BUTRANS
-
buprenorphine patch, extended release
Purdue Pharma L.P.
----------
|
||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING: IMPORTANCE OF PROPER PATIENT SELECTION, POTENTIAL FOR ABUSE, AND LIMITATIONS OF USE
Proper Patient Selection
Butrans is a transdermal formulation of buprenorphine indicated for the management of moderate to severe chronic pain in patients requiring a continuous, around-the-clock opioid analgesic for an extended period of time. (1)
Potential for Abuse
Butrans contains buprenorphine which is a mu opioid partial agonist and a Schedule III controlled substance. Butrans can be abused in a manner similar to other opioid agonists, legal or illicit. Consider the abuse potential when prescribing or dispensing Butrans in situations where the physician or pharmacist is concerned about an increased risk of misuse, abuse, or diversion. (9)
Persons at increased risk for opioid abuse include those with a personal or family history of substance abuse (including drug or alcohol abuse or addiction) or mental illness (e.g., major depression). Assess patients for their clinical risks for opioid abuse or addiction prior to being prescribed opioids. Routinely monitor all patients receiving opioids for signs of misuse, abuse, and addiction. (2.2)
Limitations of Use
Do not exceed a dose of one 20 mcg/hour Butrans system due to the risk of QTc interval prolongation. (2.3)
Avoid exposing the Butrans application site and surrounding area to direct external heat sources. Temperature-dependent increases in buprenorphine release from the system may result in overdose and death. (5.11)
1 INDICATIONS AND USAGE
Butrans is indicated for the management of moderate to severe chronic pain in patients requiring a continuous, around-the-clock opioid analgesic for an extended period of time.
2 DOSAGE AND ADMINISTRATION
2.1 General Principles
Selection of patients for treatment with Butrans is governed by the same principles that apply to the use of similar opioid analgesics. Physicians should individualize treatment in every case, using non-opioid analgesics, opioids on an as-needed basis and/or combination products, and chronic opioid therapy in a progressive plan of pain management such as outlined by the World Health Organization, the American Pain Society, and Federation of State Medical Boards Model Policy.
Butrans is for transdermal use (on intact skin) only.
Do not use Butrans if the pouch seal is broken or the patch is cut, damaged, or changed in any way. Do not cut Butrans.
Each Butrans is intended to be worn for 7 days.
Apply Butrans to the upper outer arm, upper chest, upper back or the side of the chest. These 4 sites (each present on both sides of the body) provide 8 possible application sites. Rotate Butrans among the 8 described skin sites. After Butrans removal, wait a minimum of 21 days before reapplying to the same skin site [see Clinical Pharmacology (12.3)].
Apply Butrans to a hairless or nearly hairless skin site. If none are available, the hair at the site should be clipped, not shaven. Do not apply Butrans to irritated skin. If the application site must be cleaned, clean the site with water only. Do not use soaps, alcohol, oils, lotions, or abrasive devices. Allow the skin to dry before applying Butrans.
If problems with adhesion of Butrans occur, the edges may be taped with first aid tape.
If Butrans falls off during the 7 days dosing interval, dispose of the transdermal system properly and place a new Butrans on at a different skin site [see How Supplied/Storage and Handling (16)].
2.2 Initiation of Therapy
It is critical to initiate the dosing regimen individually for each patient. Overestimating the Butrans dose when converting patients from another opioid medication can result in fatal overdose with the first dose [see Overdosage (10)]. Consider the following when selecting the initial dose of Butrans:
- The total daily dose, potency, and specific characteristics of the opioid the patient has been taking previously;
- The reliability of the relative potency estimate used to calculate the equivalent buprenorphine dose needed (when converting from other opioids or opioid-combination products);
- The patient’s degree of tolerance to the respiratory-depressant and sedating effects of opioids;
- The age, general condition, and medical status of the patient;
- Concurrent non-opioid analgesic and other medications;
- The type and severity of the patient's pain;
- The balance between pain control and adverse drug experiences;
- Risk factors for abuse, addiction, or diversion, including a prior history of abuse, addiction, or diversion.
The following dosing recommendations, therefore, can only be considered as suggested approaches to what is actually a series of clinical decisions over time in the management of the pain of each individual patient.
Opioid-Naïve Patients
For opioid-naïve patients, initiate treatment with Butrans 5 mcg/hour. Thereafter, individually titrate the dose as described in Section 2.3 Dose Titration to a level that provides adequate analgesia and minimizes side effects. Dose may be titrated to the next higher level after a minimum of 72 hours.
Conversion from Other Opioids to Butrans
There is a potential for buprenorphine to precipitate withdrawal in patients who are already on opioids. For conversion from other opioids to Butrans (see Table 1), taper the patient’s current around-the-clock opioids for up to 7 days to no more than 30 mg of morphine or equivalent per day before beginning treatment with Butrans. Patients may use short-acting analgesics as needed until analgesic efficacy with Butrans is attained.
For patients whose daily dose was less than 30 mg of oral morphine or equivalent, initiate treatment with Butrans 5 mcg/hour. For patients whose daily dose was between 30 and 80 mg morphine equivalents, initiate treatment with Butrans 10 mcg/hour (see Table 1). Thereafter, individually titrate the dose as described in Section 2.3 Dose Titration.
| Current Opioid Analgesic | Current Daily Dose | |
| Oral Morphine Equivalent | <30 mg | 30-80 mg |
 | |
|
| Recommended Butrans Starting Dose | 5 mcg/hour | 10 mcg/hour |
Use caution when prescribing Butrans to opioid-experienced patients requiring high doses of opioids (more than 80 mg/day of oral morphine equivalents). Butrans 20 mcg/hour may not provide adequate analgesia for patients requiring greater than 80 mg/day oral morphine equivalents.
2.3 Dose Titration
Based on the patient’s requirement for supplemental short-acting analgesics, upward titration may be instituted with a minimum Butrans titration interval of 72 hours, based on the pharmacokinetic profile and time to reach steady state levels [see Clinical Pharmacology (12.3)]. Individually titrate the dose, under close supervision, to a level that provides adequate analgesia with tolerable side effects.
The maximum Butrans dose is 20 mcg/hour. Do not exceed a dose of one 20 mcg/hour Butrans system due to the risk of QTc interval prolongation. In a clinical trial, Butrans 40 mcg/hour (given as two Butrans 20 mcg/hour systems) resulted in prolongation of the QTc interval [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.2)].
During periods of changing analgesic requirements, including initial titration, frequent contact is recommended between the prescriber, other members of the healthcare team, the patient, and the caregiver/family. Advise patients and caregivers/family members of the potential side effects.
2.4 Maintenance of Therapy and Supplemental Analgesia
The intent of the titration period is to establish a patient-specific weekly Butrans dose that will maintain adequate analgesia with tolerable side effects for as long as pain management is necessary. Immediate-release opioid and non-opioid medications can be used as supplemental analgesia during Butrans therapy.
During chronic opioid analgesic therapy with Butrans, reassess the continued need for around-the-clock opioid analgesic therapy periodically.
2.5 Cessation of Therapy
When the patient no longer requires therapy with Butrans, taper the dose gradually to prevent signs and symptoms of withdrawal in the physically dependent patient; consider introduction of an appropriate immediate-release opioid medication. Undertake discontinuation of therapy as part of a comprehensive treatment plan.
2.6 Patients with Hepatic Impairment
Start patients with mild to moderate hepatic impairment with the Butrans 5 mcg/hour dose. Thereafter, individually titrate the dose to a level that provides adequate analgesia and tolerable side effects, under the close supervision of the prescriber. Butrans has not been evaluated in patients with severe hepatic impairment. As Butrans is only intended for 7-day application, consider use of an alternate analgesic that may permit more flexibility with the dosing in patients with severe hepatic impairment [see Warnings and Precautions (5.1), Use In Specific Populations (8.6), and Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Butrans is available as:
- Butrans 5 mcg/hour Transdermal System (dimensions: 45 mm by 45 mm)
- Butrans 10 mcg/hour Transdermal System (dimensions: 45 mm by 68 mm)
- Butrans 20 mcg/hour Transdermal System (dimensions: 72 mm by 72 mm)
4 CONTRAINDICATIONS
Butrans is contraindicated in:
- patients who have significant respiratory depression
- patients who have severe bronchial asthma
- patients who have or are suspected of having paralytic ileus
- patients who have known hypersensitivity to any of its components or the active ingredient, buprenorphine
- the management of acute pain or in patients who require opioid analgesia for a short period of time
- the management of post-operative pain, including use after out-patient or day surgeries
- the management of mild pain
- the management of intermittent pain (e.g., use on an as-needed basis [prn])
5 WARNINGS AND PRECAUTIONS
5.1 Respiratory Depression
Respiratory depression is the chief hazard of Butrans. Respiratory depression occurs more frequently in elderly or debilitated patients as well as those suffering from conditions accompanied by hypoxia or hypercapnia when even moderate therapeutic doses may dangerously decrease pulmonary ventilation, and when opioids, including Butrans, are given in conjunction with other agents that depress respiration.
Profound sedation, unresponsiveness, infrequent deep (“sighing”) breaths or atypical snoring frequently accompany opioid-induced respiratory depression.
Use Butrans with extreme caution in patients with any of the following:
- significant chronic obstructive pulmonary disease or cor pulmonale
- other risk of substantially decreased respiratory reserve such as asthma, severe obesity, sleep apnea, myxedema, clinically-significant kyphoscoliosis, and central nervous system (CNS) depression
- hypoxia
- hypercapnia
- pre-existing respiratory depression
5.2 CNS Depression
Butrans may cause somnolence, dizziness, alterations in judgment and alterations in levels of consciousness, including coma.
5.3 Interactions with Alcohol, Central Nervous System Depressants, and Illicit Drugs
Hypotension, profound sedation, coma or respiratory depression may result if Butrans is added to a regimen that includes other CNS depressants (e.g., sedatives, anxiolytics, hypnotics, neuroleptics, muscle relaxants, other opioids). Therefore, use caution when deciding to initiate therapy with Butrans in patients who are taking other CNS depressants. Take into account the types of other medications being taken, the duration of therapy with them, and the patient’s response to those medicines, including the degree of tolerance that has developed to CNS depression. Consider the patient’s use, if any, of alcohol and/or illicit drugs that cause CNS depression. If the decision to begin Butrans is made, start with a lower Butrans dose than usual.
Consider using a lower initial dose of a CNS depressant when given to a patient currently taking Butrans due to the potential of additive CNS depressant effects.
