total radioactivity was recovered in feces, urine and expired air within 96 hours post-dose. The median recovery of the administered radioactive dose was approximately 82% in the feces, 9% in exhaled air and 1% in urine. The contribution of unchanged 14C-telaprevir and the R-diastereomer of telaprevir towards total radioactivity recovered in feces was 31.9% and 18.8%, respectively. After oral administration, the apparent total clearance (Cl/F) was estimated to be 32.4 L per hour with an inter-individual variability of 27.2%. The mean elimination half-life after single-dose oral administration of telaprevir 750 mg typically ranged from about 4.0 to 4.7 hours. At steady state, the effective half-life is about 9 to 11 hours.
Specific Populations
Hepatic Impairment
Steady-state exposure to telaprevir was reduced by 46% in HCV-negative subjects with moderate hepatic impairment (Child-Pugh Class B) compared to healthy subjects. The appropriate dose of INCIVEK in HCV-infected subjects with moderate or severe hepatic impairment has not been determined and therefore INCIVEK is not recommended in these populations.
Steady-state exposure to telaprevir was reduced by 15% in HCV-negative subjects with mild hepatic impairment (Child-Pugh Class A) compared to healthy subjects. Dose modification of INCIVEK is not required when administered to subjects with mild hepatic impairment. In previously treated subjects who had compensated liver disease and were treated with INCIVEK in combination with peginterferon alfa and ribavirin, subjects with cirrhosis had similar PK parameters compared to those without cirrhosis.
Renal Impairment
After administration of a single dose of 750 mg to HCV-negative subjects with severe renal impairment (CrCl less than 30 mL per min), the LS means of telaprevir Cmax and AUCinf were increased by 3% and 21%, respectively, compared to healthy subjects.
Gender
The effect of subject gender on telaprevir pharmacokinetics was evaluated using population pharmacokinetics of data from clinical trials of telaprevir. No dose adjustments are deemed necessary based on gender.
Race
Population pharmacokinetic analysis of telaprevir in HCV-infected subjects indicated that race had no apparent effect on the exposure to telaprevir.
Drug Interactions
In vitro studies indicated that telaprevir is a substrate and inhibitor of CYP3A and a substrate and inhibitor of P-gp. No inhibition by telaprevir of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP2E1 isozymes was observed in vitro. In vitro studies also suggest that telaprevir does not induce CYP1A, CYP3A, CYP2B6, or CYP2C. Clinical trials were conducted to evaluate the effect of drugs that can affect or be affected by telaprevir during co-administration (Tables 6 and 7).
| Drug | Dose and Schedule | N | Effect on Telaprevir PK† | LS Mean Ratio (90% CI) of Telaprevir PK With/Without Co-administered Drug | |||
|---|---|---|---|---|---|---|---|
| Drug | Telaprevir | Cmax | AUC or Cavg,ss‡ | Cmin | |||
| NA: not available/ not applicable; N = Number of subjects with data; qd = once daily; bid = twice daily; q8h = every 8 hours; q12h = every 12 hours | |||||||
|
|||||||
| Escitalopram | 10 mg qd for 7 days | 750 mg q8h for 14 days | 13 | ↔ | 1.00 (0.95; 1.05) | 0.93 (0.89; 0.97) | 0.91 (0.86; 0.97) |
| Esomeprazole | 40 mg qd for 6 days | 750 mg single dose | 24 | ↔ | 0.95 (0.86; 1.06) | 0.98 (0.91; 1.05) | NA |
| Ketoconazole | Ketoconazole 400 mg single dose | 750 mg single dose | 17 | ↑ | 1.24 (1.10; 1.41) | 1.62 (1.45; 1.81) | NA |
| Oral Contraceptive | Norethindrone/ ethinyl estradiol 0.5 mg/0.035 mg qd for 21 days | 750 mg q8h for 21 days | 23 | ↔ | 1.00 (0.93; 1.07) | 0.99 (0.93; 1.05) | 1.00 (0.93; 1.08) |
| Rifampin | 600 mg qd for 8 days | 750 mg single dose | 16 | ↓ | 0.14 (0.11; 0.18) | 0.08 (0.07; 0.11) | NA |
| Anti-HIV Drugs | |||||||
| Atazanavir (ATV)/ritonavir (rtv) | 300 mg ATV/ 100 mg rtv qd for 20 days | 750 mg q8h for 10 days | 14 | ↓ | 0.79 (0.74; 0.84) | 0.80 (0.76; 0.85) | 0.85 (0.75; 0.98) |
| Darunavir (DRV)/ritonavir (rtv) | 600 mg DRV/ 100 mg rtv bid for 20 days | 750 mg q8h for 10 days | 11 (N=14 for Cmax) | ↓ | 0.64 (0.61; 0.67) | 0.65 (0.61; 0.69) | 0.68 (0.63; 0.74) |
| Efavirenz | 600 mg qd for 20 days | 750 mg q8h for 10 days | 21 | ↓ | 0.91 (0.82; 1.02) | 0.74 (0.65; 0.84) | 0.53 (0.44; 0.65) |
| Fosamprenavir (fAPV)/ ritonavir (rtv) | 700 mg fAPV/ 100 mg rtv bid for 20 days | 750 mg q8h for 10 days | 18 | ↓ | 0.67 (0.63; 0.71) | 0.68 (0.63; 0.72) | 0.70 (0.64; 0.77) |
| Lopinavir (LPV)/ritonavir (rtv) | 400 mg LPV/ 100 mg rtv bid for 20 days | 750 mg q8h for 10 days | 12 | ↓ | 0.47 (0.41; 0.52) | 0.46 (0.41; 0.52) | 0.48 (0.40; 0.56) |
| Raltegravir | 400 mg bid for 11 days | 750 mg q8h for 7 days | 20 | ↔ | 1.07 (0.98; 1.16) | 1.07 (1.00; 1.15) | 1.14 (1.04; 1.26) |
| Ritonavir | 100 mg single dose | 750 mg single dose | 14 | ↑ | 1.30 (1.15; 1.47) | 2.00 (1.72; 2.33) | NA |
| Ritonavir | 100 mg q12h for 14 days | 750 mg q12h for 14 days | 5 | ↓ | 0.85 (0.63; 1.13) | 0.76‡,§
(0.60; 0.97) | 0.68 (0.57; 0.82) |
| Tenofovir disoproxil fumarate (TDF) | 300 mg qd TDF for 7 days | 750 mg q8h for 7 days | 16 | ↔ | 1.01 (0.96; 1.05) | 1.00 (0.94; 1.07) | 1.03 (0.93; 1.14) |
