DOCEFREZ
-
docetaxel
Sun Pharma Global FZE
----------
|
||||||||||||||||||||
FULL PRESCRIBING INFORMATION
WARNING: TOXIC DEATHS, HEPATOTOXICITY, NEUTROPENIA, HYPERSENSITIVITY REACTIONS, and FLUID RETENTION
The incidence of treatment-related mortality associated with docetaxel therapy is increased in patients with abnormal liver function, in patients receiving higher doses, and in patients with non-small cell lung carcinoma and a history of prior treatment with platinum-based chemotherapy who receive docetaxel as a single agent at a dose of 100 mg/m2 [see Warnings and Precautions (5.1)].
DOCEFREZ should not be given to patients with bilirubin > upper limit of normal (ULN), or to patients with AST and/or ALT >1.5 x ULN concomitant with alkaline phosphatase >2.5 x ULN. Patients with elevations of bilirubin or abnormalities of transaminase concurrent with alkaline phosphatase are at increased risk for the development of grade 4 neutropenia, febrile neutropenia, infections, severe thrombocytopenia, severe stomatitis, severe skin toxicity, and toxic death. Patients with isolated elevations of transaminase >1.5 x ULN also had a higher rate of febrile neutropenia grade 4 but did not have an increased incidence of toxic death. Bilirubin, AST or ALT, and alkaline phosphatase values should be obtained prior to each cycle of DOCEFREZ therapy [see Warnings and Precautions (5.2)].
DOCEFREZ therapy should not be given to patients with neutrophil counts of <1500 cells/mm3. In order to monitor the occurrence of neutropenia, which may be severe and result in infection, frequent blood cell counts should be performed on all patients receiving DOCEFREZ [see Warnings and Precautions (5.3)].
Severe hypersensitivity reactions characterized by generalized rash/erythema, hypotension and/or bronchospasm, or very rarely fatal anaphylaxis, have been reported in patients who received a 3-day dexamethasone premedication. Hypersensitivity reactions require immediate discontinuation of the DOCEFREZ infusion and administration of appropriate therapy [see Warnings and Precautions (5.4)]. DOCEFREZ must not be given to patients who have a history of severe hypersensitivity reactions to docetaxel or to other drugs formulated with polysorbate 80 [see Contraindications (4)].
Severe fluid retention occurred in 6.5% (6/92) of patients despite use of a 3-day dexamethasone premedication regimen. It was characterized by one or more of the following events: poorly tolerated peripheral edema, generalized edema, pleural effusion requiring urgent drainage, dyspnea at rest, cardiac tamponade, or pronounced abdominal distention (due to ascites) [see Warnings and Precautions (5.5)].
1 INDICATIONS AND USAGE
1.1 Breast Cancer
DOCEFREZ is indicated for the treatment of patients with locally advanced or metastatic breast cancer after failure of prior chemotherapy.
1.2 Non-Small Cell Lung Cancer
DOCEFREZ as a single agent is indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of prior platinum-based chemotherapy.
1.3 Prostate Cancer
DOCEFREZ in combination with prednisone is indicated for the treatment of patients with androgen independent (hormone refractory) metastatic prostate cancer.
2 DOSAGE AND ADMINISTRATION
For all indications, toxicities may warrant dosage adjustments [see Dosage and Administration (2.7)].
Administer in a facility equipped to manage possible complications (e.g. anaphylaxis).
2.1 Breast Cancer
- For locally advanced or metastatic breast cancer after failure of prior chemotherapy, the recommended dose of DOCEFREZ is 60 mg/m2 to 100 mg/m2 administered intravenously over 1 hour every 3 weeks.
2.2 Non-Small Cell Lung Cancer
- For treatment after failure of prior platinum-based chemotherapy, docetaxel was evaluated as monotherapy, and the recommended dose is 75 mg/m2 administered intravenously over 1 hour every 3 weeks. A dose of 100 mg/m2 in patients previously treated with chemotherapy was associated with increased hematologic toxicity, infection, and treatment-related mortality in randomized, controlled trials [see Boxed Warning, Dosage and Administration (2.7), Warnings and Precautions (5), Clinical Studies (14)].
2.3 Prostate Cancer
- For hormone-refractory metastatic prostate cancer, the recommended dose of DOCEFREZ is 75 mg/m2 every 3 weeks as a 1 hour intravenous infusion. Prednisone 5 mg orally twice daily is administered continuously [see Dosage and Administration (2.7)].
2.6 Premedication Regimen
All patients should be premedicated with oral corticosteroids (see below for prostate cancer) such as dexamethasone 16 mg per day (e.g., 8 mg twice daily) for 3 days starting 1 day prior to DOCEFREZ administration in order to reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reactions [see Boxed Warning, Warnings and Precautions (5.4)]. This regimen was evaluated in 92 patients with metastatic breast cancer previously treated with chemotherapy given docetaxel at a dose of 100 mg/m2 every 3 weeks.
For hormone-refractory metastatic prostate cancer, given the concurrent use of prednisone, the recommended premedication regimen is oral dexamethasone 8 mg, at 12 hours, 3 hours and 1 hour before the DOCEFREZ infusion [see Warnings and Precautions (5.4)].
2.7 Dosage Adjustments During Treatment
Breast Cancer
Patients who are dosed initially at 100 mg/m2 and who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than 1 week, or severe or cumulative cutaneous reactions during DOCEFREZ therapy should have the dosage adjusted from 100 mg/m2 to 75 mg/m2. If the patient continues to experience these reactions, the dosage should either be decreased from 75 mg/m2 to 55 mg/m2 or the treatment should be discontinued. Conversely, patients who are dosed initially at 60 mg/m2 and who do not experience febrile neutropenia, neutrophils <500 cells/mm3 for more than 1 week, severe or cumulative cutaneous reactions, or severe peripheral neuropathy during DOCEFREZ therapy may tolerate higher doses. Patients who develop ≥grade 3 peripheral neuropathy should have DOCEFREZ treatment discontinued entirely.
Non-Small Cell Lung Cancer
Monotherapy with DOCEFREZ for NSCLC treatment after failure of prior platinum-based chemotherapy
Patients who are dosed initially at 75 mg/m2 and who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than one week, severe or cumulative cutaneous reactions, or other grade 3/4 non-hematological toxicities during DOCEFREZ treatment should have treatment withheld until resolution of the toxicity and then resumed at 55 mg/m2. Patients who develop ≥grade 3 peripheral neuropathy should have DOCEFREZ treatment discontinued entirely.
Prostate Cancer
Combination therapy with DOCEFREZ for hormone-refractory metastatic prostate cancer
DOCEFREZ should be administered when the neutrophil count is ≥1,500 cells/mm3. Patients who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than one week, severe or cumulative cutaneous reactions or moderate neurosensory signs and/or symptoms during DOCEFREZ therapy should have the dosage of DOCEFREZ reduced from 75 to 60 mg/m². If the patient continues to experience these reactions at 60 mg/m², the treatment should be discontinued.
Combination Therapy with Strong CYP3A4 inhibitors:
Avoid using concomitant strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin and voriconazole). There are no clinical data with a dose adjustment in patients receiving strong CYP3A4 inhibitors. Based on extrapolation from a pharmacokinetic study with ketoconazole in 7 patients, consider a 50% docetaxel dose reduction if patients require co-administration of a strong CYP3A4 inhibitor. [see Drug Interactions (7), Clinical Pharmacology (12.3)].
2.8 Administration Precautions
DOCEFREZ is a cytotoxic anticancer drug and, as with other potentially toxic compounds, caution should be exercised when handling and preparing DOCEFREZ solutions. The use of gloves is recommended. Please refer to [see How Supplied/ Storage and Handling (16.3)].
If DOCEFREZ lyophilized powder, reconstituted solution, or infusion solution should come into contact with the skin, immediately and thoroughly wash with soap and water. If DOCEFREZ lyophilized powder, reconstituted solution or infusion solution should come into contact with mucosa, immediately and thoroughly wash with water.
