amaryl (glimepiride) tablet
[sanofi-aventis U.S. LLC]
Rx Only
DESCRIPTION
AMARYL® (glimepiride tablets) is an oral blood-glucose-lowering drug of the sulfonylurea class. Glimepiride is a white to yellowish-white, crystalline, odorless to practically odorless powder formulated into tablets of 1-mg, 2-mg, and 4-mg strengths for oral administration. AMARYL Tablets contain the active ingredient glimepiride and the following inactive ingredients: lactose (hydrous), sodium starch glycolate, povidone, microcrystalline cellulose, and magnesium stearate. In addition, AMARYL 1-mg tablets contain Ferric Oxide Red, AMARYL 2-mg tablets contain Ferric Oxide Yellow and FD&C Blue #2 Aluminum Lake, and AMARYL 4-mg tablets contain FD&C Blue #2 Aluminum Lake.
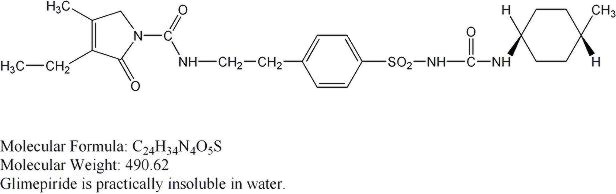
Chemically, glimepiride is identified as 1-[[p-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)ethyl]phenyl]sulfonyl]-3-(trans-4-methylcyclohexyl)urea. The CAS Registry Number is 93479-97-1
The structural formula is:
CLINICAL PHARMACOLOGY
Mechanism of Action
The primary mechanism of action of glimepiride in lowering blood glucose appears to be dependent on stimulating the release of insulin from functioning pancreatic beta cells. In addition, extrapancreatic effects may also play a role in the activity of sulfonylureas such as glimepiride. This is supported by both preclinical and clinical studies demonstrating that glimepiride administration can lead to increased sensitivity of peripheral tissues to insulin. These findings are consistent with the results of a long-term, randomized, placebo-controlled trial in which AMARYL therapy improved postprandial insulin/C-peptide responses and overall glycemic control without producing clinically meaningful increases in fasting insulin/C-peptide levels. However, as with other sulfonylureas, the mechanism by which glimepiride lowers blood glucose during long-term administration has not been clearly established.
AMARYL is effective as initial drug therapy. In patients where monotherapy with AMARYL or metformin has not produced adequate glycemic control, the combination of AMARYL and metformin may have a synergistic effect, since both agents act to improve glucose tolerance by different primary mechanisms of action. This complementary effect has been observed with metformin and other sulfonylureas, in multiple studies.
Pharmacodynamics
A mild glucose-lowering effect first appeared following single oral doses as low as 0.5–0.6 mg in healthy subjects. The time required to reach the maximum effect (i.e., minimum blood glucose level [Tmin]) was about 2 to 3 hours. In noninsulin-dependent (Type 2) diabetes mellitus (NIDDM) patients, both fasting and 2-hour postprandial glucose levels were significantly lower with glimepiride (1, 2, 4, and 8 mg once daily) than with placebo after 14 days of oral dosing. The glucose-lowering effect in all active treatment groups was maintained over 24 hours.
In larger dose-ranging studies, blood glucose and HbA1c were found to respond in a dose-dependent manner over the range of 1 to 4 mg/day of AMARYL. Some patients, particularly those with higher fasting plasma glucose (FPG) levels, may benefit from doses of AMARYL up to 8 mg once daily. No difference in response was found when AMARYL was administered once or twice daily.
In two 14-week, placebo-controlled studies in 720 subjects, the average net reduction in HbA1c for AMARYL (glimepiride tablets) patients treated with 8 mg once daily was 2.0% in absolute units compared with placebo-treated patients. In a long-term, randomized, placebo-controlled study of Type 2 diabetic patients unresponsive to dietary management, AMARYL therapy improved postprandial insulin/C-peptide responses, and 75% of patients achieved and maintained control of blood glucose and HbA1c. Efficacy results were not affected by age, gender, weight, or race.
In long-term extension trials with previously-treated patients, no meaningful deterioration in mean fasting blood glucose (FBG) or HbA1c levels was seen after 2 1/2 years of AMARYL therapy.
Combination therapy with AMARYL and insulin (70% NPH/30% regular) was compared to placebo/insulin in secondary failure patients whose body weight was >130% of their ideal body weight. Initially, 5–10 units of insulin were administered with the main evening meal and titrated upward weekly to achieve predefined FPG values. Both groups in this double-blind study achieved similar reductions in FPG levels but the AMARYL/insulin therapy group used approximately 38% less insulin.
AMARYL therapy is effective in controlling blood glucose without deleterious changes in the plasma lipoprotein profiles of patients treated for Type 2 diabetes.