| Tenofovir disoproxil fumarate (TDF) and efavirenz (EFV) | 600 mg EFV /300 mg TDF qd for 7 days | 1125 mg q8h for 7 days | 15 | ↓ | 0.86§
(0.76; 0.97) | 0.82§
(0.73; 0.92) | 0.75§
(0.66; 0.86) |
| 600 mg EFV /300 mg TDF qd for 7 days | 1500 mg q12h for 7 days | 16 | ↓ | 0.97§
(0.88; 1.06) | 0.80‡,§
(0.73; 0.88) | 0.52§
(0.42; 0.64) |
|
| Drug | Dose and Schedule | N | Effect on Drug PK* | LS Mean Ratio (90% CI) of Drug PK With/Without Telaprevir | |||
|---|---|---|---|---|---|---|---|
| Drug | Telaprevir | Cmax | AUC | Cmin | |||
|
|||||||
| Alprazolam | 0.5 mg single dose | 750 mg q8h for 10 days | 17 | ↑ | 0.97 (0.92; 1.03) | 1.35 (1.23; 1.49) | NA |
| Amlodipine | 5 mg single dose | 750 mg q8h for 7 days | 19 | ↑ | 1.27 (1.21; 1.33) | 2.79 (2.58; 3.01) | NA |
| Atorvastatin | 20 mg single dose | 750 mg q8h for 7 days | 19 | ↑ | 10.60 (8.74; 12.85) | 7.88 (6.84; 9.07) | NA |
| Buprenorphine | Buprenorphine maintenance therapy (4 to 24 mg/daily in combination with naloxone) | 750 mg q8h for 7 days | 14 | ↔ | 0.80 (0.69; 0.93) | 0.96 (0.84; 1.10) | 1.06 (0.87; 1.30) |
| Cyclosporine A (CsA) | 100 mg single dose when administered alone; 10 mg single dose when co-administered with telaprevir (D8) | 750 mg q8h for 11 days | 9 | ↑ | 0.13 (0.11; 0.16) Dose norm.: 1.32 (1.08; 1.60) | 0.46 (0.39; 0.55) Dose norm.: 4.64 (3.90; 5.51) | NA |
| Digoxin | 0.5 mg single dose | 750 mg q8h for 11 days | 20 | ↑ | 1.50 (1.36; 1.65) | 1.85 (1.70; 2.00) | NA |
| Escitalopram | 10 mg qd, for 7 days | 750 mg q8h for 14 days | 13 | ↓ | 0.70 (0.65; 0.76) | 0.65 (0.60; 0.70) | 0.58 (0.52; 0.64) |
| Ethinyl estradiol (EE), co-administered with norethindrone (NE) | 0.035 mg qd EE/ 0.5 mg qd NE for 21 days | 750 mg q8h for 21 days | 24 | ↓ | 0.74 (0.68; 0.80) | 0.72 (0.69; 0.75) | 0.67 (0.63; 0.71) |
| Ketoconazole | 400 mg single dose | 1250 mg q8h for 4 doses | 81 | ↑ | 1.23 (1.14; 1.33) | 1.46 (1.35; 1.58) | NA |
| 200 mg single dose | 1250 mg q8h for 4 doses | 28 | ↑ | 1.75 (1.51; 2.03) | 2.25 (1.93; 2.61) | NA | |
| R-Methadone | Methadone maintenance therapy (40 to 120 mg/daily) | 750 mg q8h for 7 days | 15 | ↓ | 0.71 (0.66; 0.76) | 0.71 (0.66; 0.76) | 0.69 (0.64; 0.75) |
| S-Methadone | Methadone maintenance therapy (40 to 120 mg/daily) | 750 mg q8h for 7 days | 15 | ↓ | 0.65 (0.60; 0.71) | 0.64 (0.58; 0.70) | 0.60 (0.54; 0.67) |
| Midazolam (iv) | 0.5 mg iv single dose | 750 mg q8h for 9 days | 22 | ↑ | 1.02 (0.8; 1.31) | 3.40 (3.04; 3.79) | NA |
| Midazolam (oral) | 2 mg oral single dose | 750 mg q8h for 11 days | 21 | ↑ | 2.86 (2.52; 3.25) | 8.96 (7.75; 10.35) | NA |
| Norethindrone (NE), co-administered with EE | 0.035 mg qd EE/ 0.5 mg qd NE for 21 days | 750 mg q8h for 21 days | 24 | ↔ | 0.85 (0.81; 0.89) | 0.89 (0.86; 0.93) | 0.94 (0.87; 1.0) |
| Tacrolimus | 2 mg single dose when administered alone; 0.5 mg single dose when co-administered with telaprevir (D8) | 750 mg q8h for 13 days | 9 | ↑ | 2.34 (1.68; 3.25) Dose norm.: 9.35 (6.73; 13.0) | 17.6 (13.2; 23.3) Dose norm.: 70.3 (52.9; 93.4) | NA |
| Zolpidem | 5 mg single dose | 750 mg q8h for 10 days | 19 | ↓ | 0.58 (0.52; 0.66) | 0.53 (0.45; 0.64) | NA |
| Anti-HIV Drugs | |||||||
| Atazanavir (ATV), boosted with ritonavir (rtv) | 300 mg ATV/ 100 mg rtv qd for 20 days | 750 mg q8h for 10 days | 7 | ↑ | 0.85 (0.73; 0.98) | 1.17 (0.97; 1.43) | 1.85 (1.40; 2.44) |
| Darunavir (DRV), boosted with ritonavir (rtv) | 600 mg DRV/ 100 mg rtv bid for 20 days | 750 mg q8h for 10 days | 11 (N=14 for Cmax) | ↓ | 0.60 (0.56; 0.64) | 0.60 (0.57; 0.63) | 0.58 (0.52; 0.64) |
| 600 mg DRV/ 100 mg rtv bid for 24 days | 1125 mg q12h for 4 days | 15 | ↓ | 0.53 (0.47; 0.59) | 0.49 (0.43; 0.55) | 0.42 (0.35; 0.51) |
|
| Efavirenz | 600 mg qd for 20 days | 750 mg q8h for 10 days | 21 | ↔ | 0.84 (0.76; 0.93) | 0.93 (0.87; 0.98) | 0.98 (0.94; 1.02) |
| Efavirenz (EFV), co-administered with tenofovir disoproxil fumarate (TDF) | 600 mg EFV /300 mg TDF qd for 7 days | 1125 mg q8h for 7 days | 15 | ↓ | 0.76 (0.68; 0.85) | 0.82 (0.74; 0.90) | 0.90 (0.81; 1.01) |
| 600 mg EFV /300 mg TDF qd for 7 days | 1500 mg q12h for 7 days | 16 | ↓ | 0.80 (0.74; 0.86) | 0.85 (0.79; 0.91) | 0.89 (0.82; 0.96) |
|
| Fosamprenavir (fAPV), boosted with ritonavir (rtv) | 700 mg fAPV/ 100 mg bid rtv for 20 days | 750 mg q8h for 10 days | 18 | ↓ | 0.65 (0.59; 0.70) | 0.53 (0.49; 0.58) | 0.44 (0.40; 0.50) |
| 700 mg fAPV/ 100 mg bid rtv for 24 days | 1125 mg q12h for 4 days | 17 (N=18 for Cmin) | ↓ | 0.60 (0.55; 0.67) | 0.51 (0.47; 0.55) | 0.42 (0.37; 0.47) |
|
| Lopinavir (LPV), boosted with ritonavir (rtv) | 400 mg LPV/ 100 mg rtv bid for 20 days | 750 mg q8h for 10 days | 12 | ↔ | 0.96 (0.87; 1.05) | 1.06 (0.96; 1.17) | 1.14 (0.96; 1.36) |
| Raltegravir | 400 mg bid for 11 days | 750 mg q8h for 7 days | 20 | ↑ | 1.26 (0.97; 1.62) | 1.31 (1.03; 1.67) | 1.78 (1.26; 2.53) |
| Tenofovir disoproxil fumarate | 300 mg qd for 7 days | 750 mg q8h for 7 days | 16 | ↑ | 1.30 (1.16; 1.45) | 1.30 (1.22; 1.39) | 1.41 (1.29; 1.54) |
