NAVELBINE
-
vinorelbine tartrate injection, solution, concentrate
Pierre Fabre Médicament
----------
WARNING
NAVELBINE (vinorelbine tartrate) Injection should be administered under the supervision of a physician experienced in the use of cancer chemotherapeutic agents. This product is for intravenous (IV) use only. Intrathecal administration of other vinca alkaloids has resulted in death. Syringes containing this product should be labeled "WARNING - FOR IV USE ONLY. FATAL if given intrathecally."
Severe granulocytopenia resulting in increased susceptibility to infection may occur. Granulocyte counts should be ≥1,000 cells/mm3 prior to the administration of NAVELBINE. The dosage should be adjusted according to complete blood counts with differentials obtained on the day of treatment.
Caution - It is extremely important that the intravenous needle or catheter be properly positioned before NAVELBINE is injected. Administration of NAVELBINE may result in extravasation causing local tissue necrosis and/or thrombophlebitis (see DOSAGE AND ADMINISTRATION: Administration Precautions).
DESCRIPTION
NAVELBINE (vinorelbine tartrate) Injection is for intravenous administration. Each vial contains vinorelbine tartrate equivalent to 10 mg (1-mL vial) or 50 mg (5-mL vial) in Water for Injection. No preservatives or other additives are present. The aqueous solution is sterile and nonpyrogenic. Vinorelbine tartrate is a semi-synthetic vinca alkaloid with antitumor activity.
The chemical name is 3',4'-didehydro-4'-deoxy-C'-norvincaleukoblastine [R-(R*,R*)-2, 3-dihydroxybutanedioate (1:2)(salt)].
Vinorelbine tartrate has the following structure:
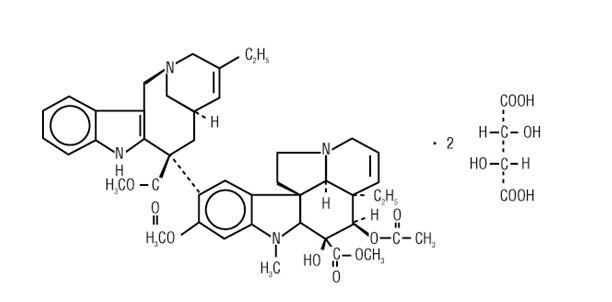
vinorelbine tartrate is a white to yellow or light brown amorphous powder with the molecular formula C45H54N4O8•2C4H6O6 and molecular weight of 1079.12. The aqueous solubility is >1,000 mg/mL in distilled water. The pH of NAVELBINE Injection is approximately 3.5.
CLINICAL PHARMACOLOGY
Vinorelbine is a vinca alkaloid that interferes with microtubule assembly. The vinca alkaloids are structurally similar compounds comprised of 2 multiringed units, vindoline and catharanthine. Unlike other vinca alkaloids, the catharanthine unit is the site of structural modification for vinorelbine. The antitumor activity of vinorelbine is thought to be due primarily to inhibition of mitosis at metaphase through its interaction with tubulin. Like other vinca alkaloids, vinorelbine may also interfere with: 1) amino acid, cyclic AMP, and glutathione metabolism, 2) calmodulin-dependent Ca++-transport ATPase activity, 3) cellular respiration, and 4) nucleic acid and lipid biosynthesis. In intact tectal plates from mouse embryos, vinorelbine, vincristine and vinblastine inhibited mitotic microtubule formation at the same concentration (2 µM), inducing a blockade of cells at metaphase. Vincristine produced depolymerization of axonal microtubules at 5 µM, but vinblastine and vinorelbine did not have this effect until concentrations of 30 µM and 40 µM, respectively. These data suggest relative selectivity of vinorelbine for mitotic microtubules.
Pharmacokinetics
The pharmacokinetics of vinorelbine were studied in 49 patients who received doses of 30 mg/m2 in 4 clinical trials. Doses were administered by 15- to 20-minute constant-rate infusions. Following intravenous administration, vinorelbine concentration in plasma decays in a triphasic manner. The initial rapid decline primarily represents distribution of drug to peripheral compartments followed by metabolism and excretion of the drug during subsequent phases. The prolonged terminal phase is due to relatively slow efflux of vinorelbine from peripheral compartments. The terminal phase half-life averages 27.7 to 43.6 hours and the mean plasma clearance ranges from 0.97 to 1.26 L/hr/kg. Steady-state volume of distribution (VSS) values range from 25.4 to 40.1 L/kg.
Vinorelbine demonstrated high binding to human platelets and lymphocytes. The free fraction was approximately 0.11 in pooled human plasma over a concentration range of 234 to 1,169 ng/mL. The binding to plasma constituents in cancer patients ranged from 79.6% to 91.2%. Vinorelbine binding was not altered in the presence of cisplatin, 5-fluorouracil, or doxorubicin.
Vinorelbine undergoes substantial hepatic elimination in humans, with large amounts recovered in feces after intravenous administration to humans. Two metabolites of vinorelbine have been identified in human blood, plasma, and urine; vinorelbine N-oxide and deacetylvinorelbine. Deacetylvinorelbine has been demonstrated to be the primary metabolite of vinorelbine in humans, and has been shown to possess antitumor activity similar to vinorelbine. Therapeutic doses of vinorelbine (30 mg/m2) yield very small, if any, quantifiable levels of either metabolite in blood or urine. The metabolism of vinca alkaloids has been shown to be mediated by hepatic cytochrome P450 isoenzymes in the CYP3A subfamily. This metabolic pathway may be impaired in patients with hepatic dysfunction or who are taking concomitant potent inhibitors of these isoenzymes (see PRECAUTIONS).
The effects of renal or hepatic dysfunction on the disposition of vinorelbine have not been assessed, but based on experience with other anticancer vinca alkaloids, dose adjustments are recommended for patients with impaired hepatic function (see DOSAGE AND ADMINISTRATION).
