BERINERT
-
human c1-esterase inhibitor
CSL Behring GmbH
----------
|
|||||||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Berinert is a plasma-derived concentrate of C1 Esterase Inhibitor (Human) indicated for the treatment of acute abdominal or facial attacks of hereditary angioedema (HAE) in adult and adolescent patients.
The safety and efficacy of Berinert for prophylactic therapy have not been established.
2 DOSAGE AND ADMINISTRATION
For Intravenous Use Only.
Administer Berinert at a dose of 20 units per kg body weight by intravenous injection.
Berinert is provided as a freeze-dried powder for reconstitution with the diluent (sterile water) provided. Store the vial in the original carton in order to protect from light. Do not freeze.
2.1 Preparation and Handling
- Check the expiration date on the product vial label. Do not use beyond the expiration date.
- Use aseptic technique when preparing and administering Berinert (see Reconstitution and Administration [2.2]).
- After reconstitution and prior to administration, inspect Berinert visually for particulate matter and discoloration. The reconstituted solution should be colorless, clear, and free from visible particles. Do not use if the solution is cloudy, discolored, or contains particulates.
- The Berinert vial is for single use only. Berinert contains no preservative. Any product that has been reconstituted should be used promptly. The reconstituted solution must be used within 8 hours. Discard partially used vials.
- Do not freeze the reconstituted solution.
2.2 Reconstitution and Administration
Each Berinert kit consists of one carton containing one single-use vial of Berinert, one 10 mL vial of diluent (sterile water), one Mix2Vial™ transfer set, and one alcohol swab.
Use either the Mix2Vial transfer set provided with Berinert (see How Supplied [16.1]) or a commercially available double-ended needle and vented filter spike.
Reconstitution
The procedures below are provided as general guidelines for the reconstitution and administration of Berinert.
| |
| |
| |
| 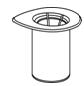 Fig. 1 |
| 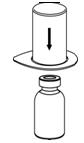 Fig. 2 |
| 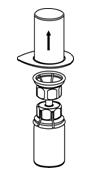 Fig. 3 |
| 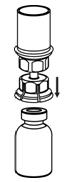 Fig. 4 |
| 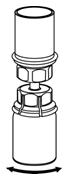 Fig. 5 |
| 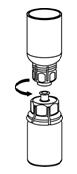 Fig. 6 |
| 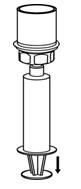 Fig. 7 |
| 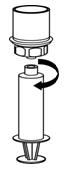 Fig. 8 |
| |
|
3 DOSAGE FORMS AND STRENGTHS
- Berinert is available in a single-use vial that contains 500 units of C1 esterase inhibitor as a lyophilized concentrate.
- Each vial must be reconstituted with 10 mL of diluent (sterile water) provided.
4 CONTRAINDICATIONS
Berinert is contraindicated in individuals who have experienced life-threatening hypersensitivity reactions, including anaphylaxis, to C1 esterase inhibitor preparations.
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur. Epinephrine should be immediately available for treatment of acute severe hypersensitivity reaction (see Patient Counseling Information [17]). The signs and symptoms of hypersensitivity reactions may include hives, generalized urticaria, tightness of the chest, wheezing, hypotension, and/or anaphylaxis during or after injection of Berinert.
Because hypersensitivity reactions may have symptoms similar to HAE attacks, treatment methods should be carefully considered. In case of suspected hypersensitivity, immediately discontinue administration of Berinert and institute appropriate treatment.
5.2 Thrombotic Events
Thrombotic events have been reported in association with Berinert when used off-label and at higher than labeled doses.1 Animal studies have confirmed the risk of thrombosis from intravenous administration of C1 esterase inhibitor products2 (see Overdosage [10] and Animal Toxicology and/or Pharmacology [13.2]).
5.3 Transmission of Infectious Agents
Because Berinert is made from human blood, it may contain infectious agents (eg, viruses and, theoretically, the Creutzfeldt-Jakob disease [CJD] agent) that can cause disease. The risk that such products will transmit an infectious agent has been reduced by screening plasma donors for prior exposure to certain viruses, by testing for the presence of certain current virus infections, and by processes demonstrated to inactivate and/or remove certain viruses during manufacturing (see Description [11] and Patient Counseling Information [17]).
Despite these measures, such products may still potentially transmit disease. There is also the possibility that unknown infectious agents may be present in such products.
The physician should discuss the risks and benefits of this product with the patient before prescribing or administering it to the patient. (See Patient Counseling Information [17.1]).
All infections thought by a physician possibly to have been transmitted by Berinert should be reported by lot number, by the physician, or other healthcare provider to the CSL Behring Pharmacovigilance Department at 1-866-915-6958.
6 ADVERSE REACTIONS
The most serious adverse reaction reported in subjects enrolled in clinical studies who received Berinert was an increase in the severity of pain associated with HAE.
The most common adverse reactions that have been reported in greater than 4% of the subjects who received Berinert in clinical studies were subsequent HAE attack, headache, abdominal pain, nausea, muscle spasm, pain, diarrhea and vomiting.
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Placebo-controlled Clinical Study
In the placebo-controlled clinical study, referred to as the randomized clinical trial (RCT) (see Clinical Studies [14]), 124 subjects experiencing an acute moderate to severe abdominal or facial HAE attack were treated with Berinert (either a 10 unit per kg body weight or a 20 unit per kg body weight dose), or placebo (physiological saline solution).