5.4 QTc Prolongation
A positive-controlled study of the effects of Butrans on the QTc interval in healthy subjects demonstrated no clinically meaningful effect at a Butrans dose of 10 mcg/hour; however, a Butrans dose of 40 mcg/hour (given as two Butrans 20 mcg/hour Transdermal Systems) was observed to prolong the QTc interval [see Clinical Pharmacology (12.2)].
Consider these observations in clinical decisions when prescribing Butrans to patients with hypokalemia or clinically unstable cardiac disease, including: unstable atrial fibrillation, symptomatic bradycardia, unstable congestive heart failure, or active myocardial ischemia. Avoid the use of Butrans in patients with a history of Long QT Syndrome or an immediate family member with this condition, or those taking Class IA antiarrhythmic medications (e.g., quinidine, procainamide, disopyramide) or Class III antiarrhythmic medications (e.g., sotalol, amiodarone, dofetilide).
5.5 Head Injury
The respiratory depressant effects of opioids, including Butrans, include carbon dioxide retention, which can lead to an elevation of cerebrospinal fluid pressure. This effect may be exaggerated in the presence of head injury, intracranial lesions, or other sources of pre-existing increased intracranial pressure. Butrans may produce miosis that is independent of ambient light, and altered consciousness, either of which may obscure neurologic signs associated with increased intracranial pressure in persons with head injuries.
5.6 Hypotensive Effects
Butrans may cause severe hypotension. There is an added risk to individuals whose ability to maintain blood pressure has been compromised by a depleted blood volume, or after concurrent administration with drugs such as phenothiazines or other agents which compromise vasomotor tone. Buprenorphine may produce orthostatic hypotension in ambulatory patients. Administer Butrans with caution to patients in circulatory shock, since vasodilation produced by the drug may further reduce cardiac output and blood pressure.
5.7 Misuse, Abuse, and Diversion of Opioids
Butrans contains buprenorphine, a partial agonist at the mu opioid receptor and a Schedule III controlled substance. Opioid agonists have potential for being abused, are sought by drug abusers and people with addiction disorders, and are subject to criminal diversion.
Butrans can be abused in a manner similar to other opioid agonists, legal or illicit. Consider this potential for abuse when prescribing or dispensing Butrans in situations where the prescriber or pharmacist is concerned about an increased risk of misuse, abuse, or diversion. Monitor all patients receiving opioids for signs of abuse, misuse, and addiction. Furthermore, assess patients for their potential for opioid abuse prior to being prescribed opioid therapy. Persons at increased risk for opioid abuse include those with a personal or family history of substance abuse (including drug or alcohol abuse) or mental illness (e.g., depression). Opioids may still be appropriate for use in these patients; however, they will require intensive monitoring for signs of abuse.
Notwithstanding concerns about abuse, addiction, and diversion, provide proper management of pain. However, all patients treated with opioid agonists require careful monitoring for signs of abuse and addiction, since use of opioid agonist analgesic products carries the risk of addiction even under appropriate medical use [see Drug Abuse and Dependence (9.2)]. Data are not available to establish the true incidence of addiction in patients with chronic pain treated with opioids.
Abuse of Butrans poses a significant risk to the abuser that could potentially result in overdose or death [see Drug Abuse and Dependence (9)].
Contact your state professional licensing board or state controlled substances authority for information on how to prevent and detect abuse or diversion of this product.
5.8 Hepatotoxicity
Although not observed in Butrans chronic pain clinical trials, cases of cytolytic hepatitis and hepatitis with jaundice have been observed in individuals receiving sublingual buprenorphine for the treatment of opioid dependence, both in clinical trials and through post-marketing adverse event reports. The spectrum of abnormalities ranges from transient asymptomatic elevations in hepatic transaminases to case reports of hepatic failure, hepatic necrosis, hepatorenal syndrome, and hepatic encephalopathy. In many cases, the presence of pre-existing liver enzyme abnormalities, infection with hepatitis B or hepatitis C virus, concomitant usage of other potentially hepatotoxic drugs, and ongoing injection drug abuse may have played a causative or contributory role. In other cases, insufficient data were available to determine the etiology of the abnormality. The possibility exists that buprenorphine had a causative or contributory role in the development of the hepatic abnormality in some cases. For patients at increased risk of hepatotoxicity (e.g., patients with a history of excessive alcohol intake, intravenous drug abuse or liver disease), baseline and periodic monitoring of liver function during treatment with Butrans is recommended. A biological and etiological evaluation is recommended when a hepatic event is suspected.
5.9 Application Site Skin Reactions
In rare cases, severe application site skin reactions with signs of marked inflammation including “burn,” “discharge,” and “vesicles” have occurred. Time of onset varies, ranging from days to months following the initiation of Butrans treatment. Instruct patients to promptly report the development of severe application site reactions and discontinue therapy.
5.10 Anaphylactic/Allergic Reactions
Cases of acute and chronic hypersensitivity to buprenorphine have been reported both in clinical trials and in the post-marketing experience. The most common signs and symptoms include rashes, hives, and pruritus. Cases of bronchospasm, angioneurotic edema, and anaphylactic shock have been reported. A history of hypersensitivity to buprenorphine is a contraindication to the use of Butrans.
5.11 Application of External Heat
Advise patients and their caregivers to avoid exposing the Butrans application site and surrounding area to direct external heat sources, such as heating pads or electric blankets, heat or tanning lamps, saunas, hot tubs, and heated water beds, etc., while wearing the system because an increase in absorption of buprenorphine may occur [see Clinical Pharmacology (12.3)]. Advise patients against exposure of the Butrans application site and surrounding area to hot water or prolonged exposure to direct sunlight. There is a potential for temperature-dependent increases in buprenorphine released from the system resulting in possible overdose and death.
5.12 Patients with Fever
Patients wearing Butrans systems who develop fever or increased core body temperature due to strenuous exertion should be monitored for opioid side effects and the Butrans dose should be adjusted if necessary [see Dosage and Administration (2.4)].
5.13 Driving and Operating Machinery
Butrans may impair the mental and physical abilities needed to perform potentially hazardous activities such as driving a car or operating machinery. Caution patients accordingly.
5.14 Seizures
Butrans, as with other opioids, may aggravate seizure disorders, may lower seizure threshold, and therefore, may induce seizures in some clinical settings. Use Butrans with caution in patients with a history of seizure disorders.
5.15 Special Risk Groups
Use Butrans with caution in the following conditions, due to increased risk of adverse reactions: alcoholism; delirium tremens; adrenocortical insufficiency; CNS depression; debilitation; kyphoscoliosis associated with respiratory compromise; myxedema or hypothyroidism; prostatic hypertrophy or urethral stricture; severe impairment of hepatic, pulmonary or renal function; and toxic psychosis.
5.16 Use in Pancreatic/Biliary Tract Disease and Other Gastrointestinal Conditions
Butrans may cause spasm of the sphincter of Oddi. Use with caution in patients with biliary tract disease, including acute pancreatitis. Opioids, including Butrans, may cause increased serum amylase.
The administration of Butrans may obscure the diagnosis or clinical course in patients with acute abdominal conditions. Use Butrans with caution in patients who are at risk of developing ileus.
6 ADVERSE REACTIONS
The following adverse reactions described elsewhere in the labeling include:
- Respiratory Depression [see Warnings and Precautions (5.1)]
- CNS Depression [see Warnings and Precautions (5.2)]
- QTc Prolongation [see Warnings and Precautions (5.4)]
- Hypotensive Effects [see Warnings and Precautions (5.6)]
- Application Site Skin Reactions [see Warnings and Precautions (5.9)]
- Anaphylactic/Allergic Reactions [see Warnings and Precautions (5.10)]
- Seizures [see Warnings and Precautions (5.14)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 5,415 patients were treated with Butrans in controlled and open-label chronic pain clinical trials. Nine hundred twenty-four subjects were treated for approximately six months and 183 subjects were treated for approximately one year. The clinical trial population consisted of patients with persistent moderate to severe pain.
The most common adverse reactions (≥5%) reported by patients in clinical trials comparing Butrans 10 or 20 mcg/hour to placebo are shown in Table 2, and comparing Butrans 20 mcg/hour to Butrans 5 mcg/hour are shown in Table 3 below:
| Open-Label Titration Period | Double-Blind Treatment Period | ||
| Butrans | Butrans | Placebo | |
| MedDRA Preferred Term | (N = 1024) | (N = 256) | (N = 283) |
| Nausea | 23% | 13% | 11% |
| Dizziness | 10% | 4% | 1% |
| Headache | 10% | 5% | 5% |
| Application site pruritus | 8% | 4% | 7% |
| Somnolence | 8% | 2% | 2% |
| Vomiting | 8% | 4% | 2% |
| Constipation | 7% | 4% | 1% |
| Open-Label Titration Period | Double-Blind Treatment Period | ||
| Butrans | Butrans 20 | Butrans 5 | |
| MedDRA Preferred Term | (N = 1160) | (N = 219) | (N = 221) |
| Nausea | 15% | 12% | 8% |
| Headache | 11% | 11% | 5% |
| Application site pruritus | 9% | 13% | 5% |
| Somnolence | 6% | 5% | 2% |
| Vomiting | 5% | 5% | 2% |
| Dizziness | 5% | 5% | 2% |
| Constipation | 4% | 6% | 3% |
| Application site erythema | 3% | 10% | 5% |
| Application site rash | 3% | 9% | 6% |
| Application site irritation | 2% | 5% | 3% |
The following table lists adverse events that were reported in at least 2.0% of patients in four placebo/active-controlled titration-to-effect trials.
| MedDRA Preferred Term
| Butrans
(N = 392) | Placebo
(N = 261) |
| Nausea | 23% | 8% |
| Dizziness | 16% | 8% |
| Headache | 16% | 11% |
| Application site pruritus | 15% | 12% |
| Constipation | 14% | 5% |
| Somnolence | 14% | 5% |
| Vomiting | 11% | 2% |
| Peripheral edema | 7% | 3% |
| Dry mouth | 7% | 2% |
| Application site erythema | 7% | 2% |
| Application site rash | 6% | 6% |
| Fatigue | 5% | 1% |
| Hyperhidrosis | 4% | 1% |
| Pruritus | 4% | 1% |
| Fall | 4% | 2% |
| Diarrhea | 3% | 2% |
| Pain in extremity | 3% | 2% |
| Insomnia | 3% | 2% |
| Dyspnea | 3% | 1% |
| Dyspepsia | 3% | 3% |
| Urinary tract infection | 3% | 2% |
| Back pain | 3% | 2% |
| Joint swelling | 3% | 1% |
| Hypoesthesia | 2% | 1% |
| Arthralgia | 2% | 2% |
| Stomach discomfort | 2% | 1% |
| Rash | 2% | 1% |
| Anorexia | 2% | 1% |
| Paraesthesia | 2% | 1% |
| Tremor | 2% | <1% |
| Confusional State | 2% | 3% |
The adverse events seen in controlled and open-label studies are presented below in the following manner: most common (≥5%), common (≥1% to <5%), and less common (<1%).
