glyset (miglitol) tablet, film coated
[Pharmacia and Upjohn Company]
DESCRIPTION
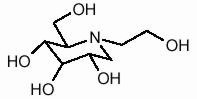
GLYSET Tablets contain miglitol, an oral alpha-glucosidase inhibitor for use in the management of non-insulin-dependent diabetes mellitus (NIDDM). Miglitol is a desoxynojirimycin derivative, and is chemically known as 3,4,5-piperidinetriol, 1-(2-hydroxyethyl)-2-(hydroxymethyl)-, [2R-(2α,3β,4α, 5β)]-. It is a white to pale-yellow powder with a molecular weight of 207.2. Miglitol is soluble in water and has a pKa of 5.9. Its empirical formula is C8H17NO5 and its chemical structure is as follows:
GLYSET is available as 25 mg, 50 mg and 100 mg tablets for oral use. The inactive ingredients are starch, microcrystalline cellulose, magnesium stearate, hypromellose, polyethylene glycol, titanium dioxide, and polysorbate 80.
CLINICAL PHARMACOLOGY
Miglitol is a desoxynojirimycin derivative that delays the digestion of ingested carbohydrates, thereby resulting in a smaller rise in blood glucose concentration following meals. As a consequence of plasma glucose reduction, GLYSET Tablets reduce levels of glycosylated hemoglobin in patients with Type II (non-insulin-dependent) diabetes mellitus. Systemic nonenzymatic protein glycosylation, as reflected by levels of glycosylated hemoglobin, is a function of average blood glucose concentration over time.
Mechanism of Action
In contrast to sulfonylureas, GLYSET does not enhance insulin secretion. The antihyperglycemic action of miglitol results from a reversible inhibition of membrane-bound intestinal α-glucoside hydrolase enzymes. Membrane-bound intestinal α-glucosidases hydrolyze oligosaccharides and disaccharides to glucose and other monosaccharides in the brush border of the small intestine. In diabetic patients, this enzyme inhibition results in delayed glucose absorption and lowering of postprandial hyperglycemia.
Because its mechanism of action is different, the effect of GLYSET to enhance glycemic control is additive to that of sulfonylureas when used in combination. In addition, GLYSET diminishes the insulinotropic and weight-increasing effects of sulfonylureas.
Miglitol has minor inhibitory activity against lactase and consequently, at the recommended doses, would not be expected to induce lactose intolerance.
Pharmacokinetics
Absorption
Absorption of miglitol is saturable at high doses: a dose of 25 mg is completely absorbed, whereas a dose of 100 mg is only 50%– 70% absorbed. For all doses, peak concentrations are reached in 2–3 hours. There is no evidence that systemic absorption of miglitol contributes to its therapeutic effect.
Distribution
The protein binding of miglitol is negligible (<4.0%). Miglitol has a volume of distribution of 0.18 L/kg, consistent with distribution primarily into the extracellular fluid.
Metabolism
Miglitol is not metabolized in man or in any animal species studied. No metabolites have been detected in plasma, urine, or feces, indicating a lack of either systemic or pre-systemic metabolism.
Excretion
Miglitol is eliminated by renal excretion as unchanged drug. Thus, following a 25-mg dose, over 95% of the dose is recovered in the urine within 24 hours. At higher doses, the cumulative recovery of drug from urine is somewhat lower due to the incomplete bioavailability. The elimination half-life of miglitol from plasma is approximately 2 hours.
Special Populations
Renal Impairment
Because miglitol is excreted primarily by the kidneys, accumulation of miglitol is expected in patients with renal impairment. Patients with creatinine clearance <25 mL/min taking 25 mg 3 times daily exhibited a greater than two-fold increase in miglitol plasma levels as compared to subjects with creatinine clearance >60 mL/min. Dosage adjustment to correct the increased plasma concentrations is not feasible because miglitol acts locally. Little information is available on the safety of miglitol in patients with creatinine clearance <25 mL/min.