Pharmacokinetics
Absorption
After oral administration, glimepiride is completely (100%) absorbed from the GI tract. Studies with single oral doses in normal subjects and with multiple oral doses in patients with Type 2 diabetes have shown significant absorption of glimepiride within 1 hour after administration and peak drug levels (Cmax) at 2 to 3 hours. When glimepiride was given with meals, the mean Tmax (time to reach Cmax) was slightly increased (12%) and the mean Cmax and AUC (area under the curve) were slightly decreased (8% and 9%, respectively).
Distribution
After intravenous (IV) dosing in normal subjects, the volume of distribution (Vd) was 8.8 L (113 mL/kg), and the total body clearance (CL) was 47.8 mL/min. Protein binding was greater than 99.5%.
Metabolism
Glimepiride is completely metabolized by oxidative biotransformation after either an IV or oral dose. The major metabolites are the cyclohexyl hydroxy methyl derivative (M1) and the carboxyl derivative (M2). Cytochrome P450 2C9 has been shown to be involved in the biotransformation of glimepiride to M1. M1 is further metabolized to M2 by one or several cytosolic enzymes. M1, but not M2, possesses about 1/3 of the pharmacological activity as compared to its parent in an animal model; however, whether the glucose-lowering effect of M1 is clinically meaningful is not clear.
Excretion
When 14C-glimepiride was given orally, approximately 60% of the total radioactivity was recovered in the urine in 7 days and M1 (predominant) and M2 accounted for 80–90% of that recovered in the urine. Approximately 40% of the total radioactivity was recovered in feces and M1 and M2 (predominant) accounted for about 70% of that recovered in feces. No parent drug was recovered from urine or feces. After IV dosing in patients, no significant biliary excretion of glimepiride or its M1 metabolite has been observed.
Pharmacokinetic Parameters
The pharmacokinetic parameters of glimepiride obtained from a single-dose, crossover, dose-proportionality (1, 2, 4, and 8 mg) study in normal subjects and from a single- and multiple-dose, parallel, dose-proportionality (4 and 8 mg) study in patients with Type 2 diabetes are summarized below:
| Volunteers | Patients with Type 2 diabetes | ||
|---|---|---|---|
| Single Dose Mean±SD | Single Dose (Day 1) Mean±SD | Multiple Dose (Day 10) Mean±SD |
|
| ( ) = No. of subjects | |||
| CL/f=Total body clearance after oral dosing | |||
| Vd/f=Volume of distribution calculated after oral dosing | |||
| Cmax (ng/mL) | |||
| 1 mg | 103 ± 34 (12) | — | — |
| 2 mg | 177 ± 44 (12) | — | — |
| 4 mg | 308 ± 69 (12) | 352 ± 222 (12) | 309 ± 134 (12) |
| 8 mg | 551± 152 (12) | 591 ± 232 (14) | 578 ± 265 (11) |
| Tmax (h) | 2.4 ± 0.8 (48) | 2.5 ± 1.2 (26) | 2.8 ± 2.2 (23) |
| CL/f (mL/min) | 52.1 ± 16.0 (48) | 48.5 ± 29.3 (26) | 52.7 ± 40.3 (23) |
| Vd/f (L) | 21.8 ± 13.9 (48) | 19.8 ± 12.7 (26) | 37.1 ± 18.2 (23) |
| T1/2 (h) | 5.3 ± 4.1 (48) | 5.0 ± 2.5 (26) | 9.2 ± 3.6 (23) |
These data indicate that glimepiride did not accumulate in serum, and the pharmacokinetics of glimepiride were not different in healthy volunteers and in Type 2 diabetes patients. Oral clearance of glimepiride did not change over the 1–8-mg dose range, indicating linear pharmacokinetics.
Variability
In normal healthy volunteers, the intra-individual variabilities of Cmax, AUC, and CL/f for glimepiride were 23%, 17%, and 15%, respectively, and the inter-individual variabilities were 25%, 29%, and 24%, respectively.
Special Populations
Geriatric
Comparison of glimepiride pharmacokinetics in Type 2 diabetic patients ≤65 years and those >65 years was performed in a study using a dosing regimen of 6 mg daily. There were no significant differences in glimepiride pharmacokinetics between the two age groups. The mean AUC at steady state for the older patients was about 13% lower than that for the younger patients; the mean weight-adjusted clearance for the older patients was about 11% higher than that for the younger patients.
Pediatric
The pharmacokinetics of glimepiride (1 mg) were evaluated in a single dose study conducted in 30 Type 2 diabetic patients (Male = 7; Female = 23) between ages 10 and 17 years. The mean AUC(0–last)(338.8±203.1 ng∙hr/mL), Cmax (102.4±47.7 ng/mL) and T1/2(3.1±1.7 hours) were comparable to those previously reported in adults (AUC(0–last) 315.2±95.9 ng∙hr/mL, Cmax 103.2±34.3 ng/mL and T1/2 5.3±4.1 hours).
Gender
There were no differences between males and females in the pharmacokinetics of glimepiride when adjustment was made for differences in body weight.