Contact of the DOCEFREZ reconstituted solution with plasticized PVC equipment or devices used to prepare solutions for infusion is not recommended. In order to minimize patient exposure to the plasticizer DEHP (di-2-ethylhexyl phthalate), which may be leached from PVC infusion bags or sets, the DOCEFREZ infusion solution should be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.
DOCEFREZ (Lyophilized Powder for Injection and Diluent)
DOCEFREZ for Injection requires reconstitution with Diluent and one further dilution with infusion solution prior to administration. Please follow the preparation instructions provided below.
The table below provides the fill range of the Diluent, the volume of Diluent to be added for the reconstitution and the concentration of the reconstituted solution for DOCEFREZ 20 mg and DOCEFREZ 80 mg (See Table 1).
| Product | Fill Range of the Diluent (35.4% w/w ethanol in polysorbate 80) | Volume of Diluent to be added for the reconstitution | Concentration of the reconstituted solution |
|---|---|---|---|
| Docetaxel 20 mg vial | 1.10 – 1.15 mL | 1 mL | 20 mg/0.8 mL |
| Docetaxel 80 mg vial | 4.13 – 4.29 mL | 4 mL | 24 mg/mL |
2.9 Preparation and Administration
DOCEFREZ (Lyophilized Powder for Injection and Diluent)
A. Preparation of the Reconstituted Solution
- DOCEFREZ vials should be stored between 2°C and 8°C (36°F and 47°F). Allow the appropriate number of vials of DOCEFREZ (docetaxel) for Injection and diluent (35.4% ethanol in polysorbate 80) vials to stand at room temperature for approximately 5 minutes.
- Aseptically withdraw 1 mL from the diluent vial for DOCEFREZ 20 mg or 4 ml from the diluent vial for DOCEFREZ 80 mg into a syringe by partially inverting the vial, and transfer it to the appropriate vial of DOCEFREZ (docetaxel) for Injection. Shake the reconstituted vial well in order to completely dissolve the docetaxel powder present in the vial. For the 20 mg vial, the resultant concentration is 20 mg/0.8 mL. For the 80 mg vial, the resultant concentration is 24 mg/mL.
- The reconstituted DOCEFREZ solution should be clear; however, there may be some air bubbles in the solution due to the polysorbate 80. Allow the solution to stand for a few minutes to allow any air bubbles to dissipate.
B. Preparation of the Infusion Solution
- Aseptically withdraw the required amount of reconstituted DOCEFREZ solution with a calibrated syringe and inject into a 250 ml infusion bag or bottle of either 0.9% Sodium Chloride solution or 5% Dextrose solution to produce a final concentration of 0.3 to 0.74 mg/ml. If a dose greater than 200 mg of DOCEFREZ is required, use a larger volume of the infusion vehicle so that a concentration of 0.74 mg/ml docetaxel is not exceeded.
- Thoroughly mix the infusion by manual rotation.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If the DOCEFREZ reconstituted solution or infusion solution is not clear or appears to have precipitation, these should be discarded.
2.10 Stability
DOCEFREZ infusion solution, if stored between 2°C and 25°C (36°F and 77°F) is stable for 4 hours. DOCEFREZ infusion solution (in either 0.9% Sodium Chloride solution or 5% Dextrose solution) should be used within 4 hours (including the 1 hour intravenous administration).
3 DOSAGE FORMS AND STRENGTHS
DOCEFREZ (Lyophilized Powder for Injection and Diluent)
DOCEFREZ 80 mg
DOCEFREZ (docetaxel) for Injection 80 mg: 80 mg docetaxel and Diluent for DOCEFREZ 80 mg (35.4% (w/w) ethanol in polysorbate 80). Both items are in a tray in one carton.
DOCEFREZ 20 mg
DOCEFREZ (docetaxel) for Injection 20 mg: 20 mg docetaxel and Diluent for DOCEFREZ 20 mg (35.4% (w/w) ethanol in polysorbate 80). Both items are in a tray in one carton.
4 CONTRAINDICATIONS
- DOCEFREZ is contraindicated in patients who have a history of severe hypersensitivity reactions to docetaxel or to other drugs formulated with polysorbate 80. Severe reactions, including anaphylaxis, have occurred [see Warnings and Precautions (5.4)].
- DOCEFREZ should not be used in patients with neutrophil counts of <1500 cells/mm3.
5 WARNINGS AND PRECAUTIONS
5.1 Toxic Deaths
Breast Cancer
DOCEFREZ administered at 100 mg/m2 was associated with deaths considered possibly or probably related to treatment in 2.0% (19/965) of metastatic breast cancer patients, both previously treated and untreated, with normal baseline liver function and in 11.5% (7/61) of patients with various tumor types who had abnormal baseline liver function (AST and/or ALT >1.5 times ULN together with AP >2.5 times ULN). Among patients dosed at 60 mg/m2, mortality related to treatment occurred in 0.6% (3/481) of patients with normal liver function, and in 3 of 7 patients with abnormal liver function. Approximately half of these deaths occurred during the first cycle. Sepsis accounted for the majority of the deaths.
Non-Small Cell Lung Cancer
DOCEFREZ administered at a dose of 100 mg/m2 in patients with locally advanced or metastatic non-small cell lung cancer who had a history of prior platinum-based chemotherapy was associated with increased treatment-related mortality (14% and 5% in two randomized, controlled studies). There were 2.8% treatment-related deaths among the 176 patients treated at the 75 mg/m2 dose in the randomized trials. Among patients who experienced treatment-related mortality at the 75 mg/m2 dose level, 3 of 5 patients had an ECOG PS of 2 at study entry [see Dosage and Administration (2.2), Clinical Studies (14)].
5.2 Hepatic Impairment
Patients with combined abnormalities of transaminases and alkaline phosphatase should not be treated with DOCEFREZ [see Boxed Warning, Use in Specific Populations (8.6), Clinical studies (14)].
5.3 Hematologic Effects
Perform frequent peripheral blood cell counts on all patients receiving DOCEFREZ. Patients should not be retreated with subsequent cycles of DOCEFREZ until neutrophils recover to a level > 1500 cells/mm3 and platelets recover to a level > 100,000 cells/mm3.
A 25% reduction in the dose of DOCEFREZ is recommended during subsequent cycles following severe neutropenia (<500 cells/mm3) lasting 7 days or more, febrile neutropenia, or a grade 4 infection in a DOCEFREZ cycle [see Dosage and Administration (2.7)].
Neutropenia (<2000 neutrophils/mm3) occurs in virtually all patients given 60 mg/m2 to 100 mg/m2 of docetaxel and grade 4 neutropenia (<500 cells/mm3) occurs in 85% of patients given 100 mg/m2 and 75% of patients given 60 mg/m2. Frequent monitoring of blood counts is, therefore, essential so that dose can be adjusted. DOCEFREZ should not be administered to patients with neutrophils <1500 cells/mm3.
Febrile neutropenia occurred in about 12% of patients given 100 mg/m2 but was very uncommon in patients given 60 mg/m2. Hematologic responses, febrile reactions and infections, and rates of septic death for different regimens are dose related [see Adverse Reactions (6.1), Clinical Studies (14)].
Three breast cancer patients with severe liver impairment (bilirubin >1.7 times ULN) developed fatal gastrointestinal bleeding associated with severe drug-induced thrombocytopenia. [see Dosage and Administration (2.7), Adverse Reactions (6)].
5.4 Hypersensitivity Reactions
Patients should be observed closely for hypersensitivity reactions, especially during the first and second infusions. Severe hypersensitivity reactions characterized by generalized rash/erythema, hypotension and/or bronchospasm, or very rarely fatal anaphylaxis, have been reported in patients premedicated with 3 days of corticosteroids. Severe hypersensitivity reactions require immediate discontinuation of the DOCEFREZ infusion and aggressive therapy. Patients with a history of severe hypersensitivity reactions should not be rechallenged with DOCEFREZ.
Hypersensitivity reactions may occur within a few minutes following initiation of a DOCEFREZ infusion. If minor reactions such as flushing or localized skin reactions occur, interruption of therapy is not required. All patients should be premedicated with an oral corticosteroid prior to the initiation of the infusion of DOCEFREZ [see Dosage and Administration (2.6)].