The disposition of radiolabeled vinorelbine given intravenously was studied in a limited number of patients. Approximately 18% and 46% of the administered dose was recovered in the urine and in the feces, respectively. Incomplete recovery in humans is consistent with results in animals where recovery is incomplete, even after prolonged sampling times.
A separate study of the urinary excretion of vinorelbine using specific chromatographic analytical methodology showed that 10.9% ± 0.7% of a 30-mg/m2 intravenous dose was excreted unchanged in the urine.
The influence of age on the pharmacokinetics of vinorelbine was examined using data from 44 cancer patients (average age, 56.7 ± 7.8 years; range, 41 to 74 years; with 12 patients ≥60 years and 6 patients ≥65 years) in 3 studies. CL (the mean plasma clearance), t1/2 (the terminal phase half-life), and Vz (the volume of distribution during terminal phase) were independent of age. A separate pharmacokinetic study was conducted in 10 elderly patients with metastatic breast cancer (age range, 66 to 81 years; 3 patients >75 years; normal liver function tests) receiving vinorelbine 30 mg/m2 intravenously. CL, Vss, and t1/2 were similar to those reported for younger adult patients in previous studies. No relationship between age, systemic exposure (AUC0-∞), and hematological toxicity was observed.
The pharmacokinetics of vinorelbine are not influenced by the concurrent administration of cisplatin with NAVELBINE (see PRECAUTIONS: Drug Interactions).
Clinical Trials
Data from 1 randomized clinical study (211 evaluable patients) with single-agent NAVELBINE and 2 randomized clinical trials (1,044 patients) using NAVELBINE combined with cisplatin support the use of NAVELBINE in patients with advanced non-small cell lung cancer (NSCLC).
Single-Agent NAVELBINE
Single-agent NAVELBINE was studied in a North American, randomized clinical trial in which patients with Stage IV NSCLC, no prior chemotherapy, and Karnofsky Performance Status ≥70 were treated with NAVELBINE (30 mg/m2) weekly or 5-fluorouracil (5-FU) (425 mg/m2 IV bolus) plus leucovorin (LV) 20 mg/m2 IV bolus) daily for 5 days every 4 weeks. A total of 211 patients were randomized at a 2:1 ratio to NAVELBINE (143) or 5-FU/LV (68). NAVELBINE showed improved survival time compared to 5-FU/LV.
In an intent-to-treat analysis, the median survival time was 30 weeks versus 22 weeks for patients receiving NAVELBINE versus 5-FU/LV, respectively (P=0.06). The 1-year survival rates were 24% (±4% SE) for NAVELBINE and 16% (±5% SE) for the 5-FU/LV group, using the Kaplan-Meier product-limit estimates. The median survival time with 5-FU/LV was similar to or slightly better than that usually observed in untreated patients with advanced NSCLC, suggesting that the difference was not related to some unknown detrimental effect of 5-FU/LV therapy. The response rates (all partial responses) for NAVELBINE and 5-FU/LV were 12% and 3%, respectively.
NAVELBINE in Combination with Cisplatin: NAVELBINE plus Cisplatin versus Single-Agent Cisplatin
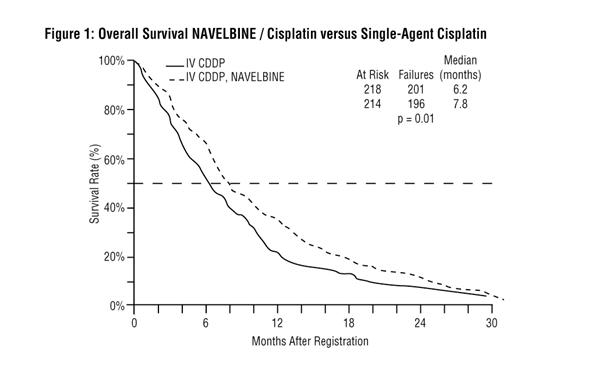
A Phase III open-label, randomized study was conducted which compared NAVELBINE (25 mg/m2/week) plus cisplatin (100 mg/m2 every 4 weeks) to single-agent cisplatin (100 mg/m2 every 4 weeks) in patients with Stage IV or Stage IIIb NSCLC patients with malignant pleural effusion or multiple lesions in more than one lobe who were not previously treated with chemotherapy. Patients included in the study had a performance status of 0 or 1, and 34% had received prior surgery and/or radiotherapy. Characteristics of the 432 randomized patients are provided in Table 1. Two hundred and twelve patients received NAVELBINE plus cisplatin and 210 received single-agent cisplatin. The primary objective of this trial was to compare survival between the 2 treatment groups. Survival (Figure 1) for patients receiving NAVELBINE plus cisplatin was significantly better compared to the patients who received single-agent cisplatin. The results of this trial are summarized in Table 1
NAVELBINE plus Cisplatin versus Vindesine plus Cisplatin versus Single-Agent NAVELBINE
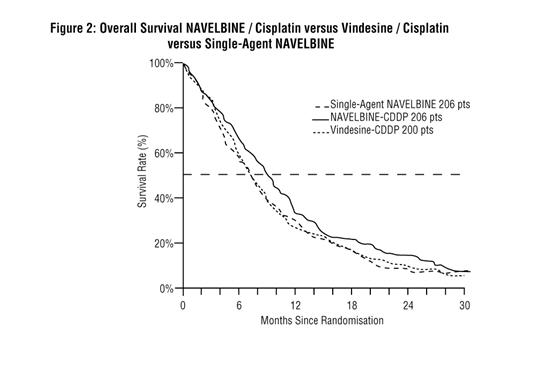
In a large European clinical trial, 612 patients with Stage III or IV NSCLC, no prior chemotherapy, and WHO Performance Status of 0, 1, or 2 were randomized to treatment with single-agent NAVELBINE (30 mg/m2/week), NAVELBINE (30 mg/m2/week) plus cisplatin (120 mg/m2 days 1 and 29, then every 6 weeks), and vindesine (3 mg/m2/week for 7 weeks, then every other week) plus cisplatin (120 mg/m2 days 1 and 29, then every 6 weeks). Patient characteristics are provided in Table 1. Survival was longer in patients treated with NAVELBINE plus cisplatin compared to those treated with vindesine plus cisplatin (Figure 2). Study results are summarized in Table 1.
