avastin (bevacizumab) injection, solution
[Genentech, Inc.]
WARNINGS
GASTROINTESTINAL PERFORATIONS
AVASTIN administration can result in the development of gastrointestinal perforation, in some instances resulting in fatality. Gastrointestinal perforation, sometimes associated with intra‑abdominal abscess, occurred throughout treatment with AVASTIN (i.e., was not correlated to duration of exposure). The incidence of gastrointestinal perforation (gastrointestinal perforation, fistula formation, and/or intra‑abdominal abscess) in patients with colorectal cancer and in patients with non‑small cell lung cancer (NSCLC) receiving AVASTIN was 2.4% and 0.9%, respectively. The typical presentation was reported as abdominal pain associated with symptoms such as constipation and vomiting. Gastrointestinal perforation should be included in the differential diagnosis of patients presenting with abdominal pain on AVASTIN. AVASTIN therapy should be permanently discontinued in patients with gastrointestinal perforation. (See WARNINGS: Gastrointestinal Perforations and DOSAGE AND ADMINISTRATION: Dose Modifications .)
WOUND HEALING COMPLICATIONS
AVASTIN administration can result in the development of wound dehiscence, in some instances resulting in fatality. AVASTIN therapy should be permanently discontinued in patients with wound dehiscence requiring medical intervention. The appropriate interval between termination of AVASTIN and subsequent elective surgery required to avoid the risks of impaired wound healing⁄wound dehiscence has not been determined. (See WARNINGS: Wound Healing Complications and DOSAGE AND ADMINISTRATION: Dose Modifications .)
Hemorrhage
Fatal pulmonary hemorrhage can occur in patients with NSCLC treated with chemotherapy and AVASTIN. The incidence of severe or fatal hemoptysis was 31% in patients with squamous histology and 2.3% in patients with NSCLC excluding predominant squamous histology. Patients with recent hemoptysis (≥1⁄2 tsp of red blood) should not receive AVASTIN. (See WARNINGS: Hemorrhage , ADVERSE REACTIONS: Hemorrhage , and DOSAGE AND ADMINISTRATION: Dose Modifications .)
DESCRIPTION
AVASTIN® (Bevacizumab) is a recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF) in in vitro and in vivo assay systems. Bevacizumab contains human framework regions and the complementarity‑determining regions of a murine antibody that binds to VEGF ( 1). Bevacizumab is produced in a Chinese Hamster Ovary mammalian cell expression system in a nutrient medium containing the antibiotic gentamicin and has a molecular weight of approximately 149 kilodaltons. AVASTIN is a clear to slightly opalescent, colorless to pale brown, sterile, pH 6.2 solution for intravenous (IV) infusion. AVASTIN is supplied in 100 mg and 400 mg preservative‑free, single‑use vials to deliver 4 mL or 16 mL of AVASTIN (25 mg/mL). The 100 mg product is formulated in 240 mg α,α‑trehalose dihydrate, 23.2 mg sodium phosphate (monobasic, monohydrate), 4.8 mg sodium phosphate (dibasic, anhydrous), 1.6 mg polysorbate 20, and Water for Injection, USP. The 400 mg product is formulated in 960 mg α,α‑trehalose dihydrate, 92.8 mg sodium phosphate (monobasic, monohydrate), 19.2 mg sodium phosphate (dibasic, anhydrous), 6.4 mg polysorbate 20, and Water for Injection, USP.
CLINICAL PHARMACOLOGY
MECHANISM OF ACTION
Bevacizumab binds VEGF and prevents the interaction of VEGF to its receptors (Flt‑1 and KDR) on the surface of endothelial cells. The interaction of VEGF with its receptors leads to endothelial cell proliferation and new blood vessel formation in in vitro models of angiogenesis. Administration of Bevacizumab to xenotransplant models of colon cancer in nude (athymic) mice caused reduction of microvascular growth and inhibition of metastatic disease progression.
PHARMACOKINETICS
The pharmacokinetic profile of Bevacizumab was assessed using an assay that measures total serum Bevacizumab concentrations (i.e., the assay did not distinguish between free Bevacizumab and Bevacizumab bound to VEGF ligand). Based on a population pharmacokinetic analysis of 491 patients who received 1 to 20 mg/kg of AVASTIN weekly, every 2 weeks, or every 3 weeks, the estimated half‑life of Bevacizumab was approximately 20 days (range 11–50 days). The predicted time to reach steady state was 100 days. The accumulation ratio following a dose of 10 mg/kg of Bevacizumab every 2 weeks was 2.8.
The clearance of Bevacizumab varied by body weight, by gender, and by tumor burden. After correcting for body weight, males had a higher Bevacizumab clearance (0.262 L/day vs. 0.207 L/day) and a larger Vc (3.25 L vs. 2.66 L) than females. Patients with higher tumor burden (at or above median value of tumor surface area) had a higher Bevacizumab clearance (0.249 L/day vs. 0.199 L/day) than patients with tumor burdens below the median. In a randomized study of 813 patients (Study 1), there was no evidence of lesser efficacy (hazard ratio for overall survival) in males or patients with higher tumor burden treated with AVASTIN as compared to females and patients with low tumor burden. The relationship between Bevacizumab exposure and clinical outcomes has not been explored.
SPECIAL POPULATIONS
Analyses of demographic data suggest that no dose adjustments are necessary for age or sex.
Patients with renal impairment. No studies have been conducted to examine the pharmacokinetics of Bevacizumab in patients with renal impairment.
Patients with hepatic dysfunction. No studies have been conducted to examine the pharmacokinetics of Bevacizumab in patients with hepatic impairment.
CLINICAL STUDIES
AVASTIN® In Metastatic Colorectal Cancer (mCRC)
The safety and efficacy of AVASTIN in the treatment of patients with metastatic carcinoma of the colon or rectum were studied in three randomized, controlled clinical trials in combination with intravenous 5‑fluorouracil‑based chemotherapy. The activity of AVASTIN in patients with metastatic colorectal cancer that progressed on or after receiving both irinotecan based- and oxaliplatin based‑chemotherapy regimens was evaluated in an open‑access trial in combination with intravenous 5‑fluorouracil‑based chemotherapy.
AVASTIN in Combination with Bolus‑IFL
Study 1 was a randomized, double‑blind, active‑controlled clinical trial evaluating AVASTIN as first‑line treatment of metastatic carcinoma of the colon or rectum. Patients were randomized to bolus‑IFL (irinotecan 125 mg/m2 IV, 5‑fluorouracil 500 mg/m2 IV, and leucovorin 20 mg/m2 IV given once weekly for 4 weeks every 6 weeks) plus placebo (Arm 1), bolus‑IFL plus AVASTIN (5 mg/kg every 2 weeks) (Arm 2), or 5‑FU/LV plus AVASTIN (5 mg/kg every 2 weeks) (Arm 3). Enrollment in Arm 3 was discontinued, as pre specified, when the toxicity of AVASTIN in combination with the bolus‑IFL regimen was deemed acceptable.