The treatment-emergent serious adverse reactions/events that occurred in 5 subjects in the RCT were laryngeal edema, facial attack with laryngeal edema, swelling (shoulder and chest), exacerbation of hereditary angioedema, and laryngospasm.
| Adverse Reactions | Number (%) of Subjects Reporting Adverse Reactions Berinert 20 units/kg (n = 43) | Number (%) of Subjects Reporting Adverse Reactions Placebo Group (n = 42) |
|---|---|---|
|
||
| Nausea† | 3 (7%) | 5 (11.9%) |
| Dysgeusia | 2 (4.7%) | 0 (0) |
| Abdominal Pain† | 2 (4.7%) | 3 (7.1%) |
| Vomiting† | 1 (2.3%) | 3 (7.1%) |
| Diarrhea† | 0 (0) | 4 (9.5%) |
| Headache | 0 (0) | 2 (4.8%) |
| Adverse Reactions | Number (%) of Subjects Reporting Adverse Reactions†‡
Berinert 20 units/kg (n = 43) | Number (%) of Subjects Reporting Adverse Reactions†‡
Placebo Group (n = 42) |
|---|---|---|
|
||
| Nausea | 3 (7%) | 11 (26.2%) |
| Headache | 3 (7%) | 5 (11.9%) |
| Abdominal Pain | 3 (7%) | 5 (11.9%) |
| Dysgeusia | 2 (4.7%) | 1 (2.4%) |
| Vomiting | 1 (2.3%) | 7 (16.7%) |
| Pain | 1 (2.3%) | 4 (9.5%) |
| Muscle spasms | 1 (2.3%) | 4 (9.5%) |
| Diarrhea | 0 (0) | 8 (19%) |
| Back pain | 0 (0) | 2 (4.8%) |
| Facial pain | 0 (0) | 2 (4.8%) |
Table 3 lists the adverse events that occurred in more than 4% of the subjects 7 to 9 days after the end of a Berinert infusion, irrespective of causality.
| Table 3: Adverse Events Occurring in More Than 4% of Subjects* Receiving Berinert at Either 10 Units/kg or 20 units/kg 7 to 9 Days after Infusion, Irrespective of Causality | ||
|---|---|---|
| Adverse Events | Number (%) of Subjects Reporting Adverse Events (n=108) |
|
|
||
| Hereditary angioedema | 12 (11.1%) | |
| Headache | 12 (11.1%) | |
| Abdominal pain† | 7 (6.5%) | |
| Nausea† | 7 (6.5%) | |
| Muscle spasms | 6 (5.6%) | |
| Pain | 6 (5.6%) | |
| Diarrhea† | 5 (4.6%) | |
| Vomiting† | 5 (4.6%) | |
Subjects were tested at baseline and after 3 months for possible exposure to Parvovirus B19, hepatitis B, hepatitis C, and HIV-1 and HIV-2. No subject who underwent testing evidenced seroconversion or treatment-emergent positive polymerase chain reaction testing for these pathogens.
Extension Study
In an interim safety analysis, of the ongoing open-label extension study, 56 subjects with 559 acute moderate to severe abdominal, facial, peripheral, and/or laryngeal attacks received a 20 unit/kg body weight dose of Berinert (see Clinical Studies [14]). This study provides additional safety data in subjects who received multiple infusions of the product for sequential HAE attacks (one infusion per attack).
Table 4 lists the adverse events that occurred in this interim safety analysis of the ongoing open-label extension study in more than 4% of subjects up to 72 hours or 9 days after the end of a Berinert infusion, irrespective of causality.
| Adverse Events | Number (%) of Subjects Reporting Adverse Events up to 72 hours (n=56) | Number (%) of Subjects Reporting Adverse Events up to 9 Days (n=56) |
|---|---|---|
| Headache | 3 (5.4%) | 4 (7.1%) |
| Abdominal pain | 3 (5.4%) | 3 (5.4%) |
| Hereditary angioedema | 2 (3.6%) | 4 (7.1%) |
| Nasopharyngitis | 2 (3.6%) | 3 (5.4%) |
6.2 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
Adverse reactions reported in Europe since 1979 in patients receiving Berinert for treatment of HAE include hypersensitivity/anaphylactic reactions, a few suspected cases of viral transmission, including cases of acute hepatitis C, injection-site pain, injection-site redness, chills, and fever.
The following adverse reactions, identified by system organ class, have been attributed to Berinert during post-approval use outside the US.
- Immune System Disorder: Hypersensitivity/anaphylactic reactions, and shock
- General/Body as a Whole: Pain on injection, redness at injection site, chills, and fever
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Animal reproduction studies have not been conducted with Berinert. It is not known whether Berinert can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Berinert should be given to a pregnant woman only if clearly needed. In a retrospective case collection study, 20 pregnant women ranging in age from 20 to 35 years received Berinert with repeated doses up to 3,500 units per attack; these women reported no complications during delivery and no harmful effects on their 34 neonates.
8.2 Labor and Delivery
The safety and effectiveness of Berinert administration prior to or during labor and delivery have not been established. Use only if clearly needed.
8.3 Nursing Mothers
It is not known whether Berinert is excreted in human milk. Because many drugs are excreted in human milk, use only if clearly needed when treating a nursing woman.
8.4 Pediatric Use
Safety and efficacy of Berinert in children (ages 0 through 12) have not been established. The clinical studies included an insufficient number of subjects in this age group to determine whether they respond differently from older subjects. The safety and efficacy of Berinert were evaluated in 5 children (ages 3 through 12) and in 8 adolescent subjects (ages 13 through 16) (see Pharmacokinetics [12.3]).