Hepatic impairment
Miglitol pharmacokinetics were not altered in cirrhotic patients relative to healthy control subjects. Since miglitol is not metabolized, no influence of hepatic function on the kinetics of miglitol is expected.
Gender
No significant difference in the pharmacokinetics of miglitol was observed between elderly men and women when body weight was taken into account.
Race
Several pharmacokinetic studies were conducted in Japanese volunteers, with results similar to those observed in Caucasians. A study comparing the pharmacodynamic response to a single 50-mg dose in Black and Caucasian healthy volunteers indicated similar glucose and insulin responses in both populations.
CLINICAL STUDIES
Clinical Experience in Non-Insulin-Dependent Diabetes Mellitus (NIDDM) Patients on Dietary Treatment Only
GLYSET Tablets were evaluated in two U.S. and three non-U.S. controlled, fixed-dose, monotherapy studies, in which 735 patients treated with GLYSET were evaluated for efficacy analyses (see Table 1).
In Study 1, a one-year study in which GLYSET was evaluated as monotherapy and also as combination therapy, there was a statistically significantly smaller increase in mean glycosylated hemoglobin (HbA1c) over time in the miglitol 50 mg 3 times daily monotherapy arm compared to placebo. Significant reductions in mean fasting and postprandial plasma glucose levels and in mean postprandial insulin levels were observed in patients treated with GLYSET compared with the placebo group.
In Study 2, a 14-week study, there was a significant decrease in HbA1c in patients receiving GLYSET 50 mg 3 times daily or 100 mg 3 times daily compared to placebo. In addition, there were significant reductions in postprandial plasma glucose and postprandial serum insulin levels compared to placebo.
Study 3 was a 6-month dose-ranging trial evaluating GLYSET at doses from 25 mg 3 times daily to 200 mg 3 times daily. GLYSET produced a greater reduction in HbA1c than placebo at all doses, although the effect was statistically significant only at the 100 mg 3 times daily and 200 mg 3 times daily doses. In addition, all doses of GLYSET produced significant reductions in postprandial plasma glucose and postprandial insulin levels compared to placebo.
Studies 4 and 5 were 6-month studies evaluating GLYSET at 50 and 100 mg 3 times daily, and 100 mg 3 times daily, respectively. As compared to placebo, GLYSET produced significant reductions in HbA1c, as well as a significant reduction in postprandial plasma glucose in both studies at the doses employed.
| HbA1c (%) | 1-hour Postprandial Glucose (mg/dL) |
||||
|---|---|---|---|---|---|
| Study | Treatment | Mean Change from Baseline* | Treatment Effect† | Mean Change from Baseline | Treatment Effect† |
| 1 (U.S.) | Placebo | +0.71 | --- | +24 | --- |
| GLYSET 50 mg t.i.d.‡ | +0.13 | -0.58§ | -39 | -63§ | |
| 2 (U.S.) | Placebo | +0.47 | --- | +15 | --- |
| GLYSET 50 mg t.i.d. | -0.22 | -0.69§ | -52 | -67§ | |
| GLYSET 100 mg t.i.d. | -0.28 | -0.75§ | -59 | -74§ | |
| 3 (non-U.S.) | Placebo | +0.18 | --- | +2 | --- |
| GLYSET 25 mg t.i.d. | -0.08 | -0.26 | -33 | -35§ | |
| GLYSET 50 mg t.i.d. | -0.22 | -0.40 | -45 | -47§ | |
| GLYSET 100 mg t.i.d. | -0.63 | -0.81§ | -62 | -64§ | |
| GLYSET 200 mg t.i.d. ¶ | -0.84 | -1.02§ | -85 | -87§ | |
| 4 (non-U.S.) | Placebo | +0.01 | --- | +8 | --- |
| GLYSET 50 mg t.i.d. | -0.35 | -0.36§ | -20 | -28§ | |
| GLYSET 100 mg t.i.d. | -0.57 | -0.58§ | -25 | -33§ | |
| 5 (non-U.S.) | Placebo | +0.32 | --- | +17 | --- |
| GLYSET 100 mg t.i.d. | -0.43 | -0.75§ | -38 | -55§ | |
Clinical Experience in NIDDM Patients Receiving Sulfonylureas
GLYSET was studied as adjunctive therapy to a background of maximal or near-maximal sulfonylurea (SFU) treatment in three large, double-blind, randomized studies (two U.S. and one non-U.S.) in which 471 patients treated with GLYSET were evaluated for efficacy (see Table 2).
