DETROL LA
-
tolterodine tartrate capsule, extended release
Cardinal Health
----------
DESCRIPTION
DETROL LA Capsules contain tolterodine tartrate. The active moiety, tolterodine, is a muscarinic receptor antagonist. The chemical name of tolterodine tartrate is (R)-N,N-diisopropyl-3-(2-hydroxy-5-methylphenyl)-3-phenylpropanamine L-hydrogen tartrate. The empirical formula of tolterodine tartrate is C26H37NO7, and its molecular weight is 475.6. The structural formula of tolterodine tartrate is represented below.
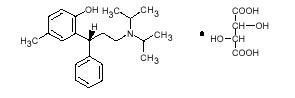
Tolterodine tartrate is a white, crystalline powder. The pKa value is 9.87 and the solubility in water is 12 mg/mL. It is soluble in methanol, slightly soluble in ethanol, and practically insoluble in toluene. The partition coefficient (Log D) between n-octanol and water is 1.83 at pH 7.3.
DETROL LA for oral administration contains 2 mg or 4 mg of tolterodine tartrate. Inactive ingredients are sucrose, starch, hypromellose, ethylcellulose, medium chain triglycerides, oleic acid, gelatin, and FD&C Blue #2. The 2 mg capsules also contain yellow iron oxide. Both capsule strengths are imprinted with a pharmaceutical grade printing ink that contains shellac glaze, titanium dioxide, propylene glycol, and simethicone.
CLINICAL PHARMACOLOGY
Tolterodine is a competitive muscarinic receptor antagonist. Both urinary bladder contraction and salivation are mediated via cholinergic muscarinic receptors.
After oral administration, tolterodine is metabolized in the liver, resulting in the formation of the 5-hydroxymethyl derivative, a major pharmacologically active metabolite. The 5-hydroxymethyl metabolite, which exhibits an antimuscarinic activity similar to that of tolterodine, contributes significantly to the therapeutic effect. Both tolterodine and the 5-hydroxymethyl metabolite exhibit a high specificity for muscarinic receptors, since both show negligible activity or affinity for other neurotransmitter receptors and other potential cellular targets, such as calcium channels.
Tolterodine has a pronounced effect on bladder function. Effects on urodynamic parameters before and 1 and 5 hours after a single 6.4-mg dose of tolterodine immediate release were determined in healthy volunteers. The main effects of tolterodine at 1 and 5 hours were an increase in residual urine, reflecting an incomplete emptying of the bladder, and a decrease in detrusor pressure. These findings are consistent with an antimuscarinic action on the lower urinary tract.
Pharmacokinetics
Absorption
In a study with 14C-tolterodine solution in healthy volunteers who received a 5-mg oral dose, at least 77% of the radiolabeled dose was absorbed. Cmax and area under the concentration-time curve (AUC) determined after dosage of tolterodine immediate release are dose-proportional over the range of 1 to 4 mg. Based on the sum of unbound serum concentrations of tolterodine and the 5-hydroxymethyl metabolite ("active moiety"), the AUC of tolterodine extended release 4 mg daily is equivalent to tolterodine immediate release 4 mg (2 mg bid). Cmax and Cmin levels of tolterodine extended release are about 75% and 150% of tolterodine immediate release, respectively. Maximum serum concentrations of tolterodine extended release are observed 2 to 6 hours after dose administration.
Distribution
Tolterodine is highly bound to plasma proteins, primarily α1-acid glycoprotein. Unbound concentrations of tolterodine average 3.7% ± 0.13% over the concentration range achieved in clinical studies. The 5-hydroxymethyl metabolite is not extensively protein bound, with unbound fraction concentrations averaging 36% ± 4.0%. The blood to serum ratio of tolterodine and the 5-hydroxymethyl metabolite averages 0.6 and 0.8, respectively, indicating that these compounds do not distribute extensively into erythrocytes. The volume of distribution of tolterodine following administration of a 1.28-mg intravenous dose is 113 ± 26.7 L.
Metabolism
Tolterodine is extensively metabolized by the liver following oral dosing. The primary metabolic route involves the oxidation of the 5-methyl group and is mediated by the cytochrome P450 2D6 (CYP2D6) and leads to the formation of a pharmacologically active 5-hydroxymethyl metabolite. Further metabolism leads to formation of the 5-carboxylic acid and N-dealkylated 5-carboxylic acid metabolites, which account for 51% ± 14% and 29% ± 6.3% of the metabolites recovered in the urine, respectively.