Dose-Ranging Study
A dose-ranging study of NAVELBINE (20, 25, or 30 mg/m2/week) plus cisplatin (120 mg/m2 days 1 and 29, then every 6 weeks) in 32 patients with NSCLC demonstrated a median survival of 10.2 months. There were no responses at the lowest dose level; the response rate was 33% in the 21 patients treated at the 2 highest dose levels.
| NAVELBINE
Cisplatin vs. Single-Agent Cisplatin | NAVELBINE/Cisplatin vs. Vindesine/Cisplatin vs. Single-Agent NAVELBINE |
||||
| NAVELBINE/ Cisplatin |
Cisplatin | NAVELBINE/ Cisplatin | Vindesine/ Cisplatin | NAVELBINE
|
|
| Demographics
Number of patients Number of males Number of females |
214 146 68 |
218 141 77 |
206 182 24 |
200 179 21 |
206 188 18 |
| Median age (years) Range (years) | 63 33-84 | 64 37-81 | 59 32-75 | 59 31-75 | 60 30-74 |
| Stage of disease Stage IIIA Stage IIIB Stage IV Local recurrence Metastatic after surgery |
NA 8% 92% NA NA |
NA 8% 92% NA NA |
11% 28% 50% 2% 9% |
11% 25% 55% 3% 8% |
10% 32% 47% 3 % 9 % |
| Histology Adenocarcinoma Squamous Large cell Unspecified |
54% 19% 14% 13% |
52% 22% 14 % 13% |
32% 56% 13% NA |
40% 50% 11% NA |
28% 56% 16% NA |
| Results
Median survival (months) |
7.8 |
6.2 |
9.2*† |
7.4 |
7.2 |
| P value | P = 0.01 | *P = 0.09 vs. vindesine/cisplatin † = 0.05 vs. single-agent NAVELBINE |
|||
| 12-Month survival rate | 38% | 22% | 35% | 27% | 30% |
| Overall response | 19% | 8% | 28%ठ| 19% | 14% |
| P value | P<0.001 | ‡P = 0.03 vs. vindesine/cisplatin §P<0.001 vs. single-agent NAVELBINE |
|||
INDICATIONS AND USAGE
NAVELBINE is indicated as a single agent or in combination with cisplatin for the first-line treatment of ambulatory patients with unresectable, advanced non-small cell lung cancer (NSCLC). In patients with Stage IV NSCLC, NAVELBINE is indicated as a single agent or in combination with cisplatin. In Stage III NSCLC, NAVELBINE is indicated in combination with cisplatin
CONTRAINDICATIONS
Administration of NAVELBINE is contraindicated in patients with pretreatment granulocyte counts <1,000 cells/mm3 (see WARNINGS).
WARNINGS
NAVELBINE should be administered in carefully adjusted doses by or under the supervision of a physician experienced in the use of cancer chemotherapeutic agents. Patients treated with NAVELBINE should be frequently monitored for myelosuppression both during and after therapy. Granulocytopenia is dose-limiting. Granulocyte nadirs occur between 7 and 10 days after dosing with granulocyte count recovery usually within the following 7 to 14 days. Complete blood counts with differentials should be performed and results reviewed prior to administering each dose of NAVELBINE. NAVELBINE should not be administered to patients with granulocyte counts <1,000 cells/mm3. Patients developing severe granulocytopenia should be monitored carefully for evidence of infection and/or fever. See DOSAGE AND ADMINISTRATION for recommended dose adjustments for granulocytopenia. Acute shortness of breath and severe bronchospasm have been reported infrequently, following the administration of NAVELBINE and other vinca alkaloids, most commonly when the vinca alkaloid was used in combination with mitomycin. These adverse events may require treatment with supplemental oxygen, bronchodilators, and/or corticosteroids, particularly when there is pre-existing pulmonary dysfunction. Reported cases of interstitial pulmonary changes and acute respiratory distress syndrome (ARDS), most of which were fatal, occurred in patients treated with single-agent NAVELBINE. The mean time to onset of these symptoms after vinorelbine administration was 1 week (range 3 to 8 days). Patients with alterations in their baseline pulmonary symptoms or with new onset of dyspnea, cough, hypoxia, or other symptoms should be evaluated promptly.
NAVELBINE has been reported to cause severe constipation (e.g., Grade 3-4), paralytic ileus, intestinal obstruction, necrosis, and/or perforation. Some events have been fatal.
Pregnancy
Pregnancy Category D. NAVELBINE may cause fetal harm if administered to a pregnant woman. A single dose of vinorelbine has been shown to be embryo- and/or fetotoxic in mice and rabbits at doses of 9 mg/m2 and 5.5 mg/m2, respectively (one third and one sixth the human dose). At nonmaternotoxic doses, fetal weight was reduced and ossification was delayed. There are no studies in pregnant women. If NAVELBINE is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with NAVELBINE.
PRECAUTIONS
General
Most drug-related adverse events of NAVELBINE are reversible. If severe adverse events occur, NAVELBINE should be reduced in dosage or discontinued and appropriate corrective measures taken. Reinstitution of therapy with NAVELBINE should be carried out with caution and alertness as to possible recurrence of toxicity. NAVELBINE should be used with extreme caution in patients whose bone marrow reserve may have been compromised by prior irradiation or chemotherapy, or whose marrow function is recovering from the effects of previous chemotherapy (see DOSAGE AND ADMINISTRATION). Administration of NAVELBINE to patients with prior radiation therapy may result in radiation recall reactions (see ADVERSE REACTIONS and Drug Interactions). Patients with a prior history or pre-existing neuropathy, regardless of etiology, should be monitored for new or worsening signs and symptoms of neuropathy while receiving NAVELBINE.