Of the 813 patients randomized to Arms 1 and 2, the median age was 60, 40% were female, and 79% were Caucasian. Fifty‑seven percent had an ECOG performance status of 0. Twenty‑one percent had a rectal primary and 28% received prior adjuvant chemotherapy. In the majority of patients, 56%, the dominant site of disease was extra abdominal, while the liver was the dominant site in 38% of patients. Results are presented in Table 1 andFigure 1.
| IFL + Placebo | IFL +
AVASTIN 5 mg/kg q 2 wks |
|
|---|---|---|
| Number of Patients | 411 | 402 |
| Overall Survival* | ||
| Median (months) | 15.6 | 20.3 |
| Hazard ratio | 0.66 | |
| Progression‑free Survival* | ||
| Median (months) | 6.2 | 10.6 |
| Hazard ratio | 0.54 | |
| Overall Response Rate† | ||
| Rate (percent) | 35% | 45% |
| Duration of Response | ||
| Median (months) | 7.1 | 10.4 |
| Error bars represent 95% confidence intervals. |
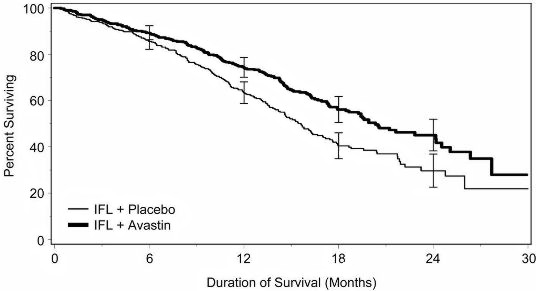 |
The clinical benefit of AVASTIN, as measured by survival in the two principal arms, was seen in subgroups defined by age (<65 yrs, ≥65 yrs) and gender.
Among the 110 patients enrolled in Arm 3, median overall survival was 18.3 months, median progression free survival was 8.8 months, overall response rate was 39%, and median duration of response was 8.5 months.
AVASTIN in Combination with 5‑FU/LV Chemotherapy
Study 2 was a randomized, active‑controlled clinical trial testing AVASTIN in combination with 5‑FU/LV as first‑line treatment of metastatic colorectal cancer. Patients were randomized to receive 5‑FU/LV (5‑fluorouracil 500 mg/m2, leucovorin 500 mg/m2 weekly for 6 weeks every 8 weeks) or 5‑FU/LV plus AVASTIN (5 mg/kg every 2 weeks) or 5‑FU/LV plus AVASTIN (10 mg/kg every 2 weeks). The primary endpoints of the trial were objective response rate and progression‑free survival. Results are presented in Table 2.
| 5-FU/LV | 5‑FU/LV + AVASTIN 5 mg/kg | 5‑FU/LV + AVASTIN 10 mg/kg | |
| Number of Patients | 36 | 35 | 33 |
| Overall Survival | |||
| Median (months) | 13.6 | 17.7 | 15.2 |
| Progression‑free Survival | |||
| Median (months) | 5.2 | 9.0 | 7.2 |
| Overall Response Rate | |||
| Rate (percent) | 17 | 40 | 24 |
Progression‑free survival was significantly longer in patients receiving 5‑FU/LV plus AVASTIN at 5 mg/kg when compared to those not receiving AVASTIN. However, overall survival and overall response rate were not significantly different. Outcomes for patients receiving 5‑FU/LV plus AVASTIN at 10 mg/kg were not significantly different than for patients who did not receive AVASTIN.
AVASTIN in Combination with 5‑FU/LV and Oxaliplatin Chemotherapy
Study 3 was an open label, randomized, 3‑arm, active‑controlled, multicenter clinical trial evaluating AVASTIN alone, AVASTIN in combination with 5‑FU/LV and oxaliplatin (FOLFOX4), and FOLFOX4 alone in the second line treatment of metastatic carcinoma of the colon or rectum. Patients were previously treated with irinotecan and 5‑FU for initial therapy for metastatic disease or as adjuvant therapy. Patients were randomized to FOLFOX4 (Day 1: oxaliplatin 85 mg/m2 and leucovorin 200 mg/m2 concurrently IV, then 5‑FU 400 mg/m2 IV bolus followed by 600 mg/m2 continuously IV; Day 2: leucovorin 200 mg/m2 IV, then 5‑FU 400 mg/m2 IV bolus followed by 600 mg/m2 continuously IV; repeated every 2 weeks), FOLFOX4 plus AVASTIN, or AVASTIN monotherapy. AVASTIN was administered at a dose of 10 mg/kg every 2 weeks and for patients in the FOLFOX4 plus AVASTIN arm, prior to the FOLFOX4 chemotherapy on Day 1.
Of the 829 patients randomized to the three arms, the median age was 61 years, 40% were female, 87% were Caucasian, and 49% had an ECOG performance status of 0. Twenty-six percent had received prior radiation therapy, and 80% received prior adjuvant chemotherapy. Ninety‑nine percent received prior irinotecan, with or without 5‑FU for metastatic colorectal cancer, and 1% received prior irinotecan and 5‑FU as adjuvant therapy.
The AVASTIN monotherapy arm of Study 3 was closed to accrual after enrollment of 244 of the planned 290 patients following a planned interim analysis by the data monitoring committee (DMC), based on evidence of decreased survival in the AVASTIN alone arm as compared to the FOLFOX4 alone arm. In the two remaining study arms, overall survival (OS) was significantly longer in patients receiving AVASTIN in combination with FOLFOX4 as compared to those receiving FOLFOX4 alone (median OS 13.0 mos vs. 10.8 mos; hazard ratio 0.75 [95% CI 0.63, 0.89], p=0.001 stratified logrank test). In addition, patients treated with AVASTIN in combination with FOLFOX4 were reported to have significantly longer progression-free survival and a higher overall response rate based on investigator assessment. The clinical benefit of AVASTIN, as measured by survival, was seen in the subgroups defined by age (<65 yrs, ≥65 yrs) and gender.
AVASTIN in Third‑Line Metastatic Colorectal Cancer
Study 4 was an open access, multicenter, single arm study that evaluated the activity of AVASTIN in combination with bolus or infusional 5‑FU/LV in 339 patients with metastatic colorectal cancer with disease progression following both irinotecan and oxaliplatin containing chemotherapy regimens. The majority (73%) of patients received concurrent 5‑FU/LV according to a bolus regimen.
There was one objective partial response in the first 100 evaluable patients for an overall response rate of 1% (95% CI 0–5.5%).
AVASTIN® In Unresectable Non‑Squamous, Non‑Small Cell Lung Cancer (NSCLC)
The safety and efficacy of AVASTIN as first line treatment of patients with locally advanced, metastatic, or recurrent non‑squamous, NSCLC was studied in a single, large, randomized, active‑controlled, open‑label, multicenter study (Study 5, n=878), supported by a randomized, dose ranging, active controlled Phase 2 study (Study 6, n=98).
In Study 5, chemotherapy‑naïve patients with locally advanced, metastatic or recurrent non‑squamous NSCLC were randomized (1:1) to receive six cycles of paclitaxel 200 mg⁄m2 and carboplatin AUC=6.0, both by IV infusion on day 1 (PC) or PC in combination with AVASTIN at a dose of 15 mg⁄kg by IV infusion on day 1 (PC plus AVASTIN). After completion or upon discontinuation of chemotherapy, patients in the PC plus AVASTIN arm continued to receive AVASTIN alone until disease progression or until unacceptable toxicity. Cycles were repeated every 21 days. Patients with predominant squamous histology (mixed cell type tumors only), central nervous system (CNS) metastasis, gross hemoptysis (≥1⁄2 tsp of red blood), or unstable angina and those receiving therapeutic anticoagulation were excluded. The main outcome measure of the study was duration of survival.