Race
No pharmacokinetic studies to assess the effects of race have been performed, but in placebo-controlled studies of AMARYL (glimepiride tablets) in patients with Type 2 diabetes, the antihyperglycemic effect was comparable in whites (n = 536), blacks (n = 63), and Hispanics (n = 63).
Renal Insufficiency
A single-dose, open-label study was conducted in 15 patients with renal impairment. AMARYL (3 mg) was administered to 3 groups of patients with different levels of mean creatinine clearance (CLcr); (Group I, CLcr = 77.7 mL/min, n = 5), (Group II, CLcr = 27.7 mL/min, n = 3), and (Group III, CLcr = 9.4 mL/min, n = 7). AMARYL was found to be well tolerated in all 3 groups. The results showed that glimepiride serum levels decreased as renal function decreased. However, M1 and M2 serum levels (mean AUC values) increased 2.3 and 8.6 times from Group I to Group III. The apparent terminal half-life (T1/2) for glimepiride did not change, while the half-lives for M1 and M2 increased as renal function decreased. Mean urinary excretion of M1 plus M2 as percent of dose, however, decreased (44.4%, 21.9%, and 9.3% for Groups I to III).
A multiple-dose titration study was also conducted in 16 Type 2 diabetic patients with renal impairment using doses ranging from 1–8 mg daily for 3 months. The results were consistent with those observed after single doses. All patients with a CLcr less than 22 mL/min had adequate control of their glucose levels with a dosage regimen of only 1 mg daily. The results from this study suggested that a starting dose of 1 mg AMARYL may be given to Type 2 diabetic patients with kidney disease, and the dose may be titrated based on fasting blood glucose levels.
Hepatic Insufficiency
No studies were performed in patients with hepatic insufficiency.
Other Populations
There were no important differences in glimepiride metabolism in subjects identified as phenotypically different drug-metabolizers by their metabolism of sparteine. The pharmacokinetics of glimepiride in morbidly obese patients were similar to those in the normal weight group, except for a lower Cmax and AUC. However, since neither Cmax nor AUC values were normalized for body surface area, the lower values of Cmax and AUC for the obese patients were likely the result of their excess weight and not due to a difference in the kinetics of glimepiride.
Drug Interactions
The hypoglycemic action of sulfonylureas may be potentiated by certain drugs, including nonsteroidal anti-inflammatory drugs and other drugs that are highly protein bound, such as salicylates, sulfonamides, chloramphenicol, coumarins, probenecid, monoamine oxidase inhibitors, and beta adrenergic blocking agents. When these drugs are administered to a patient receiving AMARYL, the patient should be observed closely for hypoglycemia. When these drugs are withdrawn from a patient receiving AMARYL, the patient should be observed closely for loss of glycemic control.
Certain drugs tend to produce hyperglycemia and may lead to loss of control. These drugs include the thiazides and other diuretics, corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, and isoniazid. When these drugs are administered to a patient receiving AMARYL, the patient should be closely observed for loss of control. When these drugs are withdrawn from a patient receiving AMARYL, the patient should be observed closely for hypoglycemia.
Coadministration of aspirin (1 g tid) and AMARYL led to a 34% decrease in the mean glimepiride AUC and, therefore, a 34% increase in the mean CL/f. The mean Cmax had a decrease of 4%. Blood glucose and serum C-peptide concentrations were unaffected and no hypoglycemic symptoms were reported. Pooled data from clinical trials showed no evidence of clinically significant adverse interactions with uncontrolled concurrent administration of aspirin and other salicylates.
Coadministration of either cimetidine (800 mg once daily) or ranitidine (150 mg bid) with a single 4-mg oral dose of AMARYL did not significantly alter the absorption and disposition of glimepiride, and no differences were seen in hypoglycemic symptomatology. Pooled data from clinical trials showed no evidence of clinically significant adverse interactions with uncontrolled concurrent administration of H2-receptor antagonists.
Concomitant administration of propranolol (40 mg tid) and AMARYL significantly increased Cmax, AUC, and T1/2 of glimepiride by 23%, 22%, and 15%, respectively, and it decreased CL/f by 18%. The recovery of M1 and M2 from urine, however, did not change. The pharmacodynamic responses to glimepiride were nearly identical in normal subjects receiving propranolol and placebo. Pooled data from clinical trials in patients with Type 2 diabetes showed no evidence of clinically significant adverse interactions with uncontrolled concurrent administration of beta-blockers. However, if beta-blockers are used, caution should be exercised and patients should be warned about the potential for hypoglycemia.
Concomitant administration of AMARYL (glimepiride tablets) (4 mg once daily) did not alter the pharmacokinetic characteristics of R-and S-warfarin enantiomers following administration of a single dose (25 mg) of racemic warfarin to healthy subjects. No changes were observed in warfarin plasma protein binding. AMARYL treatment did result in a slight, but statistically significant, decrease in the pharmacodynamic response to warfarin. The reductions in mean area under the prothrombin time (PT) curve and maximum PT values during AMARYL treatment were very small (3.3% and 9.9%, respectively) and are unlikely to be clinically important.