The most common adverse events (≥5%) reported by patients treated with Butrans in the clinical trials were nausea, headache, application site pruritus, dizziness, constipation, somnolence, vomiting, application site erythema, dry mouth, and application site rash.
The common (≥1% to <5%) adverse events reported by patients treated with Butrans in the clinical trials organized by MedDRA (Medical Dictionary for Regulatory Activities) System Organ Class were:
Gastrointestinal disorders: diarrhea, dyspepsia, and upper abdominal pain
General disorders and administration site conditions: fatigue, peripheral edema, application site irritation, pain, pyrexia, chest pain, and asthenia
Infections and infestations: urinary tract infection, upper respiratory tract infection, nasopharyngitis, influenza, sinusitis, and bronchitis
Injury, poisoning and procedural complications: fall
Metabolism and nutrition disorders: anorexia
Musculoskeletal and connective tissue disorders: back pain, arthralgia, pain in extremity, muscle spasms, musculoskeletal pain, joint swelling, neck pain, and myalgia
Nervous system disorders: hypoesthesia, tremor, migraine, and paresthesia
Psychiatric disorders: insomnia, anxiety, and depression
Respiratory, thoracic and mediastinal disorders: dyspnea, pharyngolaryngeal pain, and cough
Skin and subcutaneous tissue disorders: pruritus, hyperhidrosis, rash, and generalized pruritus
Vascular disorders: hypertension
Other less common adverse events, including those known to occur with opioid treatment, that were seen in <1% of the patients in the Butrans trials include the following in alphabetical order:
Abdominal distention, abdominal pain, accidental injury, affect lability, agitation, alanine aminotransferase increased, angina pectoris, angioedema, apathy, application site dermatitis, asthma aggravated, bradycardia, chills, confusional state, contact dermatitis, coordination abnormal, dehydration, depersonalization, depressed level of consciousness, depressed mood, disorientation, disturbance in attention, diverticulitis, drug hypersensitivity, drug withdrawal syndrome, dry eye, dry skin, dysarthria, dysgeusia, dysmenorrhea, dysphagia, euphoric mood, face edema, flatulence, flushing, gait disturbance, hallucination, hiccups, hot flush, hyperventilation, hypotension, hypoventilation, ileus, insomnia, libido decreased, loss of consciousness, malaise, memory impairment, mental impairment, mental status changes, miosis, muscle weakness, nervousness, nightmare, orthostatic hypotension, palpitations, psychotic disorder, respiration abnormal, respiratory depression, respiratory distress, respiratory failure, restlessness, rhinitis, sedation, sexual dysfunction, syncope, tachycardia, tinnitus, urinary hesitation, urinary incontinence, urinary retention, urticaria, vasodilatation, vertigo, vision blurred, visual disturbance, weight decreased, and wheezing.
7 DRUG INTERACTIONS
7.1 Metabolic Drug Interactions
CYP3A4 Inhibitors
Co-administration of ketoconazole, a strong CYP3A4 inhibitor, with Butrans, did not have any effect on Cmax and AUC of buprenorphine. Based on this observation, pharmacokinetics of Butrans is not expected to be affected by co-administration of CYP3A4 inhibitors.
However, certain protease inhibitors (PIs) with CYP3A4 inhibitory activity such as atazanavir and atazanavir/ritonavir resulted in elevated levels of buprenorphine and norbuprenorphine following sublingual administration of buprenorphine and naloxone. Patients in this study reported increased sedation, and symptoms of opiate excess have been found in post-marketing reports of patients receiving sublingual buprenorphine and atazanavir with and without ritonavir concomitantly. It should be noted that atazanavir is both a CYP3A4 and UGT1A1 inhibitor. As such, the drug-drug interaction potential for buprenorphine with CYP3A4 inhibitors is likely to be dependent on the route of administration as well as the specificity of enzyme inhibition [see Clinical Pharmacology (12.3)].
CYP3A4 Inducers
The interaction between buprenorphine and CYP3A4 enzyme inducers has not been studied; therefore it is recommended that patients receiving Butrans be closely monitored for reduced efficacy if inducers of CYP3A4 (e.g. phenobarbital, carbamazepine, phenytoin, rifampin) are co-administered [see Clinical Pharmacology (12.3)].
7.2 Non-Metabolic Drug Interactions
Benzodiazepines
There have been a number of reports regarding coma and death associated with the misuse and abuse of the combination of buprenorphine and benzodiazepines. In many, but not all of these cases, buprenorphine was misused by self-injection of crushed buprenorphine tablets. Preclinical studies have shown that the combination of benzodiazepines and buprenorphine altered the usual ceiling effect on buprenorphine-induced respiratory depression, making the respiratory effects of buprenorphine appear similar to those of full opioid agonists. Prescribe Butrans with caution to patients taking benzodiazepines or other drugs that act on the central nervous system regardless of whether these drugs are taken on the advice of a physician or are being abused/misused. Warn patients that it is extremely dangerous to self-administer benzodiazepines while taking Butrans, and caution patients to use benzodiazepines concurrently with Butrans only as directed by their physician.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C.
There are no adequate and well-controlled studies with Butrans in pregnant women. Butrans should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and the fetus. In animal studies, buprenorphine caused an increase in the number of stillborn offspring, reduced litter size, and reduced offspring growth in rats at maternal exposure levels that were approximately 10 times that of human subjects who received one Butrans 20 mcg/hour, the maximum recommended human dose (MRHD).
Teratogenic Effects
Studies in rats and rabbits demonstrated no evidence of teratogenicity following Butrans or subcutaneous (SC) administration of buprenorphine during the period of major organogenesis. Rats were administered up to one Butrans 20 mcg/hour every 3 days (gestation days 6, 9, 12, & 15) or received daily SC buprenorphine up to 5 mg/kg (gestation days 6-17). Rabbits were administered four Butrans 20 mcg/hour every 3 days (gestation days 6, 9, 12, 15, 18, & 19) or received daily SC buprenorphine up to 5 mg/kg (gestation days 6-19). No teratogenicity was observed at any dose. Area under the curve (AUC) values for buprenorphine with Butrans application and SC injection were approximately 140 and 110 times that of human subjects who received the MRHD of one Butrans 20 mcg/hour.
Non-Teratogenic Effects
In a peri- and post-natal study conducted in pregnant and lactating rats, administration of buprenorphine either as Butrans or SC buprenorphine was associated with toxicity to offspring. Buprenorphine was present in maternal milk. Pregnant rats were administered 1/4 of one Butrans 5 mcg/hour every 3 days or received daily SC buprenorphine at doses of 0.05, 0.5, or 5 mg/kg from gestation day 6 to lactation day 21 (weaning). Administration of Butrans or SC buprenorphine at 0.5 or 5 mg/kg caused maternal toxicity and an increase in the number of stillborns, reduced litter size, and reduced offspring growth at maternal exposure levels that were approximately 10 times that of human subjects who received the MRHD of one Butrans 20 mcg/hour. Maternal toxicity was also observed at the no observed adverse effect level (NOAEL) for offspring.
8.2 Labor and Delivery
The safety of Butrans given during labor and delivery has not been established.
Opioids cross the placenta and may produce respiratory depression and psychophysiologic effects in neonates. Butrans is not recommended for use in women immediately prior to and during labor, when use of shorter-acting analgesics or other analgesic techniques are more appropriate. Occasionally, opioid analgesics may prolong labor through actions which temporarily reduce the strength, duration and frequency of uterine contractions. However this effect is not consistent and may be offset by an increased rate of cervical dilatation, which tends to shorten labor.
Closely observe neonates whose mothers received opioid analgesics during labor for signs of respiratory depression. Have a specific opioid antagonist, such as naloxone or nalmefene, available for reversal of opioid-induced respiratory depression in the neonate.
Neonates whose mothers have been taking opioids chronically may also exhibit withdrawal signs, either at birth and/or in the nursery, because they have developed physical dependence. This is not, however, synonymous with addiction. Neonatal opioid withdrawal syndrome, unlike opioid withdrawal syndrome in adults, may be life-threatening and should be treated according to protocols developed by neonatology experts.
8.3 Nursing Mothers
Buprenorphine has been detected in low concentrations in human milk. Breast-feeding is not advised in mothers treated with Butrans.
8.4 Pediatric Use
The safety and efficacy of Butrans in patients under 18 years of age has not been established. Butrans is not recommended for use in pediatric patients.
8.5 Geriatric Use
Of the total number of subjects in the clinical trials (5,415), Butrans was administered to 1,377 patients aged 65 years and older. Of those, 457 patients were 75 years of age and older. In the clinical program, the incidences of selected Butrans-related AEs were higher in older subjects. The incidences of application site AEs were slightly higher among subjects <65 years of age than those ≥ 65 years of age for both Butrans and placebo treatment groups.
In a single-dose study of healthy elderly and healthy young subjects treated with Butrans 10 mcg/hour, the pharmacokinetics and safety outcomes were similar. In a separate dose-escalation safety study, the pharmacokinetics in the healthy elderly and hypertensive elderly subjects taking thiazide diuretics were similar to those in the healthy young adults. In the elderly groups evaluated, adverse event rates were similar to or lower than rates in healthy young adult subjects, except for constipation and urinary retention, which were more common in the elderly. Although specific dose adjustments on the basis of advanced age are not required for pharmacokinetic reasons, use caution in the elderly population to ensure safe use [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
In a study utilizing intravenous buprenorphine, peak plasma levels (Cmax) and exposure (AUC) of buprenorphine in patients with mild and moderate hepatic impairment did not increase as compared to those observed in subjects with normal hepatic function. Butrans has not been evaluated in patients with severe hepatic impairment and should be administered with caution [see Dosage and Administration (2.6), and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
The pharmacokinetics of buprenorphine is not altered during the course of renal failure [see Clinical Pharmacology (12.3)].
8.8 Gender Differences
There was no significant gender effect observed for Butrans with respect to either the incidence of adverse events or pharmacokinetics [see Clinical Pharmacology (12.3)].
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
Butrans contains buprenorphine, a mu opioid partial agonist and Schedule III controlled substance. Butrans can be abused and is subject to misuse, abuse, addiction and criminal diversion.
9.2 Abuse
Abuse of Butrans poses a hazard of overdose and death. This risk is increased with compromise of the Butrans Transdermal System and with concurrent abuse of alcohol or other substances. Butrans has been diverted for non-medical use.