8.5 Geriatric Use
Safety and efficacy of Berinert in the geriatric population have not been established. Clinical studies with Berinert included four subjects older than 65 years. The clinical studies included an insufficient number of subjects in this age group to determine whether they respond differently from younger subjects.
10 OVERDOSAGE
The development of thrombosis has been reported after doses exceeding 20 units/kg body weight of Berinert when used off-label1 in newborns and young children with congenital heart anomalies during or after cardiac surgery under extracorporeal circulation.
The maximum dose administered in clinical studies in hereditary angioedema was 20 units/kg body weight. Overdosage did not occur in connection with treatment of HAE.
11 DESCRIPTION
Berinert is a human plasma-derived, purified, pasteurized, lyophilized concentrate of C1 esterase inhibitor to be reconstituted for intravenous administration. Berinert is prepared from large pools of human plasma from US donors. One standard unit of C1 esterase inhibitor concentrate is equal to the amount of C1 esterase inhibitor in 1 mL of fresh citrated human plasma, which is equivalent to 270 mg/L or 2.5 µM/L. No international laboratory standard for quantifying C1 esterase inhibitor. An in-house standard is used to assure lot-to-lot consistency in product potency.
C1 esterase inhibitor is a soluble, single-chain glycoprotein containing 478 amino acid residues organized into three beta-sheets and eight or nine alpha-helices.3 The heavily glycosylated molecule has an apparent molecular weight of 105 kD, of which the carbohydrate chains comprise 26% to 35%.4
Each vial of Berinert contains 500 units C1 esterase inhibitor, 50 to 80 mg total protein, 85 to 115 mg glycine, 70 to 100 mg sodium chloride, and 25 to 35 mg sodium citrate.
All plasma used in the manufacture of Berinert is obtained from US donors and is tested using serological assays for hepatitis B surface antigen and antibodies to HIV-1/2 and HCV. Additionally, the plasma is tested with Nucleic Acid Testing (NAT) for HCV and HIV-1 and found to be non-reactive (negative). In addition, the plasma is tested by NAT for HAV and Human Parvovirus B19. Only plasma that has passed virus screening is used for production, and the limit for Parvovirus B19 in the fractionation pool is set not to exceed 104 IU of Parvovirus B19 DNA per mL.
The manufacturing process for Berinert includes multiple steps that reduce the risk of virus transmission. The virus inactivation/reduction capacity consists of three steps:
- Pasteurization in aqueous solution at 60°C for 10 hours,
- Hydrophobic interaction chromatography, and
- Virus filtration (also called nanofiltration) by two filters, 20 nm and 15 nm, in series.
This was evaluated in a series of in vitro spiking experiments. The total mean cumulative virus inactivation/reduction is shown in Table 5.
| Virus Studied | Pasteurization [log10] | Hydrophobic Interaction Chromatography [log10] | Virus Filtration [log10] | Total Cumulative [log10] |
|---|---|---|---|---|
| HIV-1, Human immunodeficiency virus type 1, a model for HIV-1 and HIV-2 BVDV, Bovine viral diarrhea virus, a model for HCV PRV, Pseudorabies virus, a model for large enveloped DNA viruses WNV, West Nile virus HAV, Hepatitis A virus CPV, Canine parvovirus B19V, Human Parvovirus B19 ND, Not determined NA, Not applicable |
||||
| Enveloped Viruses | ||||
| HIV-1 | ≥6.6 | ≥4.5 | ≥5.1 | ≥16.2 |
| BVDV | ≥9.2 | ≥4.7 | ≥5.3 | ≥19.2 |
| PRV | 6.3 | ≥6.5 | ≥7.1 | ≥19.9 |
| WNV | ≥7.0 | ND | ≥8.0 | ≥15.0 |
| Non-Enveloped Viruses | ||||
| HAV | ≥6.4 | 2.8 | ≥5.3 | ≥14.5 |
| CPV | 1.4 | 6.4 | ≥7.2 | ≥15.0 |
| B19V | 3.9 | ND | ND | NA |
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
C1 esterase inhibitor is a normal constituent of human plasma and belongs to the group of serine protease inhibitors (serpins) that includes antithrombin III, alpha1-protease inhibitor, alpha2-antiplasmin, and heparin cofactor II. As with the other inhibitors in this group, C1 esterase inhibitor has an important inhibiting potential on several of the major cascade systems of the human body, including the complement system, the intrinsic coagulation (contact) system, the fibrinolytic system, and the coagulation cascade. Regulation of these systems is performed through the formation of complexes between the proteinase and the inhibitor, resulting in inactivation of both and consumption of the C1 esterase inhibitor.
C1 esterase inhibitor, which is usually activated during the inflammatory process, inactivates its substrate by covalently binding to the reactive site. C1 esterase inhibitor is the only known inhibitor for the subcomponent of the complement component 1 (C1r), C1s, coagulation factor XIIa, and kallikrein. Additionally, C1 esterase inhibitor is the main inhibitor for coagulation factor XIa of the intrinsic coagulation cascade.