Among the 878 patients randomized to the two treatment arms, the median age was 63, 46% were female, 43% were ≥ age 65, and 28% had ≥ 5% weight loss at study entry. Eleven percent had recurrent disease and of the remaining 89% with newly diagnosed NSCLC, 12% had Stage IIIB with malignant pleural effusion and 76% had Stage IV disease. The survival curves are presented in Figure 2. Overall survival was statistically significantly higher among patients receiving PC plus AVASTIN compared with those receiving PC alone; median OS was 12.3 mos vs. 10.3 mos (hazard ratio 0.80 [repeated 95% CI 0.68, 0.94], final p‑value 0.013, stratified log‑rank test). Based on investigator assessment which was not independently verified, patients were reported to have longer progression‑free survival with AVASTIN in combination with PC compared to PC alone.
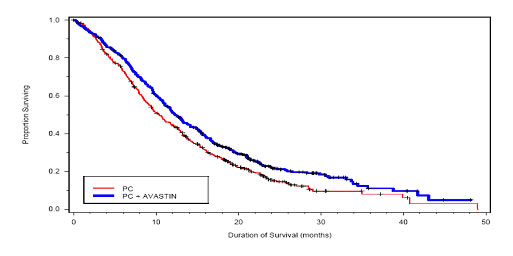 |
INDICATIONS AND USAGE
AVASTIN®, in combination with intravenous 5‑fluorouracil‑based chemotherapy, is indicated for first- or second‑line treatment of patients with metastatic carcinoma of the colon or rectum.
AVASTIN®, in combination with carboplatin and paclitaxel, is indicated for first‑line treatment of patients with unresectable, locally advanced, recurrent or metastatic non-squamous, non-small cell lung cancer.
CONTRAINDICATIONS
None.
WARNINGS
Gastrointestinal Perforations
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
Gastrointestinal perforation complicated by intra‑abdominal abscesses or fistula formation and in some instances with a fatal outcome, occurs at an increased incidence in patients receiving AVASTIN as compared to controls. In Studies 1, 2, and 3, the incidence of gastrointestinal perforation (gastrointestinal perforation, fistula formation, and/or intra-abdominal abscess) in patients receiving AVASTIN was 2.4%. These episodes occurred with or without intra‑abdominal abscesses and at various time points during treatment. The typical presentation was reported as abdominal pain associated with symptoms such as constipation and emesis.
In post‑marketing clinical studies and reports, gastrointestinal perforation, fistula formation in the gastrointestinal tract (eg. gastrointestinal, enterocutaneous, esophageal, duodenal, rectal), and/or intra‑abdominal abscess occurred in patients receiving AVASTIN for colorectal and for other types of cancer. The overall incidence in clinical studies was 1%, but may be higher in some cancer settings. Of the reported events, approximately 30% were fatal. Patients with gastrointestinal perforation, regardless of underlying cancer, typically present with abdominal pain, nausea and fever. Events were reported at various time points during treatment ranging from one week to greater than 1 year from initiation of AVASTIN, with most events occurring within the first 50 days.
Permanently discontinue AVASTIN therapy in patients with gastrointestinal perforation (gastrointestinal perforation, fistula formation, and/or intra‑abdominal abscess).
Non‑Gastrointestinal Fistula Formation
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
Non-gastrointestinal fistula formation has been reported in patients treated with AVASTIN in controlled clinical studies (with an incidence of <0.3%) and in post-marketing experience, in some cases with fatal outcome. Fistula formation involving the following areas of the body other than the gastrointestinal tract have been reported: (tracheo‑esophageal, bronchopleural, biliary, vagina and bladder). Events were reported throughout treatment with Avastin, with most events occurring within the first 6 months.
Permanently discontinue AVASTIN in patients with fistula formation involving an internal organ.
Wound Healing Complications
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
AVASTIN impairs wound healing in animal models. In clinical studies of AVASTIN, patients were not allowed to receive AVASTIN until at least 28 days had elapsed following surgery. In clinical studies of AVASTIN in combination with chemotherapy, there were 6 instances of dehiscence among 788 patients (0.8%).
The appropriate interval between discontinuation of AVASTIN and subsequent elective surgery required to avoid the risks of impaired wound healing has not been determined. In Study 1, 39 patients who received bolus‑IFL plus AVASTIN underwent surgery following AVASTIN therapy; of these patients, six (15%) had wound healing/bleeding complications. In the same study, 25 patients in the bolus‑IFL arm underwent surgery; of these patients, one of 25 (4%) had wound healing/bleeding complications. The longest interval between the last dose of study drug and dehiscence was 56 days; this occurred in a patient on the bolus‑IFL plus AVASTIN arm.
The interval between termination of AVASTIN and subsequent elective surgery should take into consideration the calculated half life of AVASTIN (approximately 20 days).
Discontinue AVASTIN in patients with wound healing complications requiring medical intervention.
Hemorrhage
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
Two distinct patterns of bleeding have occurred in patients receiving AVASTIN. The first is minor hemorrhage, most commonly NCI‑CTC Grade 1 epistaxis. The second is serious, and in some cases fatal, hemorrhagic events.
In Study 6, four of 13 (31%) AVASTIN‑treated patients with squamous cell histology and two of 53 (4%) AVASTIN‑treated patients with histology other than squamous cell, experienced serious or fatal pulmonary hemorrhage as compared to none of the 32 (0%) patients receiving chemotherapy alone. Of the patients experiencing pulmonary hemorrhage requiring medical intervention, many had cavitation and/or necrosis of the tumor, either pre‑existing or developing during AVASTIN therapy. In Study 5, the rate of pulmonary hemorrhage requiring medical intervention for the PC plus AVASTIN arm was 2.3% (10 of 427) compared to 0.5% (2 of 441) for the PC alone arm. There were seven deaths due to pulmonary hemorrhage reported by investigators in the PC plus AVASTIN arm as compared to one in the PC alone arm. Generally, these serious hemorrhagic events presented as major or massive hemoptysis without an antecedent history of minor hemoptysis during Avastin therapy. Do not administer AVASTIN to patients with recent history of hemoptysis of ≥1/2 tsp of red blood. Other serious bleeding events occurring in patients receiving AVASTIN across all indications include gastrointestinal hemorrhage, subarachnoid hemorrhage, and hemorrhagic stroke. Some of these events were fatal. (See ADVERSE REACTIONS: Hemorrhage .)
The risk of central nervous system (CNS) bleeding in patients with CNS metastases receiving AVASTIN has not been evaluated because these patients were excluded from late stage clinical studies following development of CNS hemorrhage in a patient with a CNS metastasis in a Phase 1 study.
Discontinue AVASTIN in patients with serious hemorrhage (i.e., requiring medical intervention) and initiate aggressive medical management. (See ADVERSE REACTIONS: Hemorrhage .)
Arterial Thromboembolic Events
(See DOSAGE AND ADMINISTRATION: Dose Modifications and PRECAUTIONS: Geriatric Use )
Arterial thromboembolic events (ATE) occurred at a higher incidence in patients receiving AVASTIN in combination with chemotherapy as compared to those receiving chemotherapy alone. ATE included cerebral infarction, transient ischemic attacks (TIAs), myocardial infarction (MI), angina, and a variety of other ATE. These events were fatal in some instances.