Study 6 included patients under treatment with maximal doses of SFU at entry. At the end of this 14-week study, the mean treatment effects on glycosylated hemoglobin (HbA1c) were -0.82% and -0.74% for patients receiving GLYSET 50 mg 3 times daily plus SFU, and GLYSET 100 mg 3 times daily plus SFU, respectively.
Study 7 was a one-year study in which GLYSET at 25, 50 or 100 mg 3 times daily was added to a maximal dose of glyburide (10 mg twice daily). At the end of this study, the mean treatment effects on HbA1c of GLYSET when added to maximum glyburide therapy were -0.30%, -0.62%, and -0.73% with the 25, 50 and 100 mg 3 times daily dosages of GLYSET, respectively.
In Study 8, the addition of GLYSET 100 mg 3 times daily to a background of treatment with glyburide produced an additional mean treatment effect on HbA1c of -0.66%.
| HbA1c (%) | 1-hour Postprandial Glucose (mg/dL) |
||||
|---|---|---|---|---|---|
| Study | Treatment | Mean Change from Baseline* | Treatment Effect† | Mean Change from Baseline | Treatment Effect† |
| 6 (U.S.) | Placebo + SFU | +0.33 | --- | -1 | --- |
| GLYSET 50 mg t.i.d.‡ + SFU | -0.49 | -0.82§ | -69 | -68§ | |
| GLYSET 100 mg t.i.d. + SFU | -0.41 | -0.74§ | -73 | -72§ | |
| 7 (U.S.) | Placebo + SFU | +1.01 | --- | 48 | --- |
| GLYSET 25 mg t.i.d. + SFU | +0.71 | -0.30 | -2 | -50§ | |
| GLYSET 50 mg t.i.d. + SFU | +0.39 | -0.62§ | -13 | -61§ | |
| GLYSET 100 mg t.i.d. + SFU | +0.28 | -0.73§ | -33 | -81§ | |
| 8 (non-U.S.) | Placebo + SFU | +0.16 | --- | +10 | --- |
| GLYSET 100 mg t.i.d. + SFU | -0.50 | -0.66§ | -36 | -46§ | |
Dose-Response
Results from controlled, fixed-dose studies of GLYSET as monotherapy or as combination treatment with a sulfonylurea were combined to derive a pooled estimate of the difference from placebo in the mean change from baseline in glycosylated hemoglobin (HbA1c) and postprandial plasma glucose as shown in Figures 1 and 2:
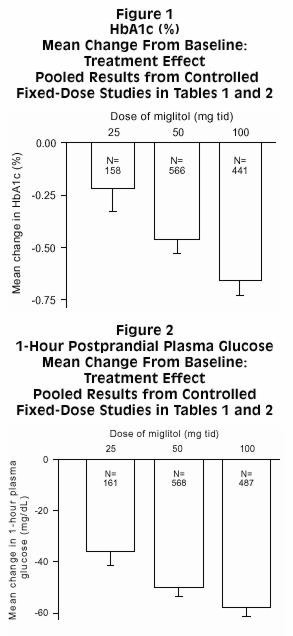
Because of its mechanism of action, the primary pharmacologic effect of miglitol is manifested as a reduction in postprandial plasma glucose, as shown previously in all of the major clinical trials. GLYSET was statistically significantly different from placebo at all doses in each of the individual studies with respect to effect on mean one-hour postprandial plasma glucose, and there is a dose response from 25 to 100 mg 3 times daily for this efficacy parameter.