| Tenofovir, on co-administration of tenofovir disoproxil fumarate (TDF) and efavirenz (EFV) | 600 mg EFV /300 mg TDF qd for 7 days | 1125 mg q8h for 7 days | 15 | ↑ | 1.22 (1.12; 1.33) | 1.10 (1.03; 1.18) | 1.17 (1.06; 1.28) |
| 600 mg EFV /300 mg TDF qd for 7 days | 1500 mg q12h for 7 days | 16 | ↑ | 1.24 (1.13; 1.37) | 1.10 (1.03; 1.17) | 1.06 (0.98; 1.15) |
|
12.4 Microbiology
Mechanism of Action
Telaprevir is an inhibitor of the HCV NS3/4A serine protease, necessary for the proteolytic cleavage of the HCV encoded polyprotein into mature forms of the NS4A, NS4B, NS5A and NS5B proteins and essential for viral replication. In a biochemical assay, telaprevir inhibited the proteolytic activity of the recombinant HCV NS3 protease domain with an IC50 value of 10 nM.
Antiviral Activity in Cell Culture
In an HCV subtype 1b replicon assay, the telaprevir EC50 value against wild-type HCV was 354 nM in a 2-day cell culture assay, and in a subtype 1a infectious virus assay, the EC50 value was 280 nM in a 5-day cell culture assay. In biochemical enzymatic assays, the median IC50 values of telaprevir against genotype 2, 3a, and 4a were 16 nM (range 6-32 nM; n=5), 40 nM (range 39-88 nM; n=5), and 130 nM (n=1), respectively, compared to a median IC50 value of 20 nM (range 16-23; n=2) for genotype 1a and 20 nM for genotype 1b (range 13-33; n=4). The presence of 40% human serum reduced the anti-HCV activity of telaprevir by approximately 10-fold. Evaluation of telaprevir in combination with interferon alfa or ribavirin showed no evidence of antagonism in reducing HCV-RNA levels in HCV replicon cells.
Resistance
In Cell Culture
HCV genotype 1b replicons with reduced susceptibility to telaprevir have been selected in cell culture and characterized for telaprevir genotypic and phenotypic resistance. Additionally, resistance to telaprevir was evaluated in both biochemical and HCV genotype 1b replicon assays using site-directed mutants and recombinant NS3/4A from telaprevir Phase 2 clinical trials isolates. Variants V36A/M, T54A/S, R155K/T, A156S, R155T+D168N, and V36A+T54A conferred 3- to 25-fold reduced susceptibility to telaprevir; and A156V/T variants and the V36M/A+R155K/T and T54S/A+A156S/T double variants conferred greater than 62-fold reduced susceptibility to telaprevir. No amino acid substitutions were observed at the proteolytic cleavage sites.
In Clinical Trials
In a pooled analysis of subjects who did not achieve SVR (on-treatment virologic failure or relapse) from the controlled Phase 3 clinical trials, NS3 amino acid substitutions V36M/A/L, T54A/S, R155K/T, and A156S/T were determined to emerge frequently on INCIVEK treatment (Table 8). Nearly all of these substitutions have been shown to reduce telaprevir anti-HCV activity in cell culture or biochemical assays. No clear evidence of treatment-emergent substitutions in the NS3 helicase domain or NS4A coding regions of the HCV genome was observed among INCIVEK-treated subjects who did not achieve SVR.
Telaprevir treatment-emergent resistance substitutions emerged in the majority of isolates from subjects who did not achieve SVR (Table 8): in almost 100% of subjects who failed during 12 weeks of T/PR and in the majority of subjects who failed on PR after Week 12 or who relapsed.
HCV genotype 1 subtype-associated patterns of INCIVEK treatment-emergent amino acid substitutions were observed. Subjects with HCV genotype 1a predominately had V36M and R155K or the combination of these variants, while subjects with HCV genotype 1b predominately had V36A, T54A/S, and A156S/T variants (Table 8). Among subjects treated with telaprevir, on-treatment virologic failure was more frequent in subjects with genotype 1a than with genotype 1b and more frequent in prior null responders [see Clinical Studies (14)].
| Emerging Substitutions* in NS3 | Percent of No SVR Subjects (n) N=525 | Percent Subtype 1a No SVR Subjects (n) N=356 | Percent Subtype 1b No SVR Subjects (n) N=169 |
|---|---|---|---|
| Any substitution at V36, T54, R155, A156 or D168 | 62% (323) | 69% (247) | 45% (76) |
| R155K/T | 38% (201) | 56% (200) | 0.6% (1) |
| V36M | 33% (178) | 49% (173) | 3% (5) |
| V36M + R155K† | 27% (142) | 40% (142) | 0% (0) |
| T54A/S | 13% (68) | 9% (31) | 22% (37) |
| V36A/L | 12% (65) | 10% (37) | 17% (28) |
| A156S/T | 9% (48) | 8% (28) | 12% (20) |
| V36G/I, I132V, R155G/M, A156V/F/N or D168N | Less than 2% | Less than 2% | Less than 2% |
Persistence of Resistance-Associated Substitutions
Persistence of telaprevir-resistant NS3 amino acid substitutions has been observed following treatment failure. Of a combined 255 treatment-naïve and previously treated subjects from Trials 108, 111, and C216 in whom telaprevir-resistant variants had emerged during treatment, 103 (40%) had detectable resistant variants by population sequencing at end of trial (follow-up range 2-70 weeks, median 45 weeks) and results for loss of variants were similar across the three trials. In the combined trials, 46% of the telaprevir-resistant substitutions in subtype 1a and 16% of the substitutions in subtype 1b were still detected by the end of trial: 29% of V36, 16% of T54, 38% of R155, 14% of A156, and 44% of V36M+R155K variants were detected at the end of trial.