In a pooled analysis of randomized, controlled clinical trials involving 1745 patients, the incidence of ATE was 4.4% among patients treated with AVASTIN in combination with chemotherapy and 1.9% among patients receiving chemotherapy alone. Fatal outcomes for these events occurred in 7 of 963 patients (0.7%) who were treated with AVASTIN in combination with chemotherapy, compared to 3 of 782 patients (0.4%) who were treated with chemotherapy alone. The incidences of both cerebrovascular arterial events (1.9% vs. 0.5%) and cardiovascular arterial events (2.1% vs. 1.0%) were increased in patients receiving AVASTIN compared to chemotherapy alone. The relative risk of ATE was greater in patients 65 and over (8.5% vs. 2.9%) as compared to those less than 65 (2.1% vs. 1.4%). (See PRECAUTIONS: Geriatric Use .)
The safety of resumption of AVASTIN therapy after resolution of an ATE has not been studied. Permanently discontinue AVASTIN in patients who experience a severe ATE during treatment. (See DOSAGE AND ADMINISTRATION: Dose Modifications and PRECAUTIONS: Geriatric Use .)
Hypertension
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
The incidence of severe hypertension was increased in patients receiving AVASTIN as compared to controls. Across clinical studies the incidence of NCI‑CTC Grade 3 or 4 hypertension ranged from 8–18%.
Medication classes used for management of patients with NCI‑CTC Grade 3 hypertension receiving AVASTIN included angiotensin‑converting enzyme inhibitors, beta blockers, diuretics, and calcium channel blockers. Development or worsening of hypertension can require hospitalization or require discontinuation of AVASTIN in up to 1.7% of patients. Hypertension can persist after discontinuation of AVASTIN. Complications can include hypertensive encephalopathy (in some cases fatal) and CNS hemorrhage.
In the post‑marketing experience, acute increases in blood pressure associated with initial or subsequent infusions of AVASTIN have been reported (see PRECAUTIONS: Infusion Reactions ). Some cases were serious and associated with clinical sequelae.
Permanently discontinue AVASTIN in patients with hypertensive crisis or hypertensive encephalopathy. Temporarily suspend AVASTIN in patients with severe hypertension that is not controlled with medical management. (See DOSAGE AND ADMINISTRATION: Dose Modifications )
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
RPLS has been reported in clinical studies (with an incidence of<0.1%) and in post‑marketing experience. RPLS is a neurological disorder which can present with headache, seizure, lethargy, confusion, blindness and other visual and neurologic disturbances. Mild to severe hypertension may be present, but is not necessary for diagnosis of RPLS. Magnetic Resonance Imaging (MRI) is necessary to confirm the diagnosis of RPLS. The onset of symptoms has been reported to occur from 16 hours to 1 year after initiation of AVASTIN.
In patients developing RPLS, discontinue AVASTIN and initiate treatment of hypertension, if present. Symptoms usually resolve or improve within days, although some patients have experienced ongoing neurologic sequelae. The safety of reinitiating AVASTIN therapy in patients previously experiencing RPLS is not known.
Neutropenia and Infection
(See PRECAUTIONS: Geriatric Use and ADVERSE REACTIONS: Neutropenia and Infection .)
Increased rates of severe neutropenia, febrile neutropenia, and infection with severe neutropenia (including some fatalities) have been observed in patients treated with myelosuppressive chemotherapy plus AVASTIN. (See PRECAUTIONS: Geriatric Use and ADVERSE REACTIONS: Neutropenia and Infection .)
Proteinuria
(See DOSAGE AND ADMINISTRATION: Dose Modifications )
The incidence and severity of proteinuria is increased in patients receiving AVASTIN as compared to control. In Studies 1, 3 and 5, the incidence of NCI‑CTC Grade 3 and 4 proteinuria, characterized as >3.5 gm/24 hours, ranged up to 3.0% in AVASTIN‑treated patients.
Nephrotic syndrome occurred in seven of 1459 (0.5%) patients receiving AVASTIN in clinical studies. One patient died and one required dialysis. In three patients, proteinuria decreased in severity several months after discontinuation of AVASTIN. No patient had normalization of urinary protein levels (by 24 hour urine) following discontinuation of AVASTIN.
The highest incidence of proteinuria was observed in a dose‑ranging, placebo‑controlled, randomized study of AVASTIN in patients with metastatic renal cell carcinoma, an indication for which AVASTIN is not approved, 24‑hour urine collections were obtained in approximately half the patients enrolled. Among patients in whom 24‑hour urine collections were obtained, four of 19 (21%) patients receiving AVASTIN at 10 mg/kg every two weeks, two of 14 (14%) patients receiving AVASTIN at 3 mg/kg every two weeks, and none of the 15 placebo patients experienced NCI‑CTC Grade 3 proteinuria (>3.5 gm protein/24 hours).
Discontinue AVASTIN in patients with nephrotic syndrome. The safety of continued AVASTIN treatment in patients with moderate to severe proteinuria has not been evaluated. In most clinical studies, AVASTIN was interrupted for ≥2 grams of proteinuria/24 hours and resumed when proteinuria was <2 gm/24 hours. Patients with moderate to severe proteinuria based on 24‑hour collections should be monitored regularly until improvement and/or resolution is observed. (See DOSAGE AND ADMINISTRATION: Dose Modifications .)
Congestive Heart Failure
Congestive heart failure (CHF), defined as NCI‑CTC Grade 2–4 left ventricular dysfunction, was reported in 25 of 1459 (1.7%) patients receiving AVASTIN in clinical studies. The risk of CHF appears to be higher in patients receiving AVASTIN who have received prior or concurrent anthracyclines. In a controlled study in patients with breast cancer (an unlabelled indication), the incidence of CHF was higher in the AVASTIN plus chemotherapy arm as compared to the chemotherapy alone arm. Congestive heart failure occurred in 13 of 299 (4%) patients who received prior anthracyclines and/or left chest wall irradiation. Congestive heart failure occurred in six of 44 (14%) patients with relapsed acute leukemia (an unlabelled indication) receiving AVASTIN and concurrent anthracyclines in a single arm study.
The safety of continuation or resumption of AVASTIN in patients with cardiac dysfunction has not been studied.
PRECAUTIONS
GENERAL
Use AVASTIN with caution in patients with known hypersensitivity to AVASTIN or any component of this drug product.
INFUSION REACTIONS
In clinical studies, infusion reactions with the first dose of AVASTIN were uncommon (<3%) and severe reactions occurred in 0.2% of patients. Infusion reactions reported in the clinical trials and postmarketing experience include hypertension, hypertensive crises associated with neurologic signs and symptoms, wheezing, oxygen desaturation, NCI‑CTC Grade 3 hypersensitivity, chest pain, headaches, rigors, and diaphoresis. Adequate information on rechallenge is not available. AVASTIN infusion should be interrupted in all patients with severe infusion reactions and appropriate medical therapy administered.
There are no data regarding the most appropriate method of identification of patients who may safely be retreated with AVASTIN after experiencing a severe infusion reaction.
SURGERY
AVASTIN therapy should not be initiated for at least 28 days following major surgery. The surgical incision should be fully healed prior to initiation of AVASTIN. Because of the potential for impaired wound healing, AVASTIN should be suspended prior to elective surgery. The appropriate interval between the last dose of AVASTIN and elective surgery is unknown; however, the half‑life of AVASTIN is estimated to be 20 days (see CLINICAL PHARMACOLOGY: Pharmacokinetics ) and the interval chosen should take into consideration the half‑life of the drug. (See WARNINGS: Gastrointestinal Perforations and Wound Healing Complications .)