The responses of serum glucose, insulin, C-peptide, and plasma glucagon to 2 mg AMARYL were unaffected by coadministration of ramipril (an ACE inhibitor) 5 mg once daily in normal subjects. No hypoglycemic symptoms were reported. Pooled data from clinical trials in patients with Type 2 diabetes showed no evidence of clinically significant adverse interactions with uncontrolled concurrent administration of ACE inhibitors.
A potential interaction between oral miconazole and oral hypoglycemic agents leading to severe hypoglycemia has been reported. Whether this interaction also occurs with the intravenous, topical, or vaginal preparations of miconazole is not known. There is a potential interaction of glimepiride with inhibitors (e.g. fluconazole) and inducers (e.g. rifampicin) of cytochrome P450 2C9.
Although no specific interaction studies were performed, pooled data from clinical trials showed no evidence of clinically significant adverse interactions with uncontrolled concurrent administration of calcium-channel blockers, estrogens, fibrates, NSAIDS, HMG CoA reductase inhibitors, sulfonamides, or thyroid hormone.
INDICATIONS AND USAGE
AMARYL is indicated as an adjunct to diet and exercise to lower the blood glucose in patients with noninsulin-dependent (Type 2) diabetes mellitus (NIDDM) whose hyperglycemia cannot be controlled by diet and exercise alone. AMARYL may be used concomitantly with metformin when diet, exercise, and AMARYL or metformin alone do not result in adequate glycemic control.
AMARYL is also indicated for use in combination with insulin to lower blood glucose in patients whose hyperglycemia cannot be controlled by diet and exercise in conjunction with an oral hypoglycemic agent. Combined use of glimepiride and insulin may increase the potential for hypoglycemia.
In initiating treatment for noninsulin-dependent diabetes, diet and exercise should be emphasized as the primary form of treatment. Caloric restriction, weight loss, and exercise are essential in the obese diabetic patient. Proper dietary management and exercise alone may be effective in controlling the blood glucose and symptoms of hyperglycemia. In addition to regular physical activity, cardiovascular risk factors should be identified and corrective measures taken where possible.
If this treatment program fails to reduce symptoms and/or blood glucose, the use of an oral sulfonylurea or insulin should be considered. Use of AMARYL must be viewed by both the physician and patient as a treatment in addition to diet and exercise and not as a substitute for diet and exercise or as a convenient mechanism for avoiding dietary restraint. Furthermore, loss of blood glucose control on diet and exercise alone may be transient, thus requiring only short-term administration of AMARYL.
During maintenance programs, AMARYL monotherapy should be discontinued if satisfactory lowering of blood glucose is no longer achieved. Judgments should be based on regular clinical and laboratory evaluations. Secondary failures to AMARYL monotherapy can be treated with AMARYL-insulin combination therapy.
In considering the use of AMARYL in asymptomatic patients, it should be recognized that blood glucose control in Type 2 diabetes has not definitely been established to be effective in preventing the long-term cardiovascular and neural complications of diabetes. However, the Diabetes Control and Complications Trial (DCCT) demonstrated that control of HbA1c and glucose was associated with a decrease in retinopathy, neuropathy, and nephropathy for insulin-dependent diabetic (IDDM) patients.
CONTRAINDICATIONS
AMARYL is contraindicated in patients with
- Known hypersensitivity to the drug.
- Diabetic ketoacidosis, with or without coma. This condition should be treated with insulin.
WARNINGS
SPECIAL WARNING ON INCREASED RISK OF CARDIOVASCULAR MORTALITY
The administration of oral hypoglycemic drugs has been reported to be associated with increased cardiovascular mortality as compared to treatment with diet alone or diet plus insulin. This warning is based on the study conducted by the University Group Diabetes Program (UGDP), a long-term, prospective clinical trial designed to evaluate the effectiveness of glucose-lowering drugs in preventing or delaying vascular complications in patients with non-insulin-dependent diabetes. The study involved 823 patients who were randomly assigned to one of four treatment groups (Diabetes, 19 supp. 2: 747–830, 1970).
UGDP reported that patients treated for 5 to 8 years with diet plus a fixed dose of tolbutamide (1.5 grams per day) had a rate of cardiovascular mortality approximately 2-1/2 times that of patients treated with diet alone. A significant increase in total mortality was not observed, but the use of tolbutamide was discontinued based on the increase in cardiovascular mortality, thus limiting the opportunity for the study to show an increase in overall mortality. Despite controversy regarding the interpretation of these results, the findings of the UGDP study provide an adequate basis for this warning. The patient should be informed of the potential risks and advantages of AMARYL (glimepiride tablets) and of alternative modes of therapy.
