integrilin (Eptifibatide) injection
[Schering Corporation]
For Intravenous Administration
DESCRIPTION
Eptifibatide is a cyclic heptapeptide containing six amino acids and one mercaptopropionyl (des-amino cysteinyl) residue. An interchain disulfide bridge is formed between the cysteine amide and the mercaptopropionyl moieties. Chemically it is N6-(aminoiminomethyl)-N2-(3-mercapto-1-oxopropyl-L-lysylglycyl-L-α-aspartyl-L-tryptophyl-L-prolyl-L-cysteinamide,cyclic (1→6)-disulfide. Eptifibatide binds to the platelet receptor glycoprotein (GP) IIb/IIIa of human platelets and inhibits platelet aggregation.
The eptifibatide peptide is produced by solution-phase peptide synthesis, and is purified by preparative reverse-phase liquid chromatography and lyophilized. The structural formula is:
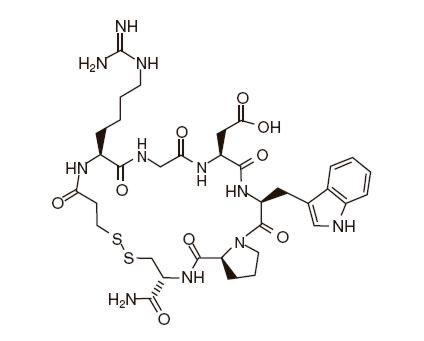
INTEGRILIN® (eptifibatide) Injection is a clear, colorless, sterile, non-pyrogenic solution for intravenous (IV) use with an empirical formula of C35H49N11O9S2 and a molecular weight of 831.96. Each 10-mL vial contains 2 mg/mL of eptifibatide and each 100-mL vial contains either 0.75 mg/mL of eptifibatide or 2 mg/mL of eptifibatide. Each vial of either size also contains 5.25 mg/mL citric acid and sodium hydroxide to adjust the pH to 5.35.
CLINICAL PHARMACOLOGY
Mechanism of Action
Eptifibatide reversibly inhibits platelet aggregation by preventing the binding of fibrinogen, von Willebrand factor, and other adhesive ligands to GP IIb/IIIa. When administered intravenously, eptifibatide inhibits ex vivo platelet aggregation in a dose- and concentration-dependent manner. Platelet aggregation inhibition is reversible following cessation of the eptifibatide infusion; this is thought to result from dissociation of eptifibatide from the platelet.
Pharmacodynamics
Infusion of eptifibatide into baboons caused a dose-dependent inhibition of ex vivo platelet aggregation, with complete inhibition of aggregation achieved at infusion rates greater than 5.0 µg/kg/min. In a baboon model that is refractory to aspirin and heparin, doses of eptifibatide that inhibit aggregation prevented acute thrombosis with only a modest prolongation (2- to 3-fold) of the bleeding time. Platelet aggregation in dogs was also inhibited by infusions of eptifibatide, with complete inhibition at 2.0 µg/kg/min. This infusion dose completely inhibited canine coronary thrombosis induced by coronary artery injury (Folts model).
Human pharmacodynamic data were obtained in healthy subjects and in patients presenting with unstable angina (UA) or non-ST-segment elevation myocardial infarction (NSTEMI) and/or undergoing percutaneous coronary interventions. Studies in healthy subjects enrolled only males; patient studies enrolled approximately one-third women. In these studies, eptifibatide inhibited ex vivo platelet aggregation induced by adenosine diphosphate (ADP) and other agonists in a dose- and concentration-dependent manner. The effect of eptifibatide was observed immediately after administration of a 180-µg/kg intravenous bolus. Table 1 shows the effects of dosing regimens of eptifibatide used in the IMPACT II and PURSUIT studies on ex vivo platelet aggregation induced by 20 µM ADP in PPACK-anticoagulated platelet-rich plasma and on bleeding time. The effects of the dosing regimen used in ESPRIT on platelet aggregation have not been studied.
| IMPACT II 135/0.5* | PURSUIT 180/2.0† |
|
|---|---|---|
| Inhibition of platelet aggregation 15 min. after bolus | 69% | 84% |
| Inhibition of platelet aggregation at steady state | 40–50% | >90% |
| Bleeding-time prolongation at steady state | <5× | <5× |
| Inhibition of platelet aggregation 4h after infusion discontinuation | <30% | <50% |
| Bleeding-time prolongation 6h after infusion discontinuation | 1× | 1.4× |
The eptifibatide dosing regimen used in the ESPRIT study included two 180-µg/kg bolus doses given 10 minutes apart combined with a continuous 2.0 µg/kg/min infusion.
When administered alone, eptifibatide has no measurable effect on prothrombin time (PT) or activated partial thromboplastin time (aPTT). (See also PRECAUTIONS: Drug Interactions.)
There were no important differences between men and women or between age groups in the pharmacodynamic properties of eptifibatide. Differences among ethnic groups have not been assessed.
Pharmacokinetics
The pharmacokinetics of eptifibatide are linear and dose-proportional for bolus doses ranging from 90 to 250 µg/kg and infusion rates from 0.5 to 3.0 µg/kg/min. Plasma elimination half-life is approximately 2.5 hours. Administration of a single 180-µg/kg bolus combined with an infusion produces an early peak level, followed by a small decline prior to attaining steady state (within 4–6 hours). This decline can be prevented by administering a second 180-µg/kg bolus 10 minutes after the first. The extent of eptifibatide binding to human plasma protein is about 25%. Clearance in patients with coronary artery disease is about 55 mL/kg/h. In healthy subjects, renal clearance accounts for approximately 50% of total body clearance, with the majority of the drug excreted in the urine as eptifibatide, deaminated eptifibatide, and other, more polar metabolites. No major metabolites have been detected in human plasma.
In patients with moderate to severe renal insufficiency (creatinine clearance <50 mL/min using the Cockcroft-Gault equation), the clearance of eptifibatide is reduced by approximately 50% and steady-state plasma levels approximately doubled (see WARNINGS and DOSAGE AND ADMINISTRATION).
Special Populations
Patients in clinical studies were older (range 20–94 years) than those in the clinical pharmacology studies. Elderly patients with coronary artery disease demonstrated higher plasma levels and lower total body clearance of eptifibatide when given the same dose as younger patients. Limited data are available on lighter weight (<50 kg) patients over 75 years of age.
No studies have been conducted in patients with hepatic impairment.
Males and females have not demonstrated any clinically significant differences in the pharmacokinetics of eptifibatide.