Variability in Metabolism
A subset (about 7%) of the Caucasian population is devoid of CYP2D6, the enzyme responsible for the formation of the 5-hydroxymethyl metabolite of tolterodine. The identified pathway of metabolism for these individuals ("poor metabolizers") is dealkylation via cytochrome P450 3A4 (CYP3A4) to N-dealkylated tolterodine. The remainder of the population is referred to as "extensive metabolizers." Pharmacokinetic studies revealed that tolterodine is metabolized at a slower rate in poor metabolizers than in extensive metabolizers; this results in significantly higher serum concentrations of tolterodine and in negligible concentrations of the 5-hydroxymethyl metabolite.
Excretion
Following administration of a 5-mg oral dose of 14C-tolterodine solution to healthy volunteers, 77% of radioactivity was recovered in urine and 17% was recovered in feces in 7 days. Less than 1% (< 2.5% in poor metabolizers) of the dose was recovered as intact tolterodine, and 5% to 14% (<1% in poor metabolizers) was recovered as the active 5-hydroxymethyl metabolite.
A summary of mean (± standard deviation) pharmacokinetic parameters of tolterodine extended release and the 5-hydroxymethyl metabolite in extensive (EM) and poor (PM) metabolizers is provided in Table 1. These data were obtained following single and multiple doses of tolterodine extended release administered daily to 17 healthy male volunteers (13 EM, 4 PM).
| Tolterodine | 5-hydroxymethyl metabolite | |||||||
| tmax*
(h) | Cmax
(µg/L) | Cavg
(µg/L) | t½ (h) | tmax*
(h) | Cmax
(µg/L) | Cavg
(µg/L) | t½ (h) |
|
| Cmax = Maximum serum concentration; tmax = Time of occurrence of Cmax; | ||||||||
| C avg = Average serum concentration; t1/2 = Terminal elimination half-life. | ||||||||
| Single dose 4 mg† EM | 4(2–6) | 1.3(0.8) | 0.8(0.57) | 8.4(3.2) | 4(3–6) | 1.6 (0.5) | 1.0(0.32) | 8.8(5.9) |
| Multiple dose 4 mg EM PM | 4(2–6) 4(3–6) | 3.4(4.9) 19(16) | 1.7(2.8) 13(11) | 6.9(3.5) 18(16) | 4(2–6) —‡ | 2.7(0.90) — | 1.4(0.6) — | 9.9(4.0) — |
Pharmacokinetics in Special Populations
Age
In Phase 1, multiple-dose studies in which tolterodine immediate release 4 mg (2 mg bid) was administered, serum concentrations of tolterodine and of the 5-hydroxymethyl metabolite were similar in healthy elderly volunteers (aged 64 through 80 years) and healthy young volunteers (aged less than 40 years). In another Phase 1 study, elderly volunteers (aged 71 through 81 years) were given tolterodine immediate release 2 or 4 mg (1 or 2 mg bid). Mean serum concentrations of tolterodine and the 5-hydroxymethyl metabolite in these elderly volunteers were approximately 20% and 50% higher, respectively, than reported in young healthy volunteers. However, no overall differences were observed in safety between older and younger patients on tolterodine in the Phase 3, 12-week, controlled clinical studies; therefore, no tolterodine dosage adjustment for elderly patients is recommended (see PRECAUTIONS, Geriatric Use).
Pediatric
Efficacy in the pediatric population has not been demonstrated.
The pharmacokinetics of tolterodine extended release capsules have been evaluated in pediatric patients ranging in age from 11–15 years. The dose-plasma concentration relationship was linear over the range of doses assessed. Parent/metabolite ratios differed according to CYP2D6 metabolizer status: EMs had low serum concentrations of tolterodine and high concentrations of the active 5-hydroxymethyl metabolite, while PMs had high concentrations of tolterodine and negligible active metabolite concentrations.
Gender
The pharmacokinetics of tolterodine immediate release and the 5-hydroxymethyl metabolite are not influenced by gender. Mean Cmax of tolterodine immediate release (1.6 µg/L in males versus 2.2 µg/L in females) and the active 5-hydroxymethyl metabolite (2.2 µg/L in males versus 2.5 µg/L in females) are similar in males and females who were administered tolterodine immediate release 2 mg. Mean AUC values of tolterodine (6.7 µg∙h/L in males versus 7.8 µg∙h/L in females) and the 5-hydroxymethyl metabolite (10 µg∙h/L in males versus 11 µg∙h/L in females) are also similar. The elimination half-life of tolterodine immediate release for both males and females is 2.4 hours, and the half-life of the 5-hydroxymethyl metabolite is 3.0 hours in females and 3.3 hours in males.