Although only one drug in the sulfonylurea class (tolbutamide) was included in this study, it is prudent from a safety standpoint to consider that this warning may also apply to other oral hypoglycemic drugs in this class, in view of their close similarities in mode of action and chemical structure.
PRECAUTIONS
General
Hypoglycemia
All sulfonylurea drugs are capable of producing severe hypoglycemia. Proper patient selection, dosage, and instructions are important to avoid hypoglycemic episodes. Patients with impaired renal function may be more sensitive to the glucose-lowering effect of AMARYL. A starting dose of 1 mg once daily followed by appropriate dose titration is recommended in those patients. Debilitated or malnourished patients, and those with adrenal, pituitary, or hepatic insufficiency are particularly susceptible to the hypoglycemic action of glucose-lowering drugs. Hypoglycemia may be difficult to recognize in the elderly and in people who are taking beta-adrenergic blocking drugs or other sympatholytic agents. Hypoglycemia is more likely to occur when caloric intake is deficient, after severe or prolonged exercise, when alcohol is ingested, or when more than one glucose-lowering drug is used. Combined use of glimepiride with insulin or metformin may increase the potential for hypoglycemia.
Loss of control of blood glucose
When a patient stabilized on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a loss of control may occur. At such times, it may be necessary to add insulin in combination with AMARYL or even use insulin monotherapy. The effectiveness of any oral hypoglycemic drug, including AMARYL, in lowering blood glucose to a desired level decreases in many patients over a period of time, which may be due to progression of the severity of the diabetes or to diminished responsiveness to the drug. This phenomenon is known as secondary failure, to distinguish it from primary failure in which the drug is ineffective in an individual patient when first given. Should secondary failure occur with AMARYL or metformin monotherapy, combined therapy with AMARYL and metformin or AMARYL and insulin may result in a response. Should secondary failure occur with combined AMARYL/metformin therapy, it may be necessary to initiate insulin therapy.
Information For Patients
Patients should be informed of the potential risks and advantages of AMARYL and of alternative modes of therapy. They should also be informed about the importance of adherence to dietary instructions, of a regular exercise program, and of regular testing of blood glucose. The risks of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development should be explained to patients and responsible family members. The potential for primary and secondary failure should also be explained.
Laboratory Tests
Fasting blood glucose should be monitored periodically to determine therapeutic response. Glycosylated hemoglobin should also be monitored, usually every 3 to 6 months, to more precisely assess long-term glycemic control.
Drug Interactions
(See CLINICAL PHARMACOLOGY, Drug Interactions)
Carcinogenesis, Mutagenesis, and Impairment Of Fertility
Studies in rats at doses of up to 5000 ppm in complete feed (approximately 340 times the maximum recommended human dose, based on surface area) for 30 months showed no evidence of carcinogenesis. In mice, administration of glimepiride for 24 months resulted in an increase in benign pancreatic adenoma formation which was dose related and is thought to be the result of chronic pancreatic stimulation. The no-effect dose for adenoma formation in mice in this study was 320 ppm in complete feed, or 46–54 mg/kg body weight/day. This is about 35 times the maximum human recommended dose of 8 mg once daily based on surface area.
Glimepiride was non-mutagenic in a battery of in vitro and in vivo mutagenicity studies (Ames test, somatic cell mutation, chromosomal aberration, unscheduled DNA synthesis, mouse micronucleus test).
There was no effect of glimepiride on male mouse fertility in animals exposed up to 2500 mg/kg body weight (>1,700 times the maximum recommended human dose based on surface area). Glimepiride had no effect on the fertility of male and female rats administered up to 4000 mg/kg body weight (approximately 4,000 times the maximum recommended human dose based on surface area).
Pregnancy
Teratogenic Effects
Pregnancy Category C. Glimepiride did not produce teratogenic effects in rats exposed orally up to 4000 mg/kg body weight (approximately 4,000 times the maximum recommended human dose based on surface area) or in rabbits exposed up to 32 mg/kg body weight (approximately 60 times the maximum recommended human dose based on surface area). Glimepiride has been shown to be associated with intrauterine fetal death in rats when given in doses as low as 50 times the human dose based on surface area and in rabbits when given in doses as low as 0.1 times the human dose based on surface area. This fetotoxicity, observed only at doses inducing maternal hypoglycemia, has been similarly noted with other sulfonylureas, and is believed to be directly related to the pharmacologic (hypoglycemic) action of glimepiride.
There are no adequate and well-controlled studies in pregnant women. On the basis of results from animal studies, Amaryl (glimepiride tablets) should not be used during pregnancy. Because recent information suggests that abnormal blood glucose levels during pregnancy are associated with a higher incidence of congenital abnormalities, many experts recommend that insulin be used during pregnancy to maintain glucose levels as close to normal as possible.