CLINICAL STUDIES
Eptifibatide was studied in three placebo-controlled, randomized studies. PURSUIT evaluated patients with acute coronary syndromes: unstable angina (UA) or non-ST-segment elevation MI (NSTEMI). Two other studies, ESPRIT and IMPACT II, evaluated patients about to undergo a percutaneous coronary intervention (PCI). Patients underwent primarily balloon angioplasty in IMPACT II and intracoronary stent placement, with or without angioplasty, in ESPRIT.
Non-ST-segment Elevation Acute Coronary Syndrome
Non-ST-segment elevation acute coronary syndrome is defined as prolonged (≥10 minutes) symptoms of cardiac ischemia within the previous 24 hours associated with either ST-segment changes (elevation between 0.6 mm and 1 mm or depression >0.5 mm), T-wave inversion (>1 mm), or positive CK-MB. This definition includes "unstable angina" and "NSTEMI" but excludes myocardial infarction that is associated with Q waves or greater degrees of ST-segment elevation.
PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using INTEGRILIN® Therapy)
PURSUIT was a 726-center, 27-country, double-blind, randomized, placebo-controlled study in 10,948 patients presenting with UA or NSTEMI. Patients could be enrolled only if they had experienced cardiac ischemia at rest (≥10 minutes) within the previous 24 hours and had either ST-segment changes (elevations between 0.6 mm and 1 mm or depression >0.5 mm), T-wave inversion (>1 mm), or increased CK-MB. Important exclusion criteria included a history of bleeding diathesis, evidence of abnormal bleeding within the previous 30 days, uncontrolled hypertension, major surgery within the previous 6 weeks, stroke within the previous 30 days, any history of hemorrhagic stroke, serum creatinine >2.0 mg/dL, dependency on renal dialysis, or platelet count <100,000/mm3.
Patients were randomized to either placebo, eptifibatide 180-µg/kg bolus followed by a 2.0-µg/kg/min infusion (180/2.0), or eptifibatide 180-µg/kg bolus followed by a 1.3-µg/kg/min infusion (180/1.3). The infusion was continued for 72 hours, until hospital discharge, or until the time of coronary artery bypass grafting (CABG), whichever occurred first, except that if PCI was performed, the eptifibatide infusion was continued for 24 hours after the procedure, allowing for a duration of infusion up to 96 hours.
The lower-infusion-rate arm was stopped after the first interim analysis when the two active-treatment arms appeared to have the same incidence of bleeding.
Patient age ranged from 20 to 94 (mean 63) years, and 65% were male. The patients were 89% Caucasian, 6% Hispanic, and 5% Black, recruited in the United States and Canada (40%), Western Europe (39%), Eastern Europe (16%), and Latin America (5%).
This was a "real world" study; each patient was managed according to the usual standards of the investigational site; frequencies of angiography, PCI, and CABG therefore differed widely from site to site and from country to country. Of the patients in PURSUIT, 13% were managed with PCI during drug infusion, of whom 50% received intracoronary stents; 87% were managed medically (without PCI during drug infusion).
The majority of patients received aspirin (75–325 mg once daily). Heparin was administered intravenously or subcutaneously, at the physician's discretion, most commonly as an intravenous bolus of 5000 U followed by a continuous infusion of 1000 U/h. For patients weighing less than 70 kg, the recommended heparin bolus dose was 60 U/kg followed by a continuous infusion of 12 U/kg/h. A target aPTT of 50–70 seconds was recommended. A total of 1250 patients underwent PCI within 72 hours after randomization, in which case they received intravenous heparin to maintain an activated clotting time (ACT) of 300–350 seconds.
The primary endpoint of the study was the occurrence of death from any cause or new myocardial infarction (MI) (evaluated by a blinded Clinical Endpoints Committee) within 30 days of randomization.
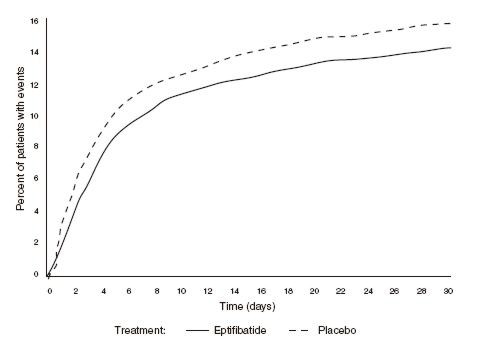
Compared to placebo, eptifibatide administered as a 180-µg/kg bolus followed by a 2.0-µg/kg/min infusion significantly (P=0.042) reduced the incidence of endpoint events (see Table 2). The reduction in the incidence of endpoint events in patients receiving eptifibatide was evident early during treatment, and this reduction was maintained through at least 30 days (see Figure 1). Table 2 also shows the incidence of the components of the primary endpoint, death (whether or not preceded by an MI) and new MI in surviving patients at 30 days.
| Death or MI | Placebo (n = 4739) n | Eptifibatide (180/2.0) (n = 4722) (%) | P-value |
|---|---|---|---|
| 3 days | 359 (7.6%) | 279 (5.9%) | 0.001 |
| 7 days | 552 (11.6%) | 477 (10.1%) | 0.016 |
| 30 days | |||
| Death or MI (Primary Endpoint) | 745 (15.7%) | 672 (14.2%) | 0.042 |
| Death | 177 (3.7%) | 165 (3.5%) | |
| Nonfatal MI | 568 (12.0%) | 507 (10.7%) |
Figure 1: Kaplan-Meier Plot of Time to Death or Myocardial Infarction Within 30 Days of Randomization
Treatment with eptifibatide prior to determination of patient management strategy reduced clinical events regardless of whether patients ultimately underwent diagnostic catheterization, revascularization (ie, PCI or CABG surgery) or continued to receive medical management alone. Table 3 shows the incidence of death or MI within 72 hours.
| Placebo | Eptifibatide 180/2.0 | |
|---|---|---|
| Overall Patient Population | n=4739 | n=4722 |
| – At 72 hours | 7.6% | 5.9% |
| Patients undergoing early PCI | n=631 | n=619 |
| – Pre-procedure (nonfatal MI only) | 5.5% | 1.8% |
| – At 72 hours | 14.4% | 9.0% |
| Patients not undergoing early PCI | n=4108 | n=4103 |
| – At 72 hours | 6.5% | 5.4% |
All of the effect of eptifibatide was established within 72 hours (during the period of drug infusion), regardless of management strategy. Moreover, for patients undergoing early PCI, a reduction in events was evident prior to the procedure.