In a 3-year follow-up trial of 56 treatment-naïve and prior treatment-failure subjects who did not achieve SVR with a telaprevir regimen in a Phase 2 trial and had telaprevir-resistant variants after treatment failure, variants were detected by population sequencing in 11% (6/56) of subjects (median follow-up of 25 months). Telaprevir-resistant variants V36L/M, T54S, and R155K were detectable (present at greater than 25% of the viral population) in some subjects at 24 months. By 36 months, V36M, T54A/S, and A156N/S/T variants had fallen below the level of detection by population sequencing in all subjects. At 36 months, 3% of the subject isolates that had the R155K variant still had detectable R155K variants by population sequencing.
The lack of detection of a substitution based on a population-based assay does not necessarily indicate the substitution has declined to the pre-treatment level. The long-term clinical impact of the emergence or persistence of detectable INCIVEK resistance-associated substitutions is unknown. No data are available regarding INCIVEK efficacy among patients who were previously exposed to INCIVEK, or who previously failed treatment with an INCIVEK-containing regimen.
Effect of Baseline HCV Substitutions/Polymorphisms on Treatment Response
A pooled analysis was conducted to explore the association between the detection (population-based assay) of baseline NS3/4A amino acid substitutions/polymorphisms and treatment outcome in Trials 108, 111, and C216. Baseline polymorphisms at NS3 position Q80 (Q80K, Q80L, Q80R), which are frequently observed in HCV genotype 1a-infected subjects and have been reported to reduce the activity of some HCV NS3/4A protease inhibitors, were not associated with reduced INCIVEK efficacy.
Telaprevir-associated resistance substitutions (substitutions at positions V36, T54, R155 or D168) were present at baseline in 5% (117/2217) of the available subject samples in the combined clinical trials. Given the small number of subjects with baseline telaprevir resistance substitutions, conclusions about their effect on response outcomes when these substitutions are present at baseline cannot be determined.
Cross-Resistance
Treatment-emergent NS3 amino acid substitutions detected in INCIVEK-treated subjects who did not achieve SVR in the clinical trials (substitutions at positions V36, T54, R155, A156 or D168) have been demonstrated to reduce the anti-HCV activity of boceprevir and other HCV NS3/4A protease inhibitors. The impact of prior INCIVEK exposure or treatment failure on the efficacy of boceprevir or other HCV NS3/4A protease inhibitors has not been studied. INCIVEK efficacy has not been established for patients with a history of exposure to NS3/4A protease inhibitors.
Cross-resistance is not expected between INCIVEK and interferons, or INCIVEK and ribavirin. HCV replicons expressing telaprevir-associated resistance substitutions remained fully sensitive to interferon-alfa and ribavirin, as well as other direct-acting antivirals with different mechanisms of action, such as NS5B polymerase inhibitors.
12.5 Pharmacogenomics
A genetic variant near the gene encoding interferon-lambda-3 (IL28B rs12979860, a C to T change) is a strong predictor of response to peginterferon alfa and ribavirin (PR). rs12979860 was genotyped in 454 of 1088 subjects in Study Trial 108 (treatment-naïve) and 527 of 662 subjects in Trial C216 (previously treated) [see Clinical Studies (14.2 and 14.3) for trial descriptions]. SVR rates tended to be lower in subjects with the CT and TT genotypes compared to those with the CC genotype, particularly among treatment-naïve subjects receiving PR48 (Table 9). Among both treatment-naïve and previous treatment failures, subjects of all IL28B genotypes appeared to have higher SVR rates with INCIVEK-containing regimens. The results of this retrospective subgroup analysis should be viewed with caution because of the small sample size and potential differences in demographic or clinical characteristics of the subtrial population relative to the overall trial population.
| Trial | rs12979860 Genotype | SVR, n/N (%) | |
|---|---|---|---|
|
|||
| T12/PR | Pbo/PR48 | ||
| 108 (treatment-naïve) | C/C | 45/50 (90%) | 35/55 (64%) |
| C/T | 48/68 (71%) | 20/80 (25%) | |
| T/T | 16/22 (73%) | 6/26 (23%) | |
| T12 /PR48* | Pbo/PR48 | ||
| C216 (previously treated) | C/C | 60/76 (79%) | 5/17 (29%) |
| C/T | 160/266 (60%) | 9/58 (16%) | |
| T/T | 49/80 (61%) | 4/30 (13%) | |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Impairment of Fertility
INCIVEK /Peginterferon Alfa/Ribavirin Combination Treatment
Animal studies have shown that ribavirin induced reversible toxicity in males while peginterferon alfa may impair female fertility. See the prescribing information for ribavirin and peginterferon alfa.
INCIVEK (telaprevir) Tablets
Telaprevir treatment alone had effects on fertility parameters in rats. The no observed adverse effect level (NOAEL) for degenerative testicular toxicity was established at exposures 0.17-fold the human exposures at the recommended clinical dose. Potential effects on sperm (e.g., decreased % motile sperm and increased non-motile sperm count) were observed in a rat fertility study at exposures 0.30-fold the human exposures at the recommended clinical dose. Additional effects on fertility include minor increases in percent preimplantation loss, the percent of dams with nonviable embryos and percent of nonviable conceptuses per litter. These effects are likely associated with testicular toxicity in male rats but contributions of the female cannot be ruled out. Degenerative testicular toxicity was not observed in chronic toxicity studies in the dog. Furthermore, mean changes in proposed hormonal biomarkers of testicular toxicity among subjects who received telaprevir were comparable to placebo.
14 CLINICAL STUDIES
14.1 Description of Adult Clinical Trials
The efficacy and safety of INCIVEK in subjects with genotype 1 chronic hepatitis C were evaluated in three adequate and well-controlled clinical trials: two in treatment-naïve subjects and one in previously treated subjects (relapsers, partial responders, and null responders). Subjects in these trials had compensated liver disease, detectable HCV RNA, and liver histopathology consistent with chronic hepatitis C. In all three trials, INCIVEK was administered at a dosage of 750 mg every 8 hours; the peginterferon alfa-2a (Peg-IFN-alfa-2a) dose was 180 micrograms per week, and the ribavirin (RBV) dose was 1000 mg per day (subjects weighing less than 75 kg) or 1200 mg per day (subjects weighing greater than or equal to 75 kg). Plasma HCV-RNA values were measured during the clinical trials using the COBAS® TaqMan® HCV test (version 2.0), for use with the High Pure System. The assay had a lower limit of quantitation of 25 IU per mL. SVR was defined as HCV RNA less than 25 IU per mL at last observation within the SVR visit window (i.e., weeks 32-78 for patients assigned to 24 weeks of treatment and weeks 56-78 for patients assigned to 48 weeks of treatment).