5.5 Fluid Retention
Severe fluid retention has been reported following docetaxel therapy. Patients should be premedicated with oral corticosteroids prior to each DOCEFREZ administration to reduce the incidence and severity of fluid retention [see Dosage and Administration (2.6)]. Patients with pre-existing effusions should be closely monitored from the first dose for the possible exacerbation of the effusions.
When fluid retention occurs, peripheral edema usually starts in the lower extremities and may become generalized with a median weight gain of 2 kg.
Among 92 breast cancer patients premedicated with 3-day corticosteroids, moderate fluid retention occurred in 27.2% and severe fluid retention in 6.5%. The median cumulative dose to onset of moderate or severe fluid retention was 819 mg/m2. Nine of 92 patients (9.8%) of patients discontinued treatment due to fluid retention: 4 patients discontinued with severe fluid retention; the remaining 5 had mild or moderate fluid retention. The median cumulative dose to treatment discontinuation due to fluid retention was 1021 mg/m2.
Fluid retention was completely, but sometimes slowly, reversible with a median of 16 weeks from the last infusion of docetaxel to resolution (range: 0 to 42+ weeks). Patients developing peripheral edema may be treated with standard measures, e.g., salt restriction, oral diuretic(s).
5.6 Acute Myeloid Leukemia
Treatment-related acute myeloid leukemia (AML) or myelodysplasia has occurred in patients given anthracyclines and/or cyclophosphamide, including use in adjuvant therapy for breast cancer. The risk of delayed myelodysplasia or myeloid leukemia requires hematological follow-up.
5.7 Cutaneous Reactions
Localized erythema of the extremities with edema followed by desquamation has been observed. In case of severe skin toxicity, an adjustment in dosage is recommended [see Dosage and Administration (2.7)]. The discontinuation rate due to skin toxicity was 1.6% (15/965) for metastatic breast cancer patients. Among 92 breast cancer patients premedicated with 3-day corticosteroids, there were no cases of severe skin toxicity reported and no patient discontinued docetaxel due to skin toxicity.
5.8 Neurologic Reactions
Severe neurosensory symptoms (e.g.paresthesia, dysesthesia, pain) were observed in 5.5% (53/965) of metastatic breast cancer patients, and resulted in treatment discontinuation in 6.1%. When these symptoms occur, dosage must be adjusted. If symptoms persist, treatment should be discontinued [see Dosage and Administration (2.7)]. Patients who experienced neurotoxicity in clinical trials and for whom follow-up information on the complete resolution of the event was available had spontaneous reversal of symptoms with a median of 9 weeks from onset (range: 0 to 106 weeks). Severe peripheral motor neuropathy mainly manifested as distal extremity weakness occurred in 4.4% (42/965).
5.9 Asthenia
Severe asthenia has been reported in 14.9% (144/965) of metastatic breast cancer patients but has led to treatment discontinuation in only 1.8%. Symptoms of fatigue and weakness may last a few days up to several weeks and may be associated with deterioration of performance status in patients with progressive disease.
5.10 Use in Pregnancy
DOCEFREZ can cause fetal harm when administered to a pregnant woman. Docetaxel caused embryofetal toxicities including intrauterine mortality when administered to pregnant rats and rabbits during the period of organogenesis. Embryofetal effects in animals occurred at doses as low as 1/50 and 1/300 the recommended human dose on a body surface area basis.
There are no adequate and well-controlled studies in pregnant women using DOCEFREZ. If DOCEFREZ is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with DOCEFREZ [see Use in Specific Populations (8.1)].
6 ADVERSE REACTIONS
The most serious adverse reactions from docetaxel are:
- Toxic Deaths [see Boxed Warning, Warnings and Precautions (5.1)]
- Hepatotoxicity [see Boxed Warning, Warnings and Precautions (5.2)]
- Neutropenia [see Boxed Warning, Warnings and Precautions (5.3)]
- Hypersensitivity [see Boxed Warning, Warnings and Precautions (5.4)]
- Fluid Retention [see Boxed Warning, Warnings and Precautions (5.5)]
Adverse reactions are described for docetaxel according to indication. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Responding patients may not experience an improvement in performance status on therapy and may experience worsening. The relationship between changes in performance status, response to therapy, and treatment-related side effects has not been established.
6.1 Clinical Trials Experience
Breast Cancer
Monotherapy with docetaxel for locally advanced or metastatic breast cancer after failure of prior chemotherapy
Docetaxel 100 mg/m2: Adverse drug reactions occurring in at least 5% of patients are compared for three populations who received docetaxel administered at 100 mg/m2 as a 1-hour infusion every 3 weeks: 2045 patients with various tumor types and normal baseline liver function tests; the subset of 965 patients with locally advanced or metastatic breast cancer, both previously treated and untreated with chemotherapy, who had normal baseline liver function tests; and an additional 61 patients with various tumor types who had abnormal liver function tests at baseline. These reactions were described using COSTART terms and were considered possibly or probably related to docetaxel. At least 95% of these patients did not receive hematopoietic support. The safety profile is generally similar in patients receiving docetaxel for the treatment of breast cancer and in patients with other tumor types (See Table 2).
| Adverse Reaction | All Tumor Types Normal LFTs*
n=2045 % | All Tumor Types Elevated LFTs†
n=61 % | Breast Cancer Normal LFTs*
n=965 % |
|---|---|---|---|
|
|||
| Hematologic
|
|||
| Neutropenia |
|||
| <2000 cells/mm3
| 96 | 96 | 99 |
| <500 cells/mm3
| 75 | 88 | 86 |
| Leukopenia |
|||
| <4000 cells/mm3
| 96 | 98 | 99 |
| <1000 cells/mm3
| 32 | 47 | 44 |
| Thrombocytopenia |
|||
| <100,000 cells/mm3
| 8 | 25 | 9 |
| Anemia |
|||
| <11 g/dL | 90 | 92 | 94 |
| <8 g/dL | 9 | 31 | 8 |
| Febrile Neutropenia‡
| 11 | 26 | 12 |
| Septic Death
| 2 | 5 | 1 |
| Non-Septic Death
| 1 | 7 | 1 |
| Infections
|
|||
| Any | 22 | 33 | 22 |
| Severe | 6 | 16 | 6 |
| Fever in Absence of Infection
|
|||
| Any | 31 | 41 | 35 |
| Severe | 2 | 8 | 2 |
| Hypersensitivity Reactions
|
|||
| Regardless of Premedication |
|||
| Any | 21 | 20 | 18 |
| Severe | 4 | 10 | 3 |
| With 3-day Premedication | n=92 | n=3 | n=92 |
| Any | 15 | 33 | 15 |
| Severe | 2 | 0 | 2 |
| Fluid Retention
|
|||
| Regardless of Premedication |
|||
| Any | 47 | 39 | 60 |
| Severe | 7 | 8 | 9 |
| With 3-day Premedication | n=92 | n=3 | n=92 |
| Any | 64 | 67 | 64 |
| Severe | 7 | 33 | 7 |
| Neurosensory
|
|||
| Any | 49 | 34 | 58 |
| Severe | 4 | 0 | 6 |
| Cutaneous
|
|||
| Any | 48 | 54 | 47 |
| Severe | 5 | 10 | 5 |
| Nail Changes
|
|||
| Any | 31 | 23 | 41 |
| Severe | 3 | 5 | 4 |
| Gastrointestinal
|
|||
| Nausea | 39 | 38 | 42 |
| Vomiting | 22 | 23 | 23 |
| Diarrhea | 39 | 33 | 43 |
| Severe | 5 | 5 | 6 |
| Stomatitis
|
|||
| Any | 42 | 49 | 52 |
| Severe | 6 | 13 | 7 |
| Alopecia
| 76 | 62 | 74 |
| Asthenia
|
|||
| Any | 62 | 53 | 66 |
| Severe | 13 | 25 | 15 |
| Myalgia
|
|||
| Any | 19 | 16 | 21 |
| Severe | 2 | 2 | 2 |
| Arthralgia
| 9 | 7 | 8 |
| Infusion Site Reactions
| 4 | 3 | 4 |
Reversible marrow suppression was the major dose-limiting toxicity of docetaxel [see Warnings and Precautions (5.3)]. The median time to nadir was 7 days, while the median duration of severe neutropenia (<500 cells/mm3) was 7 days. Among 2045 patients with solid tumors and normal baseline LFTs, severe neutropenia occurred in 75.4% and lasted for more than 7 days in 2.9% of cycles.