Follow-up data were available through 165 days for 10,611 patients enrolled in the PURSUIT trial (96.9 percent of the initial enrollment). This follow-up included 4566 patients who received eptifibatide at the 180/2.0 dose. As reported by the investigators, the occurrence of death from any cause or new myocardial infarction for patients followed for at least 165 days was reduced from 13.6 percent with placebo to 12.1 percent with eptifibatide 180/2.0.
Percutaneous Coronary Intervention
IMPACT II (INTEGRILIN® to Minimize Platelet Aggregation and Prevent Coronary Thrombosis II)
IMPACT II was a multicenter, double-blind, randomized, placebo-controlled study conducted in the United States in 4010 patients undergoing PCI. Major exclusion criteria included a history of bleeding diathesis, major surgery within 6 weeks of treatment, gastrointestinal bleeding within 30 days, any stroke or structural CNS abnormality, uncontrolled hypertension, PT >1.2 times control, hematocrit <30%, platelet count <100,000/mm3, and pregnancy.
Patient age ranged from 24 to 89 (mean 60) years, and 75% were male. The patients were 92% Caucasian, 5% Black, and 3% Hispanic. Forty-one percent of the patients underwent PCI for ongoing ACS. Patients were randomly assigned to one of three treatment regimens, each incorporating a bolus dose initiated immediately prior to PCI followed by a continuous infusion lasting 20 to 24 hours: 1) 135-µg/kg bolus followed by a continuous infusion of 0.5 µg/kg/min of eptifibatide (135/0.5); 2) 135-µg/kg bolus followed by a continuous infusion of 0.75-µg/kg/min of eptifibatide (135/0.75); or 3) a matching placebo bolus followed by a matching placebo continuous infusion. Each patient received aspirin and an intravenous heparin bolus of 100 U/kg, with additional bolus infusions of up to 2000 additional units of heparin every 15 minutes to maintain an activated clotting time (ACT) of 300 to 350 seconds.
The primary endpoint was the composite of death, MI, or urgent revascularization, analyzed at 30 days after randomization in all patients who received at least one dose of study drug.
As shown in Table 4, each eptifibatide regimen reduced the rate of death, MI, or urgent intervention, although at 30 days, this finding was statistically significant only in the lower dose eptifibatide group. As in the PURSUIT study, the effects of eptifibatide were seen early and persisted throughout the 30-day period.
| Placebo n (%) | Eptifibatide (135/0.5) n (%) | Eptifibatide (135/0.75) n (%) |
|
|---|---|---|---|
|
|||
| Patients | 1285 | 1300 | 1286 |
| Abrupt Closure | 65 (5.1%) | 36 (2.8%) | 43 (3.3%) |
| P-value vs placebo | 0.003 | 0.030 | |
| Death, MI, or Urgent Intervention | |||
| 24 hours | 123 (9.6%) | 86 (6.6%) | 89 (6.9%) |
| P-value vs placebo | 0.006 | 0.014 | |
| 48 hours | 131 (10.2%) | 99 (7.6%) | 102 (7.9%) |
| P-value vs placebo | 0.021 | 0.045 | |
| 30 days (primary endpoint) | 149 (11.6%) | 118 (9.1%) | 128 (10.0%) |
| P-value vs placebo | 0.035 | 0.179 | |
| Death or MI | |||
| 30 days | 110 (8.6%) | 89 (6.8%) | 95 (7.4%) |
| P-value vs placebo | 0.102 | 0.272 | |
| 6 months | 151 (11.9%)* | 136 (10.6%)* | 130 (10.3%)* |
| P-value vs placebo | 0.297 | 0.182 | |
ESPRIT (Enhanced Suppression of the Platelet IIb/IIIa Receptor with INTEGRILIN® Therapy)
The ESPRIT study was a multicenter, double-blind, randomized, placebo-controlled study conducted in the United States and Canada that enrolled 2064 patients undergoing elective or urgent PCI with intended intracoronary stent placement. Exclusion criteria included MI within the previous 24 hours, ongoing chest pain, administration of any oral antiplatelet or oral anticoagulant other than aspirin within 30 days of PCI (although loading doses of thienopyridine on the day of PCI were encouraged), planned PCI of a saphenous vein graft or subsequent "staged" PCI, prior stent placement in the target lesion, PCI within the previous 90 days, a history of bleeding diathesis, major surgery within 6 weeks of treatment, gastrointestinal bleeding within 30 days, any stroke or structural CNS abnormality, uncontrolled hypertension, PT >1.2 times control, hematocrit <30%, platelet count <100,000/mm3, and pregnancy.
Patient age ranged from 24 to 93 (mean 62) years and 73% of patients were male. The study enrolled 90% Caucasian, 5% African American, 2% Hispanic and 1% Asian patients. Patients received a wide variety of stents. Patients were randomized either to placebo or eptifibatide administered as an intravenous bolus of 180 µg/kg followed immediately by a continuous infusion of 2.0 µg/kg/min, and a second bolus of 180 µg/kg administered 10 minutes later (180/2.0/180). Eptifibatide infusion was continued for 18 to 24 hours after PCI or until hospital discharge, whichever came first. Each patient received at least one dose of aspirin (162–325 mg) and 60 U/kg of heparin as a bolus (not to exceed 6000 Units) if not already receiving a heparin infusion. Additional boluses of heparin (10–40 U/kg) could be administered in order to reach a target ACT between 200 and 300 seconds.
The primary endpoint of the ESPRIT study was the composite of death, MI, urgent target vessel revascularization (UTVR) and "bailout" to open label eptifibatide due to a thrombotic complication of PCI (TBO) (eg, visible thrombus, "no reflow," or abrupt closure) at 48 hours. MI, UTVR and TBO were evaluated by a blinded Clinical Events Committee.