Renal Insufficiency
Renal impairment can significantly alter the disposition of tolterodine immediate release and its metabolites. In a study conducted in patients with creatinine clearance between 10 and 30 mL/min, tolterodine immediate release and the 5-hydroxymethyl metabolite levels were approximately 2–3 fold higher in patients with renal impairment than in healthy volunteers. Exposure levels of other metabolites of tolterodine (e.g., tolterodine acid, N-dealkylated tolterodine acid, N-dealkylated tolterodine and N-dealkylated hydroxy tolterodine) were significantly higher (10–30 fold) in renally impaired patients as compared to the healthy volunteers. The recommended dose for patients with significantly reduced renal function is tolterodine 2 mg daily (see PRECAUTIONS, General and DOSAGE AND ADMINISTRATION).
Hepatic Insufficiency
Liver impairment can significantly alter the disposition of tolterodine immediate release. In a study of tolterodine immediate release conducted in cirrhotic patients, the elimination half-life of tolterodine immediate release was longer in cirrhotic patients (mean, 7.8 hours) than in healthy, young, and elderly volunteers (mean, 2 to 4 hours). The clearance of orally administered tolterodine immediate release was substantially lower in cirrhotic patients (1.0 ± 1.7 L/h/kg) than in the healthy volunteers (5.7 ± 3.8 L/h/kg). The recommended dose for patients with significantly reduced hepatic function is tolterodine 2 mg daily (see PRECAUTIONS, General and DOSAGE AND ADMINISTRATION).
Drug-Drug Interactions
Fluoxetine
Fluoxetine is a selective serotonin reuptake inhibitor and a potent inhibitor of CYP2D6 activity. In a study to assess the effect of fluoxetine on the pharmacokinetics of tolterodine immediate release and its metabolites, it was observed that fluoxetine significantly inhibited the metabolism of tolterodine immediate release in extensive metabolizers, resulting in a 4.8-fold increase in tolterodine AUC. There was a 52% decrease in Cmax and a 20% decrease in AUC of the 5-hydroxymethyl metabolite. Fluoxetine thus alters the pharmacokinetics in patients who would otherwise be extensive metabolizers of tolterodine immediate release to resemble the pharmacokinetic profile in poor metabolizers. The sums of unbound serum concentrations of tolterodine immediate release and the 5-hydroxymethyl metabolite are only 25% higher during the interaction. No dose adjustment is required when tolterodine and fluoxetine are coadministered.
Other Drugs Metabolized by Cytochrome P450 Isoenzymes
Tolterodine immediate release does not cause clinically significant interactions with other drugs metabolized by the major drug metabolizing CYP enzymes. In vivo drug-interaction data show that tolterodine immediate release does not result in clinically relevant inhibition of CYP1A2, 2D6, 2C9, 2C19, or 3A4 as evidenced by lack of influence on the marker drugs caffeine, debrisoquine, S-warfarin, and omeprazole. In vitro data show that tolterodine immediate release is a competitive inhibitor of CYP2D6 at high concentrations (Ki 1.05 µM), while tolterodine immediate release as well as the 5-hydroxymethyl metabolite are devoid of any significant inhibitory potential regarding the other isoenzymes.
CYP3A4 Inhibitors
The effect of a 200-mg daily dose of ketoconazole on the pharmacokinetics of tolterodine immediate release was studied in 8 healthy volunteers, all of whom were poor metabolizers (see Pharmacokinetics, Variability in Metabolism for discussion of poor metabolizers). In the presence of ketoconazole, the mean Cmax and AUC of tolterodine increased by 2 and 2.5 fold, respectively. Based on these findings, other potent CYP3A4 inhibitors such as other azole antifungals (eg, itraconazole, miconazole) or macrolide antibiotics (eg, erythromycin, clarithromycin) or cyclosporine or vinblastine may also lead to increases of tolterodine plasma concentrations (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Warfarin
In healthy volunteers, coadministration of tolterodine immediate release 4 mg (2 mg bid) for 7 days and a single dose of warfarin 25 mg on day 4 had no effect on prothrombin time, Factor VII suppression, or on the pharmacokinetics of warfarin.
Oral Contraceptives
Tolterodine immediate release 4 mg (2 mg bid) had no effect on the pharmacokinetics of an oral contraceptive (ethinyl estradiol 30 µg/levo-norgestrel 150 µg) as evidenced by the monitoring of ethinyl estradiol and levo-norgestrel over a 2-month period in healthy female volunteers.