Nonteratogenic Effects
In some studies in rats, offspring of dams exposed to high levels of glimepiride during pregnancy and lactation developed skeletal deformities consisting of shortening, thickening, and bending of the humerus during the postnatal period. Significant concentrations of glimepiride were observed in the serum and breast milk of the dams as well as in the serum of the pups. These skeletal deformations were determined to be the result of nursing from mothers exposed to glimepiride.
Prolonged severe hypoglycemia (4 to 10 days) has been reported in neonates born to mothers who were receiving a sulfonylurea drug at the time of delivery. This has been reported more frequently with the use of agents with prolonged half-lives. Patients who are planning a pregnancy should consult their physician, and it is recommended that they change over to insulin for the entire course of pregnancy and lactation.
Nursing Mothers
In rat reproduction studies, significant concentrations of glimepiride were observed in the serum and breast milk of the dams, as well as in the serum of the pups. Although it is not known whether AMARYL is excreted in human milk, other sulfonylureas are excreted in human milk. Because the potential for hypoglycemia in nursing infants may exist, and because of the effects on nursing animals, AMARYL should be discontinued in nursing mothers. If AMARYL is discontinued, and if diet and exercise alone are inadequate for controlling blood glucose, insulin therapy should be considered. (See above Pregnancy, Nonteratogenic Effects.)
Pediatric Use
The safety and efficacy of AMARYL were evaluated in an active-controlled, single-blind (patients only), 24-week trial involving 272 pediatric patients, ranging from 8 to 17 years of age, with Type 2 diabetes. AMARYL (n=135) was administered at 1mg initially, and then titrated up to 2, 4 or 8 mg (mean last dose 4 mg) until the therapeutic goal of self-monitored fasting blood glucose < 7.0 mmol/L (< 126 mg/dl) was achieved. The active comparator metformin (n=137) was administered at 500 mg twice daily initially and titrated up to 1000 mg twice daily (mean last dose 1365 mg).
| HbA1c (%) | Naïve Patients* | Previously Treated Patients* | ||
|---|---|---|---|---|
| Metformin | AMARYL | Metformin | AMARYL | |
| 69 | 72 | 57 | 55 | |
| Baseline (mean) | 8.2 | 8.3 | 9.0 | 8.7 |
| Change from baseline (mean)† | -1.2 | -1.0 | -0.2 | 0.2 |
| Adjusted Treatment Difference‡ | 0.2 | 0.4 | ||
| (95% CI) | (-0.3; 0.7) | (-0.4; 1.2) | ||
The profile of adverse reactions in pediatric patients treated with Amaryl was similar to that observed in adults.
Hypoglycemic events, as documented by blood glucose values <36 mg/dL, were observed in 4% of patients treated with AMARYL and in 1% of patients treated with metformin.
| Weight (kg) | Metformin* | AMARYL* |
|---|---|---|
| Baseline (mean) | 67.3 | 66.5 |
| Change from baseline (mean)† | 0.7 | 2.0 |
| Adjusted Treatment Difference‡ | 1.3 | |
| (95% CI) | (0.3; 2.3) | |
Geriatric Use
In US clinical studies of AMARYL, 608 of 1986 patients were 65 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
Comparison of glimepiride pharmacokinetics in Type 2 diabetic patients ≤65 years (n=49) and those >65 years (n=42) was performed in a study using a dosing regimen of 6 mg daily. There were no significant differences in glimepiride pharmacokinetics between the two age groups (see CLINICAL PHARMACOLOGY, Special Populations, Geriatric).
The drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
Elderly patients are particularly susceptible to hypoglycemic action of glucose-lowering drugs. In elderly, debilitated, or malnourished patients, or in patients with renal and hepatic insufficiency, the initial dosing, dose increments, and maintenance dosage should be conservative based upon blood glucose levels prior to and after initiation of treatment to avoid hypoglycemic reactions. Hypoglycemia may be difficult to recognize in the elderly and in people who are taking beta-adrenergic blocking drugs or other sympatholytic agents (see CLINICAL PHARMACOLOGY, Special Populations, Renal Insufficiency; PRECAUTIONS, General; and DOSING AND ADMINISTRATION, Special Patient Population).
ADVERSE REACTIONS
Adult Patients
The incidence of hypoglycemia with AMARYL, as documented by blood glucose values <60 mg/dL, ranged from 0.9–1.7% in two large, well controlled, 1-year studies. (See WARNINGS and PRECAUTIONS.)