As shown in Table 5, the incidence of the primary endpoint and selected secondary endpoints was significantly reduced in patients who received eptifibatide. A treatment benefit in patients who received eptifibatide was seen by 48 hours and at the end of the 30-day observation period.
| Placebo (n=1024) | Eptifibatide 180/2.0/180 (n=1040) | Relative Risk (95% CI) | P-Value | |
|---|---|---|---|---|
| Death, MI, Urgent Target Vessel Revascularization, or Thrombotic "Bailout" | ||||
| 48 Hours (primary endpoint) | 108 (10.5%) | 69 (6.6%) | 0.629 (0.471, 0.840) | 0.0015 |
| 30 Days | 120 (11.7%) | 78 (7.5%) | 0.640 (0.488, 0.840) | 0.0011 |
| Death, MI, or Urgent Target Vessel Revascularization | ||||
| 48 Hours | 95 (9.3%) | 62 (6.0%) | 0.643 (0.472, 0.875) | 0.0045 |
| 30 Days (key secondary endpoint) | 107 (10.4%) | 71 (6.8%) | 0.653 (0.490, 0.871) | 0.0034 |
| Death or MI | ||||
| 48 Hours | 94 (9.2%) | 57 (5.5%) | 0.597 (0.435, 0.820) | 0.0013 |
| 30 Days | 104 (10.2%) | 66 (6.3%) | 0.625 ( 0.465, 0.840) | 0.0016 |
The need for thrombotic "bailout" was significantly reduced with eptifibatide at 48 hours (2.1% for placebo, 1.0% for eptifibatide; P=0.029). Consistent with previous studies of GP IIb/IIIa inhibitors, most of the benefit achieved acutely with eptifibatide was in the reduction of MI. Eptifibatide reduced the occurrence of MI at 48 hours from 9.0% for placebo to 5.4% (P=0.0015) and maintained that effect with significance at 30 days.
Follow-up (12 month) mortality data were available for 2024 patients (1017 on eptifibatide) enrolled in the ESPRIT trial (98.1% of the initial enrollment). Twelve-month clinical event data were available for 1964 patients (988 on eptifibatide) representing 95.2% of the initial enrollment. As shown in Table 6, the treatment effect of eptifibatide seen at 48 hours and 30 days appeared preserved at 6 months and 1 year. Most of the benefit was in reduction of MI.
| Placebo (n=1024) | Eptifibatide 180/2.0/180 (n=1040) | Hazard Ratio (95% CI) |
|
|---|---|---|---|
| Percentages are Kaplan-Meier event rates. | |||
| Death, MI, or Target Vessel Revascularization | |||
| 6 Months | 187 (18.5%) | 146 (14.3%) | 0.744 (0.599, 0.924) |
| 1 Year | 222 (22.1%) | 178 (17.5%) | 0.762 (0.626, 0.929) |
| Death, MI | |||
| 6 Months | 117 (11.5%) | 77 (7.4%) | 0.631 (0.473, 0.841) |
| 1 Year | 126 (12.4%) | 83 (8.0%) | 0.630 (0.478, 0.832) |
INDICATIONS AND USAGE
INTEGRILIN® is indicated:
- For the treatment of patients with acute coronary syndrome (unstable angina/non-ST-segment elevation myocardial infarction), including patients who are to be managed medically and those undergoing percutaneous coronary intervention (PCI). In this setting, INTEGRILIN has been shown to decrease the rate of a combined endpoint of death or new myocardial infarction.
- For the treatment of patients undergoing PCI, including those undergoing intracoronary stenting. In this setting, INTEGRILIN has been shown to decrease the rate of a combined endpoint of death, new myocardial infarction, or need for urgent intervention.
In the IMPACT II, PURSUIT and ESPRIT studies of eptifibatide, most patients received heparin and aspirin, as described in CLINICAL TRIALS.
CONTRAINDICATIONS
Treatment with eptifibatide is contraindicated in patients with:
- A history of bleeding diathesis, or evidence of active abnormal bleeding within the previous 30 days.
- Severe hypertension (systolic blood pressure >200 mm Hg or diastolic blood pressure >110 mm Hg) not adequately controlled on antihypertensive therapy.
- Major surgery within the preceding 6 weeks.
- History of stroke within 30 days or any history of hemorrhagic stroke.
- Current or planned administration of another parenteral GP IIb/IIIa inhibitor.
- Dependency on renal dialysis.
- Known hypersensitivity to any component of the product.
WARNINGS
Bleeding
Bleeding is the most common complication encountered during eptifibatide therapy. Administration of eptifibatide is associated with an increase in major and minor bleeding, as classified by the criteria of the Thrombolysis in Myocardial Infarction Study group (TIMI), (see ADVERSE REACTIONS). Most major bleeding associated with eptifibatide has been at the arterial access site for cardiac catheterization or from the gastrointestinal or genitourinary tract.
In patients undergoing percutaneous coronary interventions, patients receiving eptifibatide experience an increased incidence of major bleeding compared to those receiving placebo without a significant increase in transfusion requirement. Special care should be employed to minimize the risk of bleeding among these patients (see PRECAUTIONS). If bleeding cannot be controlled with pressure, infusion of eptifibatide and concomitant heparin should be stopped immediately.
Renal Insufficiency
Approximately 50% of eptifibatide is cleared by the kidney in patients with normal renal function. Total drug clearance is decreased by approximately 50% and steady-state plasma eptifibatide concentrations are doubled in patients with an estimated creatinine clearance <50 mL/min (using the Cockcroft-Gault equation). Therefore, the infusion dose should be reduced to 1 µg/kg/min in such patients (see DOSAGE AND ADMINISTRATION). There has been no clinical experience in patients dependent on dialysis.
Platelet Count <100,000/mm3
Because it is an inhibitor of platelet aggregation, caution should be exercised when administering eptifibatide to patients with a platelet count <100,000/mm3; there has been no clinical experience with eptifibatide initiated in patients with a platelet count <100,000/mm3.
PRECAUTIONS
Bleeding Precautions Care of the Femoral Artery Access Site in Patients Undergoing Percutaneous Coronary Intervention (PCI)
In patients undergoing PCI, treatment with eptifibatide is associated with an increase in major and minor bleeding at the site of arterial sheath placement. After PCI, eptifibatide infusion should be continued until hospital discharge or up to 18 to 24 hours, whichever comes first. Heparin use is discouraged after the PCI procedure. Early sheath removal is encouraged while eptifibatide is being infused. Prior to removing the sheath, it is recommended that heparin be discontinued for 3 to 4 hours and an aPTT of <45 seconds or ACT <150 seconds be achieved. In any case, both heparin and eptifibatide should be discontinued and sheath hemostasis should be achieved at least 2 to 4 hours before hospital discharge.
Use of Thrombolytics, Anticoagulants, and Other Antiplatelet Agents
In the IMPACT II, PURSUIT and ESPRIT studies, eptifibatide was used concomitantly with unfractionated heparin and aspirin (see CLINICAL STUDIES). In the ESPRIT study, clopidogrel or ticlopidine were used routinely starting the day of PCI. Because eptifibatide inhibits platelet aggregation, caution should be employed when it is used with other drugs that affect hemostasis, including thrombolytics, oral anticoagulants, nonsteroidal anti-inflammatory drugs, and dipyridamole. To avoid potentially additive pharmacologic effects, concomitant treatment with other inhibitors of platelet receptor GP IIb/IIIa should be avoided.