Diuretics
Coadministration of tolterodine immediate release up to 8 mg (4 mg bid) for up to 12 weeks with diuretic agents, such as indapamide, hydrochlorothiazide, triamterene, bendroflumethiazide, chlorothiazide, methylchlorothiazide, or furosemide, did not cause any adverse electrocardiographic (ECG) effects.
Cardiac Electrophysiology
The effect of 2 mg BID and 4 mg BID of Detrol immediate release (tolterodine IR) tablets on the QT interval was evaluated in a 4-way crossover, double-blind, placebo- and active-controlled (moxifloxacin 400 mg QD) study in healthy male (N=25) and female (N=23) volunteers aged 18–55 years. Study subjects [approximately equal representation of CYP2D6 extensive metabolizers (EMs) and poor metabolizers (PMs)] completed sequential 4-day periods of dosing with moxifloxacin 400 mg QD, tolterodine 2 mg BID, tolterodine 4 mg BID, and placebo. The 4 mg BID dose of tolterodine IR (two times the highest recommended dose) was chosen because this dose results in tolterodine exposure similar to that observed upon coadministration of tolterodine 2 mg BID with potent CYP3A4 inhibitors in patients who are CYP2D6 poor metabolizers (see PRECAUTIONS, Drug Interactions). QT interval was measured over a 12-hour period following dosing, including the time of peak plasma concentration (Tmax) of tolterodine and at steady state (Day 4 of dosing).
Table 2 summarizes the mean change from baseline to steady state in corrected QT interval (QTc) relative to placebo at the time of peak tolterodine (1 hour) and moxifloxacin (2 hour) concentrations. Both Fridericia's (QTcF) and a population-specific (QTcP) method were used to correct QT interval for heart rate. No single QT correction method is known to be more valid than others. QT interval was measured manually and by machine, and data from both are presented. The mean increase of heart rate associated with a 4 mg/day dose of tolterodine in this study was 2.0 beats/minute and 6.3 beats/minute with 8 mg/day tolterodine. The change in heart rate with moxifloxacin was 0.5 beats/minute.
| Drug/Dose | N | QTcF (msec) (manual) | QTcF (msec) (machine) | QTcP (msec) (manual) | QTcP (msec) (machine) |
| Tolterodine 2 mg BID* | 48 | 5.01 (0.28, 9.74) | 1.16 (-2.99, 5.30) | 4.45 (-0.37, 9.26) | 2.00 (-1.81, 5.81) |
| Tolterodine 4 mg BID* | 48 | 11.84 (7.11, 16.58) | 5.63 (1.48, 9.77) | 10.31 (5.49, 15.12) | 8.34 (4.53, 12.15) |
| Moxifloxacin 400 mg QD † | 45 | 19.26‡
(15.49, 23.03) | 8.90 (4.77, 13.03) | 19.10‡
(15.32, 22.89) | 9.29 (5.34, 13.24) |
The reason for the difference between machine and manual read of QT interval is unclear.
The QT effect of tolterodine immediate release tablets appeared greater for 8 mg/day (two times the therapeutic dose) compared to 4 mg/day. The effect of tolterodine 8 mg/day was not as large as that observed after four days of therapeutic dosing with the active control moxifloxacin. However, the confidence intervals overlapped.
Tolterodine's effect on QT interval was found to correlate with plasma concentration of tolterodine. There appeared to be a greater QTc interval increase in CYP2D6 poor metabolizers than in CYP2D6 extensive metabolizers after tolterodine treatment in this study.
This study was not designed to make direct statistical comparisons between drugs or dose levels. There has been no association of Torsade de Pointes in the international post-marketing experience with DETROL or DETROL LA (see PRECAUTIONS, Patients with Congenital or Acquired QT Prolongation).
CLINICAL STUDIES
DETROL LA Capsules 2 mg were evaluated in 29 patients in a Phase 2 dose-effect study. DETROL LA 4 mg was evaluated for the treatment of overactive bladder with symptoms of urge urinary incontinence and frequency in a randomized, placebo-controlled, multicenter, double-blind, Phase 3, 12-week study. A total of 507 patients received DETROL LA 4 mg once daily in the morning and 508 received placebo. The majority of patients were Caucasian (95%) and female (81%), with a mean age of 61 years (range, 20 to 93 years). In the study, 642 patients (42%) were 65 to 93 years of age. The study included patients known to be responsive to tolterodine immediate release and other anticholinergic medications, however, 47% of patients never received prior pharmacotherapy for overactive bladder. At study entry, 97% of patients had at least 5 urge incontinence episodes per week and 91% of patients had 8 or more micturitions per day. The primary efficacy endpoint was change in mean number of incontinence episodes per week at week 12 from baseline. Secondary efficacy endpoints included change in mean number of micturitions per day and mean volume voided per micturition at week 12 from baseline.