INDICATIONS AND USAGE
GLYSET Tablets, as monotherapy, are indicated as an adjunct to diet to improve glycemic control in patients with non-insulin-dependent diabetes mellitus (NIDDM) whose hyperglycemia cannot be managed with diet alone. GLYSET may also be used in combination with a sulfonylurea when diet plus either GLYSET or a sulfonylurea alone do not result in adequate glycemic control. The effect of GLYSET to enhance glycemic control is additive to that of sulfonylureas when used in combination, presumably because its mechanism of action is different.
In initiating treatment for NIDDM, diet should be emphasized as the primary form of treatment. Caloric restriction and weight loss are essential in the obese diabetic patient. Proper dietary management alone may be effective in controlling blood glucose and symptoms of hyperglycemia. The importance of regular physical activity when appropriate should also be stressed. If this treatment program fails to result in adequate glycemic control, the use of GLYSET should be considered. The use of GLYSET must be viewed by both the physician and patient as a treatment in addition to diet and not as a substitute for diet or as a convenient mechanism for avoiding dietary restraint.
CONTRAINDICATIONS
GLYSET Tablets are contraindicated in patients with:
- Diabetic ketoacidosis
- Inflammatory bowel disease, colonic ulceration, or partial intestinal obstruction, and in patients predisposed to intestinal obstruction
- Chronic intestinal diseases associated with marked disorders of digestion or absorption, or with conditions that may deteriorate as a result of increased gas formation in the intestine
- Hypersensitivity to the drug or any of its components.
PRECAUTIONS
General
Hypoglycemia
Because of its mechanism of action, GLYSET when administered alone should not cause hypoglycemia in the fasted or postprandial state. Sulfonylurea agents may cause hypoglycemia. Because GLYSET Tablets given in combination with a sulfonylurea will cause a further lowering of blood glucose, it may increase the hypoglycemic potential of the sulfonylurea, although this was not observed in clinical trials. Oral glucose (dextrose), whose absorption is not delayed by GLYSET, should be used instead of sucrose (cane sugar) in the treatment of mild-to-moderate hypoglycemia. Sucrose, whose hydrolysis to glucose and fructose is inhibited by GLYSET, is unsuitable for the rapid correction of hypoglycemia. Severe hypoglycemia may require the use of either intravenous glucose infusion or glucagon injection.
Loss of Control of Blood Glucose
When diabetic patients are exposed to stress such as fever, trauma, infection, or surgery, a temporary loss of control of blood glucose may occur. At such times, temporary insulin therapy may be necessary.
Renal Impairment
Plasma concentrations of GLYSET in renally impaired volunteers were proportionally increased relative to the degree of renal dysfunction. Long-term clinical trials in diabetic patients with significant renal dysfunction (serum creatinine >2.0 mg/dL) have not been conducted. Therefore, treatment of these patients with GLYSET is not recommended.
Information for Patients
The following information should be provided to patients:
- GLYSET should be taken orally three times a day at the start (with the first bite) of each main meal. It is important to continue to adhere to dietary instructions, a regular exercise program, and regular testing of urine and/or blood glucose.
- GLYSET itself does not cause hypoglycemia even when administered to patients in the fasted state. Sulfonylurea drugs and insulin, however, can lower blood sugar levels enough to cause symptoms or sometimes life-threatening hypoglycemia. Because GLYSET given in combination with a sulfonylurea or insulin will cause a further lowering of blood sugar, it may increase the hypoglycemic potential of these agents. The risk of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development should be well understood by patients and responsible family members. Because GLYSET prevents the breakdown of table sugar, a source of glucose (dextrose, D-glucose) should be readily available to treat symptoms of low blood sugar when taking GLYSET in combination with a sulfonylurea or insulin.
- If side effects occur with GLYSET, they usually develop during the first few weeks of therapy. They are most commonly mild-to-moderate dose-related gastrointestinal effects, such as flatulence, soft stools, diarrhea, or abdominal discomfort, and they generally diminish in frequency and intensity with time. Discontinuation of drug usually results in rapid resolution of these gastrointestinal symptoms.