CARDIOVASCULAR DISEASE
Patients were excluded from participation in AVASTIN clinical trials if, in the previous year, they had experienced clinically significant cardiovascular disease. In an exploratory analysis pooling the data from five randomized, placebo‑controlled, clinical trials conducted in patients without a recent history of clinically significant cardiovascular disease, the overall incidence of arterial thromboembolic events, the incidence of fatal arterial thromboembolic events, and the incidence of cardiovascular thromboembolic events were increased in patients receiving AVASTIN plus chemotherapy as compared to chemotherapy alone.
LABORATORY TESTS
Blood pressure monitoring should be conducted every two to three weeks during treatment with AVASTIN. Patients who develop hypertension on AVASTIN may require blood pressure monitoring at more frequent intervals. Patients with AVASTIN‑induced or ‑exacerbated hypertension who discontinue AVASTIN should continue to have their blood pressure monitored at regular intervals.
Patients receiving AVASTIN should be monitored for the development or worsening of proteinuria with serial urinalyses. Patients with a 2+ or greater urine dipstick reading should undergo further assessment, e.g., a 24‑hour urine collection. (See WARNINGS: Proteinuria and DOSAGE AND ADMINISTRATION: Dose Modifications .)
DRUG INTERACTIONS
No formal drug interaction studies with anti‑neoplastic agents have been conducted. In Study 1, patients with colorectal cancer were given irinotecan/5‑FU/leucovorin (bolus‑IFL) with or without AVASTIN. Irinotecan concentrations were similar in patients receiving bolus‑IFL alone and in combination with AVASTIN. The concentrations of SN38, the active metabolite of irinotecan, were on average 33% higher in patients receiving bolus‑IFL in combination with AVASTIN when compared with bolus‑IFL alone. In Study 1, patients receiving bolus‑IFL plus AVASTIN had a higher incidence of NCI‑CTC Grade 3–4 diarrhea and neutropenia. Due to high inter‑patient variability and limited sampling, the extent of the increase in SN38 levels in patients receiving concurrent irinotecan and AVASTIN is uncertain.
In Study 6, based on limited data, there did not appear to be a difference in the mean exposure of either carboplatin or paclitaxel when each was administered alone or in combination with AVASTIN. However, 3 of the 8 patients receiving AVASTIN plus paclitaxel/carboplatin had substantially lower paclitaxel exposure after four cycles of treatment (at Day 63) than those at Day 0, while patients receiving paclitaxel/carboplatin without AVASTIN had a greater paclitaxel exposure at Day 63 than at Day 0.
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
No carcinogenicity data are available for AVASTIN in animals or humans.
AVASTIN may impair fertility. Dose‑related decreases in ovarian and uterine weights, endometrial proliferation, number of menstrual cycles, and arrested follicular development or absent corpora lutea were observed in female cynomolgus monkeys treated with 10 or 50 mg/kg of AVASTIN for 13 or 26 weeks. Following a 4‑ or 12‑week recovery period, which examined only the high–dose group, trends suggestive of reversibility were noted in the two females for each regimen that were assigned to recover. After the 12‑week recovery period, follicular maturation arrest was no longer observed, but ovarian weights were still moderately decreased. Reduced endometrial proliferation was no longer observed at the 12‑week recovery time point, but uterine weight decreases were still notable, corpora lutea were absent in 1 out of 2 animals, and the number of menstrual cycles remained reduced (67%).
PREGNANCY CATEGORY C
AVASTIN has been shown to be teratogenic in rabbits when administered in doses that approximate the human dose on a mg/kg basis. Observed effects included decreases in maternal and fetal body weights, an increased number of fetal resorptions, and an increased incidence of specific gross and skeletal fetal alterations. Adverse fetal outcomes were observed at all doses tested.
Angiogenesis is critical to fetal development and the inhibition of angiogenesis following administration of AVASTIN is likely to result in adverse effects on pregnancy. There are no adequate and well‑controlled studies in pregnant women. AVASTIN should be used during pregnancy or in any woman not employing adequate contraception only if the potential benefit justifies the potential risk to the fetus. All patients should be counseled regarding the potential risk of AVASTIN to the developing fetus prior to initiation of therapy. If the patient becomes pregnant while receiving AVASTIN, she should be apprised of the potential hazard to the fetus and/or the potential risk of loss of pregnancy. Patients who discontinue AVASTIN should also be counseled concerning the prolonged exposure following discontinuation of therapy (half‑life of approximately 20 days) and the possible effects of AVASTIN on fetal development.
NURSING MOTHERS
It is not known whether AVASTIN is secreted in human milk. Because human IgG1 is secreted into human milk, the potential for absorption and harm to the infant after ingestion is unknown. Women should be advised to discontinue nursing during treatment with AVASTIN and for a prolonged period following the use of AVASTIN, taking into account the half‑life of the product, approximately 20 days [range 11– 50 days]. (See CLINICALPHARMACOLOGY: Pharmacokineticss.)
PEDIATRIC USE
The safety and effectiveness of AVASTIN in pediatric patients has not been studied. However, physeal dysplasia was observed in juvenile cynomolgus monkeys with open growth plates treated for four weeks with doses that were less than the recommended human dose based on mg/kg and exposure. The incidence and severity of physeal dysplasia were dose‑related and were at least partially reversible upon cessation of treatment.
GERIATRIC USE
In Study 1, NCI‑CTC Grade 3–4 adverse events were collected in all patients receiving study drug (396 bolus‑IFL plus placebo; 392 bolus‑IFL plus AVASTIN; 109 5‑FU/LV plus AVASTIN), while NCI‑CTC Grade 1 and 2 adverse events were collected in a subset of 309 patients. There were insufficient numbers of patients 65 years and older in the subset in which NCI‑CTC Grade 1–4 adverse events were collected to determine whether the overall adverse event profile was different in the elderly as compared to younger patients. Among the 392 patients receiving bolus‑IFL plus AVASTIN, 126 were at least 65 years of age. Severe adverse events that occurred at a higher incidence (≥2%) in the elderly when compared to those less than 65 years were asthenia, sepsis, deep thrombophlebitis, hypertension, hypotension, myocardial infarction, congestive heart failure, diarrhea, constipation, anorexia, leukopenia, anemia, dehydration, hypokalemia, and hyponatremia. The effect of AVASTIN on overall survival was similar in elderly patients as compared to younger patients.
In Study 3, patients age 65 and older receiving AVASTIN plus FOLFOX4 had a greater relative risk as compared to younger patients for the following adverse events: nausea, emesis, ileus, and fatigue.
In Study 5, patients age 65 and older receiving carboplatin, paclitaxel, and AVASTIN had a greater relative risk for proteinuria as compared to younger patients.
Of the 742 patients enrolled in Genentech‑sponsored clinical studies in which all adverse events were captured, 212 (29%) were age 65 or older and 43 (6%) were age 75 or older. Adverse events of any severity that occurred at a higher incidence in the elderly as compared to younger patients, in addition to those described above, were dyspepsia, gastrointestinal hemorrhage, edema, epistaxis, increased cough, and voice alteration.