All patients treated with opioids, including Butrans, require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carries the risk of addiction even under appropriate medical use.
Addiction is a primary, chronic, neurobiologic disease, with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Opioid drugs are sought by people with substance use disorders (abuse or addiction, the latter of which is also called “substance dependence”) and criminals who supply them by diverting medicines out of legitimate distribution channels. Butrans is a target for theft and diversion.
“Drug-seeking” behavior is very common in persons with substance use disorders. Drug-seeking tactics include, but are not limited to, emergency calls or visits near the end of office hours, refusal to undergo appropriate examination, testing or referral, repeated “loss” of prescriptions, altering or forging of prescriptions and reluctance to provide prior medical records or contact information for other treating physician(s). “Doctor shopping” to obtain additional prescriptions is common among people with untreated substance use disorders, and criminals who divert controlled substances.
Abuse and addiction are separate and distinct from physical dependence and tolerance. Physicians should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all addicts. In addition, abuse of opioids can occur in the absence of true addiction and is characterized by misuse for nonmedical purposes, often in combination with other psychoactive substances. Since Butrans may be diverted for non-medical use, careful record-keeping of prescribing information, including quantity, frequency, and renewal requests is strongly advised.
The risks of misuse and abuse should be considered when prescribing or dispensing Butrans. Concerns about abuse and addiction, should not prevent the proper management of pain, however. Treatment of pain should be individualized, balancing the potential benefits and risks for each patient.
Butrans is intended for transdermal use only. Compromising the transdermal delivery system will result in the uncontrolled delivery of buprenorphine and pose a significant risk to the abuser that could result in overdose and death [see Warnings and Precautions (5.1)]. The risk of fatal overdose is further increased when buprenorphine is abused concurrently with alcohol or other CNS depressants, including other opioids and benzodiazepines [see Warnings and Precautions (5.3)]. Abuse may occur by applying the transdermal system in the absence of legitimate purpose, or by swallowing, snorting or injecting buprenorphine extracted from the transdermal system.
Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, proper dispensing and correct storage and handling are appropriate measures that help to limit misuse and abuse of opioid drugs. Careful record-keeping of prescribing information, including quantity, frequency, and renewal requests is strongly advised.
Healthcare professionals should contact their State Professional Licensing Board or State Controlled Substances Authority for information on how to prevent and detect abuse or diversion of this product.
9.3 Physical Dependence and Tolerance
Tolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s effects over time. Tolerance could occur to both the desired and undesired effects of drugs, and may develop at different rates for different effects.
Physical dependence to an opioid is manifested by characteristic withdrawal signs and symptoms after abrupt discontinuation of a drug, significant dose reduction, or upon administration of an antagonist. Physical dependence and tolerance are not unusual during chronic opioid analgesic therapy.
The opioid abstinence or withdrawal syndrome in adults is characterized by some or all of the following: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, piloerection, myalgia, mydriasis, irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, or increased blood pressure, respiratory rate, or heart rate [see Use In Specific Populations (8.2)]
Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory difficulties and withdrawal symptoms.
In general, opioids should not be abruptly discontinued [see Dosage and Administration (2.5)].
10 OVERDOSAGE
10.1 Symptoms
Acute overdosage with Butrans can be manifested by respiratory depression, somnolence progressing to stupor or coma, skeletal muscle flaccidity, cold and clammy skin, constricted pupils, bradycardia, hypotension, partial or complete airway obstruction, atypical snoring and death.
Deaths due to overdose have been reported with abuse and misuse of buprenorphine. Review of case reports has indicated that the risk of fatal overdose is further increased when Butrans is abused concurrently with alcohol or other CNS depressants, including other opioids.
10.2 Treatment
In cases of overdose, remove Butrans immediately. It is important to take the pharmacokinetic profile of Butrans into account when treating overdose. Even in the face of improvement, continued medical monitoring is required because of the possibility of extended effects as opioid continues to be absorbed from the skin. After removal of Butrans, the mean buprenorphine concentrations decrease approximately 50% in 12 hours (range 10-24 hours) with an apparent terminal half-life of approximately 26 hours. Due to this long apparent terminal half-life, patients may require monitoring and treatment for at least 24 hours.
In the treatment of Butrans overdosage, primary attention should be given to the maintenance of a patent airway, and of effective ventilation (clearance of CO2) and oxygenation, whether by spontaneous, assisted or controlled respiration. Supportive measures (including oxygen and vasopressors) should be employed in the management of circulatory shock and pulmonary edema accompanying overdose as indicated. Cardiac arrest or arrhythmias may require cardiac massage or defibrillation.
Naloxone may not be effective in reversing any respiratory depression produced by buprenorphine. High doses of naloxone, 10-35 mg/70 kg, may be of limited value in the management of buprenorphine overdose. The onset of naloxone effect may be delayed by 30 minutes or more. Doxapram hydrochloride (a respiratory stimulant) has also been used. Since the duration of action of Butrans may exceed that of the antagonist, keep the patient under continued surveillance and administer repeated doses of the antagonist according to the antagonist labeling as needed to maintain adequate respiration. Maintenance of adequate ventilation is essential when managing Butrans overdose and more important than specific antidote treatment with an opioid antagonist such as naloxone.
Do not administer opioid antagonists in the absence of clinically significant respiratory or circulatory depression secondary to buprenorphine overdose. In patients who are physically dependent on any opioid agonist including Butrans, an abrupt partial or complete reversal of opioid effects may precipitate an acute abstinence or withdrawal syndrome. The severity of the withdrawal syndrome produced will depend on the degree of physical dependence and the dose of the antagonist administered. See the prescribing information for the specific opioid antagonist for details of its proper use.
11 DESCRIPTION
Butrans is a transdermal system providing systemic delivery of buprenorphine, a mu opioid partial agonist analgesic, continuously for 7 days. The chemical name of buprenorphine is 6,14-ethenomorphinan-7-methanol, 17-(cyclopropylmethyl)- α-(1,1-dimethylethyl)-4, 5-epoxy-18, 19-dihydro-3-hydroxy-6-methoxy-α-methyl-, [5α, 7α, (S)]. The structural formula is:
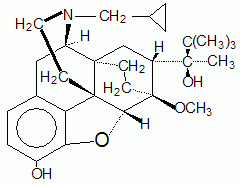
The molecular weight of buprenorphine is 467.6; the empirical formula is C29H41NO4. Buprenorphine occurs as a white or almost white powder and is very slightly soluble in water, freely soluble in acetone, soluble in methanol and ether, and slightly soluble in cyclohexane. The pKa is 8.5 and the melting point is about 217°C.
System Components and Structure
Three different strengths of Butrans are available: 5, 10, and 20 mcg/hour (Table 5). The active component of the system is buprenorphine. The remaining components are pharmacologically inactive. The proportion of buprenorphine mixed in the adhesive matrix is the same in each of the 3 strengths. The amount of buprenorphine released from each system per hour is proportional to the active surface area of the system. The skin is the limiting barrier to diffusion from the system into the bloodstream.
| Buprenorphine Delivery Rate (mcg/hour) | Active Surface Area (cm2) | Total Buprenorphine Content (mg) |
| Butrans 5 | 6.25 | 5 |
| Butrans 10 | 12.5 | 10 |
| Butrans 20 | 25 | 20 |
Butrans is a rectangular or square, beige-colored system consisting of a protective liner and functional layers. Proceeding from the outer surface toward the surface adhering to the skin, the layers are (1) a beige-colored web backing layer; (2) an adhesive rim without buprenorphine; (3) a separating layer over the buprenorphine-containing adhesive matrix; (4) the buprenorphine-containing adhesive matrix; and (5) a peel-off release liner. Before use, the release liner covering the adhesive layer is removed and discarded.
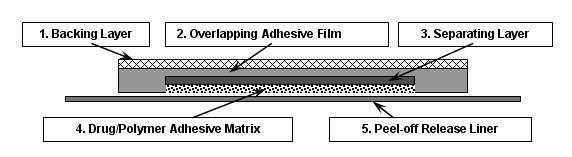
Figure 1 Cross-Section Diagram of Butrans (not to scale).
The active ingredient in Butrans is buprenorphine. The inactive ingredients in each system are: levulinic acid, oleyl oleate, povidone, and polyacrylate cross-linked with aluminum.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Buprenorphine is a partial agonist at mu opioid receptors. Buprenorphine is also an antagonist at kappa opioid receptors, an agonist at delta opioid receptors, and a partial agonist at ORL-1 (nociceptin) receptors. Its clinical actions result from binding to the opioid receptors.
12.2 Pharmacodynamics
Central Nervous System Effects
Buprenorphine binds to and dissociates from the mu opioid receptor slowly.
Electrophysiology
The effect of Butrans 10 mcg/hour and 2 x Butrans 20 mcg/hour on QTc interval was evaluated in a double-blind (Butrans vs. placebo), randomized, placebo and active-controlled (moxifloxacin 400 mg, open label), parallel-group, dose-escalating, single-dose study in 132 healthy male and female subjects aged 18 to 55 years. The dose escalation sequence for Butrans during the titration period was: Butrans 5 mcg/hour for 3 days, then Butrans 10 mcg/hour for 3 days, then Butrans 20 mcg/hour for 3 days, then 2 x Butrans 20 mcg/hour for 4 days. The QTc evaluation was performed during the third day of Butrans 10 mcg/hour and the fourth day of 2 x Butrans 20 mcg/hour when the plasma levels of buprenorphine were at steady state for the corresponding doses [see Warnings and Precautions (5.4)].
There was no clinically meaningful effect on mean QTc with a Butrans dose of 10 mcg/hour. A Butrans dose of 40 mcg/hour (given as two 20 mcg/hour Butrans Transdermal Systems) prolonged mean QTc by a maximum of 9.2 (90% CI: 5.2-13.3) msec across the 13 assessment time points.
Endocrine Effects
Opioids may influence the hypothalamic-pituitary-adrenal or -gonadal axes. Some changes that can be seen include an increase in serum prolactin, and decreases in plasma cortisol and testosterone. Clinical symptoms may be manifest from these hormonal changes.
Other Effects
Buprenorphine causes dose-related miosis and produces urinary retention in some patients.
In-vitro and animal studies indicate various effects of naturally occurring opioids, such as morphine, on components of the immune system. The clinical significance of these findings is unknown. Whether buprenorphine, a semi-synthetic opioid, has immunological effects similar to morphine is unknown.
12.3 Pharmacokinetics
Each Butrans system provides delivery of buprenorphine for 7 days. Steady state was achieved during the first application by Day 3 (see Figure 2).