HAE patients have low levels of endogenous or functional C1 esterase inhibitor. Although the events that induce attacks of angioedema in HAE patients are not well defined, it has been postulated that increased vascular permeability and the clinical manifestation of HAE attacks may be primarily mediated through contact system activation. Suppression of contact system activation by C1 esterase inhibitor through the inactivation of plasma kallikrein and factor XIIa is thought to modulate this vascular permeability by preventing the generation of bradykinin.5
Administration of Berinert to patients with C1 esterase inhibitor deficiency replaces the missing or malfunctioning protein in patients. The plasma concentration of C1 esterase inhibitor in healthy volunteers is approximately 270 mg/L.6
12.3 Pharmacokinetics
The pharmacokinetics of Berinert were evaluated in an open-label, uncontrolled, single-center study in 40 subjects (35 adults and 5 children under 16 years of age) with either mild or severe HAE. All subjects received a single intravenous injection of Berinert ranging from 500 units to 1500 units. Blood samples were taken during an attack-free period at baseline and for up to 72 hours after drug administration. Pharmacokinetic parameters were estimated using non-compartmental analysis (with or without baseline adjustment). Table 6 summarizes the pharmacokinetic parameters in 35 adult subjects with HAE.
| Parameters | Unadjusted for baseline | Adjusted for baseline |
|---|---|---|
| AUC: Area under the curve CL: Clearance Vss: Volume steady state MRT: Mean residence time |
||
|
||
| AUC(0-t) (hr × IU/mL)* | 27.5 ± 8.5 (15.7-44.7) | 12.8 ± 6.7 (3.9-34.7) |
| CL (mL/hr/kg) | 0.60 ± 0.17 (0.34-0.96) | 1.44 ± 0.67 (0.43-3.85) |
| Vss (mL/kg) | 18.6 ± 4.9 (11.1-27.6) | 35.4 ± 10.5 (14.1-56.1) |
| Half-life (hrs) | 21.9 ± 1.7 (16.5-24.4) | 18.4 ± 3.5 (7.4-22.8) |
| MRT (hrs) | 31.5 ± 2.4 (23.7-35.2) | 26.4 ± 5.0 (10.7-33.0) |
Table 7 summarizes the pharmacokinetic parameters in 5 pediatric subjects (ages 6 through 13) with HAE. Based on adjusted baseline, compared to adults, the half-life of Berinert was shorter and clearance was faster in this limited cohort of children. However, the clinical implication of this difference is not known.
| Parameters | Unadjusted for baseline | Adjusted for baseline |
|---|---|---|
| AUC: Area under the curve CL: Clearance Vss: Volume steady state MRT: Mean residence time |
||
|
||
| AUC(0-t) (hr × IU/mL)* | 25.45 ± 5.8 (16.8-31.7) | 9.78 ± 4.37 (4.1-15.2) |
| CL (mL/hr/kg) | 0.62 ± 0.17 (0.47-0.89) | 1.9 ± 1.1 (0.98-3.69) |
| Vss (mL/kg) | 19.8 ± 4.0 (16.7-26.1) | 38.8 ± 8.9 (31.9-54.0) |
| Half-life (hrs) | 22.4 ± 1.6 (20.3-24.4) | 16.7 ± 5.8 (7.4-22.5) |
| MRT (hrs) | 32.3 ± 2.3 (29.3-35.2) | 24.0 ± 8.3 (10.7-32.4) |
Studies have not been conducted to evaluate the pharmacokinetics of Berinert in special patient populations identified by gender, race, geriatric age, or the presence of renal or hepatic impairment.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been completed to evaluate the effects of Berinert on carcinogenesis, mutagenesis, and impairment of fertility.
13.2 Animal Toxicology and/or Pharmacology
Acute intravenous toxicity of Berinert was performed in mice at 1500, 3000, and 6000 units/kg and in rats at 1000, 2000, and 3000 units/kg. Berinert was well tolerated and no signs of toxicity were observed up to the highest dose administered.
Repeat intravenous dose toxicity was studied in a 14-day repeat dose study in rats at doses of 20, 60, and 200 units/kg/day. Berinert was well tolerated and no toxicity was observed up to the highest dose administered. No antibody response against C1 esterase inhibitor could be demonstrated in this study after multiple dosing with Berinert.
In a safety pharmacology study, Berinert was administered to beagle dogs intravenously at a cumulative dose of 3500 units/kg. No adverse effects were seen on the cardiovascular and respiratory system. There was a drop in body temperature, reduced coagulation time, and a decrease in thrombocyte aggregation.
Local intravenous tolerance of Berinert was evaluated in rabbits at 1500 units. No pathological changes were noted at the time of injection or during the following 24 hours. No pathological signs were noted during necropsy.
Thrombotic events have been reported in association with C1 esterase inhibitor products when used off-label and at higher than labeled doses1 (see Overdosage [10]). Animal studies have confirmed the risk of thrombosis from intravenous administration of C1 esterase inhibitor products.2
14 CLINICAL STUDIES
The safety and efficacy of Berinert in the treatment of acute abdominal or facial attacks in subjects with hereditary angioedema were demonstrated in a placebo-controlled, double-blind, prospective, multinational, randomized, parallel-group, dose-finding, three-arm, clinical study, referred to as the randomized clinical trial (RCT). The RCT assessed the efficacy and safety of Berinert in 124 adult and pediatric subjects with C1 esterase inhibitor deficiency who were experiencing an acute moderate to severe attack of abdominal or facial HAE. Subjects ranged in age from six to 72 years of age; 67.7% were female and 32.3% were male; and approximately 90% were Caucasian.
The study objectives were to evaluate whether Berinert shortens the time to onset of relief of symptoms of an abdominal or facial attack compared to placebo and to compare the efficacy of two different doses of Berinert. The time to onset of relief of symptoms was determined by the subject's response to a standard question posed at appropriate time intervals for as long as 24 hours after start of treatment, taking into account all single HAE symptoms. In addition the severity of the single HAE symptoms was assessed over time.