14.2 Treatment-Naïve Adults
Trial 108 (ADVANCE)
Trial 108 was a randomized, double-blind, parallel-group, placebo-controlled trial conducted in treatment-naïve subjects (had received no prior therapy for HCV, including interferon or pegylated interferon monotherapy). INCIVEK was given for the first 8 weeks of treatment (T8/PR regimen) or the first 12 weeks of treatment (T12/PR regimen) in combination with Peg-IFN-alfa-2a/RBV for either 24 or 48 weeks. Subjects who had undetectable HCV RNA (Target Not Detected) at weeks 4 and 12 (extended Rapid Virologic Response [eRVR]) received 24 weeks of Peg-IFN-alfa-2a/RBV treatment, and subjects who did not have undetectable HCV RNA at weeks 4 and 12 (no eRVR) received 48 weeks of Peg-IFN-alfa-2a/RBV treatment. The control regimen (Pbo/PR48) had a fixed treatment duration, with telaprevir-matching placebo for the first 12 weeks and Peg-IFN-alfa-2a/RBV for 48 weeks.
The 1088 enrolled subjects had a median age of 49 years (range: 18 to 69); 59% of the subjects were male; 23% had a body mass index greater than or equal to 30 kg/m2; 9% were Black; 11% were Hispanic or Latino; 77% had baseline HCV-RNA levels greater than 800,000 IU per mL; 15% had bridging fibrosis; 6% had cirrhosis; 59% had HCV genotype 1a; and 40% had HCV genotype 1b.
Table 10 shows the response rates for the T12/PR and Pbo/PR48 groups.
| Treatment Outcome | T12/PR N = 363 n/N (%) | Pbo/PR48 N = 361 n/N (%) |
|---|---|---|
|
||
| Overall SVR | 79% (285/363) | 46% (166/361) |
| eRVR | 58% (212/363) | 8% (29/361) |
| SVR in eRVR subjects | 92% (195/212) | 93% (27/29) |
| No eRVR | 42% (151/363) | 92% (332/361) |
| SVR in no eRVR subjects | 60% (90/151) | 42% (139/332) |
| Outcome for Subjects without SVR | ||
| On-treatment virologic failure* | 7% (26/363) | 29% (105/361) |
| Relapse† | 4% (11/298) | 24% (53/220) |
| Other‡ | 11% (41/363) | 10% (37/361) |
In the T8/PR group, the overall SVR rate was 72%. The eRVR rate was 57% and the SVR rate for eRVR subjects was 86%. The SVR rate for no eRVR subjects was 52%. More subjects in the T8/PR group experienced virologic failure after Week 12 while receiving peginterferon alfa and ribavirin alone, 7% compared to 4% in T12/PR group.
SVR rates were higher (absolute difference of at least 22%) for the T12/PR group than for the Pbo/PR48 group across subgroups by sex, age, race, ethnicity, body mass index, HCV genotype subtype, baseline HCV RNA (less than 800,000, greater than or equal to 800,000 IU per mL), and extent of liver fibrosis. However, there were small numbers of subjects enrolled in some key subgroups. In the T12/PR group:
- Twenty-one subjects had cirrhosis at baseline and the overall SVR in these subjects was 71% (15/21). Among subjects with cirrhosis, 43% (9/21) were assigned to 24 weeks of treatment and of those 78% (7/9) achieved SVR.
- Twenty-six subjects were Black/African Americans. The overall SVR among Black/African American subjects was 62% (16/26). Among these subjects, 35% (9/26) were assigned to 24 weeks of treatment and of those 89% (8/9) achieved SVR.
Trial 111 (ILLUMINATE)
Trial 111 was a randomized, open-label trial conducted in treatment-naïve subjects. The trial was designed to compare SVR rates in subjects achieving eRVR who were treated with INCIVEK for 12 weeks in combination with Peg-IFN-alfa-2a/RBV for either 24 weeks (T12/PR24 regimen) or 48 weeks (T12/PR48 regimen).
The 540 enrolled subjects had a median age of 51 years (range: 19 to 70); 60% were male; 32% had a body mass index greater than or equal to 30 kg/m2; 14% were Black; 10% were Hispanic or Latino; 82% had baseline HCV-RNA levels greater than 800,000 IU per mL; 16% had bridging fibrosis; 11% had cirrhosis; 72% had HCV genotype 1a; and 27% had HCV genotype 1b.
The SVR rate for all subjects enrolled in the trial was 74%. A total of 352 (65%) subjects achieved eRVR and of those 322 (60%) were randomized to 24 weeks (T12/PR24, n=162) or 48 weeks (T12/PR48, n=160) of treatment. The SVR rates were similar at 92% (T12/PR24) and 90% (T12/PR48), respectively. Again, small numbers of subjects were enrolled in some key subgroups:
- Sixty-one (11%) of subjects had cirrhosis at baseline. Among subjects with cirrhosis, 30 (49%) achieved an eRVR: 18 were randomized to T12/PR24 and 12 to T12/PR48. The SVR rates were 61% (11/18) for the T12/PR24 group and 92% (11/12) for the T12/PR48 group.
- Blacks/African Americans comprised 14% (73/540) of trial subjects. Thirty-four (47%) Black/African American subjects achieved an eRVR and were randomized to T12/PR24 or T12/PR48. The respective SVR rates were 88% (15/17) and 88% (15/17), compared to 92% (244/266) for Caucasians among randomized subjects.
14.3 Previously Treated Adults
Trial C216 (REALIZE)
Trial C216 was a randomized, double-blind, placebo-controlled trial conducted in subjects who did not achieve SVR with prior treatment with Peg-IFN-alfa-2a/RBV or Peg-IFN-alfa-2b/RBV. The trial enrolled prior relapsers (subjects with HCV RNA undetectable at end of treatment with a pegylated interferon-based regimen, but HCV RNA detectable within 24 weeks of treatment follow-up) and prior non-responders (subjects who did not have undetectable HCV-RNA levels during or at the end of a prior course of at least 12 weeks of treatment). The nonresponder population included 2 subgroups: prior partial responders (greater than or equal to 2-log10 reduction in HCV RNA at week 12, but not achieving HCV RNA undetectable at end of treatment with peginterferon alfa and ribavirin) and prior null responders (less than 2-log10 reduction in HCV RNA at week 12 of prior treatment with peginterferon alfa and ribavirin).
Subjects were randomized in a 2:2:1 ratio to one of two INCIVEK combination treatment groups (with and without a Peg-IFN-alfa-2a/RBV lead-in) or a control group. The T12/PR48 group received INCIVEK and Peg-IFN-alfa-2a/RBV for 12 weeks (without a lead-in), followed by placebo and Peg-IFN-alfa-2a/RBV for 4 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The T12(DS)/PR48 group had a lead-in (delayed start of INCIVEK) with placebo and Peg-IFN-alfa-2a/RBV for 4 weeks, followed by INCIVEK and Peg-IFN-alfa-2a/RBV for 12 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The Pbo/PR48 group received placebo and Peg-IFN-alfa-2a/RBV for 16 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks.