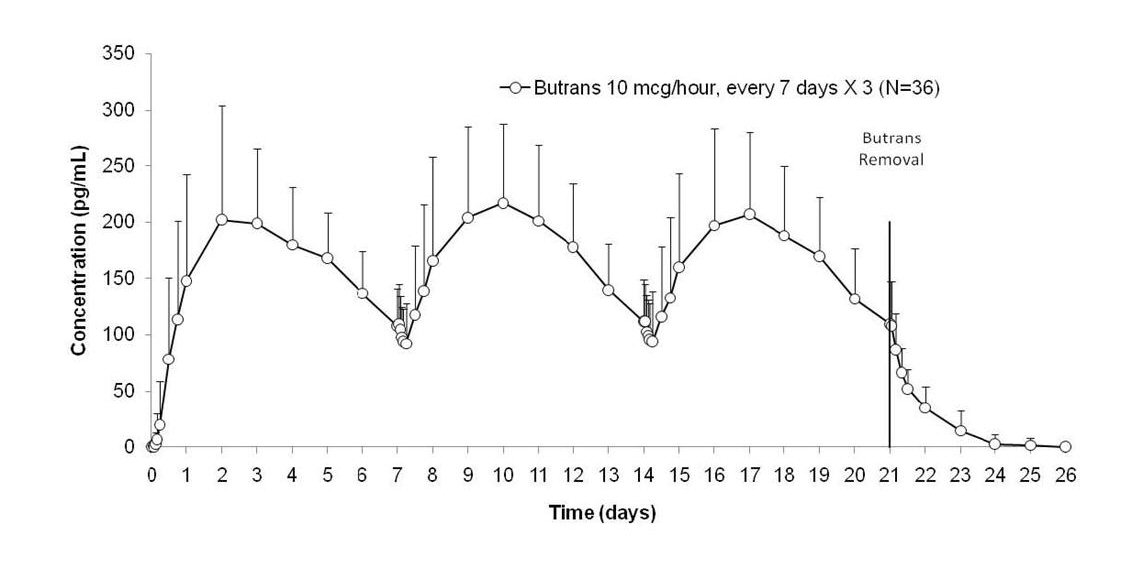
Figure 2
Mean (SD) Buprenorphine Plasma Concentrations Following Three Consecutive Applications of Butrans 10 mcg/hour (N = 36 Healthy Subjects)
Butrans 5, 10, and 20 mcg/hour provide dose-proportional total buprenorphine exposures (AUC) following 7-day applications. Butrans single 7-day application and steady-state pharmacokinetic parameters are summarized in Table 6. Plasma buprenorphine concentrations after titration showed no further change over the 60-day period studied. After removal of Butrans, mean buprenorphine concentrations decrease approximately 50% within 10 - 24 hours, followed by decline with an apparent terminal half-life of approximately 26 hours.
| Single 7-day Application | AUCinf (pg.h/mL) | Cmax (pg/mL) |
| Butrans 5 mcg/hour | 12087 (37) | 176 (67) |
| Butrans 10 mcg/hour | 27035 (29) | 191 (34) |
| Butrans 20 mcg/hour | 54294 (36) | 471 (49) |
| Multiple 7-day Applications | AUCtau,ss (pg.h/mL) | Cmax,ss (pg/mL) |
| Butrans 10 mcg/hour, steady-state | 27543 (33) | 224 (35) |
Absorption
Transdermal delivery studies showed that intact human skin is permeable to buprenorphine. In clinical pharmacology studies, the median time for Butrans 10 mcg/hour to deliver quantifiable buprenorphine concentrations (≥25 pg/mL) was approximately 17 hours. The absolute bioavailability of Butrans relative to IV administration, following a 7-day application, is approximately 15% for all doses (Butrans 5, 10, and 20 mcg/hour).
Distribution
Buprenorphine is approximately 96% bound to plasma proteins, mainly to alpha- and beta-globulin.
Studies of IV buprenorphine have shown a large volume of distribution (approximately 430 L), implying extensive distribution of buprenorphine.
Following IV administration, buprenorphine and its metabolites are secreted into bile and excreted in urine. CSF buprenorphine concentrations appear to be approximately 15-25% of concurrent plasma concentrations.
Metabolism
Buprenorphine metabolism in the skin following Butrans application is negligible. Following transdermal application, buprenorphine is eliminated via hepatic metabolism, with subsequent biliary excretion and renal excretion of soluble metabolites.
Buprenorphine primarily undergoes N-dealkylation by CYP3A4 to norbuprenorphine and glucuronidation by UGT-isoenzymes (mainly UGT1A1 and 2B7) to buprenorphine 3-O-glucuronide. Norbuprenorphine, the major metabolite, is also glucuronidated (mainly UGT1A3) prior to excretion.
Norbuprenorphine is the only known active metabolite of buprenorphine. It has been shown to be a respiratory depressant in rats, but only at concentrations at least 50-fold greater than those observed following application to humans of Butrans 20 mcg/hour.
Since metabolism and excretion of buprenorphine occur mainly via hepatic elimination, reductions in hepatic blood flow induced by some general anesthetics (e.g., halothane) and other drugs may result in a decreased rate of hepatic elimination of the drug, resulting in increased plasma concentrations.
Excretion
Following intramuscular administration of 2 mcg/kg dose of buprenorphine, approximately 70% of the dose was excreted in feces within 7 days. Approximately 27% was excreted in urine. The total clearance of buprenorphine is approximately 55 L/hour in postoperative patients.
Drug Interactions
Effect of CYP3A4 inhibitors
In a drug-drug interaction study, Butrans 10 mcg/hour (single dose x 7 days) was co-administered with 200 mg ketoconazole, a strong CYP3A4 inhibitor or ketoconazole placebo twice daily for 11 days and the pharmacokinetics of buprenorphine and its metabolites were evaluated. Plasma buprenorphine concentrations did not accumulate during co-medication with ketoconazole 200 mg twice daily. Based on the results from this study, metabolism during therapy with Butrans is not expected to be affected by co-administration of CYP3A4 inhibitors [see Drug Interactions (7.1)].
Antiretroviral agents have been evaluated for CYP3A4 mediated interactions with sublingual buprenorphine. Nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) do not appear to have clinically significant interactions with buprenorphine. However, certain protease inhibitors (PIs) with CYP3A4 inhibitory activity such as atazanavir and atazanavir/ritonavir resulted in elevated levels of buprenorphine and norbuprenorphine when buprenorphine and naloxone were administered sublingually. Cmax and AUC for buprenorphine increased by up to 1.6 and 1.9 fold, and Cmax and AUC for norbuprenorphine increased by up to 1.6 and 2.0 fold respectively, when sublingual buprenorphine was administered with these PIs. Patients in this study reported increased sedation, and symptoms of opiate excess have been found in post-marketing reports of patients receiving buprenorphine and atazanavir with and without ritonavir concomitantly. It should be noted that atazanavir is both a CYP3A4 and UGT1A1 inhibitor. As such, the drug-drug interaction potential for buprenorphine with CYP3A4 inhibitors is likely to be dependent on the route of administration as well as the specificity of enzyme inhibition [see Drug Interactions (7.1)].
Effect of CYP3A4 inducers on buprenorphine
The interaction between buprenorphine and CYP3A4 inducers has not been studied.
Application Site
A study in healthy subjects demonstrated that the pharmacokinetic profile of buprenorphine delivered by Butrans 10 mcg/hour is similar when applied to the upper outer arm, upper chest, upper back, or the side of the chest [see Dosage and Administration (2.3)].
The reapplication of Butrans 10 mcg/hour after various rest periods to the same application site in healthy subjects showed that the minimum rest period needed to avoid variability in drug absorption is 3 weeks (21 days) [see Dosage and Administration (2.3)].
External Heat
In a study of healthy subjects, application of a heating pad directly on the Butrans 10 mcg/hour system caused a 26% - 55% increase in blood concentrations of buprenorphine. Concentrations returned to normal within 5 hours after the heat was removed. For this reason, applying heating pads directly to the Butrans system during system wear should be avoided [see Warnings and Precautions (5.11)].
Endotoxin Challenge
Fever may increase the permeability of the skin, leading to increased buprenorphine concentrations during Butrans treatment. As a result, febrile patients are at increased risk for the possibility of Butrans-related reactions during treatment with Butrans. Monitor patients with febrile illness for adverse effects and consider dose adjustment [see Warnings and Precautions (5.10)]. In a crossover study of healthy subjects receiving endotoxin or placebo challenge during Butrans 10 mcg/hour wear, the AUC and Cmax were similar despite a physiologic response of mild fever to endotoxin.
Specific Populations
Gender
In a pooled data analysis utilizing data from several studies that administered Butrans 10 mcg/hour to healthy subjects, no differences in buprenorphine Cmax and AUC or body-weight normalized Cmax and AUC were observed between males and females treated with Butrans [see Use In Specific Populations (8.8)].
Geriatric
Following a single application of Butrans 10 mcg/hour to 12 healthy young adults (mean age 32 years) and 12 healthy elderly subjects (mean age 72 years), the pharmacokinetic profile of Butrans was similar in healthy elderly and healthy young adult subjects, though the elderly subjects showed a trend toward higher plasma concentrations immediately after Butrans removal. Both groups eliminated buprenorphine at similar rates after system removal [see Dosage and Administration (2.4) and Use In Specific Populations (8.5)].
In a study of healthy young subjects, healthy elderly subjects, and elderly subjects treated with thiazide diuretics, Butrans at a fixed dose-escalation schedule (Butrans 5 mcg/hour for 3 days, followed by Butrans 10 mcg/hour for 3 days and Butrans 20 mcg/hour for 7 days) produced similar mean plasma concentration vs. time profiles for each of the three subject groups. There were no significant differences between groups in buprenorphine Cmax or AUC [see Dosage and Administration (2.4) and Use In Specific Populations (8.5)].
Renal Impairment
No studies in patients with renal impairment have been performed with Butrans.
In an independent study, the effect of impaired renal function on buprenorphine pharmacokinetics after IV bolus and after continuous IV infusion administrations was evaluated. It was found that plasma buprenorphine concentrations were similar in patients with normal renal function and in patients with impaired renal function or renal failure. In a separate investigation of the effect of intermittent hemodialysis on buprenorphine plasma concentrations in chronic pain patients with end-stage renal disease who were treated with a transdermal buprenorphine product (marketed outside the US) up to 70 mcg/hour, no significant differences in buprenorphine plasma concentrations before or after hemodialysis were observed [see Dosage and Administration (2.4) and Use In Specific Populations (8.7)].
No notable relationship was observed between estimated creatinine clearance rates and steady-state buprenorphine concentrations among patients during Butrans therapy [see Use In Specific Populations (8.7)].