Subjects were randomized to receive a single 10 unit/kg body weight dose of Berinert (39 subjects), a single 20 unit/kg dose of Berinert (43 subjects), or a single dose of placebo (42 subjects) by slow intravenous infusion (recommended to be given at a rate of approximately 4 mL per minute) within 5 hours of an HAE attack. At least 70% of the subjects in each treatment group were required to be experiencing an abdominal attack.
If a subject experienced no relief or insufficient relief of symptoms by 4 hours after infusion, investigators had the option to administer a second infusion of Berinert (20 units/kg for the placebo group, 10 units/kg for the 10 units/kg group), or placebo (for the 20 units/kg group). This masked (blinded) "rescue study medication" was administered to subjects and they were then followed until complete resolution of symptoms was achieved. Adverse events were collected for up to 7 to 9 days following the initial administration of Berinert or placebo.
In the rare case that a subject developed life-threatening laryngeal edema after inclusion into the study, immediate start of open-label treatment with a 20 unit/kg body weight dose of Berinert was allowed.
All subjects who received confounding medication (rescue medication) before symptom relief were regarded as "non-responders." Therefore, time to onset of symptom relief was set at 24 hours if a subject received any rescue medication (ie, rescue study medication, narcotic analgesics, non-narcotic analgesics, anti-emetics, open-label C1 inhibitor, androgens at increased dose, or fresh frozen plasma) between 5 hours before administration of blinded study medication until time to onset of relief.
For the trial to be considered successful, the study protocol specified the following criteria for the differences between the Berinert 20 units/kg and the placebo group:
- The time to onset of relief of symptoms of the HAE attack had to achieve a one-sided p-value of less than 0.0249 for the final analysis, and at least one of the following criteria had to demonstrate a trend in favor of Berinert with a one-sided p-value of less than 0.1:
- The proportion of subjects with increased intensity of clinical HAE symptoms between 2 and 4 hours after start of treatment with study medication compared to baseline, or
- The number of vomiting episodes within 4 hours after start of study treatment.
Subjects treated with 20 units/kg body weight of Berinert experienced a significant reduction (p=0.0016; "Wilcoxon Rank Sum test") in time to onset of relief from symptoms of an HAE attack as compared to placebo (median of 48 minutes for Berinert 20 units/kg body weight, as compared to a median of >4 hours for placebo). The time to onset of relief from symptoms of an HAE attack for subjects in the 10 unit/kg dose of Berinert was not statistically significantly different from that of subjects in the placebo group.
Figure 9 is a Kaplan-Meier curve showing the percentage of subjects reporting onset of relief of HAE attack symptoms as a function of time. Individual time points beyond 4 hours are not presented on the graph, because the protocol permitted blinded rescue medication, analgesics, and/or anti-emetics to be administered starting 4 hours after randomized blinded study medication had been administered.
|
| Figure 9: Time to Onset of Symptom Relief With Imputation to >4 Hours for Subjects Who Received any Rescue Medication* or Non-narcotic Analgesics Before Start of Relief |
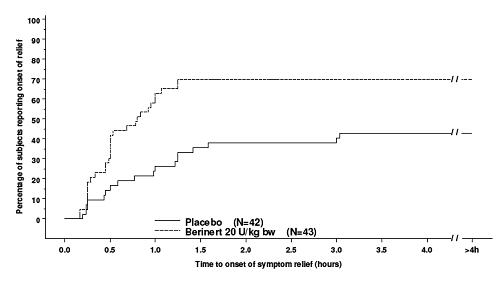 |
In addition, the efficacy of Berinert 20 units/kg body weight could be confirmed by observing a reduction in the intensity of single HAE symptoms at an earlier time compared to placebo. For abdominal attacks Figure 10a shows the time to start of relief of the last symptom to improve that was already present at baseline. Pre-defined abdominal HAE symptoms included pain, nausea, vomiting, cramps and diarrhea. Figure 10b shows the respective time to start of relief of the first symptom to improve that was already present at baseline.
|
| Figure 10a: Time to Start of Relief of the Last Symptom to Improve (Abdominal Attacks) with Imputation to >4 Hours for Subjects Who Received any Rescue Medication* Before Start of Relief |
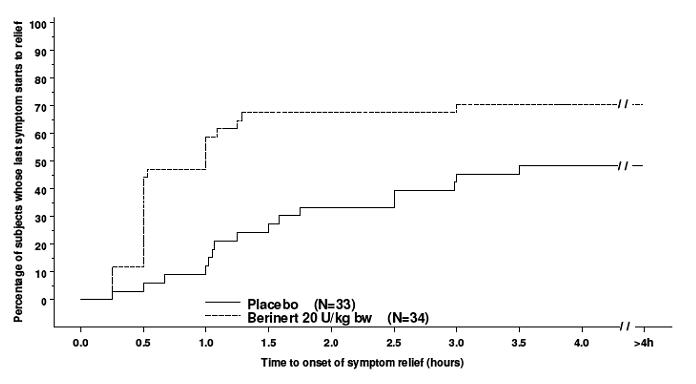 |
|
| Figure 10b: Time to Start of Relief of the First Symptom to Improve (Abdominal Attacks) With Imputation to >4 Hours for Subjects Who Received Any Rescue Medication* Before Start of Relief |
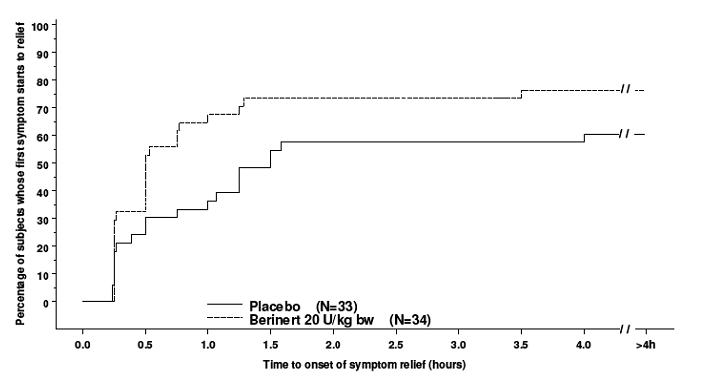 |
For facial attacks, single HAE symptoms were recorded. In addition, photos were taken at pre-determined time points and assessed by the members of an independent Data Safety Monitoring Board (DSMB), who were blinded as to treatment, center and other outcome measures. The change in the severity of the edema when compared to baseline was assessed on a scale with outcomes "no change", "better", "worse" and "resolved". Figure 11 shows the time to start of relief from serial facial photographs by DSMB assessment.