In an exploratory, pooled analysis of 1745 patients treated in five randomized, controlled studies, there were 618 (35%) patients age 65 or older and 1127 patients less than 65 years of age. The overall incidence of arterial thromboembolic events was increased in all patients receiving AVASTIN with chemotherapy as compared to those receiving chemotherapy alone, regardless of age. However, the increase in arterial thromboembolic events incidence was greater in patients 65 and over (8.5% vs. 2.9%) as compared to those less than 65 (2.1% vs. 1.4%). (See WARNINGS: Arterial Thromboembolic Events )
ADVERSE REACTIONS
The most serious adverse reactions in patients receiving AVASTIN were:
- Gastrointestinal Perforations (see WARNINGS )
- Non-Gastrointestinal Fistula Formation (see WARNINGS )
- Wound Healing Complications (see WARNINGS )
- Hemorrhage (see WARNINGS )
- Arterial Thromboembolic Events (see WARNINGS )
- Hypertensive Crises (see WARNINGS: Hypertension )
- Reversible Posterior Leukoencephalopathy Syndrome (see WARNINGS )
- Neutropenia and Infection (see WARNINGS )
- Nephrotic Syndrome (see WARNINGS: Proteinuria )
- Congestive Heart Failure (see WARNINGS )
The most common adverse events in patients receiving AVASTIN were asthenia, pain, abdominal pain, headache, hypertension, diarrhea, nausea, vomiting, anorexia, stomatitis, constipation, upper respiratory infection, epistaxis, dyspnea, exfoliative dermatitis, and proteinuria.
Adverse Reactions in Clinical Trails
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
The data described below reflect exposure to AVASTIN in 1529 patients, including 665 receiving AVASTIN for at least 6 months and 199 receiving AVASTIN for at least one year. AVASTIN was studied primarily in placebo- and active‑controlled trials (n = 501, and n = 1028, respectively).
Gastrointestinal Perforation
The incidence of gastrointestinal perforation across all studies ranged from 0–3.7%. The incidence of gastrointestinal perforation, in some cases fatal, in patients with mCRC receiving AVASTIN alone or in combination with chemotherapy was 2.4% compared to 0.3% in patients receiving only chemotherapy. The incidence of gastrointestinal perforation in NSCLC patients receiving AVASTIN was 0.9% compared to 0% in patients receiving only chemotherapy. (See WARNINGS: Gastrointestinal Perforations and DOSAGE AND ADMINISTRATION: Dose Modifications .)
Non-Gastrointestinal Fistula Formation
(See WARNINGS: Non‑Gastrointestinal Fistula Formation and DOSAGE AND ADMINISTRATION: Dose Modifications .)
Would Healing Complications
The incidence of post‑operative wound healing and/or bleeding complications was increased in patients with mCRC receiving AVASTIN as compared to patients receiving only chemotherapy. Among patients requiring surgery on or within 60 days of receiving study treatment, wound healing and/or bleeding complications occurred in 15% (6/39) of patients receiving bolus‑IFL plus AVASTIN as compared to 4% (1/25) of patients who received bolus‑IFL alone. In the same study, the incidence of wound dehiscence was also higher in the AVASTIN‑treated patients (1% vs. 0.5%).
Hemorrhage
Severe or fatal hemorrhages, including hemoptysis, gastrointestinal bleeding, hematemesis, CNS hemorrhage, epistaxis, and vaginal bleeding occurred up to five‑fold more frequently in AVASTIN‑treated patients compared to patients treated with chemotherapy alone. NCI‑CTC Grade 3–5 hemorrhagic events occurred in 4.7% of NSCLC patients and 5.2% of mCRC patients receiving AVASTIN compared to 1.1% and 0.7% for the control groups respectively. (See WARNINGS: Hemorrhage .)
The incidence of epistaxis was higher (35% vs. 10%) in patients with mCRC receiving bolus‑IFL plus AVASTIN compared with patients receiving bolus‑IFL plus placebo. These events were generally mild in severity (NCI‑CTC Grade 1) and resolved without medical intervention. Additional mild to moderate hemorrhagic events reported more frequently in patients receiving bolus‑IFL plus AVASTIN when compared to those receiving bolus‑IFL plus placebo included gastrointestinal hemorrhage (24% vs. 6%), minor gum bleeding (2% vs. 0), and vaginal hemorrhage (4% vs. 2%). (See WARNINGS: Hemorrhage and DOSAGE AND ADMINISTRATION: Dose Modifications .)
Arterial Thromboembolic Events
The incidence of arterial thromboembolic events was increased in NSCLC patients receiving PC plus AVASTIN (3.0%) compared with patients receiving PC alone (1.4%). Five events were fatal in the PC plus AVASTIN arm, compared with 1 event in the PC alone arm. This increased risk is consistent with that observed in patients with mCRC. (See WARNINGS: Arterial Thromboembolic Events , DOSAGE AND ADMINISTRATION: Dose Modifications and PRECAUTIONS: Geriatric Use .)
Venous Thromboembolic Events
The incidence of NCI‑CTC grade 3–4 venous thromboembolic events was higher in patients with mCRC or NSCLC receiving AVASTIN with chemotherapy as compared to those receiving chemotherapy alone. In addition, in patients with mCRC, the risk of developing a second subsequent thromboembolic event in patients receiving AVASTIN and chemotherapy is increased compared to patients receiving chemotherapy alone. In Study 1, 53 patients (14%) on the bolus‑IFL plus AVASTIN arm and 30 patients (8%) on the bolus‑IFL plus placebo arm received full dose warfarin following a venous thromboembolic event. Among these patients, an additional thromboembolic event occurred in 21% (11/53) of patients receiving bolus‑IFL plus AVASTIN and 3% (1/30) of patients receiving bolus‑IFL alone.
The overall incidence of NCI‑CTC Grade 3–4 venous thromboembolic events in Study 1 was 15.1% in patients receiving bolus‑IFL plus AVASTIN and 13.6% in patients receiving bolus‑IFL plus placebo. In Study 1, the incidence of the following NCI‑CTC Grade 3 and 4 venous thromboembolic events was higher in patients receiving bolus‑IFL plus AVASTIN as compared to patients receiving bolus‑IFL plus placebo: deep venous thrombosis (34 vs. 19 patients) and intra‑abdominal venous thrombosis (10 vs. 5 patients).
Hypertension
Fatal CNS hemorrhage complicating AVASTIN induced hypertension can occur.
In Study 1 the incidences of hypertension and of severe hypertension were increased in patients with mCRC receiving AVASTIN compared to those receiving chemotherapy alone (see Table 3).
| Arm 1 IFL + Placebo (n = 394) | Arm 2 IFL + AVASTIN (n = 392) | Arm 3 5‑FU/LV + AVASTIN (n = 109) |
|
|
|||
| Hypertension* (>150/100 mmHg) | 43% | 60% | 67% |
| Severe Hypertension* (>200/110 mmHg) | 2% | 7% | 10% |
Among patients with severe hypertension in the AVASTIN arms, slightly over half the patients (51%) had a diastolic reading greater than 110 mmHg associated with a systolic reading less than 200 mmHg.
Similar results were seen in patients receiving AVASTIN alone or in combination with FOLFOX4 or carboplatin and paclitaxel. (See WARNINGS: Hypertension and DOSAGE AND ADMINISTRATION: Dose Modifications .)