Hepatic Impairment
The pharmacokinetics of buprenorphine following an IV infusion of 0.3 mg of buprenorphine was compared in 8 patients with mild impairment (Child-Pugh A), 4 patients with moderate impairment (Child-Pugh B) and 12 subjects with normal hepatic function. Buprenorphine and norbuprenorphine exposure did not increase in the mild and moderate hepatic impairment patients.
Butrans has not been evaluated in patients with severe (Child-Pugh C) hepatic impairment [see Dosage and Administration (2.6), Warnings and Precautions (5.8) and Use In Specific Populations (8.6)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Buprenorphine administered daily by skin painting to Sprague Dawley rats for 100 weeks at dosages (20, 60, or 200 mg/kg) produced systemic exposures (based on area under the curve, AUC) that ranged from approximately 130 to 350 times that of human subjects administered the maximum recommended human dose (MRHD) of Butrans 20 mcg/hour. An increased incidence of benign testicular interstitial cell tumors, considered buprenorphine treatment-related, was observed in male rats compared with concurrent controls. The tumor incidence was also above the highest incidence in the historical control database of the testing facility. These tumors were noted at 60 mg/kg/day and higher at approximately 220 times the proposed MRHD based on AUC). The no observed effect level (NOEL) was 20 mg/kg/day (approximately 140 times the proposed MRHD based on AUC). The mechanism leading to the tumor findings and relevance to humans is unknown.
Buprenorphine was administered by skin painting to hemizygous Tg.AC mice over a 6-month study period. At the dosages administered daily (18.75, 37.5, 150, or 600 mg/kg/day), buprenorphine was not carcinogenic or tumorigenic at systemic exposure to buprenorphine, based on AUC, of up to approximately 1000 times that of human subjects administered Butrans 20 mcg/hour, the MRHD.
Mutagenesis
Buprenorphine was not genotoxic in 3 in-vitro genetic toxicology studies (bacterial mutagenicity test, mouse lymphoma assay, chromosomal aberration assay in human peripheral blood lymphocytes), and in one in vivo mouse micronucleus test).
Impairment of Fertility
Butrans (1/4 of a Butrans 5 mcg/hour, one Butrans 5 mcg/hour, or one Butrans 20 mcg/hour) every 3 days in males for 4 weeks prior to mating for a total of 10 weeks and in females for 2 weeks prior to mating through gestation day 7) had no effect on fertility or general reproductive performance of rats at AUC-based exposure levels as high as approximately 65 times (females) and 100 times (males) that for human subjects who received Butrans 20 mcg/hour, the MRHD.
14 CLINICAL STUDIES
The efficacy of Butrans has been evaluated in four 12-week double-blind, controlled clinical trials in opioid-naïve and opioid-experienced patients with moderate to severe chronic low back pain or osteoarthritis using pain scores as the primary efficacy variable. Two of these studies, described below, demonstrated efficacy in patients with low back pain. One study in low back pain failed to show efficacy. One study in osteoarthritis, that included an active comparator, failed to show efficacy for Butrans and the active comparator.
12-Week Study in Opioid-Naïve Patients with Chronic Low Back Pain
A total of 1,024 patients with chronic low back pain who were suboptimally responsive to their non-opioid therapy entered an open-label, dose-titration period for up to four weeks. Patients initiated therapy with three days of treatment with Butrans 5 mcg/hour. After three days, if adverse events were tolerated but the pain persisted (≥5 on an 11-point, 0 to 10 Numerical Rating Scale), the dose was increased to Butrans 10 mcg/hour. If adverse effects were tolerated but adequate analgesia was not reached, the dose was increased to Butrans 20 mcg/hour for an additional 10-12 days. Patients who achieved adequate analgesia and tolerable adverse effects on Butrans were then randomized to remain on their titrated dose of Butrans or matching placebo. Fifty-three percent of the patients who entered the open-label titration period were able to titrate to a tolerable and effective dose and were randomized into a 12-week, double-blind treatment period. Twenty three percent of patients discontinued due to an adverse event from the open-label titration period and 14% discontinued due to lack of a therapeutic effect. The remaining 10% of patients were dropped due to various administrative reasons.
During the first seven days of double-blind treatment patients were allowed up to two tablets per day of immediate-release oxycodone 5 mg as supplemental analgesia to minimize opioid withdrawal symptoms in patients randomized to placebo. Thereafter, the supplemental analgesia was limited to either acetaminophen 500 mg or ibuprofen 200 mg at a maximum of four tablets per day. Sixty-six percent of the patients treated with Butrans completed the 12-week treatment compared to 70% of the patients treated with placebo. Of the 256 patients randomized to Butrans, 9% discontinued due to lack of efficacy and 16% due to adverse events. Of the 283 patients randomized to placebo, 13% discontinued due to lack of efficacy and 7% due to adverse events.
Of the patients who were randomized, the mean pain (SE) NRS scores were 7.2 (0.08) and 7.2 (0.07) at screening and 2.6 (0.08) and 2.6 (0.07) at pre-randomization (beginning of double-blind phase) for the Butrans and placebo groups, respectively.
The score for average pain over the last 24 hours at the end of the study (Week 12/Early Termination) was statistically significantly lower for patients treated with Butrans compared with patients treated with placebo. The proportion of patients with various degrees of improvement, from screening to study endpoint, is shown in Figure 3 below.
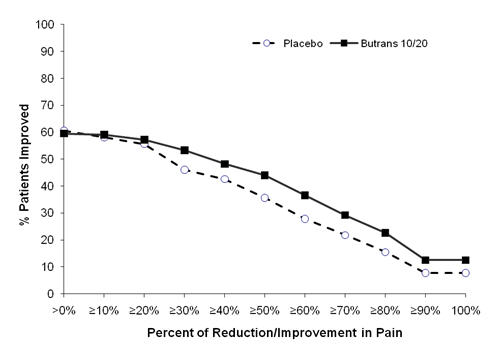
Figure 3 Percent Reduction in Pain Intensity
12-Week Study in Opioid-Experienced Patients with Chronic Low Back Pain
One thousand one hundred and sixty (1,160) patients on chronic opioid therapy (total daily dose 30-80 mg morphine equivalent) entered an open-label, dose-titration period with Butrans for up to 3 weeks, following taper of prior opioids. Patients initiated therapy with Butrans 10 mcg/hour for three days. After three days, if the patient tolerated the adverse effects, the dose was increased to Butrans 20 mcg/hour for up to 18 days. Patients with adequate analgesia and tolerable adverse effects on Butrans 20 mcg/hour were randomized to remain on Butrans 20 mcg/hour or were switched to a low-dose control (Butrans 5 mcg/hour) or an active control. Fifty-seven percent of the patients who entered the open-label titration period were able to titrate to and tolerate the adverse effects of Butrans 20 mcg/hour and were randomized into a 12-week double-blind treatment phase. Twelve percent of patients discontinued due to an adverse event and 21% discontinued due to lack of a therapeutic effect during the open-label titration period.
During the double-blind period, patients were permitted to take ibuprofen (200 mg tablets) or acetaminophen (500 mg tablets) every 4 hours as needed for supplemental analgesia (up to 3200 mg of ibuprofen and 4 grams of acetaminophen daily). Sixty seven percent of patients treated with Butrans 20 mcg/hour and 58% of patients treated with Butrans 5 mcg/hour completed the 12-week treatment. Of the 219 patients randomized to Butrans 20 mcg/hour, 11% discontinued due to lack of efficacy and 13% due to adverse events. Of the 221 patients randomized to Butrans 5 mcg/hour, 24% discontinued due to lack of efficacy and 6% due to adverse events.
Of the patients who were able to be randomized in the double-blind period, the mean pain (SE) NRS scores were 6.4 (0.08) and 6.5 (0.08) at screening and were 2.8 (0.08) and 2.9 (0.08) at pre-randomization (beginning of Double-Blind Period) for the Butrans 5 mcg/hour and Butrans 20 mcg/hour, respectively.
The score for average pain over the last 24 hours at Week 12 was statistically significantly lower for subjects treated with Butrans 20 mcg/hour compared to subjects treated with Butrans 5 mcg/hour. A higher proportion of Butrans 20 mcg/hour patients (49%) had at least a 30% reduction in pain score from screening to study endpoint when compared to Butrans 5 mcg/hour patients (33%). The proportion of patients with various degrees of improvement from screening to study endpoint is shown in Figure 4 below.
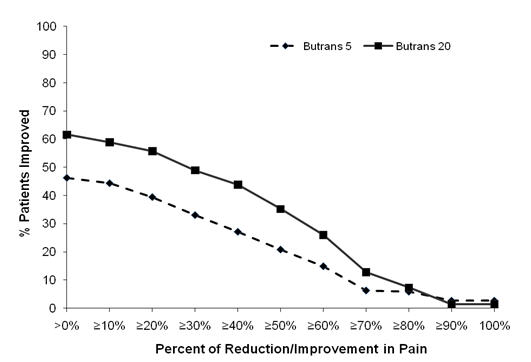
Figure 4 Percent Reduction in Pain Intensity
16 HOW SUPPLIED/STORAGE AND HANDLING
Butrans (buprenorphine) Transdermal System is supplied in cartons containing 4 individually-packaged systems and a pouch containing 4 Patch-Disposal Units.
Butrans 5 mcg/hour Transdermal System, 4-count carton
NDC 59011-750-04
Butrans 10 mcg/hour Transdermal System, 4-count carton
NDC 59011-751-04
Butrans 20 mcg/hour Transdermal System, 4-count carton
NDC 59011-752-04
Store at 25°C (77°F); excursions permitted between 15°C - 30°C (59°F - 86°F).
Special Handling Instructions
If the buprenorphine-containing adhesive matrix accidentally contacts the skin, wash the area with water. Do not use soap, alcohol, or other solvents to remove the adhesive because they may enhance the absorption of the drug.
When changing the system, remove Butrans, fold it over on itself, and flush it down the toilet. Alternatively, Butrans can be sealed in the Patch-Disposal Unit provided and then disposed of in the trash. Never throw Butrans away in the trash without sealing it in the Patch-Disposal Unit.
Apply immediately after removal from the individually sealed pouch. Do not use Butrans if the pouch seal is broken or the patch is cut, damaged, or changed in any way. Butrans is for transdermal use only.
Keep Butrans out of the reach of children and pets.
17 PATIENT COUNSELING INFORMATION
See MEDICATION GUIDE (including Instructions for Use) as appended at the end of the full prescribing information.
17.1 Information for Patients and Caregivers
Provide the following information to patients receiving Butrans or their caregivers:
- Advise patients to carefully follow instructions for the application, removal, and disposal of Butrans. Each week, apply Butrans to a different site based on the 8 described skin sites, with a minimum of 3 weeks between applications to a previously used site.