The 662 enrolled subjects had a median age of 51 years (range: 21 to 70); 70% of the subjects were male; 26% had a body mass index greater than or equal to 30 kg/m2; 5% were Black; 11% were Hispanic or Latino; 89% had baseline HCV-RNA levels greater than 800,000 IU per mL; 22% had bridging fibrosis; 26% had cirrhosis; 54% had HCV genotype 1a, and 46% had HCV genotype 1b. Null and partial responders had higher baseline HCV-RNA levels and more advanced liver disease (cirrhosis) than relapsers; other characteristics were similar across these populations.
The lead-in and immediate start regimens produced comparable SVR and no SVR rates, so data from these two groups were pooled (Table 11).
| Treatment Outcome | All T12/PR48*
% (n/N) | Pbo/PR48 % (n/N) |
|---|---|---|
|
||
| SVR rate | ||
| Prior relapsers | 86% (246/286) | 22% (15/68) |
| Prior partial responders | 59% (57/97) | 15% (4/27) |
| Prior null responders | 32% (47/147) | 5% (2/37) |
| Treatment Outcomes for Subjects Without SVR | ||
| On-treatment virologic failure† | ||
| Prior relapsers | 1% (3/286) | 26% (18/68) |
| Prior partial responders | 18% (17/97) | 70% (19/27) |
| Prior null responders | 52% (76/147) | 84% (31/37) |
| Relapse‡ | ||
| Prior relapsers | 3% (8/254) | 63% (27/43) |
| Prior partial responders | 20% (14/71) | 0% (0/4) |
| Prior null responders | 24% (15/62) | 50% (2/4) |
Among prior relapsers, 76% (218/286) achieved an eRVR and of those 95% (208/218) achieved an SVR. In an earlier, dose-finding clinical trial, 78% (52/67) of prior relapsers achieved an eRVR and were treated with 24 weeks of peginterferon alfa and ribavirin (T12/PR24); of those 94% (49/52) achieved an SVR.
For all populations in the trial (prior relapsers, prior partial responders, and prior null responders), SVR rates were higher for the T12/PR group than for the Pbo/PR48 group across subgroups by sex, age, ethnicity, body mass index, HCV genotype subtype, baseline HCV-RNA level, and extent of liver fibrosis.
Twenty-six percent (139/530) of INCIVEK-treated subjects had cirrhosis at baseline. SVR rates among cirrhotic subjects who received INCIVEK combination treatment compared to Pbo/PR48 were: 84% (48/57) compared to 7% (1/15) for prior relapsers, 34% (11/32) compared to 20% (1/5) for prior partial responders, and 14% (7/50) compared to 10% (1/10) for prior null responders.
Four percent (19/530) of treatment experienced subjects who received INCIVEK combination treatment were Black/African Americans; the SVR rate for these subjects was 63% (12/19) compared to 66% (328/498) for Caucasians.
16 HOW SUPPLIED/STORAGE AND HANDLING
INCIVEK™ (telaprevir) is supplied as purple film-coated capsule-shaped tablets containing 375 mg of telaprevir. Each tablet is debossed with the characters "V 375" on one side and is packaged as follows:
- 28-day packer contains 4 weekly cartons of 7 blister strips each (6 tablets per blister strip) NDC 51167-100-01
17 PATIENT COUNSELING INFORMATION
17.1 Pregnancy
Ribavirin must not be used by women who are pregnant or by men whose female partners are pregnant. Ribavirin therapy should not be initiated until a report of a negative pregnancy test has been obtained immediately before starting therapy. Because INCIVEK must be used in combination with ribavirin and peginterferon alfa, the contraindications and warnings applicable to those drugs are applicable to combination treatment. INCIVEK combination treatment is contraindicated in women who are pregnant and in men whose female partners are pregnant (see also the prescribing information for ribavirin).
Patients must be advised of the teratogenic/embryocidal risks of ribavirin and should be advised that extreme care must be taken to avoid pregnancy in female patients and in female partners of male patients—both during treatment and for 6 months after the completion of all treatment. Women of childbearing potential must be counseled about use of effective contraception (two methods) prior to initiating treatment. Patients (both male and female) should be advised to notify their health care provider immediately in the event of a pregnancy [see Contraindications (4), Warnings and Precautions (5.1), and Use in Specific Populations (8.1)].
Patients should also be advised that hormonal contraceptives may not be reliable during INCIVEK dosing and for up to two weeks following cessation of INCIVEK [see Drug Interactions (7)]. During this time, female patients of childbearing potential should use 2 non-hormonal methods of effective birth control. Examples of non-hormonal methods of contraception include a male condom with spermicidal jelly OR female condom with spermicidal jelly (a combination of a male condom and a female condom is not suitable), a diaphragm with spermicidal jelly, a cervical cap with spermicidal jelly, or an intrauterine device (IUD).
17.2 Serious Skin Reactions/Rash
Patients should be informed that INCIVEK combination treatment may cause rash. The rash can be severe and may be accompanied by fever and skin breakdown. Patients should promptly report any skin changes or itching to their healthcare provider. Patients should not stop INCIVEK due to rash unless instructed by their healthcare provider.
17.3 Hepatitis C Virus Transmission
Patients should be informed that the effect of treatment of hepatitis C infection on transmission is not known, and that appropriate precautions to prevent transmission of the hepatitis C virus during treatment or in the event of treatment failure should be taken.
17.4 Administration
Patients should be advised INCIVEK must be administered in combination with both peginterferon alfa and ribavirin. If peginterferon alfa and/or ribavirin is discontinued for any reason, INCIVEK must also be discontinued.
Patients should be advised that the dose of INCIVEK must not be reduced or interrupted, as it may increase the possibility of treatment failure.
The recommended dose of INCIVEK tablets is 750 mg (two 375-mg tablets) taken orally 3 times a day (7-9 hours apart) with food containing approximately 20 grams of fat. Patients should be advised that the fat content of the meal or snack is critical for the absorption of telaprevir. Food that is taken with INCIVEK should be ingested within 30 minutes prior to each INCIVEK dose. Examples of some foods that could be taken with INCIVEK include: a bagel with cream cheese, ½ cup nuts, 3 tablespoons peanut butter, 1 cup ice cream, 2 ounces American or cheddar cheese, 2 ounces potato chips, or ½ cup trail mix.
Patients should be informed about what to do in the event they miss a dose of INCIVEK:
- In case a dose of INCIVEK is missed within 4 hours of the time it is usually taken, patients should be instructed to take the prescribed dose of INCIVEK with food as soon as possible.