Febrile neutropenia (<500 cells/mm3 with fever > 38°C with intravenous antibiotics and/or hospitalization) occurred in 11% of patients with solid tumors, in 12.3% of patients with metastatic breast cancer, and in 9.8% of 92 breast cancer patients premedicated with 3-day corticosteroids.
Severe infectious episodes occurred in 6.1% of patients with solid tumors, in 6.4% of patients with metastatic breast cancer, and in 5.4% of 92 breast cancer patients premedicated with 3-day corticosteroids.
Thrombocytopenia (<100,000 cells/mm3) associated with fatal gastrointestinal hemorrhage has been reported.
Hypersensitivity Reactions
Severe hypersensitivity reactions have been reported [see Boxed Warning, Warnings and Precautions (5.4)]. Minor events, including flushing, rash with or without pruritus, chest tightness, back pain, dyspnea, drug fever, or chills, have been reported and resolved after discontinuing the infusion and instituting appropriate therapy.
Fluid Retention
Fluid retention can occur with the use of DOCEFREZ [see Boxed Warning, Dosage and Administration (2.6), Warnings and Precautions (5.5)].
Cutaneous Reactions
Severe skin toxicity is discussed elsewhere in the label [see Warnings and Precautions (5.7)]. Reversible cutaneous reactions characterized by a rash including localized eruptions, mainly on the feet and/or hands, but also on the arms, face, or thorax, usually associated with pruritus, have been observed. Eruptions generally occurred within 1 week after docetaxel infusion, recovered before the next infusion, and were not disabling.
Severe nail disorders were characterized by hypo- or hyperpigmentation, and occasionally by onycholysis (in 0.8% of patients with solid tumors) and pain.
Neurologic Reactions
Neurologic reactions are discussed elsewhere in the label [see Warnings and Precautions (5.8)].
Gastrointestinal Reactions
Nausea, vomiting, and diarrhea were generally mild to moderate. Severe reactions occurred in 3-5% of patients with solid tumors and to a similar extent among metastatic breast cancer patients. The incidence of severe reactions was 1% or less for the 92 breast cancer patients premedicated with 3-day corticosteroids.
Severe stomatitis occurred in 5.5% of patients with solid tumors, in 7.4% of patients with metastatic breast cancer, and in 1.1% of the 92 breast cancer patients premedicated with 3-day corticosteroids.
Cardiovascular Reactions
Hypotension occurred in 2.8% of patients with solid tumors; 1.2% required treatment. Clinically meaningful events such as heart failure, sinus tachycardia, atrial flutter, dysrhythmia, unstable angina, pulmonary edema, and hypertension occurred rarely. Seven of 86 patients (8.1%) of metastatic breast cancer patients receiving docetaxel 100 mg/m2 in a randomized trial and who had serial left ventricular ejection fractions assessed developed deterioration of LVEF by ≥ 10% associated with a drop below the institutional lower limit of normal.
Infusion Site Reactions
Infusion site reactions were generally mild and consisted of hyperpigmentation, inflammation, redness or dryness of the skin, phlebitis, extravasation, or swelling of the vein.
Hepatic Reactions
In patients with normal LFTs at baseline, bilirubin values greater than the ULN occurred in 8.9% of patients. Increases in AST or ALT > 1.5 times the ULN, or alkaline phosphatase > 2.5 times ULN, were observed in 18.9% and 7.3% of patients, respectively. While on docetaxel, increases in AST and/or ALT > 1.5 times ULN concomitant with alkaline phosphatase > 2.5 times ULN occurred in 4.3% of patients with normal LFTs at baseline. Whether these changes were related to the drug or underlying disease has not been established.
Hematologic and Other Toxicity: Relation to dose and baseline liver chemistry abnormalities
Hematologic and other toxicity is increased at higher doses and in patients with elevated baseline liver function tests (LFTs). In the following tables, adverse drug reactions are compared for three populations: 730 patients with normal LFTs given docetaxel at 100 mg/m2 in the randomized and single arm studies of metastatic breast cancer after failure of previous chemotherapy; 18 patients in these studies who had abnormal baseline LFTs (defined as AST and/or ALT > 1.5 times ULN concurrent with alkaline phosphatase > 2.5 times ULN); and 174 patients in Japanese studies given docetaxel at 60 mg/m2 who had normal LFTs (see Tables 3 and 4).
| Adverse Reaction | Docetaxel 100 mg/m2 | Docetaxel 60 mg/m2 | |
|---|---|---|---|
| Normal LFTs*
n=730 % | Elevated LFTs†
n=18 % | Normal LFTs*
n=174 % |
|
|
|||
| Neutropenia
| |||
| Any <2000 cells/mm3
| 98 | 100 | 95 |
| Grade 4 <500 cells/mm3
| 84 | 94 | 75 |
| Thrombocytopenia
| |||
| Any <100,000 cells/mm3
| 11 | 44 | 14 |
| Grade 4 <20,000 cells/mm3
| 1 | 17 | 1 |
| Anemia <11 g/dL | 95 | 94 | 65 |
| Infection‡
| |||
| Any | 23 | 39 | 1 |
| Grade 3 and 4 | 7 | 33 | 0 |
| Febrile Neutropenia§
| |||
| By Patient | 12 | 33 | 0 |
| By Course | 2 | 9 | 0 |
| Septic Death
| 2 | 6 | 1 |
| Non-Septic Death
| 1 | 11 | 0 |
| Adverse Reaction | Docetaxel 100 mg/m2 | Docetaxel 60 mg/m2 | |
|---|---|---|---|
| Normal LFTs*
n=730 % | Elevated LFTs†
n=18 % | Normal LFTs*
n=174 % |
|
| NA = not available |
|||
|
|||
| Acute Hypersensitivity Reaction
| |||
| Regardless of Premedication
| |||
| Any | 13 | 6 | 1 |
| Severe | 1 | 0 | 0 |
| Fluid Retention‡
| |||
| Regardless of Premedication
| |||
| Any | 56 | 61 | 13 |
| Severe | 8 | 17 | 0 |
| Neurosensory
| |||
| Any | 57 | 50 | 20 |
| Severe | 6 | 0 | 0 |
| Myalgia
| 23 | 33 | 3 |
| Cutaneous
| |||
| Any | 45 | 61 | 31 |
| Severe | 5 | 17 | 0 |
| Asthenia
| |||
| Any | 65 | 44 | 66 |
| Severe | 17 | 22 | 0 |
| Diarrhea
| |||
| Any | 42 | 28 | NA |
| Severe | 6 | 11 |
|
| Stomatitis
| |||
| Any | 53 | 67 | 19 |
| Severe | 8 | 39 | 1 |
The following adverse reactions were associated with increasing docetaxel doses: fluid retention (26%, 38%, and 46% at 60 mg/m2 , 75 mg/m2 , and 100 mg/m2 respectively), thrombocytopenia (7%, 11%, and 12% respectively), neutropenia (92%, 94%, and 97% respectively), febrile neutropenia (5%, 7%, and 14% respectively), treatment-related grade 3/4 infection (2%, 3%, and 7% respectively) and anemia (87%, 94%, and 97% respectively).