Care must be taken to avoid contamination of the eye with concentrations of NAVELBINE used clinically. Severe irritation of the eye has been reported with accidental exposure to another vinca alkaloid. If exposure occurs, the eye should immediately be thoroughly flushed with water.
Information for Patients
Patients should be informed that the major acute toxicities of NAVELBINE are related to bone marrow toxicity, specifically granulocytopenia with increased susceptibility to infection. They should be advised to report fever or chills immediately. Women of childbearing potential should be advised to avoid becoming pregnant during treatment. Patients should be advised to contact their physician it they experience increased shortness of breath, cough, or other new pulmonary symptoms, or if they experience symptoms of abdominal pain or constipation.
Laboratory Tests
Since dose-limiting clinical toxicity is the result of depression of the white blood cell count, it is imperative that complete blood counts with differentials be obtained and reviewed on the day of treatment prior to each dose of NAVELBINE (see ADVERSE REACTIONS: Hematologic).
Hepatic
There is no evidence that the toxicity of NAVELBINE is enhanced in patients with elevated liver enzymes. No data are available for patients with severe baseline cholestasis, but the liver plays an important role in the metabolism of NAVELBINE. Because clinical experience in patients with severe liver disease is limited, caution should be exercised when administering NAVELBINE to patients with severe hepatic injury or impairment (see DOSAGE AND ADMINISTRATION).
Drug Interactions
Acute pulmonary reactions have been reported with NAVELBINE and other anticancer vinca alkaloids used in conjunction with mitomycin. Although the pharmacokinetics of vinorelbine are not influenced by the concurrent administration of cisplatin, the incidence of granulocytopenia with NAVELBINE used in combination with cisplatin is significantly higher than with single-agent NAVELBINE. Patients who receive NAVELBINE and paclitaxel, either concomitantly or sequentially, should be monitored for signs and symptoms of neuropathy. Administration of NAVELBINE to patients with prior or concomitant radiation therapy may result in radio sensitizing effects. Caution should be exercised in patients concurrently taking drugs known to inhibit drug metabolism by hepatic cytochrome P450 isoenzymes in the CYP3A subfamily, or in patients with hepatic dysfunction. Concurrent administration of vinorelbine tartrate with an inhibitor of this metabolic pathway may cause an earlier onset and/or an increased severity of side effects.
Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of NAVELBINE has not been studied. Vinorelbine has been shown to affect chromosome number and possibly structure in vivo (polyploidy in bone marrow cells from Chinese hamsters and a positive micronucleus test in mice). It was not mutagenic in the Ames test and gave inconclusive results in the mouse lymphoma TK Locus assay. The significance of these or other short-term test results for human risk is unknown. Vinorelbine did not affect fertility to a statistically significant extent when administered to rats on either a once-weekly (9 mg/m2, approximately one third the human dose) or alternate-day schedule (4.2 mg/m2, approximately one seventh the human dose) prior to and during mating. However, biweekly administration for 13 or 26 weeks in the rat at 2.1 and 7.2 mg/m2 (approximately one fifteenth and one fourth the human dose) resulted in decreased spermatogenesis and prostate/seminal vesicle secretion.
Nursing Mothers
It is not known whether the drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from NAVELBINE, it is recommended that nursing be discontinued in women who are receiving therapy with NAVELBINE.
Pediatric Use
Safety and effectiveness of NAVELBINE in pediatric patients have not been established. Data from a single-arm study in 46 patients with recurrent solid malignant tumors, including rhabdomyosarcoma/undifferentiated sarcoma, neuroblastoma, and CNS tumors, at doses similar to those used in adults, showed no meaningful clinical activity. Toxicities were similar to those reported in adults.
Geriatric Use
Of the total number of patients in North American clinical studies of IV NAVELBINE, approximately one third were 65 years of age or greater. No overall differences in effectiveness or safety were observed between these patients and younger adult patients. Other reported clinical experience has not identified differences in responses between the elderly and younger adult patients, but greater sensitivity of some older individuals cannot be ruled out.
The pharmacokinetics of vinorelbine in elderly and younger adult patients are similar (see CLINICAL PHARMACOLOGY).
ADVERSE REACTIONS
The pattern of adverse reactions is similar whether NAVELBINE is used as a single agent or in combination. Adverse reactions from studies with single-agent and combination use of NAVELBINE are summarized in Tables 2-4.
Single-Agent NAVELBINE
Data in the following table are based on the experience of 365 patients (143 patients with NSCLC; 222 patients with advanced breast cancer) treated with IV NAVELBINE as a single agent in 3 clinical studies. The dosing schedule in each study was 30 mg/m2NAVELBINE on a weekly basis.