- If more than 4 hours has passed since INCIVEK is usually taken, the missed dose should NOT be taken and the patient should resume the usual dosing schedule.
- Patients should be advised to contact their health care provider if they have questions.
Patients should be advised that they can contact the local Poison Control Center in the event of an overdose.
Manufactured for
Vertex Pharmaceuticals Incorporated
Cambridge, MA 02139
U.S. Patent No. 7,820,671
©2012 Vertex Pharmaceuticals Incorporated
All rights reserved.
INCIVEK and the Blue Arrow logo are trademarks of Vertex Pharmaceuticals Incorporated. VERTEX and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
The brands listed are the registered trademarks of their respective owners and are not trademarks of Vertex Pharmaceuticals Incorporated.
60884-02
MEDICATION GUIDE
INCIVEK (in-SEE-veck)
(telaprevir)
Film-Coated Tablets
Read this Medication Guide before you start taking INCIVEK™ and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
INCIVEK is taken along with peginterferon alfa and ribavirin. You should also read those Medication Guides.
What is the most important information I should know about INCIVEK?
INCIVEK combination treatment may cause serious side effects including:
- 1.
- Birth defects or death of your unborn baby. INCIVEK in combination with peginterferon alfa and ribavirin may cause birth defects or death of your unborn baby. If you are pregnant or your sexual partner is pregnant or plans to become pregnant, do not take these medicines. You or your sexual partner should not become pregnant while taking INCIVEK with peginterferon alfa and ribavirin and for 6 months after treatment is over.
If you are a female who can become pregnant, or you are a female whose male partner takes these medicines:
- You must have a negative pregnancy test before starting treatment, each month during treatment, and for 6 months after your treatment ends.
- You must use 2 forms of effective birth control during treatment and for the 6 months after treatment with these medicines. Hormonal forms of birth control including birth control pills, vaginal rings, implants, or injections may not work during treatment with INCIVEK. You could become pregnant. Talk to your healthcare provider about other forms of birth control that may be used during this time. If your healthcare provider tells you to stop taking INCIVEK, peginterferon alfa and ribavirin, you must still use two forms of birth control for the 6 months after treatment with these medicines. You may use a hormonal form of birth control as one of your two forms of birth control after 2 weeks of stopping INCIVEK.
- If you or your female sexual partner becomes pregnant while taking INCIVEK, peginterferon alfa, and ribavirin or within 6 months after you stop taking these medicines, tell your healthcare provider right away. You or your healthcare provider should contact the Ribavirin Pregnancy Registry by calling 1-800-593-2214. The Ribavirin Pregnancy Registry collects information about what happens to mothers and their babies if the mother takes ribavirin while she is pregnant.
- 2.
-
Skin reactions. Mild skin rashes are common with INCIVEK combination treatment. Sometimes these skin rashes and other skin reactions can become severe and require treatment in a hospital.
-
Call your healthcare provider right away if you develop any skin changes with any of the symptoms below. Your healthcare provider will decide if your skin changes or symptoms may be a sign of a serious skin reaction:
- rash, with or without itching
- blisters or skin lesions
- mouth sores or ulcers
- red or inflamed eyes, like "pink eye" (conjunctivitis)
- swelling of your face
- fever
- Your healthcare provider will decide if you need treatment for your rash or if you need to stop taking INCIVEK, or any of your other medicines.
- Never stop taking INCIVEK combination treatment without talking with your healthcare provider first.
-
Call your healthcare provider right away if you develop any skin changes with any of the symptoms below. Your healthcare provider will decide if your skin changes or symptoms may be a sign of a serious skin reaction:
See "What are the possible side effects of INCIVEK?" for more information about side effects.
- 3.
- Do not take INCIVEK alone to treat chronic hepatitis C infection. INCIVEK must be used with peginterferon alfa and ribavirin to treat chronic hepatitis C infection.
What is INCIVEK?
INCIVEK is a prescription medicine used with the medicines peginterferon alfa and ribavirin to treat chronic (lasting a long time) hepatitis C genotype 1 infection in adults with stable liver problems, who have not been treated before or who have failed previous treatment.
It is not known if INCIVEK is safe and effective in children under 18 years of age.
Who should not take INCIVEK?
Do not take INCIVEK if you:
- are pregnant or may become pregnant. See "What is the most important information I should know about INCIVEK?"
- are a man with a sexual partner who is pregnant.
- take certain medicines. INCIVEK may cause serious side effects when taken with certain medicines. Read the section "What should I tell my healthcare provider before taking INCIVEK?"
Talk to your health care provider before taking INCIVEK if any of the above applies to you.
What should I tell my healthcare provider before taking INCIVEK?
Before you take INCIVEK, tell your healthcare provider if you:
- have certain blood problems, such as low red blood cell count (anemia)
- have liver problems other than hepatitis C infection
- have hepatitis B infection
- have Human Immunodeficiency Virus (HIV) infection or any other problems with your immune system
- history of gout or high uric acid levels in your blood
- have had an organ transplant
- plan to have surgery
- have any other medical condition
- are breastfeeding. It is not known if INCIVEK passes into your breast milk. You and your healthcare provider should decide if you will take INCIVEK or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
INCIVEK and other medicines can affect each other. This can cause you to have too much or not enough INCIVEK or your other medicines in your body, and cause side effects that can be serious or life-threatening. Your healthcare provider may need to change the amount of medicine you take.