Laboratory Tests
Therapeutic response to GLYSET may be monitored by periodic blood glucose tests. Measurement of glycosylated hemoglobin levels is recommended for the monitoring of long-term glycemic control.
Drug Interactions
Several studies investigated the possible interaction between miglitol and glyburide. In six healthy volunteers given a single dose of 5-mg glyburide on a background of 6 days treatment with miglitol (50 mg 3 times daily for 4 days followed by 100 mg 3 times daily for 2 days) or placebo, the mean Cmax and AUC values for glyburide were 17% and 25% lower, respectively, when glyburide was given with miglitol. In a study in diabetic patients in which the effects of adding miglitol 100 mg 3 times daily × 7 days or placebo to a background regimen of 3.5 mg glyburide daily were investigated, the mean AUC value for glyburide was 18% lower in the group treated with miglitol, although this difference was not statistically significant. Further information on a potential interaction with glyburide was obtained from one of the large U.S. clinical trials (Study 7) in which patients were dosed with either miglitol or placebo on a background of glyburide 10 mg twice daily. At the 6-month and 1-year clinic visits, patients taking concomitant miglitol 100 mg 3 times daily exhibited mean Cmax values for glyburide that were 16% and 8% lower, respectively, compared to patients taking glyburide alone. However, these differences were not statistically significant. Thus, although there was a trend toward lower AUC and Cmax values for glyburide when co-administered with GLYSET, no definitive statement regarding a potential interaction can be made based on the foregoing three studies.
The effect of miglitol (100 mg 3 times daily × 7 days) on the pharmacokinetics of a single 1000-mg dose of metformin was investigated in healthy volunteers. Mean AUC and Cmax values for metformin were 12% to 13% lower when the volunteers were given miglitol as compared with placebo, but this difference was not statistically significant.
In a healthy volunteer study, co-administration of either 50 mg or 100 mg miglitol 3 times daily together with digoxin reduced the average plasma concentrations of digoxin by 19% and 28%, respectively. However, in diabetic patients under treatment with digoxin, plasma digoxin concentrations were not altered by co-administration of miglitol 100 mg 3 times daily × 14 days.
Other healthy volunteer studies have demonstrated that miglitol may significantly reduce the bioavailability of ranitidine and propranolol by 60% and 40%, respectively. No effect of miglitol was observed on the pharmacokinetics or pharmacodynamics of either warfarin or nifedipine.
Intestinal adsorbents (e.g., charcoal) and digestive enzyme preparations containing carbohydrate-splitting enzymes (e.g., amylase, pancreatin) may reduce the effect of GLYSET and should not be taken concomitantly.
In 12 healthy males, concomitantly administered antacid did not influence the pharmacokinetics of miglitol.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Miglitol was administered to mice by the dietary route at doses as high as approximately 500 mg/kg body weight (corresponding to greater than 5 times the exposure in humans based on AUC) for 21 months. In a two-year rat study, miglitol was administered in the diet at exposures comparable to the maximum human exposures based on AUC. There was no evidence of carcinogenicity resulting from dietary treatment with miglitol.
In vitro, miglitol was found to be nonmutagenic in the bacterial mutagenesis (Ames) assay and the eukaryotic forward mutation assay (CHO/HGPRT). Miglitol did not have any clastogenic effects in vivo in the mouse micronucleus test. There were no heritable mutations detected in dominant lethal assay.
A combined male and female fertility study conducted in Wistar rats treated orally with miglitol at dose levels of 300 mg/kg body weight (approximately 8 times the maximum human exposure based on body surface area) produced no untoward effect on reproductive performance or capability to reproduce. In addition, survival, growth, development, and fertility of the offspring were not compromised.