| DETROL LA (n=507) | Placebo (n=508) † | Treatment Difference, vs. Placebo (95% Cl) |
|
| SD = Standard Deviation. | |||
| Number of incontinence episodes/ week Mean Baseline Mean Change from Baseline | 22.1 –11.8 (SD 17.8) | 23.3 –6.9 (SD 15.4) | -4.8 ‡
(–6.9, –2.8) |
| Number of micturitions/day Mean Baseline Mean Change from Baseline | 10.9 –1.8 (SD 3.4) | 11.3 –1.2 (SD 2.9) | -0.6 ‡
(–1.0, –0.2) |
| Volume Voided per micturition (mL) Mean Baseline Mean Change from Baseline | 141 34 (SD 51) | 136 14 (SD 41) | 20 ‡
(14, 26) |
INDICATIONS AND USAGE
DETROL LA Capsules are once-daily extended release capsules indicated for the treatment of overactive bladder with symptoms of urge urinary incontinence, urgency, and frequency.
CONTRAINDICATIONS
DETROL LA Capsules are contraindicated in patients with urinary retention, gastric retention, or uncontrolled narrow-angle glaucoma. DETROL LA is also contraindicated in patients who have demonstrated hypersensitivity to the drug or its ingredients.
PRECAUTIONS
General
Risk of Urinary Retention and Gastric Retention
DETROL LA Capsules should be administered with caution to patients with clinically significant bladder outflow obstruction because of the risk of urinary retention and to patients with gastrointestinal obstructive disorders, such as pyloric stenosis, because of the risk of gastric retention (see CONTRAINDICATIONS).
Decreased Gastrointestinal Motility
DETROL LA, like other antimuscarinic drugs, should be used with caution in patients with decreased gastrointestinal motility.
Controlled Narrow-Angle Glaucoma
DETROL LA should be used with caution in patients being treated for narrow-angle glaucoma.
Reduced Hepatic and Renal Function
For patients with significantly reduced hepatic function or renal function, the recommended dose for DETROL LA is 2 mg daily (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations).
Patients with Congenital or Acquired QT Prolongation
In a study of the effect of tolterodine immediate release tablets on the QT interval (see CLINICAL PHARMACOLOGY, Cardiac Electrophysiology), the effect on the QT interval appeared greater for 8 mg/day (two times the therapeutic dose) compared to 4 mg/day and was more pronounced in CYP2D6 poor metabolizers (PM) than extensive metabolizers (EMs). The effect of tolterodine 8 mg/day was not as large as that observed after four days of therapeutic dosing with the active control moxifloxacin. However, the confidence intervals overlapped. These observations should be considered in clinical decisions to prescribe DETROL LA for patients with a known history of QT prolongation or patients who are taking Class IA (e.g., quinidine, procainamide) or Class III (e.g., amiodarone, sotalol) antiarrhythmic medications (see PRECAUTIONS, Drug Interactions). There has been no association of Torsade de Pointes in the international post-marketing experience with DETROL or DETROL LA.
Information for Patients
Patients should be informed that antimuscarinic agents such as DETROL LA may produce the following effects: blurred vision, dizziness, or drowsiness. Patients should be advised to exercise caution in decisions to engage in potentially dangerous activities until the drug's effects have been determined.
Drug Interactions
CYP3A4 Inhibitors
Ketoconazole, an inhibitor of the drug metabolizing enzyme CYP3A4, significantly increased plasma concentrations of tolterodine when coadministered to subjects who were poor metabolizers (see CLINICAL PHARMACOLOGY, Variability in Metabolism and Drug-Drug Interactions). For patients receiving ketoconazole or other potent CYP3A4 inhibitors such as other azole antifungals (e.g., itraconazole, miconazole) or macrolide antibiotics (eg, erythromycin, clarithromycin) or cyclosporine or vinblastine, the recommended dose of DETROL LA is 2 mg daily (see DOSAGE AND ADMINISTRATION).
Drug-Laboratory-Test Interactions
Interactions between tolterodine and laboratory tests have not been studied.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with tolterodine immediate release were conducted in mice and rats. At the maximum tolerated dose in mice (30 mg/kg/day), female rats (20 mg/kg/day), and male rats (30 mg/kg/day), AUC values obtained for tolterodine were 355, 291, and 462 µg∙h/L, respectively. In comparison, the human AUC value for a 2-mg dose administered twice daily is estimated at 34 µg∙h/L. Thus, tolterodine exposure in the carcinogenicity studies was 9- to 14-fold higher than expected in humans. No increase in tumors was found in either mice or rats.