Lung Cancer
Monotherapy with docetaxel for unresectable, locally advanced or metastatic NSCLC previously treated with platinum-based chemotherapy
Docetaxel 75 mg/m2: Treatment emergent adverse drug reactions are shown in Table 5. Included in this table are safety data for a total of 176 patients with non-small cell lung carcinoma and a history of prior treatment with platinum-based chemotherapy who were treated in two randomized, controlled trials. These reactions were described using NCI Common Toxicity Criteria regardless of relationship to study treatment, except for the hematologic toxicities or where otherwise noted.
| Adverse Reaction | Docetaxel 75 mg/m2
n=176 % | Best Supportive Care n=49 % | Vinorelbine/Ifosfamide n=119 % |
||||
|---|---|---|---|---|---|---|---|
|
|||||||
| Neutropenia
| |||||||
| Any | 84 | 14 | 83 |
||||
| Grade 3/4 | 65 | 12 | 57 |
||||
| Leukopenia
| |||||||
| Any | 84 | 6 | 89 |
||||
| Grade 3/4 | 49 | 0 | 43 |
||||
| Thrombocytopenia
| |||||||
| Any | 8 | 0 | 8 |
||||
| Grade 3/4 | 3 | 0 | 2 |
||||
| Anemia
| |||||||
| Any | 91 | 55 | 91 |
||||
| Grade 3/4 | 9 | 12 | 14 |
||||
| Febrile Neutropenia†
| 6 | NA‡
| 1 |
||||
| Infection
| |||||||
| Any | 34 | 29 | 30 |
||||
| Grade 3/4 | 10 | 6 | 9 |
||||
| Treatment Related Mortality
| 3 | NA‡
| 3 |
||||
| Hypersensitivity Reactions
| |||||||
| Any | 6 | 0 | 1 |
||||
| Grade 3/4 | 3 | 0 | 0 |
||||
| Fluid Retention
| |||||||
| Any | 34 | ND§
| 23 |
||||
| Severe | 3 | 3 |
|||||
| Neurosensory
| |||||||
| Any | 23 | 14 | 29 |
||||
| Grade 3/4 | 2 | 6 | 5 |
||||
| Neuromotor
| |||||||
| Any | 16 | 8 | 10 |
||||
| Grade 3/4 | 5 | 6 | 3 |
||||
| Skin
| |||||||
| Any | 20 | 6 | 17 |
||||
| Grade 3/4 | 1 | 2 | 1 |
||||
| Gastrointestinal
| |||||||
| Nausea | |||||||
| Any | 34 | 31 | 31 |
||||
| Grade 3/4 | 5 | 4 | 8 |
||||
| Vomiting | |||||||
| Any | 22 | 27 | 22 |
||||
| Grade 3/4 | 3 | 2 | 6 |
||||
| Diarrhea | |||||||
| Any | 23 | 6 | 12 |
||||
| Grade 3/4 | 3 | 0 | 4 |
||||
| Alopecia
| 56 | 35 | 50 |
||||
| Asthenia
| |||||||
| Any | 53 | 57 | 54 |
||||
| Severe¶
| 18 | 39 | 23 |
||||
| Stomatitis
| |||||||
| Any | 26 | 6 | 8 |
||||
| Grade 3/4 | 2 | 0 | 1 |
||||
| Pulmonary
| |||||||
| Any | 41 | 49 | 45 |
||||
| Grade 3/4 | 21 | 29 | 19 |
||||
| Nail Disorder
| |||||||
| Any | 11 | 0 | 2 |
||||
| Severe¶
| 1 | 0 | 0 |
||||
| Myalgia
| |||||||
| Any | 6 | 0 | 3 |
||||
| Severe¶
| 0 | 0 | 0 |
||||
| Arthralgia
| |||||||
| Any | 3 | 2 | 2 |
||||
| Severe¶
| 0 | 0 | 1 |
||||
| Taste Perversion
| |||||||
| Any | 6 | 0 | 0 |
||||
| Severe¶
| 1 | 0 | 0 |
||||
Combination therapy with docetaxel in patients with prostate cancer
The following data are based on the experience of 332 patients, who were treated with docetaxel 75 mg/m² every 3 weeks in combination with prednisone 5 mg orally twice daily (see Table 6).
| Docetaxel 75 mg/m2 every 3 weeks + prednisone 5 mg twice daily n=332 % | Mitoxantrone 12 mg/m2 every 3 weeks + prednisone 5 mg twice daily n=335 % |
|||
|---|---|---|---|---|
| Adverse Reaction | Any | Grade 3/4 | Any | Grade 3/4 |
|
||||
| Anemia | 67 | 5 | 58 | 2 |
| Neutropenia | 41 | 32 | 48 | 22 |
| Thrombocytopenia | 3 | 1 | 8 | 1 |
| Febrile neutropenia | 3 | N/A | 2 | N/A |
| Infection | 32 | 6 | 20 | 4 |
| Epistaxis | 6 | 0 | 2 | 0 |
| Allergic Reactions | 8 | 1 | 1 | 0 |
| Fluid Retention*
Weight Gain* Peripheral Edema* | 24 8 18 | 1 0 0 | 5 3 2 | 0 0 0 |
| Neuropathy Sensory | 30 | 2 | 7 | 0 |
| Neuropathy Motor | 7 | 2 | 3 | 1 |
| Rash/Desquamation | 6 | 0 | 3 | 1 |
| Alopecia | 65 | N/A | 13 | N/A |
| Nail Changes | 30 | 0 | 8 | 0 |
| Nausea | 41 | 3 | 36 | 2 |
| Diarrhea | 32 | 2 | 10 | 1 |
| Stomatitis/Pharyngitis | 20 | 1 | 8 | 0 |
| Taste Disturbance | 18 | 0 | 7 | 0 |
| Vomiting | 17 | 2 | 14 | 2 |
| Anorexia | 17 | 1 | 14 | 0 |
| Cough | 12 | 0 | 8 | 0 |
| Dyspnea | 15 | 3 | 9 | 1 |
| Cardiac left ventricular function | 10 | 0 | 22 | 1 |
| Fatigue | 53 | 5 | 35 | 5 |
| Myalgia | 15 | 0 | 13 | 1 |
| Tearing | 10 | 1 | 2 | 0 |
| Arthralgia | 8 | 1 | 5 | 1 |
6.2 Post-marketing Experiences
The following adverse reactions have been identified from clinical trials and/or post-marketing surveillance. Because they are reported from a population of unknown size, precise estimates of frequency cannot be made.
Body as a whole: diffuse pain, chest pain, radiation recall phenomenon.
Cardiovascular: atrial fibrillation, deep vein thrombosis, ECG abnormalities, thrombophlebitis, pulmonary embolism, syncope, tachycardia, myocardial infarction.
Cutaneous: very rare cases of cutaneous lupus erythematosus and rare cases of bullous eruptions such as erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, and Scleroderma-like changes usually preceded by peripheral lymphedema. In some cases multiple factors may have contributed to the development of these effects. Severe hand and foot syndrome has been reported.
Gastrointestinal: abdominal pain, anorexia, constipation, duodenal ulcer, esophagitis, gastrointestinal hemorrhage, gastrointestinal perforation, ischemic colitis, colitis, intestinal obstruction, ileus, neutropenic enterocolitis and dehydration as a consequence to gastrointestinal events have been reported.
Hematologic: bleeding episodes. Disseminated intravascular coagulation (DIC), often in association with sepsis or multiorgan failure, has been reported. Cases of acute myeloid leukemia and myelodysplasic syndrome have been reported in association with docetaxel when used in combination with other chemotherapy agents and/or radiotherapy.
Hypersensitivity: rare cases of anaphylactic shock have been reported. Very rarely these cases resulted in a fatal outcome in patients who received premedication.
Hepatic: rare cases of hepatitis, sometimes fatal primarily in patients with pre-existing liver disorders, have been reported.
Neurologic: confusion, rare cases of seizures or transient loss of consciousness have been observed, sometimes appearing during the infusion of the drug.
Ophthalmologic: conjunctivitis, lacrimation or lacrimation with or without conjunctivitis. Excessive tearing which may be attributable to lacrimal duct obstruction has been reported. Rare cases of transient visual disturbances (flashes, flashing lights, scotomata) typically occurring during drug infusion and in association with hypersensitivity reactions have been reported. These were reversible upon discontinuation of the infusion.
Hearing: rare cases of ototoxicity, hearing disorders and/or hearing loss have been reported, including cases associated with other ototoxic drugs.
Respiratory: dyspnea, acute pulmonary edema, acute respiratory distress syndrome, interstitial pneumonia. Pulmonary fibrosis has been rarely reported. Rare cases of radiation pneumonitis have been reported in patients receiving concomitant radiotherapy.
Renal: renal insufficiency and renal failure have been reported, the majority of these cases were associated with concomitant nephrotoxic drugs.