- Advise patients to apply Butrans to a hairless or nearly hairless skin site. If none are available, instruct patients to clip the hair at the site and not to shave the area. Instruct patients not to apply to irritated skin. If the application site must be cleaned, use clear water only. Soaps, alcohol, oils, lotions, or abrasive devices should not be used. Allow the skin to dry before applying Butrans.
- Advise the patient to wear Butrans continuously for 7 days.
- Advise patients to talk to their doctor if they have any pain or bothersome side effects while they are using Butrans. The dose may have to be changed.
- Advise patients not to increase or decrease the Butrans dose they are using without first speaking to their doctor.
- Advise patients that Butrans may impair mental and/or physical ability required for the performance of potentially hazardous tasks (e.g., driving, operating heavy machinery).
- Advise patients who are taking Butrans not to drink alcohol. They should also avoid taking sleep aids and CNS depressants, unless a doctor prescribes them.
- Advise patients that while wearing Butrans, they should avoid exposing the Butrans site to external heat sources, such as heating pads, electric blankets, heat lamps, saunas, hot tubs, heated water beds, etc, because an increase in absorption of buprenorphine may occur that could lead to an overdose or death.
- Advise women who become pregnant, or who plan to become pregnant, to ask their doctor about the effects that Butrans may have on themselves and their pregnancy.
- Advise patients that buprenorphine is a drug that some people may abuse. They should use Butrans only as directed, and not give it to anyone other than the individual for whom it was prescribed. Protect it from theft. Be especially careful to keep this medication away from children and pets.
- Advise patients to tell their doctor if they have a history of serious skin reactions to adhesives, as they may not be able to use Butrans.
- Advise patients who must stop using Butrans that they should speak with their doctor to manage the transition to other pain medications.
Healthcare professionals can telephone Purdue Pharma’s Medical Services Department
(1-888-726-7535) for information on this product.
Distributed by: Purdue Pharma L.P., Stamford, CT 06901-3431
Manufactured by: LTS Lohmann Therapie-Systeme AG, Andernach, Germany
U.S. Patent Numbers 5681413; 5804215; 6264980; 6315854; 6344211; RE41408; RE41489; RE41571.
© 2011, Purdue Pharma L.P.
302546-0B
MEDICATION GUIDE
Butrans™ (BYOO-trans) CIII
(buprenorphine)
Transdermal System
| Keep Butrans in a safe place away from children. Accidental use by a child is a medical emergency and can result in death. If a child accidentally uses Butrans, get emergency help right away. |
Read the Medication Guide that comes with Butrans before you start using it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or your treatment. Make sure you read and understand all the instructions for using Butrans. Talk to your doctor if you have questions.
What is the most important information I should know about Butrans?
- Butrans overdose can cause serious and life threatening breathing problems.
- Butrans is a skin patch that contains the strong opioid pain medicine (narcotic) buprenorphine.
- Butrans is used to treat moderate to severe chronic pain that continues around-the-clock and is expected to last for a long period of time.
- Butrans is not for pain that:
- you only have once in while (“as needed”)
- is expected to last for only a short time or pain due to surgery
- Serious and life-threatening breathing problems can happen with Butrans, especially during the first 24 to 72 hours after you apply a new patch. This can happen because of an overdose or if the dose you are using is too high for you.
-
Call your doctor right away or get emergency medical help if you:
- have trouble breathing
- have changes in breathing
- unusual deep “sighing” breathing
- slow or shallow breathing
- new or unusual snoring
- have a slow heartbeat
- have severe sleepiness
- have cold, clammy skin
- feel faint, dizzy, confused, or cannot think, walk, or talk normally
-
Do not place direct heat on Butrans. Exposure of Butrans to direct heat may cause too much of the medicine in Butrans to pass into your body. This can lead to overdose and death. Keep the Butrans system away from:
- heating pads
- electric blankets
- heaters
- tanning lamps
- saunas
- hot tubs
- heated waterbeds
- hot baths
- sunbathing
- Place the Butrans patch only on clean skin. Do not use Butrans on broken, irritated and cracked skin.
- Do not use Butrans if the seal on the protective pouch is broken or if the patch is cut, damaged or changed. Do not cut the patch.
What is Butrans?
Butrans is a prescription medicine used to treat moderate to severe chronic pain that continues around-the-clock and is expected to last for a long period of time.
| Butrans is a controlled substance (CIII) because it contains buprenorphine that can be a target for people who abuse prescription medicines or street drugs. Prevent theft, misuse and abuse. Keep Butrans in a safe place to protect from being stolen. Never give Butrans to anyone else, even if they have the same symptoms you have. It may harm them or even cause death. Selling or giving away this medicine is against the law. |
It is not known if Butrans is safe and effective in children.
Who should not use Butrans?
Do not use Butrans if you:
- have trouble breathing, severe asthma or severe lung problems
- have a bowel blockage called paralytic ileus.
- are allergic to any of the ingredients in Butrans. See the end of this Medication Guide for a list of the ingredients in Butrans. Ask your doctor if you are not sure.
Talk to your doctor before taking this medicine if you have any of these conditions listed above.
What should I tell my doctor before using Butrans?
Butrans may not be right for you. Before taking Butrans, tell your doctor if you:
- have trouble breathing or lung problems
- have or a family member has a history of a heart problem called Long QT syndrome
- have or have had head injury or brain problems
- have low blood pressure
- have liver or kidney problems
- have hepatitis B or hepatitis C
- have or have had convulsions or seizures
- have severe scoliosis
- have thyroid problems
- have prostate problems or trouble urinating
- have adrenal gland problems, such as Addison’s disease
- have a past or present drinking problem or alcoholism, or a family history of this problem
- have mental problems including major depression or hallucinations (seeing or hearing things that are not there)
- have a past or present drug abuse or addiction problem, or a family history of this problem
- have any other medical conditions
- are pregnant or plan to become pregnant.
- are breast-feeding or plan to breast-feed. Butrans passes into your breast milk. You and your doctor should decide if you will take Butrans or breast-feed. You should not do both. Talk to your doctor about the best way to feed your baby if you take Butrans.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Some medicines may cause serious or life-threatening medical problems when taken with Butrans. Sometimes, the doses of certain medicines and Butrans need to be changed if used together.
Especially tell your doctor if you take:
- other pain medicines
- antidepressant medicines
- sleeping pills
- antihistamines
- anti-anxiety medicines
- muscle relaxants
- anti-nausea medicines
- sedative or tranquilizer medicines (medicines that make you sleepy)
- a medicine for abnormal heartbeats
You should not take Butrans if you already take a monoamine oxidase inhibitor medicine (MAOI) or within 14 days after you stop taking an MAOI medicine.
Ask your doctor if you are not sure if your medicine is one listed above.
Know the medicines you take. Keep a list of your medicines to show your doctor and pharmacist. Your doctor will tell you if it is safe to take other medicines while you are using Butrans.
How should I use Butrans?
- See the “What is the most important information I should know about Butrans?”
-
Butrans patch comes in three strengths. Each Butrans patch has the strength listed on the patch. Your doctor will prescribe the patch that is right for you.
- 5 mcg/hour
- 10 mcg/hour
- 20 mcg/hour
- Before you start using Butrans: If you already use a continuous around-the-clock medicine for your pain, your doctor will tell you how to slowly stop using it. Your doctor should prescribe a short-acting opioid pain medicine for you to use while your dose of Butrans is being adjusted to treat your moderate to severe continuous around-the-clock pain.
- Use Butrans exactly as prescribed by your doctor. Do not change your dose unless your doctor tells you to change it.
- Do not apply Butrans more often than prescribed.
- Do not use more than one patch at the same time unless your doctor tells you to do so.
- You should wear 1 Butrans patch continuously for 7 days.
- If the patch comes off and accidentally sticks to the skin of another person, take the patch off of that person right away, wash the area with clear water, and get medical care for them right away.
- Use only water to wash your skin where you apply Butrans. Do not use soap, alcohol, or other solvents to wash the area or remove any leftover adhesive from the patch.
- See the detailed Instructions for Use that comes with this Medication Guide to learn how to apply Butrans the right way. Talk to your doctor if you have any questions. Your doctor should show you how to use Butrans before you start to use it.
- If you use more Butrans than your doctor prescribed, or overdose, call your local emergency number or your local Poison Control Center right away at 1-800-222-1222 or get emergency help right away.
- Call your doctor right away if you have any swelling or blistering around a patch site.
- Do not apply any medicine, cream, or lotion on the skin at the Butrans application site before applying the patch. This might affect how the patch sticks to the skin and how the medicine is absorbed from the patch.
- Do not stop using Butrans without first talking to your doctor. Your doctor will give you instructions on how to stop using this medicine slowly to avoid uncomfortable symptoms.
- After you stop using Butrans patches flush used or unused patches down the toilet or dispose of the patches in household trash using the Patch-Disposal Unit. See the Instructions for Use that comes with this Medication Guide for disposal instructions.
What should I avoid while using Butrans?
- You should not drive, operate heavy machinery, or do other dangerous activities, until you know how you react to this medicine. Butrans can make you sleepy and cause you to feel dizzy or lightheaded. This may affect your ability to think and react. Ask your doctor when it is okay to do these activities.
- You should not drink alcohol or use prescription or non-prescription medicines that have alcohol in them while using Butrans. Alcohol can increase your chances of having serious side effects including death.
What are the possible side effects of Butrans?
Butrans can cause serious side effects that can lead to death, including:
- See “What is the most important information that I should know about Butrans?”
-
Serious breathing problems that can be life threatening.
Call your doctor or get emergency medical help right away if you:- have trouble breathing,
- have extreme drowsiness with slowed breathing
- have slow shallow breathing (little chest movement with breathing)
- feel faint, very dizzy, confused, or have any other unusual symptoms
- Severe skin reactions. Butrans can cause skin reactions at the site where the patch is applied.
-
Allergic reactions. Rash, itching, and hives are the most common symptoms of an allergic reaction. Call your doctor if you have these symptoms. Get medical help right away if you have any of these symptoms of an allergic reaction while taking Butrans:
- swelling of your lips or tongue
- breathing problems
- wheezing
- chest pain
- Butrans can cause a drop in your blood pressure. Low blood pressure can make you feel dizzy if you get up too fast from sitting or lying down. Low blood pressure is also more likely to happen if you take other medicines that can also lower your blood pressure. Severe low blood pressure can happen if you lose blood or take certain other medicines.
- Liver problems. Your skin or the white part of your eyes can turn yellow (jaundice), urine can turn dark, stools can turn light in color, you may have less of an appetite, and nausea. Your doctor may do tests before you start and while you take Butrans.