No mutagenic effects of tolterodine were detected in a battery of in vitro tests, including bacterial mutation assays (Ames test) in 4 strains of Salmonella typhimurium and in 2 strains of Escherichia coli, a gene mutation assay in L5178Y mouse lymphoma cells, and chromosomal aberration tests in human lymphocytes. Tolterodine was also negative in vivo in the bone marrow micronucleus test in the mouse.
In female mice treated for 2 weeks before mating and during gestation with 20 mg/kg/day (corresponding to AUC value of about 500 µg∙h/L), neither effects on reproductive performance or fertility were seen. Based on AUC values, the systemic exposure was about 15-fold higher in animals than in humans. In male mice, a dose of 30 mg/kg/day did not induce any adverse effects on fertility.
Pregnancy
Pregnancy Category C. At oral doses of 20 mg/kg/day (approximately 14 times the human exposure), no anomalies or malformations were observed in mice. When given at doses of 30 to 40 mg/kg/day, tolterodine has been shown to be embryolethal and reduce fetal weight, and increase the incidence of fetal abnormalities (cleft palate, digital abnormalities, intra-abdominal hemorrhage, and various skeletal abnormalities, primarily reduced ossification) in mice. At these doses, the AUC values were about 20- to 25-fold higher than in humans. Rabbits treated subcutaneously at a dose of 0.8 mg/kg/day achieved an AUC of 100 µg∙h/L, which is about 3-fold higher than that resulting from the human dose. This dose did not result in any embryotoxicity or teratogenicity. There are no studies of tolterodine in pregnant women. Therefore, DETROL LA should be used during pregnancy only if the potential benefit for the mother justifies the potential risk to the fetus.
Nursing Mothers
Tolterodine immediate release is excreted into the milk in mice. Offspring of female mice treated with tolterodine 20 mg/kg/day during the lactation period had slightly reduced body weight gain. The offspring regained the weight during the maturation phase. It is not known whether tolterodine is excreted in human milk; therefore, DETROL LA should not be administered during nursing. A decision should be made whether to discontinue nursing or to discontinue DETROL LA in nursing mothers.
Pediatric Use
Efficacy in the pediatric population has not been demonstrated.
A total of 710 pediatric patients (486 on DETROL LA, 224 on placebo) aged 5–10 with urinary frequency and urge incontinence were studied in two Phase 3 randomized, placebo-controlled, double-blind, 12-week studies. The percentage of patients with urinary tract infections was higher in patients treated with DETROL LA (6.6%) compared to patients who received placebo (4.5%). Aggressive, abnormal and hyperactive behavior and attention disorders occurred in 2.9% of children treated with DETROL LA compared to 0.9% of children treated with placebo.
Geriatric Use
No overall differences in safety were observed between the older and younger patients treated with tolterodine (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations).
ADVERSE REACTIONS
The Phase 2 and 3 clinical trial program for DETROL LA Capsules included 1073 patients who were treated with DETROL LA (n=537) or placebo (n=536). The patients were treated with 2, 4, 6, or 8 mg/day for up to 15 months. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates. The data described below reflect exposure to DETROL LA 4 mg once daily every morning in 505 patients and to placebo in 507 patients exposed for 12 weeks in the Phase 3, controlled clinical study.
Adverse events were reported in 52% (n=263) of patients receiving DETROL LA and in 49% (n=247) of patients receiving placebo. The most common adverse events reported by patients receiving DETROL LA were dry mouth, headache, constipation, and abdominal pain. Dry mouth was the most frequently reported adverse event for patients treated with DETROL LA occurring in 23.4% of patients treated with DETROL LA and 7.7% of placebo-treated patients. Dry mouth, constipation, abnormal vision (accommodation abnormalities), urinary retention, and dry eyes are expected side effects of antimuscarinic agents. A serious adverse event was reported by 1.4% (n=7) of patients receiving DETROL LA and by 3.6% (n=18) of patients receiving placebo.
The frequency of discontinuation due to adverse events was highest during the first 4 weeks of treatment. Similar percentages of patients treated with DETROL LA or placebo discontinued treatment due to adverse events. Treatment was discontinued due to adverse events and dry mouth was reported as an adverse event in 2.4% (n=12) of patients treated with DETROL LA and in 1.2% (n=6) of patients treated with placebo.