| Adverse Event | All Patients (n=365) | NSCLC (n=143) |
||
| Bone Marrow | ||||
| Granulocytopenia | <2,000 cells/mm3 | 90% | 80% | |
| <500 cells/mm3 | 36% | 29% | ||
| Leukopenia | <4,000 celIs/mm3 | 92% | 81% | |
| <1,000 cells/mm3 | 15% | 12% | ||
| Thrombocytopenia | <100,000 cells/mm3 | 5% | 4% | |
| <50,000 cells/mm3 | 1% | 1% | ||
| Anemia | <11 g/dL | 83% | 77% | |
| <8 g/dL | 9% | 1% | ||
| Hospitalizations due to granulocytopenic complications | 9% | 8% | ||
| * None of the reported toxicities were influenced by age. Grade based on modified criteria from the National Cancer Institute. † Patients with NSCLC had not received prior chemotherapy. The majority of the remaining patients had received prior chemotherapy. ‡ Incidence of paresthesia plus hypesthesia. | ||||||
| All Grades | Grade 3 | Grade 4 | ||||
| All Patients | NSCLC | All Patients | NSCLC | All Patients | NSCLC | |
| Clinical Chemistry | ||||||
| Elevations | ||||||
| Total Bilirubin (n=351) | 13% | 9% | 4% | 3% | 3% | 2% |
| SGOT (n=346) | 67% | 54% | 5% | 2% | 1% | 1% |
| General | ||||||
| Asthenia | 36% | 27% | 7% | 5% | 0% | 0% |
| Injection Site Reactions | 28% | 38% | 2% | 5% | 0% | 0% |
| Injection Site Pain | 16% | 13% | 2% | 1% | 0% | 0% |
| Phlebitis | 7% | 10% | <1% | 1% | 0% | 0% |
| Digestive | ||||||
| Nausea | 44% | 34% | 2% | 1% | 0% | 0% |
| Vomiting | 20% | 15% | 2% | 1% | 0% | 0% |
| Constipation | 35% | 29% | 3% | 2% | 0% | 0% |
| Diarrhea | 17% | 13% | 1% | 1% | 0% | 0% |
| Peripheral Neuropathy‡ | 25% | 20% | 1% | 1% | <1% | 0% |
| Dyspnea | 7% | 3% | 2% | 2% | 1% | 0% |
| Alopecia | 12% | 12% | <1% | 1% | 0% | 0% |
Hematologic
Granulocytopenia is the major dose-limiting toxicity with NAVELBINE. Dose adjustments are required for hematologic toxicity and hepatic insufficiency (see DOSAGE AND ADMINISTRATION). Granulocytopenia was generally reversible and not cumulative over time. Granulocyte nadirs occurred 7 to 10 days after the dose, with granulocyte recovery usually within the following 7 to 14 days. Granulocytopenia resulted in hospitalizations for fever and/or sepsis in 8% of patients. Septic deaths occurred in approximately 1% of patients. Prophylactic hematologic growth factors have not been routinely used with NAVELBINE.
If medically necessary, growth factors may be administered at recommended doses no earlier than 24 hours after the administration of cytotoxic chemotherapy. Growth factors should not be administered in the period 24 hours before the administration of chemotherapy. Whole blood and/or packed red blood cells were administered to 18% of patients who received NAVELBINE.
Neurologic
Loss of deep tendon reflexes occurred in less than 5% of patients. The development of severe peripheral neuropathy was infrequent (1%) and generally reversible.
Skin
Like other antirancer vinca alkaloids, NAVELBINE is a moderate vesicant. Injection site reactions, including erythema, pain at injection site, and vein discoloration, occurred in approximately one third of patients; 5% were severe. Chemical phlebitis along the vein proximal to the site of injection was reported in 10% of patients.
Gastrointestinal
Prophylactic administration of antiemetics was not routine in patients treated with single-agent NAVELBINE. Due to the low incidence of severe nausea and vomiting with single-agent NAVELBINE, the use of serotonin antagonists is generally not required.
Cardiovascular
Chest pain was reported in 5% of patients. Most reports of chest pain were in patients who had either a history of cardiovascular disease or tumor within the chest. There have been rare reports of myocardial infarction.
Pulmonary
Shortness of breath was reported in 3% of patients; it was severe in 2% (see WARNINGS). Interstitial pulmonary changes were documented.
Other
Fatigue occurred in 27% of patients. It was usually mild or moderate but tended to increase with cumulative dosing.
Other toxicities that have been reported in less than 5% of patients include jaw pain, myalgia, arthralgia, and rash. Hemorrhagic cystitis and the syndrome of inappropriate ADH secretion were each reported in <1% of patients.
NAVELBINE in Combination with Cisplatin
NAVELBINE plus Cisplatin versus Single-Agent Cisplatin (Table 3): Myelosuppression was the predominant toxicity in patients receiving combination therapy, Grade 3 and 4 granulocytopenia of 82% compared to 5% in the single-agent cisplatin arm. Fever and/or sepsis related to granulocytopenia occurred in 11% of patients on NAVELBINE and cisplatin compared to 0% on the cisplatin arm. Four patients on the combination died of granulocytopenia-related sepsis. During this study, the use of granulocyte colony-stimulating factor ([G-CSF] filgrastim) was permitted, but not mandated, after the first course of treatment for patients who experienced Grade 3 or 4 granulocytopenia (x1,000 cells/mm3) or in those who developed neutropenic fever between cycles of chemotherapy. Beginning 24 hours after completion of chemotherapy, G-CSF was started at a dose of 5 mcg/kg per day and continued until the total granulocyte count was >1,000 cells/mm3 on 2 successive determinations. G-CSF was not administered on the day of treatment. Grade 3 and 4 anemia occurred more frequently in the combination arm compared to control, 24% vs. 8%, respectively. Thrombocytopenia occurred in 6% of patients treated with NAVELBINE plus cisplatin compared to 2% of patients treated with cisplatin. The incidence of severe non-hematologic toxicity was similar among the patients in both treatment groups. Patients receiving NAVELBINE plus cisplatin compared to single-agent cisplatin experienced more Grade 3 and/or 4 peripheral numbness (2% vs. <1%), phlebitis/thrombosis/embolism (3% vs. <1%), and infection (6% vs. <1%). Grade 3-4 constipation and/or ileus occurred in 3% of patients treated with combination therapy and in 1% of patients treated with cisplatin. Seven deaths were reported on the combination arm; 2 were related to cardiac ischemia, 1 massive cerebrovascular accident, 1 multisystem failure due to an overdose of NAVELBINE, and 3 from febrile neutropenia. One death, secondary to respiratory infection unrelated to granulocytopenia, occurred with single-agent cisplatin.