Pregnancy
Teratogenic Effects
Pregnancy Category B
The safety of GLYSET in pregnant women has not been established. Developmental toxicology studies have been performed in rats at doses of 50, 150 and 450 mg/kg, corresponding to levels of approximately 1.5, 4, and 12 times the maximum recommended human exposure based on body surface area. In rabbits, doses of 10, 45, and 200 mg/kg corresponding to levels of approximately 0.5, 3, and 10 times the human exposure were examined. These studies revealed no evidence of fetal malformations attributable to miglitol. Doses of miglitol up to 4 and 3 times the human dose (based on body surface area), for rats and rabbits, respectively, did not reveal evidence of impaired fertility or harm to the fetus. The highest doses tested in these studies, 450 mg/kg in the rat and 200 mg/kg in the rabbit promoted maternal and/or fetal toxicity. Fetotoxicity was indicated by a slight but significant reduction in fetal weight in the rat study and slight reduction in fetal weight, delayed ossification of the fetal skeleton and increase in the percentage of non-viable fetuses in the rabbit study. In the peri-postnatal study in rats, the NOAEL (No Observed Adverse Effect Level) was 100 mg/kg (corresponding to approximately four times the exposure to humans, based on body surface area). An increase in stillborn progeny was noted at the high dose (300 mg/kg) in the rat peri-postnatal study, but not at the high dose (450 mg/kg) in the delivery segment of the rat developmental toxicity study. Otherwise, there was no adverse effect on survival, growth, development, behavior, or fertility in either the rat developmental toxicity or peri-postnatal studies. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Miglitol has been shown to be excreted in human milk to a very small degree. Total excretion into milk accounted for 0.02% of a 100-mg maternal dose. The estimated exposure to a nursing infant is approximately 0.4% of the maternal dose. Although the levels of miglitol reached in human milk are exceedingly low, it is recommended that GLYSET not be administered to a nursing woman.
Pediatric Use
Safety and effectiveness of GLYSET in pediatric patients have not been established.
Geriatric Use
Of the total number of subjects in clinical studies of GLYSET in the United States, patients valid for safety analyses included 24% over 65, and 3% over 75. No overall differences in safety and effectiveness were observed between these subjects and younger subjects. The pharmacokinetics of miglitol were studied in elderly and young males (n=8 per group). At the dosage of 100 mg 3 times daily for 3 days, no differences between the two groups were found.
ADVERSE REACTIONS
Gastrointestinal
Gastrointestinal symptoms are the most common reactions to GLYSET Tablets. In U.S. placebo-controlled trials, the incidences of abdominal pain, diarrhea, and flatulence were 11.7%, 28.7%, and 41.5% respectively in 962 patients treated with GLYSET 25–100 mg 3 times daily, whereas the corresponding incidences were 4.7%, 10.0%, and 12.0% in 603 placebo-treated patients. The incidence of diarrhea and abdominal pain tended to diminish considerably with continued treatment.
Dermatologic
Skin rash was reported in 4.3% of patients treated with GLYSET compared to 2.4% of placebo-treated patients. Rashes were generally transient and most were assessed as unrelated to GLYSET by physician-investigators.
Abnormal Laboratory Findings
Low serum iron occurred more often in patients treated with GLYSET (9.2%) than in placebo-treated patients (4.2%) but did not persist in the majority of cases and was not associated with reductions in hemoglobin or changes in other hematologic indices.
OVERDOSAGE
Unlike sulfonylureas or insulin, an overdose of GLYSET Tablets will not result in hypoglycemia. An overdose may result in transient increases in flatulence, diarrhea, and abdominal discomfort. Because of the lack of extra-intestinal effects seen with GLYSET, no serious systemic reactions are expected in the event of an overdose.
DOSAGE AND ADMINISTRATION
There is no fixed dosage regimen for the management of diabetes mellitus with GLYSET Tablets or any other pharmacologic agent. Dosage of GLYSET must be individualized on the basis of both effectiveness and tolerance while not exceeding the maximum recommended dosage of 100 mg 3 times daily. GLYSET should be taken three times daily at the start (with the first bite) of each main meal. GLYSET should be started at 25 mg, and the dosage gradually increased as described below, both to reduce gastrointestinal adverse effects and to permit identification of the minimum dose required for adequate glycemic control of the patient.