Table 4 lists the adverse events reported in 1% or more of patients treated with DETROL LA 4 mg once daily in the 12-week study. The adverse events were reported regardless of causality.
| Body System | Adverse Event | % DETROL LA n=505 | % Placebo n=507 |
|
|||
| Autonomic Nervous | dry mouth | 23 | 8 |
| General | headache | 6 | 4 |
| fatigue | 2 | 1 | |
| Central/Peripheral Nervous | dizziness | 2 | 1 |
| Gastrointestinal | constipation | 6 | 4 |
| abdominal pain | 4 | 2 | |
| dyspepsia | 3 | 1 | |
| Vision | xerophthalmia | 3 | 2 |
| vision abnormal | 1 | 0 | |
| Psychiatric | somnolence | 3 | 2 |
| anxiety | 1 | 0 | |
| Respiratory | sinusitis | 2 | 1 |
| Urinary | dysuria | 1 | 0 |
Post-marketing Surveillance
The following events have been reported in association with tolterodine use in worldwide post-marketing experience: General: anaphylactoid reactions, including angioedema; Cardiovascular: tachycardia, palpitations, peripheral edema; Gastrointestinal: diarrhea; Central/Peripheral Nervous: confusion, disorientation, memory impairment, hallucinations.
Reports of aggravation of symptoms of dementia (e.g. confusion, disorientation, delusion) have been reported after tolterodine therapy was initiated in patients taking cholinesterase inhibitors for the treatment of dementia.
Because these spontaneously reported events are from the worldwide post-marketing experience, the frequency of events and the role of tolterodine in their causation cannot be reliably determined.
OVERDOSAGE
A 27-month-old child who ingested 5 to 7 tolterodine immediate release tablets 2 mg was treated with a suspension of activated charcoal and was hospitalized overnight with symptoms of dry mouth. The child fully recovered.
Management of Overdosage
Overdosage with DETROL LA Capsules can potentially result in severe central anticholinergic effects and should be treated accordingly.
ECG monitoring is recommended in the event of overdosage. In dogs, changes in the QT interval (slight prolongation of 10% to 20%) were observed at a suprapharmacologic dose of 4.5 mg/kg, which is about 68 times higher than the recommended human dose. In clinical trials of normal volunteers and patients, QT interval prolongation was observed with tolterodine immediate release at doses up to 8 mg (4 mg bid) and higher doses were not evaluated (see PRECAUTIONS, Patients with Congenital or Acquired QT Prolongation).
DOSAGE AND ADMINISTRATION
The recommended dose of DETROL LA Capsules are 4 mg daily. DETROL LA should be taken once daily with liquids and swallowed whole. The dose may be lowered to 2 mg daily based on individual response and tolerability, however, limited efficacy data is available for DETROL LA 2 mg (see CLINICAL STUDIES).
For patients with significantly reduced hepatic or renal function or who are currently taking drugs that are potent inhibitors of CYP3A4, the recommended dose of DETROL LA is 2 mg daily (see CLINICAL PHARMACOLOGY and PRECAUTIONS, Drug Interactions).
HOW SUPPLIED
DETROL LA Capsules 2 mg are blue-green with symbol and 2 printed in white ink. DETROL LA Capsules 4 mg are blue with symbol and 4 printed in white ink. DETROL LA Capsules are supplied as follows:
| Bottles of 30 | Bottles of 500 | ||
| 2 mg Capsules | NDC 0009-5190-01 | 2 mg Capsules | NDC 0009-5190-03 |
| 4 mg Capsules | NDC 0009-5191-01 | 4 mg Capsules | NDC 0009-5191-03 |
| Bottles of 90 | Unit Dose Blisters | ||
| 2 mg Capsules | NDC 0009-5190-02 | 2 mg Capsules | NDC 0009-5190-04 |
| 4 mg Capsules | NDC 0009-5191-02 | 4 mg Capsules | NDC 0009-5191-04 |
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature]. Protect from light.

PATIENT INFORMATION DETROL® LA (DE-trol el-ay) (tolterodine tartrate extended release capsules)
Read the Patient Information that comes with DETROL LA before you start using it and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your doctor about your condition or your treatment. Only your doctor can determine if treatment with DETROL LA is right for you.
What is DETROL LA?
DETROL LA is a prescription medicine for adults used to treat the following symptoms due to a condition called overactive bladder:
- having a strong need to urinate with leaking or wetting accidents (urge urinary incontinence)
- having a strong need to urinate right away (urgency)
- having to urinate often (frequency)
DETROL LA did not help the symptoms of overactive bladder when studied in children.
What is overactive bladder?