There is only a small experience with concomitant use of eptifibatide and thrombolytics. In a study of 180 patients with acute myocardial infarction (AMI), eptifibatide (in regimens up to a bolus of 180 µg/kg followed by a continuous infusion of 0.75 µg/kg/min for 24 hours) was administered concomitantly with the approved "accelerated" regimen of alteplase, a thrombolytic agent. The studied regimens of eptifibatide did not increase the incidence of major bleeding or transfusion compared to the incidence seen when alteplase was given alone.
In the IMPACT II study, 15 patients received a thrombolytic agent in conjunction with the 135/0.5 dosing regimen, 2 of whom experienced a major bleed. In the PURSUIT study, 40 patients who received eptifibatide at the 180/2.0 dosing regimen received a thrombolytic agent, 10 of whom experienced a major bleed.
In another AMI study involving 181 patients, eptifibatide (in regimens up to a bolus of 180 µg/kg followed by a continuous infusion of up to 2.0 µg/kg/min for up to 72 hours) was administered concomitantly with streptokinase (1.5 million units over 60 minutes), another thrombolytic agent. At the highest studied infusion rates (1.3 µg/kg/min and 2.0 µg/kg/min), eptifibatide was associated with an increase in the incidence of bleeding and transfusions compared to the incidence seen when streptokinase was given alone.
These limited data on the use of eptifibatide in patients receiving thrombolytic agents do not allow an estimate of the bleeding risk associated with concomitant use of thrombolytics. Systemic thrombolytic therapy should be used with caution in patients who have received eptifibatide.
Minimization of Vascular and Other Trauma
Arterial and venous punctures, intramuscular injections, and the use of urinary catheters, nasotracheal intubation, and nasogastric tubes should be minimized. When obtaining intravenous access, non-compressible sites (eg, subclavian or jugular veins) should be avoided.
Laboratory Tests
Before infusion of eptifibatide, the following laboratory tests should be performed to identify preexisting hemostatic abnormalities: hematocrit or hemoglobin, platelet count, serum creatinine, and PT/aPTT. In patients undergoing PCI, the activated clotting time (ACT) should also be measured.
Maintaining Target aPTT and ACT
The aPTT should be maintained between 50 and 70 seconds unless PCI is to be performed. In patients treated with heparin, bleeding can be minimized by close monitoring of the aPTT. Table 7 displays the risk of major bleeding according to the maximum aPTT attained within 72 hours in the PURSUIT study.
| Placebo n (%) | Eptifibatide 180/1.3* n (%) | Eptifibatide 180/2.0 n (%) |
|
|---|---|---|---|
|
|||
| Maximum aPTT (seconds) | |||
| <50 | 44/721 (6.1%) | 21/244 (8.6%) | 44/743 (5.9%) |
| 50 to 70 (recommended) | 92/908 (10.1%) | 28/259 (10.8%) | 99/883 (11.2%) |
| >70 | 281/2786 (10.1%) | 99/891 (11.1%) | 345/2811 (12.3%) |
The ESPRIT study stipulated a target ACT of 200 to 300 seconds during PCI. Patients receiving eptifibatide 180/2.0/180 (mean ACT 284 seconds) experienced an increased incidence of bleeding relative to placebo (mean ACT 276 seconds), primarily at the femoral artery access site. At these lower ACTs, bleeding was less than previously reported with eptifibatide in the PURSUIT and IMPACT II studies.
The aPTT or ACT should be checked prior to arterial sheath removal. The sheath should not be removed unless the aPTT is <45 seconds or the ACT is <150 seconds.
Thrombocytopenia
If the patient experiences a confirmed platelet decrease to <100,000/mm3, INTEGRILIN® and heparin should be discontinued and the condition appropriately monitored and treated.
Drug Interactions
Enoxaparin dosed as a 1.0-mg/kg subcutaneous injection q12h for four doses did not alter the pharmacokinetics of eptifibatide or the level of platelet aggregation in healthy adults.
Geriatric Use
The PURSUIT and IMPACT II clinical studies enrolled patients up to the age of 94 years (45% were age 65 and over; 12% were age 75 and older). There was no apparent difference in efficacy between older and younger patients treated with eptifibatide. The incidence of bleeding complications was higher in the elderly in both placebo and eptifibatide groups, and the incremental risk of eptifibatide-associated bleeding was greater in the older patients. No dose adjustment was made for elderly patients, but patients over 75 years of age had to weigh at least 50 kg to be enrolled in the PURSUIT study; no such limitation was stipulated in the ESPRIT study (see also ADVERSE REACTIONS).
Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies in animals have been performed to evaluate the carcinogenic potential of eptifibatide. Eptifibatide was not genotoxic in the Ames test, the mouse lymphoma cell (L 5178Y, TK+/-) forward mutation test, the human lymphocyte chromosome aberration test, or the mouse micronucleus test. Administered by continuous intravenous infusion at total daily doses up to 72 mg/kg/day (about 4 times the recommended maximum daily human dose on a body surface area basis), eptifibatide had no effect on fertility and reproductive performance of male and female rats.
Pregnancy
Pregnancy Category B
Teratology studies have been performed by continuous intravenous infusion of eptifibatide in pregnant rats at total daily doses of up to 72 mg/kg/day (about 4 times the recommended maximum daily human dose on a body surface area basis) and in pregnant rabbits at total daily doses of up to 36 mg/kg/day (also about 4 times the recommended maximum daily human dose on a body surface area basis). These studies revealed no evidence of harm to the fetus due to eptifibatide. There are, however, no adequate and well-controlled studies in pregnant women with eptifibatide. Because animal reproduction studies are not always predictive of human response, eptifibatide should be used during pregnancy only if clearly needed.
Pediatric Use
Safety and effectiveness of eptifibatide in pediatric patients have not been studied.
Nursing Mothers
It is not known whether eptifibatide is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when eptifibatide is administered to a nursing mother.
ADVERSE REACTIONS
A total of 16,782 patients were treated in the Phase III clinical trials (PURSUIT, ESPRIT and IMPACT II). These 16,782 patients had a mean age of 62 years (range 20–94 years). Eighty-nine percent of the patients were Caucasian, with the remainder being predominantly Black (5%) and Hispanic (5%). Sixty-eight percent were men. Because of the different regimens used in PURSUIT, IMPACT II and ESPRIT, data from the three studies were not pooled.