NAVELBINE plus Cisplatin versus Vindesine plus Cisplatin versus Single-Agent Vinorelbine (Table 4)
Myelosuppression, specifically Grade 3 and 4 granulocytopenia, was significantly greater with the combination of NAVELBINE plus cisplatin (79%) than with either single-agent NAVELBINE (53%) or vindesine plus cisplatin (48%), P<0.0001. Hospitalization due to documented sepsis occurred in 4.4% of patients treated with NAVELBINE plus cisplatin: 2% of patients treated with vindesine and cisplatin, and 4% of patients treated with single-agent NAVELBINE. Grade 3 and 4 thrombocytopenia was infrequent in patients receiving combination chemotherapy and no events were reported with single-agent NAVELBINE.
The incidence of Grade 3 and/or 4 nausea and vomiting, alopecia, and renal toxicity were reported more frequently in the cisplatin-containing combinations compared to single-agent NAVELBINE. Severe localNAVELBINE® (vinorelbine tartrate) Injection reactions occurred in 2% of patients treated with combinations containing NAVELBINE; none were observed in the vindesine plus cisplatin arm. Grade 3 and 4 neurotoxicity was significantly more frequent in patients receiving vindesine plus cisplatin (17%) comparedto NAVELBINE plus cisplatin (7%) and single-agent vinorelbine (9%) (P<0.005). Cisplatin did not appear to increase the incidence of neurotoxicity observed with single-agent NAVELBINE.
| *Graded according to the standard SWOG criteria. | ||||||
| NAVELBINE 25 mg/m2 plus Cisplatin 100 mg/m2 (n=212) |
Cisplatin 100 mg/m2 (n=210) |
|||||
| Adverse Event | All Grades | Grade 3 | Grade 4 | All Grades | Grade 3 | Grade 4 |
| Bone Marrow | ||||||
| Granulocytopenia | 89% | 22% | 60% | 26% | 4% | 1% |
| Anemia | 88% | 21% | 3% | 72% | 7% | <1% |
| Leukopenia | 88% | 39% | 19% | 31% | <1% | 0% |
| Thrombocytopenia | 29% | 4% | 1% | 21% | 1% | <1% |
| Febrile neutropenia | N/A | N/A | 11% | N/A | N/A | 0% |
| Hepatic | ||||||
| Elevated transaminase | 1% | 0% | 0% | <1% | <1% | 0% |
| Renal | ||||||
| Elevated creatinine | 37% | 2% | 2% | 28% | 4% | <1% |
| Non-Laboratory | ||||||
| Malaise/fatigue/lethargy | 67% | 12% | 0% | 49% | 8% | 0% |
| Vomiting | 60% | 7% | 6% | 60% | 10% | 4% |
| Nausea | 58% | 14% | 0% | 57% | 12% | 0% |
| Anorexia | 46% | 0% | 0% | 37% | 0% | 0% |
| Constipation | 35% | 3% | 0% | 16% | 1% | 0% |
| Alopecia | 34% | 0% | 0% | 14% | 0% | 0% |
| Weight loss | 34% | 1% | 0% | 21% | <1% | 0% |
| Fever without infection | 20% | 2% | 0% | 4% | 0% | 0% |
| Hearing | 18% | 4% | 0% | 18% | 3% | <1% |
| Local (injection site reactions) | 17% | <1% | 0% | 1% | 0% | 0% |
| Diarrhea | 17% | 2% | <1% | 11% | 1% | <1% |
| Paresthesias | 17% | <1% | 0% | 10% | <1% | 0% |
| Taste alterations | 17% | 0% | 0% | 15% | 0% | 0% |
| Peripheral numbness | 11% | 2% | 0% | 7% | <1% | 0% |
| Myalgia/arthralgia | 12% | <1% | 0% | 3% | <1% | 0% |
| Phlebitis/thrombosis/embolism | 10% | 3% | 0% | <1% | 0% | <1% |
| Weakness | 12% | 2% | <1% | 7% | 2% | 0% |
| Dizziness/vertigo | 9% | <1% | 0% | 3% | <1% | 0% |
| Infection | 11% | 5% | <1% | <1% | <1% | 0% |
| Respiratory infection | 10% | 4% | <1% | 3% | 3% | 0% |
| * Grade based on criteria from the World Health Organization (WHO). † n=194 to 207; all patients receiving NAVELBINE/cisplatin with laboratory and non-laboratory data. ‡ n=173 to 192; all patients receiving vindesine/cisplatin with laboratory and non-laboratory data. § n=165 to 201; all patients receiving NAVELBINE with laboratory and non-laboratory data. € Categorical toxicity grade not specified. ¶ Neurotoxicity includes peripheral neuropathy and constipation. |
|||||||||
| NAVELBINE/Cisplatin† | Vindesine/Cisplatin‡ | NAVELBINE§ | |||||||
| Adverse Event | All Grades | Grades 3 | Grade 4 | All Grades | Grades 3 | Grade 4 | All Grades | Grades 3 | Grade 4 |
| Bone Marrow | |||||||||
| Neutropenia | 95% | 20% | 58% | 79% | 26% | 22% | 85% | 25% | 28% |
| Leukopenia | 94% | 40% | 17% | 82% | 24% | 3% | 83% | 26% | 6% |
| Thrombocytopenia | 15% | 3% | 1% | 10% | 3% | 0.50% | 3% | 0% | 0% |
| Febrile neutropenia | N/A | N/A | 4% | N/A | N/A | 2% | N/A | N/A | 4% |
| Hepatic | |||||||||
| Elevated € | 6% | N/A | N/A | 5% | NIA | N/A | 5% | N/A | N/A |
| Renal | |||||||||
| Elevated € | 46% | N/A | N/A | 37% | N/A | N/A | 13% | N/A | N/A |
| Non-Laboratory | |||||||||
| Nausea/vomiting | 74% | 27% | 3% | 72% | 24% | 1% | 31% | 1% | 1% |
| Alopecia | 51% | 7% | 0.50% | 56% | 14% | 0% | 30% | 2% | 0% |
| Ototoxicity | 10% | 1% | 1% | 14% | 1% | 0% | 1% | 0% | 0% |
| Local reactions | 17% | 2% | 0.50% | 7% | 0% | 0% | 22% | 2% | 0% |
| Diarrhea | 25% | 1.50% | 0% | 24% | 1% | 0% | 12% | 0% | 0.50% |
| Neurotoxicity ¶ | 44% | 7% | 0% | 58% | 16% | 1% | 44% | 8% | 0.50% |
Observed During Clinical Practice
In addition to the adverse events reported from clinical trials, the following events have been identified during post-approval use of NAVELBINE. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These events have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to NAVELBINE.