- Butrans can increase your chances of having a seizure if you have history of seizures. Tell your doctor if you have a seizure or convulsion while taking Butrans.
- Butrans can cause physical dependence. Do not stop using Butrans or any other opioid without talking to your doctor. You could become sick with uncomfortable withdrawal symptoms because your body has become used to these medicines. Physical dependence is not the same as drug addiction.
- There is a chance of abuse or addiction with Butrans. The chance is higher if you are or have been addicted to or abused other medicines, street drugs or alcohol in the past. You may have a greater risk of developing abuse or addiction again while using Butrans.
The most common side effects of Butrans include:
- nausea
- headache
- dizziness
- constipation
- drowsiness
- vomiting
- dry mouth and
- itching, redness, or rash at the patch site
Constipation (incomplete or hard bowel movements) is a very common side effect of all opioid medicines. Talk to your doctor about the use of laxatives (medicines to treat constipation) and stool softeners to prevent or treat constipation while using Butrans.
Talk to your doctor about any side effect that bother you or do not go away.
These are not all the possible side effects of Butrans. For a complete list, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Butrans?
- Store Butrans at room temperature, between 59°F to 86°F (15°C to 30°C).
- Keep the Butrans patch in its unopened protective pouch until you are ready to use it.
- Keep Butrans in a safe place out of the reach of children.
General information about Butrans
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Butrans for a condition for which it was not prescribed. Do not give Butrans to other people for any reason, even if they have the same symptoms you have. It may harm them and it is against the law.
This Medication Guide summarizes the most important information about Butrans. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about Butrans that is written for doctors.
For questions about Butrans, call Purdue Pharma at 1-888-726-7535 or visit www.butransrems.com or www.purduepharma.com.
What are the ingredients in Butrans?
Active ingredient: buprenorphine
Inactive ingredients: levulinic acid, oleyl oleate, povidone, and polyacrylate cross-linked with aluminum.
Distributed by:
Pharma L.P.
Stamford, CT 06901-3431
Manufactured by:
LTS Lohmann Therapie-Systeme AG
Andernach, Germany
U.S. Patent Numbers: 6,231,886 (5 and 10 mcg/hour patches only); 5,804,215; 5,968,547; 6,264,980; 6,344,211; and 6,344,212.
Issued: June 2010
©2010, Purdue Pharma L.P.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Instructions for Use
Butrans™ (BYOO-trans) CIII
(buprenorphine)
Transdermal System
Be sure that you read, understand, and follow these Instructions for Use before you use Butrans. Talk to your doctor or pharmacist if you have any questions.
Before Applying Butrans:
- Do not use soap, alcohol, lotions, oils, or other products to remove any leftover medicine gel from a patch because this may cause more Butrans to pass through the skin.
- Each patch is sealed in its own protective pouch. Do not remove a patch from the pouch until you are ready to use it.
- Do not use a patch if the seal on the protective pouch is broken or if the patch is cut, damaged or changed in any way.
- Butrans patches are available in 3 different strengths and patch sizes. Make sure you have the right strength patch that has been prescribed for you.
Where to apply Butrans:
- Butrans should be applied to the upper outer arm, upper chest, upper back, or the side of the chest (See Figure 1). These 4 sites (located on both sides of the body) provide 8 possible Butrans application sites. You should change the skin site where you apply Butrans each week, making sure that at least 3 weeks (21 days) pass before you re-use the same skin site.
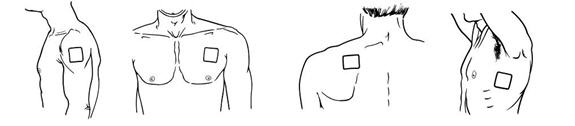
Figure 1
- Apply Butrans to a hairless or nearly hairless skin site. If needed, you can clip the hair at the skin site (See Figure 2). Do not shave the area. The skin site should not be irritated. Use only water to clean the application site. You should not use soaps, alcohol, oils, lotions, or abrasive devices. Allow the skin to dry before you apply the patch.
Figure 2
- The skin site should be free of cuts and irritation (rashes, swelling, redness, or other skin problems).
When to apply a new patch:
- When you apply a new patch, write down the date and time that the patch is applied. Use this to remember when the patch should be removed.
- Change the patch at the same time of day, one week (exactly 7 days) after you apply it.
- After removing and disposing of the patch, write down the time it was removed and how it was disposed.
How to apply Butrans:
- If you are wearing a patch, remember to remove it before applying a new one.
- Each patch is sealed in its own protective pouch.
- Use scissors to cut open the pouch along the dotted line (See Figure 3) and remove the patch. Do not remove the patch from the pouch until you are ready to use it. Do not use patches that have been cut or damaged in anyway.
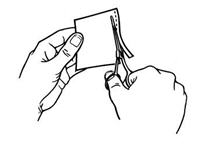
Figure 3
- Hold the patch with the protective liner facing you.
- Gently bend the patch (See Figures 4a and 4b) along the faint line and slowly peel the larger portion of the liner, which covers the sticky surface of the patch.
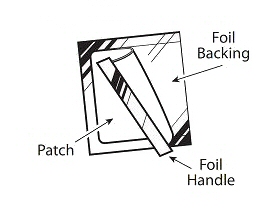
Figure 4a
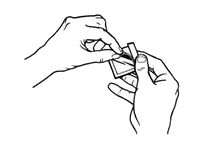
Figure 4b
- Do not touch the sticky side of the patch with your fingers.
- Using the smaller portion of the protective liner as a handle (See Figure 5), apply the sticky side of the patch to one of the 8 body locations described above (see Where to apply Butrans?).
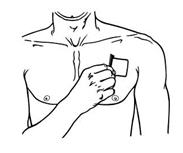
Figure 5
- While still holding the sticky side down, gently fold back the smaller portion of the patch. Grasp an edge of the remaining protective liner and slowly peel it off (See Figure 6).
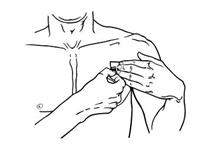
Figure 6
- Press the entire patch firmly into place with the palm (See Figure 7) of your hand over the patch, for about 15 seconds. Do not rub the patch.
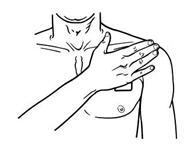
Figure 7
- Make sure that the patch firmly sticks to the skin.
- Go over the edges with your fingers to assure good contact around the patch.
- Always wash your hands after applying or handling a patch.
- After the patch is applied, write down the date and time that the patch is applied. Use this to remember when the patch should be removed.
If the patch falls off right away after applying, throw it away and put a new one on at a different skin site (see Disposing of Butrans Patch).
If a patch falls off, do not touch the sticky side of the patch with your fingers. A new patch should be applied to a different site. Patches that fall off should not be re-applied. They must be thrown away correctly.
If the edges of the Butrans patch start to loosen:
- Apply first aid tape only to the edges of the patch.
- If problems with the patch not sticking continue, cover the patch with special see-through adhesive dressings (for example Bioclusive or Tegaderm).
- Remove the backing from the transparent adhesive dressing and place it carefully and completely over the Butrans patch, smoothing it over the patch and your skin.
- Never cover a Butrans patch with any other bandage or tape. It should only be covered with a special see-through adhesive dressing. Talk to your doctor or pharmacist about the kinds of dressing that should be used.
If your patch falls off later, but before 1 week (7 days) of use, throw it away properly (see Disposing of a Butrans Patch) and apply a new patch at a different skin site. Be sure to let your doctor know that this has happened. Do not replace the new patch until 1 week (7 days) after you put it on (or as directed by your doctor).
Disposing of Butrans Patch:
Butrans patches must be disposed of by flushing them down the toilet or using the Patch-Disposal Unit.
To flush your Butrans patches down the toilet:
Remove your Butrans patch, fold the sticky sides of a used patch together (See Figure 8) and flush it down the toilet right away.
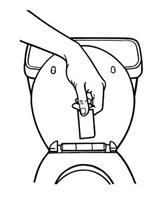
Figure 8
When disposing of unused Butrans patches you no longer need, remove the leftover patches from their protective pouch and remove the protective liner. Fold the patches in half with the sticky sides together, and flush the patches down the toilet.
Do not flush the pouch or the protective liner down the toilet. These items can be thrown away in the trash.
If you prefer not to flush the used patch down the toilet, you must use the Patch-Disposal Unit provided to you to discard the patch.
Never put used Butrans patches in the trash without first sealing them in the Patch-Disposal Unit.
To dispose of Butrans patches in household trash using the Patch-Disposal Unit:
Remove your patch and follow the directions printed on the Patch-Disposal Unit (See Figure 9) or see complete instructions below. Use one Patch-Disposal Unit for each patch.

Figure 9
- Peel back the disposal unit liner to show the sticky surface (See Figure. 10).
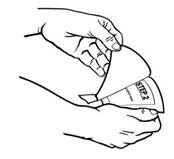
Figure 10
- Place the sticky side of the used or unused patch to the indicated area on the disposal unit (See Figure 11).

Figure 11
- Close the disposal unit by folding the sticky sides together (See Figure 12). Press firmly and smoothly over the entire disposal unit so that the patch is sealed within.
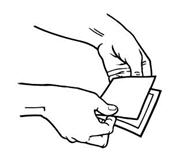
Figure 12
- The closed disposal unit, with the patch sealed inside may be thrown away in the trash (See Figure 13).

Figure 13
Do not put unused patches in household trash without first sealing them in the Patch-Disposal Unit.
Always remove the leftover patches from their protective pouch and remove the protective liner. The pouch and liner can be disposed of separately in the trash and should not be sealed in the Patch-Disposal Unit.
Distributed by:
Purdue Pharma L.P.
Stamford, CT 06901-3431
Manufactured by:
LTS Lohmann Therapie-Systeme AG
Andernach, Germany
Issued: June 2010
©2010, Purdue Pharma L.P.
Bioclusive is a trademark of Systagenix Wound Management (US), Inc.
Tegaderm is a trademark of 3M.





| BUTRANS
buprenorphine patch, extended release |
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021306 | 01/10/2011 | |
| BUTRANS
buprenorphine patch, extended release |
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021306 | 01/10/2011 | |
| BUTRANS
buprenorphine patch, extended release |
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021306 | 01/10/2011 | |
| Labeler - Purdue Pharma L.P. (932323652) |
| Registrant - Purdue Pharma L.P. (932323652) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Lohman Therapie System | 342693590 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Anderson Packaging, Inc | 053217022 | PACK | |
Revised: 09/2011 Purdue Pharma L.P.