Overactive bladder happens when you cannot control your bladder muscle. When the muscle contracts too often or cannot be controlled, you get symptoms of overactive bladder, which are leakage of urine (urge urinary incontinence), needing to urinate right away (urgency), and needing to urinate often (frequency).
Who should not take DETROL LA?
Do not take DETROL LA if:
- you have trouble emptying your bladder (also called "urinary retention")
- your stomach empties slowly (also called "gastric retention")
- you have an eye problem called "uncontrolled narrow-angle glaucoma"
- you are allergic to DETROL LA or to any of its ingredients. See the end of this leaflet for a complete list of ingredients
What should I tell my doctor before starting DETROL LA?
Before starting DETROL LA, tell your doctor about all of your medical conditions, including if you:
- have any stomach or intestinal problems
- have trouble emptying your bladder or you have a weak urine stream
- have an eye problem called narrow-angle glaucoma
- have liver problems
- have kidney problems
- have a condition called myasthenia gravis
- or any family members have a rare heart condition called QT prolongation (long QT syndrome)
- are pregnant or trying to become pregnant. It is not known if DETROL LA could harm your unborn baby
- are breastfeeding. It is not known if DETROL LA passes into your milk and if it can harm your child
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. Other drugs can affect how your body handles DETROL LA. Your doctor may use a lower dose of DETROL LA if you are taking:
- Certain medicines for fungus or yeast infections such as Nizoral® (ketoconazole), Sporanox® (itraconazole), and Monistat® (miconazole)
- Certain medicines for bacteria infections such as Biaxin® (clarithromycin), and erythromycin
- Sandimmune® (cyclosporine) or Velban® (vinblastine)
Know the medicines you take. Keep a list of them with you to show your doctor or pharmacist each time you get a new medicine.
How should I take DETROL LA?
- Take DETROL LA exactly as prescribed. Your doctor will prescribe the dose that is right for you. Do not change your dose unless told to do so by your doctor.
- Take DETROL LA capsules once a day with liquid. Swallow the whole capsule. Tell your doctor if you cannot swallow a capsule.
- DETROL LA can be taken with or without food.
- Take DETROL LA the same time each day.
- If you miss a dose of DETROL LA, begin taking DETROL LA again the next day. Do not take 2 doses of DETROL LA in the same day.
- If you took more than your prescribed dose of DETROL LA, call your doctor, or poison control center, or go to the hospital emergency room.
What are possible side effects of DETROL LA?
The most common side effects with DETROL LA are:
- dry mouth
- headache
- constipation
- stomach pain
Medicines like DETROL LA can cause blurred vision, dizziness, or drowsiness.
Use caution while driving or doing other dangerous activities until you know how DETROL LA affects you.
These are not all the side effects with DETROL LA. For a complete list, ask your doctor or pharmacist.
How do I store DETROL LA?
- Store DETROL LA at room temperature (59 to 86° F) out of the light. Keep it in a dry place.
- Keep DETROL LA and all medicines out of the reach of children.
General Information about DETROL LA
Medicines are sometimes prescribed for conditions that are not in the patient information leaflet. Only use DETROL LA the way your doctor tells you. Do not share it with other people even if they have the same symptoms you have. It may harm them.
This leaflet summarizes the most important information about DETROL LA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about DETROL LA that is written for health professionals. You can also visit www.DETROLLA.com on the Internet, or call 1-888-4-DETROL (1-888-433-8765).
What are the ingredients in DETROL LA?
Active ingredients: tolterodine tartrate
Inactive ingredients: sucrose, starch, hypromellose, ethylcellulose, medium chain triglycerides, oleic acid, gelatin, and FD&C Blue #2. 2mg capsule also contains yellow iron oxide. Capsules have pharmaceutical grade printing ink that contains shellac glaze, titanium dioxide, propylene glycol, and simethicone.
Distributed by:

LAB-0312-3.0
March 2008
Registered trademarks are the property of their respective owners.
Principal Display Panel - 2 mg Bag
Detrol LA®
Tolterodine Tartrate Extended Release Capsules
2 mg
10 Capsules

Principal Display Panel - 4 mg Bag
Detrol LA®
Tolterodine Tartrate Extended Release Capsules
4 mg
10 Capsules

| DETROL LA
tolterodine tartrate capsule, extended release |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021228 | 12/22/2000 | |
| DETROL LA
tolterodine tartrate capsule, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021228 | 12/22/2000 | |
| Labeler - Cardinal Health (188557102) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Cardinal Health | 188557102 | REPACK | |
Revised: 05/2011 Cardinal Health