Body as a Whole: Systemic allergic reactions reported as anaphylaxis, pruritus, urticaria, and angioedema; flushing; and radiation recall events such as dermatitis and esophagitis (see PRECAUTIONS) have been reported.
Hematologic: Thromboembolic events, including pulmonary embolus and deep venous thrombosis, have been reported primarily in seriously ill and debilitated patients with known predisposing risk factors for these events.
Neurologic: Peripheral neurotoxicities such as, but not limited to, muscle weakness and disturbance of gait; have been observed in patients with and without prior symptoms.
There may be increased potential for neurotoxicity in patients with pre-existing neuropathy, regardless of etiology, who receive NAVELBINE. Vestibular and auditory deficits have been observed with NAVELBINE, usually when used in combination with cisplatin.
Skin: Injection site reactions, including localized rash and urticaria, blister formation, and skin sloughing have been observed in clinical practice. Some of these reactions may be delayed in appearance.
Gastrointestinal: Dysphagia, mucositis, and pancreatitis have been reported.
Cardiovascular: Hypertension, hypotension, vasodilation, tachycardia, and pulmonary edema have been reported.
Pulmonary: Pneumonia has been reported.
Musculoskeletal: Headache has been reported, with and without other musculoskeletal aches and pains.
Other: Pain in tumor-containing tissue, back pain, and abdominal pain have been reported. Electrolyte abnormalities, including hyponatremia with or without the syndrome of inappropriate ADH secretion, have been reported in seriously ill and debilitated patients.
Combination Use
Patients with prior exposure to paclitaxel and who have demonstrated neuropathy should he monitored closely for new or worsening neuropathy. Patients who have experienced neuropathy with previous drug regimens should be monitored for symptoms of neuropathy while receiving NAVELBINE. NAVELBINE may result in radiosensitizing effects with prior or concomitant radiation therapy (see PRECAUTIONS).
OVERDOSAGE
There is no known antidote for overdoses of NAVELBINE. Overdoses involving quantities up to 10 times the recommended dose (30 mg/m2) have been reported. The toxicities described were consistent with those listed in the ADVERSE REACTIONS section including paralytic ileus, stomatitis, and esophagitis. Bone marrow aplasia, sepsis, and paresis have also been reported. Fatalities have occurred following overdose of NAVELBINE.
If overdosage occurs, general supportive measures together with appropriate blood transfusions, growth factors, and antibiotics should be instituted as deemed necessary by the physician.
DOSAGE AND ADMINISTRATION
Single-Agent NAVELBINE
The usual initial dose of single-agent NAVELBINE is 30 mg/m2 administered weekly. The recommended method of administration is an intravenous injection over 6 to 10 minutes. In controlled trials, single-agent NAVELBINE was given weekly until progression or dose-limiting toxicity.
NAVELBINE in Combination with Cisplatin
NAVELBINE may be administered weekly at a dose of 25 mg/m2 in combination with cisplatin given every 4 weeks at a dose of 100 mg/m2.
Blood counts should be checked weekly to determine whether dose reductions of vinorelbine and/or cisplatin are necessary. In the SWOG study, most patients required a 50% dose reduction of NAVELBINE at day 15 of each cycle and a 50% dose reduction of cisplatin by cycle 3.
NAVELBINE may also be administered weekly at a dose of 30 mg/m2 in combination with cisplatin, given on days 1 and 29, then every 6 weeks with cisplatin at a dose of 120 mg/m2.
Dose Modifications for NAVELBINE
The dosage should be adjusted according to hematologic toxicity or hepatic insufficiency, whichever results in the lower dose for the corresponding starling dose of NAVELBINE (see Table 5).
Dose Modifications for Hematologic Toxicity
Granulocyte counts should be ≥1,000 cells/mm3 prior to the administration of NAVELBINE. Adjustments in the dosage of NAVELBINE should be based on granulocyte counts obtained on the day of treatment according to Table 5.
| Granulocytes on Day of Treatment (Cells/mm3) | Percentage of Starting Dose of NAVELBINE |
| ≥1,500 | 100% |
| 1,000 to 1,499 | 50% |
| <1,000 count | Do not administer. Repeat granulocyte count in 1 week. If 3 consecutive weekly doses are held because granulocyte is <1,000 cells/mm3, discontinue NAVELBINE. |
| Note: For patients who, during treatment with vinorelbine, experienced fever and/or sepsis while granulocytopenic or had 2 consecutive weekly doses held due to granulocytopenia, subsequent doses of vinorelbine should be: | |
| >1,500 | 75 |
| 1,000 to 1,499 | 37.5% |
| <1,000 | See above |
Dose Modifications for Hepatic Insufficiency
NAVELBINE should be administered with caution to patients with hepatic insufficiency. In patients who develop hyperbilirubinemia during treatment with NAVELBINE, the dose should be adjusted for total bilirubin according to Table 6.
| Total Bilirubin (mg/dL) | Percentage of Starting Dose of NAVELBINE |
| ≤2.0 | 100% |
| 2.1 to 3.0 | 50% |
| >3.0 | 25% |
Dose Modifications for Concurrent Hematologic Toxicity and Hepatic Insufficiency
In patients with both hematologic toxicity and hepatic insufficiency, the lower of the doses based on the corresponding starting dose of NAVELBINE determined from Table 5 and Table 6 should be administered.