Neutropenia and Infection
An increased incidence of neutropenia has been reported in patients receiving AVASTIN and chemotherapy compared to chemotherapy alone. In Study 1, the incidence of NCI‑CTC Grade 3 or 4 neutropenia was increased in patients with mCRC receiving IFL+AVASTIN (21%) compared to patients receiving IFL alone (14%). In Study 5, the incidence of NCI‑CTC Grade 4 neutropenia was increased in patients with NSCLC receiving PC plus AVASTIN (26.2%) compared with patients receiving PC alone (17.2%). Febrile neutropenia was also increased (5.4% for PC plus AVASTIN vs. 1.8% for PC alone). There were 19 (4.5%) infections with NCI‑CTC Grade 3 or 4 neutropenia in the PC plus AVASTIN arm of which 3 were fatal compared to 9 (2%) neutropenic infections in patients receiving PC alone, of which none were fatal. During the first 6 cycles of treatment, the incidence of serious infections including pneumonia, febrile neutropenia, catheter infections and wound infections was increased in the PC plus AVASTIN arm [58 patients (13.6%)] compared to the PC alone arm [29 patients (6.6%)].
Proteinuria
(See WARNINGS: Proteinuria , DOSAGE AND ADMINISTRATION: Dose Modifications , and PRECAUTIONS: Geriatric Use .)
Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The incidence of antibody development in patients receiving AVASTIN has not been adequately determined because the assay sensitivity was inadequate to reliably detect lower titers. Enzyme linked immunosorbent assays (ELISAs) were performed on sera from approximately 500 patients treated with AVASTIN, primarily in combination with chemotherapy. High titer human anti‑AVASTIN antibodies were not detected.
Immunogenicity data are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors, including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to AVASTIN with the incidence of antibodies to other products may be misleading.
Metastatic Carcinoma of the Colon and Rectum
The data in Tables 4 and 5 were obtained in Study 1. All NCI‑CTC Grade 3 and 4 adverse events and selected NCI‑CTC Grade 1 and 2 adverse events (hypertension, proteinuria, thromboembolic events) were reported for the overall study population. The median age was 60, 60% were male, 79% were Caucasian, 78% had a colon primary lesion, 56% had extra‑abdominal disease, 29% had prior adjuvant or neoadjuvant chemotherapy, and 57% had ECOG performance status of 0. The median duration of exposure to AVASTIN was 8 months in Arm 2 and 7 months in Arm 3. Severe and life threatening (NCI‑CTC Grade 3 and 4) adverse events, which occurred at a higher incidence (≥2%) in patients receiving bolus‑IFL plus AVASTIN as compared to bolus‑IFL plus placebo, are presented in Table 4.
| Arm 1 IFL + Placebo (n = 396) | Arm 2 IFL + AVASTIN (n = 392) | |
|
||
| NCI‑CTC Grade 3–4 Events | 295 (74%) | 340 (87%) |
| Body as a Whole | ||
| Asthenia | 28 (7%) | 38 (10%) |
| Abdominal Pain | 20 (5%) | 32 (8%) |
| Pain | 21 (5%) | 30 (8%) |
| Cardiovascular | ||
| Hypertension | 10 (2%) | 46 (12%) |
| Deep Vein Thrombosis | 19 (5%) | 34 (9%) |
| Intra‑Abdominal Thrombosis | 5 (1%) | 13 (3%) |
| Syncope | 4 (1%) | 11 (3%) |
| Digestive | ||
| Diarrhea | 99 (25%) | 133 (34%) |
| Constipation | 9 (2%) | 14 (4%) |
| Hemic/Lymphatic | ||
| Leukopenia | 122 (31%) | 145 (37%) |
| Neutropenia* | 41 (14%) | 58 (21%) |
NCI‑CTC Grade 1–4 adverse events which occurred at a higher incidence (≥5%) in patients receiving bolus‑IFL plus AVASTIN as compared to the bolus‑IFL plus placebo arm, are presented in Table 5.
| Arm 1 IFL + Placebo (n = 98) | Arm 2 IFL + AVASTIN (n = 102) | Arm 3 5‑FU/LV + AVASTIN (n = 109) |
|
| Body as a Whole | |||
| Pain | 54 (55%) | 62 (61%) | 67 (62%) |
| Abdominal Pain | 54 (55%) | 62 (61%) | 55 (50%) |
| Headache | 19 (19%) | 27 (26%) | 30 (26%) |
| Cardiovascular | |||
| Hypertension | 14 (14%) | 23 (23%) | 37 (34%) |
| Hypotension | 7 (7%) | 15 (15%) | 8 (7%) |
| Deep Vein Thrombosis | 3 (3%) | 9 (9%) | 6 (6%) |
| Digestive | |||
| Vomiting | 46 (47%) | 53 (52%) | 51 (47%) |
| Anorexia | 29 (30%) | 44 (43%) | 38 (35%) |
| Constipation | 28 (29%) | 41 (40%) | 32 (29%) |
| Stomatitis | 18 (18%) | 33 (32%) | 33 (30%) |
| Dyspepsia | 15 (15%) | 25 (24%) | 19 (17%) |
| GI Hemorrhage | 6 (6%) | 25 (24%) | 21 (19%) |
| Weight Loss | 10 (10%) | 15 (15%) | 18 (16%) |
| Dry Mouth | 2 (2%) | 7 (7%) | 4 (4%) |
| Colitis | 1 (1%) | 6 (6%) | 1 (1%) |
| Hemic/Lymphatic | |||
| Thrombocytopenia | 0 | 5 (5%) | 5 (5%) |
| Nervous | |||
| Dizziness | 20 (20%) | 27 (26%) | 21 (19%) |
| Respiratory | |||
| Upper Respiratory Infection | 38 (39%) | 48 (47%) | 44 (40%) |
| Epistaxis | 10 (10%) | 36 (35%) | 35 (32%) |
| Dyspnea | 15 (15%) | 26 (26%) | 27 (25%) |
| Voice Alteration | 2 (2%) | 9 (9%) | 6 (6%) |
| Skin/Appendages | |||
| Alopecia | 25 (26%) | 33 (32%) | 6 (6%) |
| Skin Ulcer | 1 (1%) | 6 (6%) | 7 (6%) |
| Special Senses | |||
| Taste Disorder | 9 (9%) | 14 (14%) | 23 (21%) |
| Urogenital | |||
| Proteinuria | 24 (24%) | 37 (36%) | 39 (36%) |
The data in Table 6 were obtained in Study 3. Only NCI‑CTC Grade 3–5 non‑hematologic and Grade 4–5 hematologic adverse events related to treatment were reported. The median age was 61 years, 40% were female, 87% were Caucasian, 99% received prior chemotherapy for metastatic colorectal cancer, 26% had received prior radiation therapy, and the 49% had an ECOG performance status of 0. Selected NCI‑CTC Grade 3–5 non‑hematologic and Grade 4–5 hematologic adverse events which occurred at a higher incidence in patients receiving FOLFOX4 plus AVASTIN as compared to those who received FOLFOX4 alone, are presented in Table 6. These data are likely to under‑estimate the true adverse event rates due to the reporting mechanisms used in Study 3.