Bleeding
The incidences of bleeding events and transfusions in the PURSUIT, IMPACT II and ESPRIT studies are shown in Table 8. Bleeding was classified as major or minor by the criteria of the TIMI study group. Major bleeding events consisted of intracranial hemorrhage and other bleeding that led to decreases in hemoglobin greater than 5 g/dL. Minor bleeding events included spontaneous gross hematuria, spontaneous hematemesis, other observed blood loss with a hemoglobin decrease of more than 3 g/dL, and other hemoglobin decreases that were greater than 4 g/dL but less than 5 g/dL. In patients who received transfusions, the corresponding loss in hemoglobin was estimated through an adaptation of the method of Landefeld et al.
| Note: denominator is based on patients for whom data are available. | |||
|
|||
| PURSUIT | |||
| Placebo n (%) | Eptifibatide 180/1.3*
n (%) | Eptifibatide 180/2.0 n (%) |
|
| Patients | 4696 | 1472 | 4679 |
| Major bleeding† | 425 (9.3%) | 152 (10.5%) | 498 (10.8%) |
| Minor bleeding† | 347 (7.6%) | 152 (10.5%) | 604 (13.1%) |
| Requiring transfusions‡ | 490 (10.4%) | 188 (12.8%) | 601 (12.8%) |
| ESPRIT | |||
| Placebo n (%) | Eptifibatide 180/2.0/180 n (%) | ||
| Patients | 1024 | 1040 | |
| Major bleeding† | 4 (0.4%) | 13 (1.3%) | |
| Minor bleeding† | 18 (2.0%) | 29 (3.0%) | |
| Requiring transfusions‡ | 11 (1.1%) | 16 (1.5%) | |
| IMPACT II | |||
| Placebo n (%) | Eptifibatide 135/0.5 n (%) | Eptifibatide 135/0.75 n (%) |
|
| Patients | 1285 | 1300 | 1286 |
| Major bleeding† | 55 (4.5%) | 55 (4.4%) | 58 (4.7%) |
| Minor bleeding† | 115 (9.3%) | 146 (11.7%) | 177 (14.2%) |
| Requiring transfusions‡ | 66 (5.1%) | 71 (5.5%) | 74 (5.8%) |
The majority of major bleeding events in the ESPRIT study occurred at the vascular access site (1 and 8 patients, or 0.1% and 0.8% in the placebo and eptifibatide groups, respectively). Bleeding at "other" locations occurred in 0.2% and 0.4% of patients, respectively.
In the PURSUIT study, the greatest increase in major bleeding in eptifibatide-treated patients compared to placebo-treated patients was also associated with bleeding at the femoral artery access site (2.8% versus 1.3%). Oropharyngeal (primarily gingival), genitourinary, gastrointestinal, and retroperitoneal bleeding were also seen more commonly in eptifibatide-treated patients compared to placebo-treated patients.
Among patients experiencing a major bleed in the IMPACT II study, an increase in bleeding on eptifibatide versus placebo was observed only for the femoral artery access site (3.2% versus 2.8%).
Table 9 displays the incidence of TIMI major bleeding according to the cardiac procedures carried out in the PURSUIT study. The most common bleeding complications were related to cardiac revascularization (CABG-related or femoral artery access site bleeding). A corresponding table for ESPRIT is not presented as every patient underwent PCI in the ESPRIT study and only 11 patients underwent CABG.
| Placebo n (%) | Eptifibatide 180/1.3* n (%) | Eptifibatide 180/2.0 n (%) |
|
|---|---|---|---|
| Denominators are based on the total number of patients whose TIMI classification was resolved. | |||
|
|||
| Patients | 4577 | 1451 | 4604 |
| Overall Incidence of Major Bleeding | 425 (9.3%) | 152 (10.5%) | 498 (10.8%) |
| Breakdown by Procedure: | |||
| CABG | 375 (8.2%) | 123 (8.5%) | 377 (8.2%) |
| Angioplasty without CABG | 27 (0.6%) | 16 (1.1%) | 64 (1.4%) |
| Angiography without Angioplasty or CABG | 11 (0.2%) | 7 (0.5%) | 29 (0.6%) |
| Medical Therapy Only | 12 (0.3%) | 6 (0.4%) | 28 (0.6%) |
In the PURSUIT and ESPRIT studies, the risk of major bleeding with eptifibatide increased as patient weight decreased. This relationship was most apparent for patients weighing less than 70 kg.
Bleeding adverse events resulting in discontinuation of study drug were more frequent among patients receiving eptifibatide than placebo (4.6% versus 0.9% in ESPRIT, 8% versus 1% in PURSUIT, 3.5% versus 1.9% in IMPACT II).
Intracranial Hemorrhage and Stroke
Intracranial hemorrhage was rare in the PURSUIT, IMPACT II and ESPRIT clinical studies. In the PURSUIT study, 3 patients in the placebo group, 1 patient in the group treated with eptifibatide 180/1.3 and 5 patients in the group treated with eptifibatide 180/2.0 experienced a hemorrhagic stroke. The overall incidence of stroke was 0.5% in patients receiving eptifibatide 180/1.3, 0.7% in patients receiving eptifibatide 180/2.0, and 0.8% in placebo patients.
In the IMPACT II study, intracranial hemorrhage was experienced by 1 patient treated with eptifibatide 135/0.5, 2 patients treated with eptifibatide 135/0.75 and 2 patients in the placebo group. The overall incidence of stroke was 0.5% in patients receiving 135/0.5 eptifibatide, 0.7% in patients receiving eptifibatide 135/0.75 and 0.7% in the placebo group.
In the ESPRIT study, there were 3 hemorrhagic strokes, 1 in the placebo group and 2 in the eptifibatide group. In addition there was 1 case of cerebral infarction in the eptifibatide group.
Thrombocytopenia
In the PURSUIT and IMPACT II studies, the incidence of thrombocytopenia (<100,000/mm3 or ≥50% reduction from baseline) and the incidence of platelet transfusions were similar between patients treated with eptifibatide and placebo. In the ESPRIT study, the incidence was 0.6% in the placebo group and 1.2% in the eptifibatide group.