Do not take INCIVEK if you take a medicine that contains:
- alfuzosin hydrochloride (Uroxatral®)
- cisapride (Propulsid®)
- ergot, including:
- dihydroergotamine mesylate (D.H.E. 45®, Migranal®)
- ergotamine tartrate (Cafergot®, Migergot®, Ergomar®, Ergostat®, Medihaler Ergotamine, Wigraine®, Wigrettes)
- methylergonovine maleate (Ergotrate®, Methergine®)
- lovastatin (Advicor®, Altoprev®, Mevacor®)
- pimozide (Orap®)
- rifampin (Rifadin®, Rifamate®, Rifater®)
- sildenafil citrate (Revatio®) or tadalafil (Adcirca®) for the lung problem, pulmonary artery hypertension (PAH)
- simvastatin (Zocor®, Vytorin®, Simcor®)
- St. John's wort (Hypericum perforatum) or products containing St. John's wort
- triazolam (Halcion®)
Tell your healthcare provider if you are taking or starting to take medicines that contain:
- atorvastatin (Lipitor®, Caduet®)
- budesonide (Pulmicort®, Rhinocort®, Symbicort®)
- colchicine (Colcrys®)
- darunavir (Prezista®) and ritonavir (Norvir®)
- fluticasone (Advair®, Flonase®, Flovent®, Veramyst®)
- fosamprenavir (Lexiva®) and ritonavir (Norvir®)
- lopinavir and ritonavir (Kaletra®)
- methylprednisolone (Medrol®)
- prednisone
- rifabutin (Mycobutin®)
- salmeterol (Advair®, Serevent®)
Your healthcare provider may need to monitor your therapy more closely if you take INCIVEK with the following medicines. Talk to your healthcare provider if you are taking or starting to take medicines that contain:
- alprazolam (Xanax®)
- amiodarone (Cordarone®, Pacerone®)
- amlodipine (Norvasc®)
- atazanavir and ritonavir (Reyataz®, Norvir®)
- bepridil hydrochloride (Vascor®, Bepadin)
- bosentan (Tracleer®)
- carbamazepine (Carbatrol®, Equetro®, Tegretol®)
- clarithromycin (Biaxin®, Prevpac®)
- colchicine (Colcrys®)
- cyclosporine (Gengraf®, Neoral®, Sandimmune®)
- dexamethasone
- digoxin (Lanoxin®)
- diltiazem (Cardizem®, Dilacor XR®, Tiazac®)
- efavirenz (Sustiva®, Atripla®)
- erythromycin (E.E.S.®, Eryc®, Ery-Tab®, Erythrocin®, Erythrocin Stearate®)
- escitalopram (Lexapro®)
- ethinyl estradiol containing birth control methods (Lo Loestrin™ FE, Norinyl®, Ortho Tri-Cyclen Lo®)
- felodipine (Plendil®)
- flecainide (Tambocor™)
- itraconazole (Sporanox®)
- ketoconazole (Nizoral®)
- methadone (Dolophine®, Methadose™)
- nicardipine (Cardene®)
- nifedipine (Adalat®, Procardia®)
- nisoldipine (Sular®)
- phenobarbital
- phenytoin (Dilantin®, Phenytek®)
- posaconazole (Noxafil®)
- propafenone (Rythmol®)
- quinidine (Nuedexta®)
- sildenafil for the treatment of erectile dysfunction (Viagra®)
- sirolimus (Rapamune®)
- tacrolimus (Prograf®)
- tadalafil for the treatment of erectile dysfunction (Cialis®)
- telithromycin (Ketek®)
- tenofovir disoproxil fumarate (Atripla®, Complera®, Truvada®, Viread®)
- trazodone (Desyrel®, Trialodine, Oleptro™)
- vardenafil for the treatment of erectile dysfunction (Levitra®, Staxyn®)
- verapamil (Calan®, Covera-HS®, Isoptin®, Tarka®)
- voriconazole (Vfend®)
- warfarin (Coumadin®)
- zolpidem (Ambien®, Edluar®)
Know the medicines you take. Keep a list of them with you and show it to your healthcare provider and pharmacist each time you get a new medicine.
How should I take INCIVEK?
- Take INCIVEK exactly as your healthcare provider tells you. Your healthcare provider will tell you how much INCIVEK to take and when to take it.
- Take INCIVEK 3 times a day. Each dose should be taken 7 to 9 hours apart. Eat a meal or snack that contains about 20 grams of fat, within 30 minutes before you take each dose of INCIVEK. Talk to your healthcare provider about examples of foods that you can eat that contain about 20 grams of fat. Always take INCIVEK with food.
- If you miss a dose within 4 hours of when you usually take it, take your dose with food as soon as possible.
- If you miss a dose and it is more than 4 hours after the time you usually take it, skip that dose only and take the next dose at your normal dosing schedule.
- Do not stop taking INCIVEK unless your healthcare provider tells you to. If you think there is a reason to stop taking INCIVEK, talk to your healthcare provider before doing so.
- If your healthcare provider tells you to stop taking INCIVEK, you should not start taking it again even if the reason for stopping goes away.
- If you take too much INCIVEK or overdose, call your healthcare provider or local Poison Control Center, or go to the nearest hospital emergency room right away.
What are the possible side effects of INCIVEK?
INCIVEK may cause serious side effects including:
- See "What is the most important information I should know about INCIVEK?"
- Low red blood cell count (anemia), which can be severe. Tell your healthcare provider if you have any of these symptoms of anemia:
- dizziness
- shortness of breath
- tiredness
- weakness
Your healthcare provider will do blood tests regularly to check your red blood cell count during treatment. If your anemia is severe, your healthcare provider may tell you to stop taking INCIVEK. If INCIVEK is stopped for this reason, do not start taking it again.
Common side effects of INCIVEK in combination with peginterferon alfa and ribavirin include:
- itching
- nausea
- diarrhea
- vomiting
- anal or rectal problems, including:
- hemorrhoids
- discomfort or burning around or near the anus
- itching around or near the anus
- taste changes
- tiredness
Tell your healthcare provider about any side effect that bothers you or does not go away.
These are not all the possible side effects of INCIVEK. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Vertex Pharmaceuticals Incorporated at 1-877-824-4281.
How should I store INCIVEK?
- Store INCIVEK tablets at room temperature between 59°F to 86°F (15°C to 30°C).
Keep INCIVEK and all medicines out of the reach of children.
General information about INCIVEK
It is not known if treatment with INCIVEK will prevent you from infecting another person with the hepatitis C virus during treatment or if you do not respond to treatment.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use INCIVEK for a condition for which it was not prescribed. Do not give INCIVEK to other people, even if they have the same symptoms or condition you have. It may harm them.
This Medication Guide summarizes the most important information about INCIVEK. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about INCIVEK that is written for healthcare professionals.
For more information, go to www.incivek.com or call 1-877-824-4281.
What are the ingredients in INCIVEK?
Active ingredient: telaprevir
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, D&C Red No. 40, dibasic calcium phosphate (anhydrous), FD&C Blue No. 2, hypromellose acetate succinate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, sodium lauryl sulfate, sodium stearyl fumarate, talc, and titanium dioxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured for
Vertex Pharmaceuticals Incorporated
Cambridge, MA 02139
Revised June 2012
©2012 Vertex Pharmaceuticals Incorporated
All rights reserved.
INCIVEK and the Blue Arrow logo are trademarks of Vertex Pharmaceuticals Incorporated. VERTEX and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
The brands listed are trademarks of their respective owners. They are not trademarks of Vertex Pharmaceuticals Incorporated. The makers of these brands are not affiliated with and do not endorse Vertex Pharmaceuticals Incorporated or its products.
PRINCIPAL DISPLAY PANEL - 375 mg Tablet Carton
168 TABLETS
NDC 51167-100-01
INCIVEK™
(telaprevir)
Tablets
375 mg
Rx only
ATTENTION PHARMACIST: Dispense the enclosed Medication Guide to each patient.

| INCIVEK
telaprevir tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA201917 | 05/23/2011 | |
| Labeler - Vertex Pharmaceuticals Incorporated (602478257) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Vertex Pharmaceuticals Incorporated | 602478257 | MANUFACTURE | |
Revised: 07/2012 Vertex Pharmaceuticals Incorporated