AMARYL has been evaluated for safety in 2,013 patients in US controlled trials, and in 1,551 patients in foreign controlled trials. More than 1,650 of these patients were treated for at least 1 year. Adverse events, other than hypoglycemia, considered to be possibly or probably related to study drug that occurred in US placebo-controlled trials in more than 1% of patients treated with AMARYL are shown below.
| Adverse Events Occurring in ≥1% AMARYL Patients | ||||
|---|---|---|---|---|
| AMARYL | Placebo | |||
| No. | % | No. | % | |
| Total Treated | 746 | 100 | 294 | 100 |
| Dizziness | 13 | 1.7 | 1 | 0.3 |
| Asthenia | 12 | 1.6 | 3 | 1.0 |
| Headache | 11 | 1.5 | 4 | 1.4 |
| Nausea | 8 | 1.1 | 0 | 0.0 |
Gastrointestinal Reactions
Vomiting, gastrointestinal pain, and diarrhea have been reported, but the incidence in placebo-controlled trials was less than 1%. In rare cases, there may be an elevation of liver enzyme levels. In isolated instances, impairment of liver function (e.g. with cholestasis and jaundice), as well as hepatitis, which may also lead to liver failure have been reported with sulfonylureas, including AMARYL.
Dermatologic Reactions
Allergic skin reactions, e.g., pruritus, erythema, urticaria, and morbilliform or maculopapular eruptions, occur in less than 1% of treated patients. These may be transient and may disappear despite continued use of AMARYL. If those hypersensitivity reactions persist or worsen, the drug should be discontinued. Porphyria cutanea tarda, photosensitivity reactions, and allergic vasculitis have been reported with sulfonylureas, including AMARYL.
Hematologic Reactions
Leukopenia, agranulocytosis, thrombocytopenia, hemolytic anemia, aplastic anemia, and pancytopenia have been reported with sulfonylureas, including AMARYL.
Metabolic Reactions
Hepatic porphyria reactions and disulfiram-like reactions have been reported with sulfonylureas, including AMARYL. Cases of hyponatremia have been reported with glimepiride and all other sulfonylureas, most often in patients who are on other medications or have medical conditions known to cause hyponatremia or increase release of antidiuretic hormone. The syndrome of inappropriate antidiuretic hormone (SIADH) secretion has been reported with sulfonylureas, including AMARYL, and it has been suggested that certain sulfonylureas may augment the peripheral (antidiuretic) action of ADH and/or increase release of ADH.
Other Reactions
Changes in accommodation and/or blurred vision may occur with the use of AMARYL. This is thought to be due to changes in blood glucose, and may be more pronounced when treatment is initiated. This condition is also seen in untreated diabetic patients, and may actually be reduced by treatment. In placebo-controlled trials of AMARYL, the incidence of blurred vision was placebo, 0.7%, and AMARYL, 0.4%.
Pediatric Patients
In a clinical trial, 135 pediatric patients with Type 2 diabetes were treated with AMARYL. The profile of adverse reactions in these patients was similar to that observed in adults.
OVERDOSAGE
Overdosage of sulfonylureas, including AMARYL, can produce hypoglycemia. Mild hypoglycemic symptoms without loss of consciousness or neurologic findings should be treated aggressively with oral glucose and adjustments in drug dosage and/or meal patterns. Close monitoring should continue until the physician is assured that the patient is out of danger. Severe hypoglycemic reactions with coma, seizure, or other neurological impairment occur infrequently, but constitute medical emergencies requiring immediate hospitalization. If hypoglycemic coma is diagnosed or suspected, the patient should be given a rapid intravenous injection of concentrated (50%) glucose solution. This should be followed by a continuous infusion of a more dilute (10%) glucose solution at a rate that will maintain the blood glucose at a level above 100 mg/dL. Patients should be closely monitored for a minimum of 24 to 48 hours, because hypoglycemia may recur after apparent clinical recovery.
DOSAGE AND ADMINISTRATION
There is no fixed dosage regimen for the management of diabetes mellitus with AMARYL or any other hypoglycemic agent. The patient's fasting blood glucose and HbA1c must be measured periodically to determine the minimum effective dose for the patient; to detect primary failure, i.e., inadequate lowering of blood glucose at the maximum recommended dose of medication; and to detect secondary failure, i.e., loss of adequate blood glucose lowering response after an initial period of effectiveness. Glycosylated hemoglobin levels should be performed to monitor the patients response to therapy.
Short-term administration of AMARYL may be sufficient during periods of transient loss of control in patients usually controlled well on diet and exercise.
Usual Starting Dose
The usual starting dose of AMARYL as initial therapy is 1–2 mg once daily, administered with breakfast or the first main meal. Those patients who may be more sensitive to hypoglycemic drugs should be started at 1 mg once daily, and should be titrated carefully. (See PRECAUTIONS Section for patients at increased risk.)
No exact dosage relationship exists between AMARYL and the other oral hypoglycemic agents. The maximum starting dose of AMARYL should be no more than 2 mg.
Failure to follow an appropriate dosage regimen may precipitate hypoglycemia. Patients who do not adhere to their prescribed dietary and drug regimen are more prone to exhibit unsatisfactory response to therapy.
Usual Maintenance Dose
The usual maintenance dose is 1 to 4 mg once daily. The maximum recommended dose is 8 mg once daily. After reaching a dose of 2 mg, dosage increases should be made in increments of no more than 2 mg at 1–2 week intervals based upon the patient's blood glucose response. Long-term efficacy should be monitored by measurement of HbA1c levels, for example, every 3 to 6 months.