7 DRUG INTERACTIONS
Docetaxel is a CYP3A4 substrate. In vitro studies have shown that the metabolism of docetaxel may be modified by the concomitant administration of compounds that induce, inhibit, or are metabolized by cytochrome P450 3A4.
In vivo studies showed that the exposure of docetaxel increased 2.2-fold when it was coadministered with ketoconazole, a potent inhibitor of CYP3A4. Protease inhibitors, particularly ritonavir, may increase the exposure of docetaxel. Concomitant use of DOCEFREZ and drugs that inhibit CYP3A4 may increase exposure to docetaxel and should be avoided. In patients receiving treatment with DOCEFREZ, close monitoring for toxicity and a DOCEFREZ dose reduction could be considered if systemic administration of a potent CYP3A4 inhibitor cannot be avoided [see Dosage and Administration (2.7) and Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see ‘Warnings and Precautions’ section]
Based on its mechanism of action and findings in animals, DOCEFREZ can cause fetal harm when administered to a pregnant woman. If DOCEFREZ is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with DOCEFREZ.
DOCEFREZ can cause fetal harm when administered to a pregnant woman. Studies in both rats and rabbits at doses ≥0.3 and 0.03 mg/kg/day, respectively (about 1/50 and 1/300 the daily maximum recommended human dose on a mg/m2 basis), administered during the period of organogenesis, have shown that docetaxel is embryotoxic and fetotoxic (characterized by intrauterine mortality, increased resorption, reduced fetal weight, and fetal ossification delay). The doses indicated above also caused maternal toxicity.
8.3 Nursing Mothers
It is not known whether docetaxel is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from DOCEFREZ, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of docetaxel in pediatric patients have not been established.
8.5 Geriatric Use
In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy in elderly patients.
Prostate Cancer
Of the 333 patients treated with docetaxel every three weeks plus prednisone in the prostate cancer study (TAX327), 209 patients were 65 years of age or greater and 68 patients were older than 75 years. In patients treated with docetaxel every three weeks, the following treatment emergent adverse reactions occurred at rates ≥10% higher in patients 65 years of age or greater compared to younger patients: anemia (71% vs. 59%), infection (37% vs. 24%), nail changes (34% vs. 23%), anorexia (21% vs. 10%), weight loss (15% vs. 5%) respectively.
8.6 Hepatic Impairment
Patients with bilirubin > ULN should not receive docetaxel. Also, patients with AST and/or ALT > 1.5 x ULN concomitant with alkaline phosphatase > 2.5 x ULN should not receive docetaxel. [see Boxed Warning, Warnings and Precautions (5.2), Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no known antidote for DOCEFREZ overdosage. In case of overdosage, the patient should be kept in a specialized unit where vital functions can be closely monitored. Anticipated complications of overdosage include: bone marrow suppression, peripheral neurotoxicity, and mucositis. Patients should receive therapeutic G-CSF as soon as possible after discovery of overdose. Other appropriate symptomatic measures should be taken, as needed.
In two reports of overdose, one patient received 150 mg/m2 and the other received 200 mg/m2 as 1-hour infusions. Both patients experienced severe neutropenia, mild asthenia, cutaneous reactions, and mild paresthesia, and recovered without incident.
In mice, lethality was observed following single intravenous doses that were ≥154 mg/kg (about 4.5 times the human dose of 100 mg/m2 on a mg/m2 basis); neurotoxicity associated with paralysis, non-extension of hind limbs, and myelin degeneration was observed in mice at 48 mg/kg (about 1.5 times the human dose of 100 mg/m2 basis). In male and female rats, lethality was observed at a dose of 20 mg/kg (comparable to the human dose of 100 mg/m2 on a mg/m2 basis) and was associated with abnormal mitosis and necrosis of multiple organs.
11 DESCRIPTION
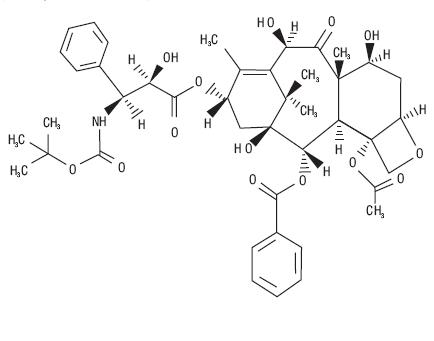
Docetaxel is an antineoplastic agent belonging to the taxoid family. It is prepared by semisynthesis beginning with a precursor extracted from the renewable needle biomass of yew plants. The chemical name for docetaxel (anhydrous) is (2R,3S)-N-carboxy-3-phenylisoserine,N-tert-butyl ester, 13-ester with 5β-20-epoxy-1,2α,4,7β,10β,13α-hexahydroxytax-11-en-9-one 4-acetate 2-benzoate. Docetaxel (anhydrous) has the following structural formula:
DOCEFREZ (Lyophilized Powder for Injection and Diluent)
DOCEFREZ (docetaxel) for injection is a sterile, lyophilized, non-pyrogenic, white powder and is available in single use vials containing 20 mg or 80 mg of docetaxel (anhydrous).
DOCEFREZ (docetaxel) for injection requires reconstitution with Diluent prior to use. For each 20 mg or 80 mg vial, a sterile, non-pyrogenic, single dose Diluent vial is co-packaged. The Diluent for DOCEFREZ (docetaxel) for injection contains 35.4 % w/w ethanol in polysorbate 80.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Docetaxel is an antineoplastic agent that acts by disrupting the microtubular network in cells that is essential for mitotic and interphase cellular functions. Docetaxel binds to free tubulin and promotes the assembly of tubulin into stable microtubules while simultaneously inhibiting their disassembly. This leads to the production of microtubule bundles without normal function and to the stabilization of microtubules, which results in the inhibition of mitosis in cells. Docetaxel’s binding to microtubules does not alter the number of protofilaments in the bound microtubules, a feature which differs from most spindle poisons currently in clinical use.12.3 Pharmacokinetics
Absorption: The pharmacokinetics of docetaxel have been evaluated in cancer patients after administration of 20 mg/m2 to 115 mg/m2 in phase 1 studies. The area under the curve (AUC) was dose proportional following doses of 70 mg/m2 to 115 mg/m2 with infusion times of 1 to 2 hours. Docetaxel’s pharmacokinetic profile is consistent with a three-compartment pharmacokinetic model, with half-lives for the α, β, and γ phases of 4 min, 36 min, and 11.1 hr, respectively. Mean total body clearance was 21 L/h/m2.
Distribution: The initial rapid decline represents distribution to the peripheral compartments and the late (terminal) phase is due, in part, to a relatively slow efflux of docetaxel from the peripheral compartment. Mean steady state volume of distribution was 113 L. In vitro studies showed that docetaxel is about 94% protein bound, mainly to a1-acid glycoprotein, albumin, and lipoproteins. In three cancer patients, the in vitro binding to plasma proteins was found to be approximately 97%. Dexamethasone does not affect the protein binding of docetaxel.
Metabolism: In vitro drug interaction studies revealed that docetaxel is metabolized by the CYP3A4 isoenzyme, and its metabolism may be modified by the concomitant administration of compounds that induce, inhibit, or are metabolized by cytochrome P450 3A4 [see Drug Interactions (7)].
Elimination: A study of 14C-docetaxel was conducted in three cancer patients. Docetaxel was eliminated in both the urine and feces following oxidative metabolism of the tert-butyl ester group, but fecal excretion was the main elimination route. Within 7 days, urinary and fecal excretion accounted for approximately 6% and 75% of the administered radioactivity, respectively. About 80% of the radioactivity recovered in feces is excreted during the first 48 hours as 1 major and 3 minor metabolites with very small amounts (less than 8%) of unchanged drug.
Effect of Age: A population pharmacokinetic analysis was carried out after docetaxel treatment of 535 patients dosed at 100 mg/m2. Pharmacokinetic parameters estimated by this analysis were very close to those estimated from phase 1 studies. The pharmacokinetics of docetaxel were not influenced by age.
Effect of Gender: The population pharmacokinetics analysis described above also indicated that gender did not influence the pharmacokinetics of docetaxel.