| FOLFOX4 (n = 285) | FOLFOX4 + AVASTIN (n = 287) | AVASTIN (n = 234) |
|
| Patients with at least one event | 171 (60%) | 219 (76%) | 87 (37%) |
| Gastrointestinal | |||
| Diarrhea | 36 (13%) | 51 (18%) | 5 (2%) |
| Nausea | 13 (5%) | 35 (12%) | 14 (6%) |
| Vomiting | 11 (4%) | 32 (11%) | 15 (6%) |
| Dehydration | 14 (5%) | 29 (10%) | 15 (6%) |
| Ileus | 4 (1%) | 10 (4%) | 11 (5%) |
| Neurology | |||
| Neuropathy–sensory | 26 (9%) | 48 (17%) | 2 (1%) |
| Neurologic–other | 8 (3%) | 15 (5%) | 3 (1%) |
| Constitutional symptoms | |||
| Fatigue | 37 (13%) | 56 (19%) | 12 (5%) |
| Pain | |||
| Abdominal pain | 13 (5%) | 24 (8%) | 19 (8%) |
| Headache | 0 (0%) | 8 (3%) | 4 (2%) |
| Cardiovascular (general) | |||
| Hypertension | 5 (2%) | 26 (9%) | 19 (8%) |
| Hemorrhage | |||
| Hemorrhage | 2 (1%) | 15 (5%) | 9 (4%) |
Non‑Squamous, Non‑Small Cell Lung Cancer
The data in Table 7 were obtained in Study 5. Only NCI‑CTC Grade 3–5 non‑hematologic and Grade 4–5 hematologic adverse events were reported. The median age was 63, 46% were female, no patients had received prior chemotherapy, 76% had Stage IV disease, 12% had Stage IIIB disease with malignant pleural effusion, 11% had recurrent disease, and 40% had an ECOG performance status of 0. The median duration of exposure to AVASTIN was 4.9 months.
NCI CTC Grade 3, 4, and 5 adverse events that occurred at a ≥2% higher incidence in patients receiving PC plus AVASTIN as compared with PC alone are presented in Table 7.
| NCI‑CTC Category Term* | No.(%) of NSCLC Patients | |
| PC (n=441) | PC + AVASTIN (n=427) | |
|
||
| Any event | 286 (65%) | 334 (78%) |
| Blood/bone marrow | ||
| Neutropenia | 76 (17%) | 113 (27%) |
| Constitutional Symptoms | ||
| Fatigue | 57 (13%) | 67 (16%) |
| Cardiovascular (general) | ||
| Hypertension | 3 (0.7%) | 33 (8%) |
| Vascular | ||
| Venous thrombus/embolism | 14 (3%) | 23 (5%) |
| Infection/febrile neutropenia | ||
| Infection without neutropenia | 12 (3%) | 30 (7%) |
| Infection with NCI‑CTC Grade 3 or 4 neutropenia | 9 (2%) | 19 (4%) |
| Febrile neutropenia | 8 (2%) | 23 (5%) |
| Pulmonary/upper respiratory | ||
| Pneumonitis/pulmonary infiltrates | 11 (3%) | 21 (5%) |
| Metabolic/laboratory | ||
| Hyponatremia | 5 (1%) | 16 (4%) |
| Pain | ||
| Headache | 2 (0.5%) | 13 (3%) |
| Renal/genitourinary | ||
| Proteinuria | 0 (0%) | 13 (3%) |
Other Serious Adverse Events
The following additional serious adverse events occurred in at least one subject treated with AVASTIN in clinical studies or post‑marketing experience.
Body as a Whole: polyserositis
Digestive: intestinal necrosis, mesenteric venous occlusion, anastomotic ulceration
Hemic and lymphatic: pancytopenia
Respiratory: nasal septum perforation
OVERDOSAGE
The highest dose tested in humans (20 mg/kg IV) was associated with headache in nine of 16 patients and with severe headache in three of 16 patients.
DOSAGE AND ADMINISTRATION
Do not initiate AVASTIN until at least 28 days following major surgery. The surgical incision should be fully healed prior to initiation of AVASTIN.
Metastatic Carcinoma of the Colon or Rectum
AVASTIN, used in combination with intravenous 5‑FU‑based chemotherapy, is administered as an intravenous infusion (5 mg/kg or 10 mg/kg) every 14 days.
The recommended dose of AVASTIN, when used in combination with bolus‑IFL, is 5 mg/kg.
The recommended dose of AVASTIN, when used in combination with FOLFOX4, is 10 mg/kg.
Non‑Squamous, Non‑Small Cell Lung Cancer
The recommended dose of AVASTIN is 15 mg/kg, as an IV infusion every 3 weeks.
Dose Modifications
There are no recommended dose reductions for the use of AVASTIN. If needed, AVASTIN should be either discontinued or temporarily suspended as described below.
AVASTIN should be permanently discontinued in patients who develop gastrointestinal perforation, (gastrointestinal perforation, fistula formation in the gastrointestinal tract, intra‑abdominal abscess), fistula formation involving an internal organ, wound dehiscence requiring medical intervention, serious bleeding, a severe arterial thromboembolic event, nephrotic syndrome, hypertensive crisis or hypertensive encephalopathy. In patients developing RPLS, discontinue AVASTIN and initiate treatment of hypertension, if present. (See WARNINGS: Reversible Posterior Leukoencephalopathy Syndrome.)
Temporary suspension of AVASTIN is recommended in patients with evidence of moderate to severe proteinuria pending further evaluation and in patients with severe hypertension that is not controlled with medical management. The risk of continuation or temporary suspension of AVASTIN in patients with moderate to severe proteinuria is unknown.
AVASTIN should be suspended at least several weeks prior to elective surgery. (See WARNINGS: Gastrointestinal Perforation and Wound Healing Complications and PRECAUTIONS: Surgery). AVASTIN should not be resumed until the surgical incision is fully healed.
Preparation for Administration
AVASTIN should be diluted for infusion by a healthcare professional using aseptic technique. Withdraw the necessary amount of AVASTIN to obtain the required dose and dilute in a total volume of 100 mL of 0.9% Sodium Chloride Injection, USP. Discard any unused portion left in a vial, as the product contains no preservatives. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
Diluted AVASTIN solutions for infusion may be stored at 2°C–8°C (36°F–46°F) for up to 8 hours. No incompatibilities between AVASTIN and polyvinylchloride or polyolefin bags have been observed.
AVASTIN infusions should not be administered or mixed with dextrose solutions.
Administration
DO NOT ADMINISTER AS AN IV PUSH OR BOLUS. The initial AVASTIN dose should be delivered over 90 minutes as an IV infusion following chemotherapy. If the first infusion is well tolerated, the second infusion may be administered over 60 minutes. If the 60‑minute infusion is well tolerated, all subsequent infusions may be administered over 30 minutes.
Stability and Storage
AVASTIN vials must be refrigerated at 2–8°C (36–46°F). AVASTIN vials should be protected from light. Store in the original carton until time of use. DO NOT FREEZE. DO NOT SHAKE.
HOW SUPPLIED
AVASTIN is supplied as 4 mL and 16 mL of a sterile solution in single‑use glass vials to deliver 100 and 400 mg of Bevacizumab per vial, respectively.
Single unit
100 mg carton: Contains one 4 mL vial of
AVASTIN
(25 mg/mL). NDC 50242‑060‑01
Single unit
400 mg carton: Contains one 16 mL vial of
AVASTIN
(25 mg/mL). NDC
50242‑061‑01
REFERENCES
- Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti‑vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res 1997;57:4593–9.
AVASTIN®
(Bevacizumab)
For Intravenous Use
Manufactured by:
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990
7455311
LV0017
4835702
Initial U.S. Approval: February 2004
Code Revision Date: September 2007
© 2007 Genentech, Inc
| Avastin (bevacizumab) | |||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Avastin (bevacizumab) | |||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
Revised: 09/2007Genentech, Inc.