AMARYL-Metformin Combination Therapy
If patients do not respond adequately to the maximal dose of AMARYL monotherapy, addition of metformin may be considered. Published clinical information exists for the use of other sulfonylureas including glyburide, glipizide, chlorpropamide, and tolbutamide in combination with metformin.
With concomitant AMARYL and metformin therapy, the desired control of blood glucose may be obtained by adjusting the dose of each drug. However, attempts should be made to identify the minimum effective dose of each drug to achieve this goal. With concomitant AMARYL and metformin therapy, the risk of hypoglycemia associated with AMARYL therapy continues and may be increased. Appropriate precautions should be taken.
AMARYL-Insulin Combination Therapy
Combination therapy with AMARYL and insulin may also be used in secondary failure patients. The fasting glucose level for instituting combination therapy is in the range of >150 mg/dL in plasma or serum depending on the patient. The recommended AMARYL dose is 8 mg once daily administered with the first main meal. After starting with low-dose insulin, upward adjustments of insulin can be done approximately weekly as guided by frequent measurements of fasting blood glucose. Once stable, combination-therapy patients should monitor their capillary blood glucose on an ongoing basis, preferably daily. Periodic adjustments of insulin may also be necessary during maintenance as guided by glucose and HbA1clevels.
Specific Patient Populations
AMARYL (glimepiride tablets) is not recommended for use in pregnancy or nursing mothers. Data are insufficient to recommend pediatric use of AMARYL. In elderly, debilitated, or malnourished patients, or in patients with renal or hepatic insufficiency, the initial dosing, dose increments, and maintenance dosage should be conservative to avoid hypoglycemic reactions (See CLINICAL PHARMACOLOGY, Special Populations and PRECAUTIONS, General).
Patients Receiving Other Oral Hypoglycemic Agents
As with other sulfonylurea hypoglycemic agents, no transition period is necessary when transferring patients to AMARYL. Patients should be observed carefully (1–2 weeks) for hypoglycemia when being transferred from longer half-life sulfonylureas (e.g., chlorpropamide) to AMARYL due to potential overlapping of drug effect.
HOW SUPPLIED
AMARYL Tablets are available in the following strengths and package sizes:
- 1 mg (pink, flat-faced, oblong with notched sides at double bisect, either imprinted with "AMA RYL" on one side) or imprinted with "AMA RYL" on one side and the Hoechst logo on both sides of the bisect on the other side)
- Bottles of 100 (NDC 0039-0221-10)
- 2 mg (green, flat-faced, oblong with notched sides at double bisect, either imprinted with "AMA RYL" on one side) or imprinted with "AMA RYL" on one side and the Hoechst logo on both sides of the bisect on the other side)
- Bottles of 100 (NDC 0039-0222-10)
- Unit Dose Cartons (100) (NDC 0039-0222-11)
- 4 mg (blue, flat-faced, oblong with notched sides at double bisect, either imprinted with "AMA RYL" on one side, or imprinted with "AMA RYL" on one side and the Hoechst logo on both sides of the bisect on the other side)
- Bottles of 100 (NDC 0039-0223-10)
- Unit Dose Cartons (100) (NDC 0039-0223-11)
Store between 59 and 86° F (15 and 30° C).
Dispense in well-closed containers with safety closures.
ANIMAL TOXICOLOGY
Reduced serum glucose values and degranulation of the pancreatic beta cells were observed in beagle dogs exposed to 320 mg glimepiride/kg/day for 12 months (approximately 1,000 times the recommended human dose based on surface area). No evidence of tumor formation was observed in any organ. One female and one male dog developed bilateral subcapsular cataracts. Non-GLP studies indicated that glimepiride was unlikely to exacerbate cataract formation. Evaluation of the co-cataractogenic potential of glimepiride in several diabetic and cataract rat models was negative and there was no adverse effect of glimepiride on bovine ocular lens metabolism in organ culture.
HUMAN OPHTHALMOLOGY DATA
Ophthalmic examinations were carried out in over 500 subjects during long-term studies using the methodology of Taylor and West and Laties et al. No significant differences were seen between AMARYL and glyburide in the number of subjects with clinically important changes in visual acuity, intra-ocular tension, or in any of the five lens-related variables examined. Ophthalmic examinations were carried out during long-term studies using the method of Chylack et al. No significant or clinically meaningful differences were seen between AMARYL and glipizide with respect to cataract progression by subjective LOCS II grading and objective image analysis systems, visual acuity, intraocular pressure, and general ophthalmic examination.
Rev. February 2006
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807 USA
www.sanofi-aventis.us
© 2006 sanofi-aventis U.S. LLC
| Amaryl (glimepiride) | ||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Amaryl (glimepiride) | ||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Amaryl (glimepiride) | ||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
Revised: 01/2008sanofi-aventis U.S. LLC