|
| Figure 11: Time to Start of Relief From Serial Facial Photographs* |
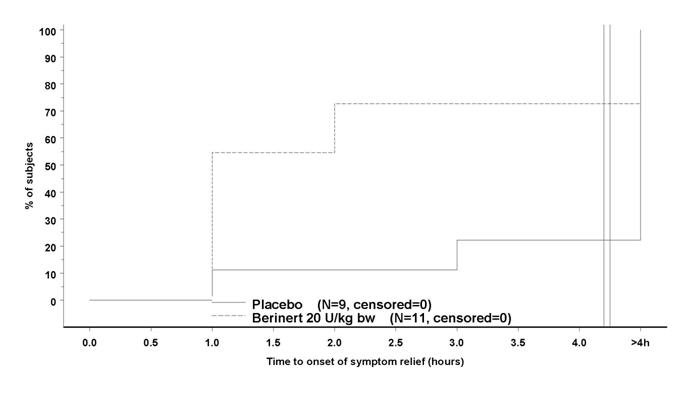 |
Table 8 compares additional endpoints, including changes in HAE symptoms and use of rescue medication in subjects receiving Berinert at 20 units/kg body weight and placebo.
| Additional Endpoints | Number (%) of Subjects Berinert 20 units/kg Body Weight Group (n=43) | Number (%) of Subjects Placebo Group (n=42) |
|---|---|---|
| Onset of symptom relief within 60 minutes after administration of study medication (post-hoc) | 27 (62.8%) | 11 (26.2%) |
| Onset of symptom relief within 4 hours after administration of study medication | 30 (69.8%) | 18 (42.9%) |
| Number of vomiting episodes within 4 hours after start of study treatment* | 6 episodes | 35 episodes |
| Worsened intensity of clinical HAE symptoms between 2 and 4 hours after administration of study medication compared to baseline† | 0 (0%) | 12 (28.6%) |
| Number (percent) of combined abdominal and facial attack subjects receiving rescue study medication, analgesics, or anti-emetics at any time prior to initial relief of symptoms | 13 (30.2%) | 23 (54.8%) |
| At least one new HAE symptom not present at baseline and starting within 4 hours after administration of study medication | 2 (4.6%) | 6 (14.3%) |
Both the proportion of subjects with increased intensity of clinical HAE symptoms between 2 and 4 hours after start of treatment compared to baseline, and the number of vomiting episodes within 4 hours after start of study treatment demonstrated trends in favor of Berinert in comparison to placebo (p-values <0.1). Tables 9 through 12 present additional information regarding responses to treatment.
| Attack Type | Berinert 20 units/kg Body Weight (Abdominal Subjects =34) (Facial Subjects = 9) (Other subjects = 0) | Placebo Group (Abdominal Subjects = 33) (Facial Subjects = 8) (Other subjects = 1)* |
|---|---|---|
|
||
| Abdominal | 24 (70.6%) | 15 (45.5%) |
| Facial | 6 (66.7%) | 3 (37.5%) |
| Attack Type | Berinert 20 units/kg Body Weight (Abdominal Subjects = 34) (Facial Subjects = 9) | Placebo Group (Abdominal Subjects = 33) (Facial Subjects = 8) |
|---|---|---|
| Abdominal | 33 (97.1%) | 29 (87.9%) |
| Facial | 6 (66.7%) | 4 (50%) |
| Attack Type | Berinert 20 units/kg Body Weight (Subjects = 9) | Placebo Group (Subjects = 8) |
|---|---|---|
|
||
| Facial | 7 (77.8%) | 2 (25%) |
| Attack Type | Berinert 20 U/kg Body Weight (Abdominal Subjects = 34) (Facial Subjects = 9) | Placebo Group (Abdominal Subjects = 33) (Facial Subjects = 8) |
|
|---|---|---|---|
| Abdominal | 7 (20.6%) | 17 (51.5%) | |
| Facial | 1 (11.1%) | 6 (75%) | |
No subjects treated with Berinert at 20 units/kg body weight reported worsening of symptoms at 4 hours after administration of study medication compared to baseline.
The study demonstrated that the Berinert 20 unit/kg body weight dose was significantly more efficacious than the Berinert 10 unit/kg body weight dose or placebo.
Open-Label Extension Study
Berinert was evaluated in a prospective, open-label, uncontrolled, multicenter extension study conducted at 10 centers in the US and Canada in subjects who had participated in the RCT study for the treatment of acute abdominal or facial attacks in subjects with hereditary angioedema.