Hepatic Impairment: The population pharmacokinetic analysis described above indicated that in patients with clinical chemistry data suggestive of mild to moderate liver impairment (AST and/or ALT >1.5 times ULN concomitant with alkaline phosphatase > 2.5 times ULN), total body clearance was lowered by an average of 27%, resulting in a 38% increase in systemic exposure (AUC). This average, however, includes a substantial range and there is, at present, no measurement that would allow recommendation for dose adjustment in such patients. Patients with combined abnormalities of transaminase and alkaline phosphatase should not be treated with DOCEFREZ. Patients with severe hepatic impairment have not been studied. [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
Effect of Race: Mean total body clearance for Japanese patients dosed at the range of 10 mg/m2 to 90 mg/m2 was similar to that of European/American populations dosed at 100 mg/m2, suggesting no significant difference in the elimination of docetaxel in the two populations.
Effect of Ketoconazole: The effect of ketoconazole (a strong CYP3A4 inhibitor) on the pharmacokinetics of docetaxel was investigated in 7 cancer patients. Patients were randomized to receive either docetaxel (100 mg/m² intravenous) alone or docetaxel (10 mg/m² intravenous) in combination with ketoconazole (200 mg orally once daily for 3 days) in a crossover design with a 3-week washout period. The results of this study indicated that the mean dose-normalized AUC of docetaxel was increased 2.2-fold and its clearance was reduced by 49% when docetaxel was co-administration with ketoconazole [see Dosage and Administration (2.7) and Drug-Drug Interactions (7)].
Effect of Combination Therapies:
Dexamethasone: Docetaxel total body clearance was not modified by pretreatment with dexamethasone.
Prednisone: A population pharmacokinetic analysis of plasma data from 40 patients with hormone-refractory metastatic prostate cancer indicated that docetaxel systemic clearance in combination with prednisone is similar to that observed following administration of docetaxel alone.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with docetaxel have not been performed.
Docetaxel was clastogenic in the in vitro chromosome aberration test in CHO-K1 cells and in the in vivo micronucleus test in mice administered doses of 0.39 to 1.56 mg/kg (about 1/60th to 1/15th the recommended human dose on a mg/m2 basis). Docetaxel was not mutagenic in the Ames test or the CHO/HGPRT gene mutation assays.
Docetaxel did not reduce fertility in rats when administered in multiple intravenous doses of up to 0.3 mg/kg (about 1/50th the recommended human dose on a mg/m2 basis), but decreased testicular weights were reported. This correlates with findings of a 10-cycle toxicity study (dosing once every 21 days for 6 months) in rats and dogs in which testicular atrophy or degeneration was observed at intravenous doses of 5 mg/kg in rats and 0.375 mg/kg in dogs (about 1/3rd and 1/15th the recommended human dose on a mg/m2 basis, respectively). An increased frequency of dosing in rats produced similar effects at lower dose levels.
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic Breast Cancer
The efficacy and safety of docetaxel have been evaluated in locally advanced or metastatic breast cancer after failure of previous chemotherapy (alkylating agent-containing regimens or anthracycline-containing regimens).
Randomized Trials
In one randomized trial, patients with a history of prior treatment with an anthracycline-containing regimen were assigned to treatment with docetaxel (100 mg/m2 every 3 weeks) or the combination of mitomycin (12 mg/m2 every 6 weeks) and vinblastine (6 mg/m2 every 3 weeks). Two hundred three patients were randomized to docetaxel and 189 to the comparator arm. Most patients had received prior chemotherapy for metastatic disease; only 27 patients on the docetaxel arm and 33 patients on the comparator arm entered the study following relapse after adjuvant therapy. Three-quarters of patients had measurable, visceral metastases. The primary endpoint was time to progression. The following table summarizes the study results (See Table 7).
|
|||
| Efficacy Parameter
| Docetaxel
(n=203) | Mitomycin/
Vinblastine (n=189) | p-value
|
| Median Survival | 11.4 months | 8.7 months | p=0.01 Log Rank |
| Risk Ratio*, Mortality (Docetaxel: Control) 95% CI (Risk Ratio) | 0.73 0.58-0.93 |
||
| Median Time to Progression | 4.3 months | 2.5 months | p=0.01 Log Rank |
| Risk Ratio*, Progression (Docetaxel: Control) 95% CI (Risk Ratio) | 0.75 0.61-0.94 |
||
| Overall Response Rate Complete Response Rate | 28.1% 3.4% | 9.5% 1.6% | p<0.0001 Chi Square |
| Efficacy Parameter | Docetaxel (n=161) | Doxorubicin (n=165) | p-value |
|---|---|---|---|
|
|||
| Median Survival | 14.7 months | 14.3 months | p=0.39 Log Rank |
| Risk Ratio*, Mortality (Docetaxel: Control) 95% CI (Risk Ratio) | 0.89 0.68-1.16 |
||
| Median Time to Progression | 6.5 months | 5.3 months | p=0.45 Log Rank |
| Risk Ratio*, Progression (Docetaxel: Control) 95% CI (Risk Ratio) | 0.93 0.71-1.16 |
||
| Overall Response Rate Complete Response Rate | 45.3% 6.8% | 29.7% 4.2% | p=0.004 Chi Square |
Single Arm Studies
Docetaxel at a dose of 100 mg/m2 was studied in six single arm studies involving a total of 309 patients with metastatic breast cancer in whom previous chemotherapy had failed. Among these, 190 patients had anthracycline-resistant breast cancer, defined as progression during an anthracycline-containing chemotherapy regimen for metastatic disease, or relapse during an anthracycline-containing adjuvant regimen. In anthracycline-resistant patients, the overall response rate was 37.9% (72/190; 95% C.I.: 31.0% to 44.8%) and the complete response rate was 2.1%.
Docetaxel was also studied in three single arm Japanese studies at a dose of 60 mg/m2, in 174 patients who had received prior chemotherapy for locally advanced or metastatic breast cancer. Among 26 patients whose best response to an anthracycline had been progression, the response rate was 34.6% (95% C.I.: 17.2% to 55.7%), similar to the response rate in single arm studies of 100 mg/m2.
14.3 Non-Small Cell Lung Cancer (NSCLC)
The efficacy and safety of docetaxel has been evaluated in patients with unresectable, locally advanced or metastatic non-small cell lung cancer whose disease has failed prior platinum-based chemotherapy.
Monotherapy with Docetaxel for NSCLC Previously Treated with Platinum-Based Chemotherapy
Two randomized, controlled trials established that a docetaxel dose of 75 mg/m2 was tolerable and yielded a favorable outcome in patients previously treated with platinum-based chemotherapy (see below). Docetaxel at a dose of 100 mg/m2, however, was associated with unacceptable hematologic toxicity, infections, and treatment-related mortality and this dose should not be used [see Boxed Warning, Dosage and Administration (2.7), Warnings and Precautions (5.3)].
One trial (TAX317), randomized patients with locally advanced or metastatic non-small cell lung cancer, a history of prior platinum-based chemotherapy, no history of taxane exposure, and an ECOG performance status ≤ 2 to docetaxel or best supportive care. The primary endpoint of the study was survival. Patients were initially randomized to docetaxel 100 mg/m2 or best supportive care, but early toxic deaths at this dose led to a dose reduction to docetaxel 75 mg/m2. A total of 104 patients were randomized in this amended study to either docetaxel 75 mg/m2 or best supportive care.