Allergic Reactions
In the PURSUIT study, anaphylaxis was reported in 7 patients receiving placebo (0.15%) and 7 patients receiving eptifibatide 180/2.0 (0.16%). In the IMPACT II study, anaphylaxis was reported in 1 patient (0.08%) on placebo and in no patients on eptifibatide. In the IMPACT II study, 2 patients (1 patient [0.04%] receiving eptifibatide and 1 patient [0.08%] receiving placebo) discontinued study drug because of allergic reactions. In the ESPRIT study, there were no cases of anaphylaxis reported. There were 3 patients who suffered an allergic reaction, 1 on placebo and 2 on eptifibatide. In addition, 1 patient in the placebo group was diagnosed with urticaria.
The potential for development of antibodies to eptifibatide has been studied in 433 subjects. Eptifibatide was non-antigenic in 412 patients receiving a single administration of eptifibatide (135-µg/kg bolus followed by a continuous infusion of either 0.5 µg/kg/min or 0.75 µg/kg/min), and in 21 subjects to whom eptifibatide (135-µg/kg bolus followed by a continuous infusion of 0.75 µg/kg/min) was administered twice, 28 days apart. In both cases, plasma for antibody detection was collected approximately 30 days after each dose. The development of antibodies to eptifibatide at higher doses has not been evaluated.
Other Adverse Reactions
In the PURSUIT and ESPRIT studies, the incidence of serious nonbleeding adverse events was similar in patients receiving placebo or eptifibatide (19% and 19%, respectively in PURSUIT; 6% and 7%, respectively in ESPRIT). In PURSUIT, the only serious nonbleeding adverse event that occurred at a rate of at least 1% and was more common with eptifibatide than placebo (7% versus 6%) was hypotension. Most of the serious nonbleeding events consisted of cardiovascular events typical of an unstable angina population. In the IMPACT II study, serious nonbleeding events that occurred in greater than 1% of patients were uncommon and similar in incidence between placebo- and eptifibatide-treated patients.
Discontinuation of study drug due to adverse events other than bleeding was uncommon in the PURSUIT, IMPACT II, and ESPRIT studies, with no single event occurring in >0.5% of the study population (except for "other" in the ESPRIT study). In the PURSUIT study, nonbleeding adverse events leading to discontinuation occurred in the eptifibatide and placebo groups in the following body systems with an incidence of ≥0.1%: cardiovascular system (0.3% and 0.3%), digestive system (0.1% and 0.1%), hemic/lymphatic system (0.1% and 0.1%), nervous system (0.3% and 0.4%), urogenital system (0.1% and 0.1%), and whole body system (0.2% and 0.2%). In the ESPRIT study, the following nonbleeding adverse events leading to discontinuation occurred in the eptifibatide and placebo groups with an incidence of ≥0.1%: "other" (1.2% and 1.1%). In the IMPACT II study, nonbleeding adverse events leading to discontinuation occurred in the 135/0.5 eptifibatide and placebo groups in the following body systems with an incidence of ≥0.1%: whole body (0.3% and 0.1%), cardiovascular system (1.4% and 1.4%), digestive system (0.2% and 0%), hemic/lymphatic system (0.2% and 0%), nervous system (0.3% and 0.2%), and respiratory system (0.1% and 0.1%).
Post-Marketing Experience
The following adverse events have been reported in post-marketing experience, primarily with eptifibatide in combination with heparin and aspirin: cerebral, GI and pulmonary hemorrhage. Fatal bleeding events have been reported. Acute profound thrombocytopenia has been reported.
OVERDOSAGE
There has been only limited experience with overdosage of eptifibatide. There were 8 patients in the IMPACT II study, 9 patients in the PURSUIT study, and no patients in the ESPRIT study who received bolus doses and/or infusion doses more than double those called for in the protocols. None of these patients experienced an intracranial bleed or other major bleeding.
Eptifibatide was not lethal to rats, rabbits, or monkeys when administered by continuous intravenous infusion for 90 minutes at a total dose of 45 mg/kg (about 2 to 5 times the recommended maximum daily human dose on a body surface area basis). Symptoms of acute toxicity were loss of righting reflex, dyspnea, ptosis, and decreased muscle tone in rabbits and petechial hemorrhages in the femoral and abdominal areas of monkeys.
From in vitro studies, eptifibatide is not extensively bound to plasma proteins and thus may be cleared from plasma by dialysis.
DOSAGE AND ADMINISTRATION
The safety and efficacy of eptifibatide has been established in clinical studies that employed concomitant use of heparin and aspirin. Different dose regimens of eptifibatide were used in the major clinical studies (see CLINICAL STUDIES).
Acute Coronary Syndrome
The recommended adult dosage of eptifibatide in patients with acute coronary syndrome and normal renal function is an intravenous bolus of 180 µg/kg as soon as possible following diagnosis, followed by a continuous infusion of 2.0 µg/kg/min until hospital discharge or initiation of CABG surgery, up to 72 hours. If a patient is to undergo a percutaneous coronary intervention (PCI) while receiving eptifibatide, the infusion should be continued up to hospital discharge, or for up to 18 to 24 hours after the procedure, whichever comes first, allowing for up to 96 hours of therapy.
Patients with Creatinine Clearance Less Than 50 mL/min
The recommended adult dosage of eptifibatide in patients with acute coronary syndrome with an estimated creatinine clearance (using the Cockcroft-Gault equation)1 <50 mL/min is an intravenous bolus of 180 µg/kg as soon as possible following diagnosis, immediately followed by a continuous infusion of 1.0 µg/kg/min.
Percutaneous Coronary Intervention (PCI)
The recommended adult dosage of eptifibatide in patients with normal renal function is an intravenous bolus of 180 µg/kg administered immediately before the initiation of PCI followed by a continuous infusion of 2.0 µg/kg/min and a second 180-µg/kg bolus 10 minutes after the first bolus. Infusion should be continued until hospital discharge, or for up to 18 to 24 hours, whichever comes first. A minimum of 12 hours of infusion is recommended.
Patients with Creatinine Clearance Less Than 50 mL/min
The recommended adult dose of eptifibatide in patients with an estimated creatinine clearance (using the Cockcroft-Gault equation)1 <50 mL/min is an intravenous bolus of 180 µg/kg administered immediately before the initiation of the procedure, immediately followed by a continuous infusion of 1.0 µg/kg/min and a second 180-µg/kg bolus administered 10 minutes after the first.
In patients who undergo coronary artery bypass graft surgery, eptifibatide infusion should be discontinued prior to surgery.