The purpose of this ongoing extension study is to provide Berinert to subjects who had participated in the RCT study and who experienced any type of subsequent HAE attack (ie, abdominal, facial, peripheral, or laryngeal).
In a non-pre-specified interim safety analysis of the ongoing open-label extension study, a total of 56 subjects (19 males and 37 females, age range: 10 to 53 years) with 559 HAE attacks treated with 20 unit/kg body weight dose of Berinert per attack, were observed at the study site until onset of relief of HAE symptoms, and were followed up for adverse events for 7 to 9 days following treatment of each HAE attack (see Adverse Reactions, Clinical Trials Experience [6.1]). There were 49 subjects with abdominal attacks, 11 subjects with facial attacks, 28 subjects with peripheral attacks, and 12 subjects with laryngeal attacks.
15 REFERENCES
- German Medical Profession's Drugs Committee. Severe thrombus formation of Berinert® HS. Deutsches Ärzteblatt. 2000;97:B-864.
- Horstick, G et al. Application of C1-Esterase Inhibitor During Reperfusion of Ischemic Myocardium: Dose-Related Beneficial Versus Detrimental Effects. Circulation. 2002;104:3125-3131.
- Carrell RW, Boswell DR. Serpins: the superfamily of plasma serine proteinase inhibitors. In: Barrett A, Salvesen G, eds. Proteinase Inhibitors. Amsterdam: Elsevier. 1986;12:403-420.
- Harrison RA. Human C1 inhibitor: Improved isolation and preliminary structural characterization. Biochemistry 1983;22:5001-5007.
- Davis AE, The pathophysiology of hereditary angioedema. Clin Immunol. 2005; 114:3-9.
- Nuijens JH, Eerenberg-Belmer AJM, Huijbregts CCM, et al. Proteolytic inactivation of plasma C1 inhibitor in sepsis. J Clin Invest. 1989;84:443-450.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
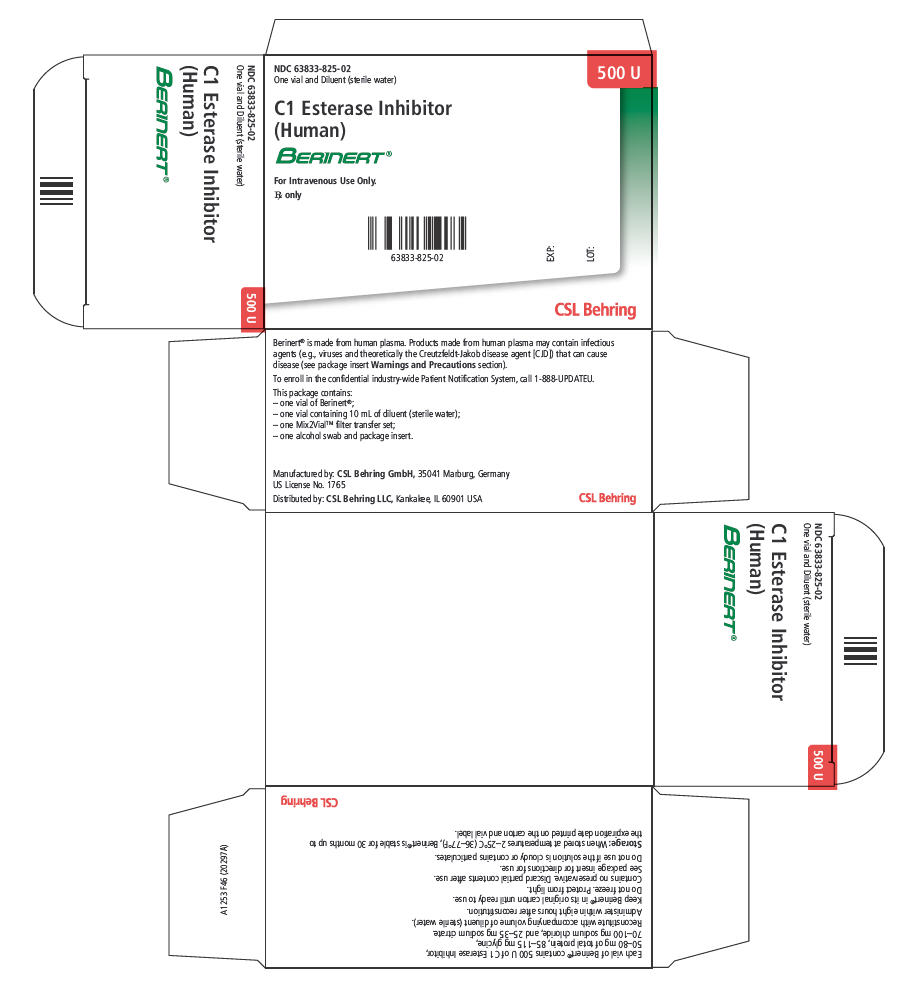
Berinert is supplied in a single-use vial. Each carton contains a 500 unit vial of Berinert for reconstitution with 10 mL of diluent containing sterile water (meets USP chemistry requirements except for pH; pH 4.5-8.5). The components used in the packaging for Berinert are latex-free.