Dose Modifications for Renal Insufficiency
No dose adjustments for NAVELBINE are required for renal insufficiency. Appropriate dose reductions for cisplatin should be made when NAVELBINE is used in combination.
Dose Modifications for Neurotoxicity
If Grade ≥2 neurotoxicity develops, NAVELBINE should be discontinued.
Administration Precautions
Caution - NAVELBINE must be administered intravenously. It is extremely important that the intravenous needle or catheter be properly positioned before any NAVELBINE is injected. Leakage into surrounding tissue during intravenous administration of NAVELBINE may cause considerable irritation, local tissue necrosis, and/or thrombophlebitis. If extravasation occurs, the injection should be discontinued immediately, and any remaining portion of the dose should then be introduced into another vein. Since there are no established guidelines for the treatment of extravasation injuries with NAVELBINE, institutional guidelines may be used. The ONS Chemotherapy Guidelines provide additional recommendations for the prevention of extravasation injuries.1
As with other toxic compounds, caution should be exercised in handling and preparing the solution of NAVELBINE. Skin reactions may occur with accidental exposure. The use of gloves is recommended. If the solution of NAVELBINE contacts the skin or mucosa, immediately wash the skin or mucosa thoroughly with soap and water. Severe irritation of the eye has been reported with accidental contamination of the eye with another vinca alkaloid. If this happens with NAVELBINE, the eye should be flushed with water immediately and thoroughly.
Procedures for proper handling and disposal of anticancer drugs should be used. Several guidelines on this subject have been published.2-8
There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
NAVELBINE Injection is a clear, colorless to pale yellow solution. Parenteral drug products should be visually inspected for particulate matter and discoloration prior to administration whenever solution and container permit. If particulate matter is seen, NAVELBINE should not be administered.
Preparation for Administration
NAVELBINE Injection must be diluted in either a syringe or IV bag using one of the recommended solutions. The diluted NAVELBINE should be administered over 6 to 10 minutes into the side port of a free-flowing IV closest to the IV bag followed by flushing with at least 75 to 125 mL of one of the solutions. Diluted vinorelbine may be used for up to 24 hours under normal room light when stored in polypropylene syringes or polyvinyl chloride bags at 5° to 30°C (41° to 86°F).
Syringe: The calculated dose of NAVELBINE should be diluted to a concentration between 1.5 and 3.0 mg/mL.
The following solutions may be used for dilution:
- 5% Dextrose Injection, USP
- 0.9% Sodium Chloride Injection, USP
IV Bag:The calculated dose of NAVELBINE should be diluted to a concentration between 0.5 and 2 mg/mL.
The following solutions may be used for dilution:
- 5% Dextrose Injection, USP
- 0.9% Sodium Chloride Injection, USP
- 0.45% Sodium Chloride Injection, USP
- 5% Dextrose and 0.45% Sodium Chloride Injection, USP
- Ringer's Injection, USP
- Lactated Ringer's Injection, USP
Stability
Unopened vials of NAVELBINE are stable until the date indicated on the package when stored under refrigeration at 2° to 8°C (36° to 46°F) and protected from light in the carton. Unopened vials of NAVELBINE are stable at temperatures up to 25°C (77°F) for up to 72 hours. This product should not be frozen.
HOW SUPPLIED
NAVELBINE Injection is a clear, colorless to pale yellow solution in Water for Injection, containing 10 mg vinorelbine per mL. Vinorelbine Injection is available in single-use, clear glass vials with elastomeric stoppers and royal blue caps, individually packaged in a carton in the following vial sizes:
10 mg/1 mL Single-Use Vial, Carton of 1 (NDC 64370-532-01).
50 mg/5 mL Single-Use Vial, Carton of 1 (NDC 64370-532-02).
Store the vials under refrigeration at 2° to 8°C (36° to 46°F) in the carton. Protect from light. DO NOT FREEZE.
REFERENCES
- ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, Pa: Oncology Nursing Society; 1999: 32-41.
- Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC: Division of Safety, National Institutes of Health; 1983. US Dept of Health and Human Services, Public Health Service publication NIH 83-2621.
- AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA.1985; 253: 1590-1591.
- National Study Commission on Cytotoxic Exposure. Recommendations for handling cytotoxic agents. 1987. Available from Louis P Jeffrey, Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia. 1983; 1: 426-428.
- Jones RB, Frank R, Mass T Safe handling of chemotherapeutic agents: a report from the Mount Sinai Medical Center. CA-A Cancer J for Clin. 1983; 33: 258-263.
- American Society of Hospital Pharmacists. ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm. 1990; 47: 1033-1049.
- Controlling Occupational Exposure to Hazardous Drugs. (OSHA Work-Practice Guidelines.) Am J Health-Syst Pharm. 1996; 53: 1669-1685
Manufactured by Pierre Fabre Médicament
45 place Abel Gance - 92100 Boulogne - FRANCE
For further information please contact
Pierre Fabre Pharmaceuticals Inc.
9 campus Drive - Parsippany, NJ 07054
October 2007




| NAVELBINE
vinorelbine tartrate injection, solution, concentrate |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020388 | 11/15/2005 | |
| Labeler - Pierre Fabre Médicament (504638276) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Pierre Fabre Médicament | 504638276 | analysis, MANUFACTURE | |
Revised: 08/2011 Pierre Fabre Médicament