During treatment initiation and dose titration (see below), one-hour postprandial plasma glucose may be used to determine the therapeutic response to GLYSET and identify the minimum effective dose for the patient. Thereafter, glycosylated hemoglobin should be measured at intervals of approximately three months. The therapeutic goal should be to decrease both postprandial plasma glucose and glycosylated hemoglobin levels to normal or near normal by using the lowest effective dose of GLYSET, either as monotherapy or in combination with a sulfonylurea.
Initial Dosage
The recommended starting dosage of GLYSET is 25 mg, given orally three times daily at the start (with the first bite) of each main meal. However, some patients may benefit by starting at 25 mg once daily to minimize gastrointestinal adverse effects, and gradually increasing the frequency of administration to 3 times daily.
Maintenance Dosage
The usual maintenance dose of GLYSET is 50 mg 3 times daily, although some patients may benefit from increasing the dose to 100 mg 3 times daily. In order to allow adaptation to potential gastrointestinal adverse effects, it is recommended that GLYSET therapy be initiated at a dosage of 25 mg 3 times daily, the lowest effective dosage, and then gradually titrated upward to allow adaptation. After 4 – 8 weeks of the 25 mg 3 times daily regimen, the dosage should be increased to 50 mg 3 times daily for approximately three months, following which a glycosylated hemoglobin level should be measured to assess therapeutic response. If, at that time, the glycosylated hemoglobin level is not satisfactory, the dosage may be further increased to 100 mg 3 times daily, the maximum recommended dosage. Pooled data from controlled studies suggest a dose-response for both HbA1c and one-hour postprandial plasma glucose throughout the recommended dosage range. However, no single study has examined the effect on glycemic control of titrating patients' doses upwards within the same study. If no further reduction in postprandial glucose or glycosylated hemoglobin levels is observed with titration to 100 mg 3 times daily, consideration should be given to lowering the dose. Once an effective and tolerated dosage is established, it should be maintained.
Maximum Dosage
The maximum recommended dosage of GLYSET is 100 mg 3 times daily. In one clinical trial, 200 mg 3 times daily gave additional improved glycemic control but increased the incidence of the gastrointestinal symptoms described above.
Patients Receiving Sulfonylureas
Sulfonylurea agents may cause hypoglycemia. There was no increased incidence of hypoglycemia in patients who took GLYSET in combination with sulfonylurea agents compared to the incidence of hypoglycemia in patients receiving sulfonylureas alone in any clinical trial. However, GLYSET given in combination with a sulfonylurea will cause a further lowering of blood glucose and may increase the risk of hypoglycemia due to the additive effects of the two agents. If hypoglycemia occurs, appropriate adjustments in the dosage of these agents should be made.
HOW SUPPLIED
GLYSET Tablets are available as 25 mg, 50 mg, and 100 mg white, round, film-coated tablets. The tablets are debossed with the word "GLYSET" on one side and the strength on the other side, as indicated below.
| Tablet Identification |
|||
|---|---|---|---|
| Strength | NDC | Front | Back |
| Bottles of 100: | |||
| 25 mg | 0009-5012-01 | GLYSET | 25 |
| 50 mg | 0009-5013-01 | GLYSET | 50 |
| 100 mg | 0009-5014-01 | GLYSET | 100 |
Store at 25°C (77°F); excursions permitted to 15°–30°C (59°–86°F) [see USP Controlled Room Temperature].
Rx only
U.S. Patent No. 4,639,436

Manufactured by:
Bayer HealthCare AG
Leverkusen, Germany
GLYSET is a registered trademark of Bayer Pharmaceuticals Corporation used under license.
LAB-0167-4.0
| Glyset (miglitol) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
| Glyset (miglitol) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
| Glyset (miglitol) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
Revised: 03/2006