Each product package consists of the following:
| NDC Number | Component |
|---|---|
| 63833-825-02 | Carton (kit) containing one 500 unit vial of Berinert [NDC 63833-835-01], one 10 mL vial of diluent (sterile water) [NDC 63833-765-15], one Mix2Vial filter transfer set, and one alcohol swab. |
17 PATIENT COUNSELING INFORMATION
Inform patients to immediately report the following to their physician:
- Signs and symptoms of allergic hypersensitivity reactions, such as hives, urticaria, tightness of the chest, wheezing, hypotension and/or anaphylaxis experienced during or after injection of Berinert (see WARNINGS AND PRECAUTIONS/Hypersensitivity [5.1])
- Signs and symptoms of thrombosis, such as new onset swelling and pain in the limbs or abdomen, new onset chest pain, shortness of breath, loss of sensation or motor power, or altered consciousness, vision, or speech (see WARNINGS AND PRECAUTIONS/Thrombotic Events [5.2])
Advise female patients to notify their physician if they become pregnant or intend to become pregnant during the treatment of acute abdominal or facial attacks of HAE with Berinert.
Advise patients to notify their physician if they are breastfeeding or plan to breastfeed.
Advise patients to consult with their healthcare professional prior to travel.
Advise patients that, because Berinert is made from human blood, it may carry a risk of transmitting infectious agents, eg, viruses, and, theoretically, the Creutzfeldt-Jakob (CJD) agent (see WARNINGS AND PRECAUTIONS/Transmission of Infectious Agents [5.3] and Description [11]). Inform patients of the risks and benefits of Berinert before prescribing or administering it to the patient.
Berinert (BEAR-ĭ-nert)
C1 Esterase Inhibitor (Human)
Freeze-Dried Powder for Reconstitution
This leaflet summarizes important information about BERINERT. Please read it carefully before using Berinert and each time you get a refill. There may be new information provided. This information does not take the place of talking with your healthcare provider, and it does not include all of the important information about BERINERT. If you have any questions after reading this, ask your healthcare provider.
What is BERINERT?
BERINERT is an injectable medicine used to treat swelling and/or painful attacks in adults and adolescents with hereditary angioedema (HAE). HAE is caused by the poor functioning or lack of a protein called C1 that is present in your blood and helps control inflammation (swelling) and parts of the immune system. Berinert contains C1 esterase inhibitor, a protein that helps control C1.
Who should not use BERINERT?
You should not use BERINERT if you have experienced life-threatening immediate hypersensitivity reactions, including anaphylaxis to the product.
What should I tell my healthcare provider before BERINERT is given?
Tell your healthcare provider about all of your medical conditions, including if you:
- Are pregnant or planning to become pregnant. It is not known if BERINERT can harm your unborn baby.
- Are breastfeeding or plan to breastfeed. It is not known if BERINERT passes into your milk and if it can harm your baby.
- Have a history of blood clotting problems. Blood clots (thrombosis) have occurred in patients receiving large amounts of Berinert. Very high doses of C1 esterase inhibitor could increase the risk of blood clots.
Tell your healthcare provider and pharmacist about all of the medicines you take, including all prescription and non-prescription medicines such as over-the-counter medicines, supplements, or herbal remedies.
How is BERINERT given?
Your healthcare provider will infuse BERINERT into your vein (intravenous injection). Before infusing, he or she must dissolve the BERINERT powder using the sterile water provided. Your healthcare provider will prescribe the dose that you should be given.
What are the possible side effects of BERINERT?
Allergic reactions may occur with BERINERT. Call your healthcare provider or the emergency department right away if you have any of the following symptoms after using BERINERT:
- wheezing
- difficulty breathing
- chest tightness
- turning blue (look at lips and gums)
- fast heartbeat
- swelling of the face
- faintness
- rash
- hives
Signs of a blood clot include:
- new onset of swelling and pain in the limbs or abdomen
- new onset of chest pain
- shortness of breath
- loss of sensation or control of muscles/muscle weakness on one side of the body
- altered consciousness, vision, or speech.
In clinical studies, the most severe side effect reported in subjects who received BERINERT was an increase in the severity of pain associated with HAE.
Other side effects patients experienced during clinical research studies include:
- subsequent HAE attack
- headache
- abdominal pain
- nausea
- muscle spasms
- pain
- diarrhea
- vomiting
Because BERINERT is made from human blood, it may carry a risk of transmitting infectious agents, eg, viruses, and, theoretically, the Creutzfeldt-Jakob (CJD) agent.
These are not all the possible side effects of BERINERT.
Tell your healthcare provider about any side effect that bothers you or that does not go away. You can also report side effects to the FDA at 1-800-FDA-1088.
What else should I know about BERINERT?
Medicines are sometimes prescribed for purposes other than those listed here. Do not use BERINERT for a condition for which it is not prescribed. Do not share BERINERT with other people, even if they have the same symptoms that you have.
This leaflet summarizes the most important information about BERINERT. If you would like more information, talk to your healthcare provider. You can ask your healthcare provider or pharmacist for information about BERINERT that was written for healthcare professionals.
Talk to your healthcare provider before traveling.
This Patient Package Insert has been approved by the US Food and Drug Administration.
Manufactured by:
CSL Behring GmbH
35041 Marburg, Germany
US License No. 1765
Distributed by:
CSL Behring LLC
Kankakee, IL 60901 USA
Mix2Vial is a trademark of West Pharmaceuticals Services, Inc.
PRINCIPAL DISPLAY PANEL
NDC 63833-825-02
One vial and Diluent (sterile water)
500 U
C1 Esterase Inhibitor
(Human)
BERINERT®
For Intravenous Use Only.
Rx only
EXP:
LOT:
CSL Behring
| BERINERT
human c1-esterase inhibitor kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA125287 | 07/29/2011 | |
| Labeler - CSL Behring GmbH (326530474) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| CSL Behring GmbH | 326530474 | MANUFACTURE | |
Revised: 08/2011 CSL Behring GmbH