- 1
- Use the Cockcroft-Gault equation with actual body weight to calculate creatinine clearance:

Aspirin and Heparin Dosing Recommendations
In the clinical trials that showed eptifibatide to be effective, most patients received concomitant aspirin and heparin. The recommended aspirin and heparin doses to be used are as follows:
Acute Coronary Syndrome
Aspirin
160 to 325 mg orally initially and daily thereafter
Heparin
Target aPTT 50 to 70 seconds during medical management
- -
- If weight ≥70 kg, 5000 U bolus followed by infusion of 1000 U/hr.
- -
- If weight <70 kg, 60 U/kg bolus followed by infusion of 12 U/kg/hr.
Target ACT 200 to 300 seconds during PCI
- -
- If heparin is initiated prior to PCI, additional boluses during PCI to maintain an ACT target of 200 to 300 seconds.
- -
- Heparin infusion after the PCI is discouraged.
PCI
Aspirin
160 to 325 mg orally 1 to 24 hours prior to PCI and daily thereafter
Heparin
Target ACT 200 to 300 seconds
- -
- 60 U/kg bolus initially in patients not treated with heparin within 6 hours prior to PCI.
- -
- Additional boluses during PCI to maintain ACT within target.
- -
- Heparin infusion after the PCI is strongly discouraged.
Patients requiring thrombolytic therapy should have eptifibatide infusions stopped.
Instructions for Administration
- Like other parenteral drug products, INTEGRILIN® solutions should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- INTEGRILIN® may be administered in the same intravenous line as alteplase, atropine, dobutamine, heparin, lidocaine, meperidine, metoprolol, midazolam, morphine, nitroglycerin, or verapamil. INTEGRILIN® should not be administered through the same intravenous line as furosemide.
- INTEGRILIN® may be administered in the same IV line with 0.9% NaCl or 0.9% NaCl/5% dextrose. With either vehicle, the infusion may also contain up to 60 mEq/L of potassium chloride. No incompatibilities have been observed with intravenous administration sets. No compatibility studies have been performed with PVC bags.
- The bolus dose(s) of INTEGRILIN® should be withdrawn from the 10-mL vial into a syringe. The bolus dose(s) should be administered by IV push.
- Immediately following the bolus dose administration, a continuous infusion of INTEGRILIN® should be initiated. When using an intravenous infusion pump, INTEGRILIN® should be administered undiluted directly from the 100-mL vial. The 100-mL vial should be spiked with a vented infusion set. Care should be taken to center the spike within the circle on the stopper top.
INTEGRILIN® is to be administered by volume according to patient weight.Patients should receive INTEGRILIN® according to the following table:
| Patient Weight | 180 µg/kg Bolus Volume | 2.0 µg/kg/min Infusion Volume | 1.0 µg/kg/min Infusion Volume |
|||
|---|---|---|---|---|---|---|
| (kg) | (lb) | (from 2 mg/mL vial) | (from 2 mg/mL 100-mL vial) | (from 0.75 mg/mL 100-mL vial) | (from 2 mg/mL 100-mL vial) | (from 0.75 mg/mL 100-mL vial) |
| 37–41 | 81–91 | 3.4 mL | 2.0 mL/h | 6.0 mL/h | 1.0 mL/h | 3.0 mL/h |
| 42–46 | 92–102 | 4.0 mL | 2.5 mL/h | 7.0 mL/h | 1.3 mL/h | 3.5 mL/h |
| 47–53 | 103–117 | 4.5 mL | 3.0 mL/h | 8.0 mL/h | 1.5 mL/h | 4.0 mL/h |
| 54–59 | 118–130 | 5.0 mL | 3.5 mL/h | 9.0 mL/h | 1.8 mL/h | 4.5 mL/h |
| 60–65 | 131–143 | 5.6 mL | 3.8 mL/h | 10.0 mL/h | 1.9 mL/h | 5.0 mL/h |
| 66–71 | 144–157 | 6.2 mL | 4.0 mL/h | 11.0 mL/h | 2.0 mL/h | 5.5 mL/h |
| 72–78 | 158–172 | 6.8 mL | 4.5 mL/h | 12.0 mL/h | 2.3 mL/h | 6.0 mL/h |
| 79–84 | 173–185 | 7.3 mL | 5.0 mL/h | 13.0 mL/h | 2.5 mL/h | 6.5 mL/h |
| 85–90 | 186–198 | 7.9 mL | 5.3 mL/h | 14.0 mL/h | 2.7 mL/h | 7.0 mL/h |
| 91–96 | 199–212 | 8.5 mL | 5.6 mL/h | 15.0 mL/h | 2.8 mL/h | 7.5 mL/h |
| 97–103 | 213–227 | 9.0 mL | 6.0 mL/h | 16.0 mL/h | 3.0 mL/h | 8.0 mL/h |
| 104–109 | 228–240 | 9.5 mL | 6.4 mL/h | 17.0 mL/h | 3.2 mL/h | 8.5 mL/h |
| 110–115 | 241–253 | 10.2 mL | 6.8 mL/h | 18.0 mL/h | 3.4 mL/h | 9.0 mL/h |
| 116–121 | 254–267 | 10.7 mL | 7.0 mL/h | 19.0 mL/h | 3.5 mL/h | 9.5 mL/h |
| >121 | >267 | 11.3 mL | 7.5 mL/h | 20.0 mL/h | 3.7 mL/h | 10.0 mL/h |
HOW SUPPLIED
INTEGRILIN® (eptifibatide) Injection is supplied as a sterile solution in 10-mL vials containing 20 mg of eptifibatide (NDC 0085-1177-01) and 100-mL vials containing either 75 mg of eptifibatide (NDC 0085-1136-01) or 200 mg of eptifibatide (NDC 0085-1177-02).
Vials should be stored refrigerated at 2–8°C (36–46°F). Vials may be transferred to room temperature storage2 for a period not to exceed 2 months. Upon transfer, vial cartons must be marked by the dispensing pharmacist with a "DISCARD BY" date (2 months from the transfer date or the labeled expiration date, whichever comes first).
Do not use beyond the labeled expiration date. Protect from light until administration. Discard any unused portion left in the vial.
- 2
- Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature].
Schering Corporation
Kenilworth, NJ 07033 USA
INTEGRILIN® is a registered trademark of Millennium Pharmaceuticals, Inc.
Manufactured for Schering Corporation, Kenilworth, NJ 07033 USA.
U.S. Patent Nos. 5,686,570; 5,747,447; 5,756,451; 5,807,825; and 5,968,902.
B-30359402 9/06
| INTEGRILIN (Eptifibatide) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| INTEGRILIN (Eptifibatide) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Revised: 11/2007Schering Corporation