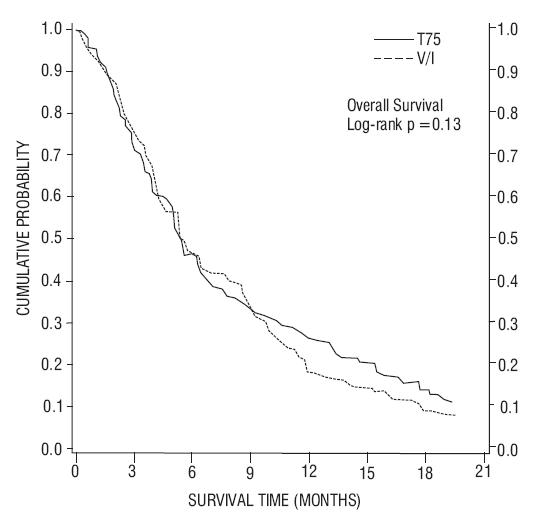
In a second randomized trial (TAX320), 373 patients with locally advanced or metastatic non-small cell lung cancer, a history of prior platinum-based chemotherapy, and an ECOG performance status ≤ 2 were randomized to docetaxel 75 mg/m2, docetaxel 100 mg/m2 and a treatment in which the investigator chose either vinorelbine 30 mg/m2 days 1, 8, and 15 repeated every 3 weeks or ifosfamide 2 g/m2 days 1-3 repeated every 3 weeks. Forty percent of the patients in this study had a history of prior paclitaxel exposure. The primary endpoint was survival in both trials. The efficacy data for the docetaxel 75 mg/m2 arm and the comparator arms are summarized in Table 9 and Figures 1 and 2 showing the survival curves for the two studies.
| TAX317 | TAX320 | |||
|---|---|---|---|---|
| Docetaxel 75 mg/m2 n=55 | Best Supportive Care n=49 | Docetaxel 75 mg/m2 n=125 | Control (V/I*) n=123 |
|
| Overall Survival Log-rank Test | p=0.01 | p=0.13 |
||
| Risk Ratio†, Mortality (Docetaxel: Control) 95% CI (Risk Ratio) | 0.56 (0.35, 0.88) | 0.82 (0.63, 1.06) |
||
| Median Survival 95% CI | 7.5 months‡
(5.5, 12.8) | 4.6 months (3.7, 6.1) | 5.7 months (5.1, 7.1) | 5.6 months (4.4, 7.9) |
| % 1-year Survival 95% CI | 37%‡§
(24, 50) | 12% (2, 23) | 30%‡§
(22, 39) | 20% (13, 27) |
| Time to Progression 95% CI | 12.3 weeks‡
(9.0, 18.3) | 7.0 weeks (6.0, 9.3) | 8.3 weeks (7.0, 11.7) | 7.6 weeks (6.7, 10.1) |
| Response Rate 95% CI | 5.5% (1.1, 15.1) | Not Applicable | 5.7% (2.3, 11.3) | 0.8% (0.0, 4.5) |
Figure 1- TAX317 Survival K-M Curves - Docetaxel 75 mg/m2 vs. Best Supportive Care
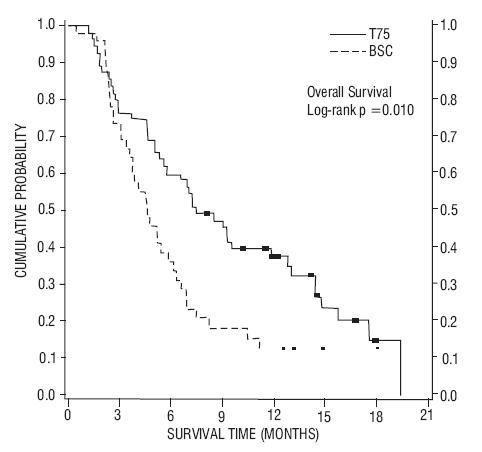
Figure 2 - TAX320 Survival K-M Curves – Docetaxel 75 mg/m2 vs. Vinorelbine or Ifosfamide Control
Patients treated with docetaxel at a dose of 75 mg/m2 experienced no deterioration in performance status and body weight relative to the comparator arms used in these trials.
14.4 Hormone Refractory Prostate Cancer
The safety and efficacy of docetaxel in combination with prednisone in patients with androgen independent (hormone refractory) metastatic prostate cancer were evaluated in a randomized multicenter active control trial. A total of 1006 patients with Karnofsky Performance Status (KPS) ≥60 were randomized to the following treatment groups:
- Docetaxel 75 mg/m2 every 3 weeks for 10 cycles.
- Docetaxel 30 mg/m2 administered weekly for the first 5 weeks in a 6-week cycle for 5 cycles.
- Mitoxantrone 12 mg/m2 every 3 weeks for 10 cycles.
In the docetaxel every three week arm, a statistically significant overall survival advantage was demonstrated compared to mitoxantrone. In the docetaxel weekly arm, no overall survival advantage was demonstrated compared to the mitoxantrone control arm. Efficacy results for the docetaxel every 3 week arm versus the control arm are summarized in Table 10 and Figure 3.
| Docetaxel+Prednisone every 3 weeks | Mitoxantrone+Prednisone every 3 weeks |
|
|---|---|---|
|
||
| Number of patients Median survival (months) 95% CI Hazard ratio 95% CI p-value* | 335 18.9 (17.0-21.2) 0.761 (0.619-0.936) 0.0094 | 337 16.5 (14.4-18.6) -- -- -- |
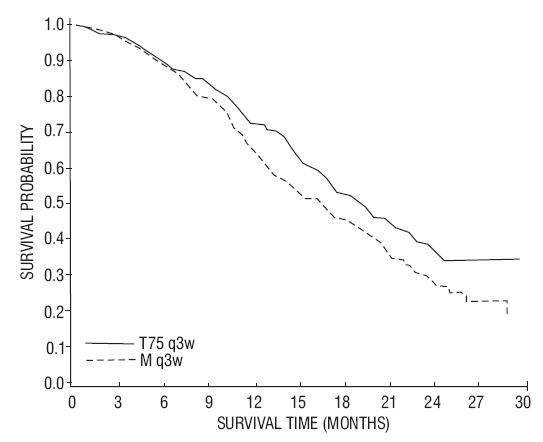
15 REFERENCES
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- American Society of Health-System Pharmacists. (2006) ASHP Guidelines on Handling Hazardous Drugs. Am J Health-Syst Pharm. 2006;63:1172-1193
- Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh , PA : Oncology Nursing Society.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
DOCEFREZ (Lyophilized Powder for Injection and Diluent)
DOCEFREZ (docetaxel) for Injection is supplied in a single use vial as a sterile, lyophilized powder with an accompanying sterile, non-pyrogenic, Diluent (35.4% w/w ethanol in polysorbate 80) vial.
DOCEFREZ (docetaxel) for Injection, 80 mg (NDC 47335-286-41)
DOCEFREZ (docetaxel) for Injection 80 mg: 80 mg docetaxel and Diluent for docetaxel 80 mg (35.4% (w/w) ethanol in polysorbate 80). Both items are in a tray in one carton.
DOCEFREZ (docetaxel) for Injection 20 mg (NDC 47335-285-41)
DOCEFREZ (docetaxel) for Injection 20 mg: 20 mg docetaxel and Diluent for docetaxel 20 mg (35.4% (w/w) ethanol in polysorbate 80). Both items are in a tray in one carton.
16.2 Storage
Store between 2°C and 8°C (36°F and 46°F). Retain in the original package to protect from bright light.
16.3 Handling and Disposal
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published [see References (15)].
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling
- DOCEFREZ may cause fetal harm. Advise patients to avoid becoming pregnant while receiving this drug. Women of childbearing potential should use effective contraceptives if receiving DOCEFREZ [see Warnings and Precautions (5.10) and Use in Specific Populations (8.1)].
- Obtain detailed allergy and concomitant drug information from the patient prior to DOCEFREZ administration.
- Explain the significance of oral corticosteroids such as dexamethasone administration to the patient to help facilitate compliance. Instruct patients to report if they were not compliant with oral corticosteroid regimen.
- Instruct patients to immediately report signs of a hypersensitivity reaction.
- Tell patients to watch for signs of fluid retention such as peripheral edema in the lower extremities, weight gain and dyspnea.
- Explain the significance of routine blood cell counts. Instruct patients to monitor their temperature frequently and immediately report any occurrence of fever.
- Instruct patients to report myalgia, cutaneous, or neurologic reactions.
- Explain to patients that side effects such as nausea, vomiting, diarrhea, constipation, fatigue, excessive tearing, infusion site reactions, and hair loss are associated with docetaxel administration.
Patient Information
DOCEFREZ™ (‘dō-sə-‘frāz)
(docetaxel)
for Injection
Read this Patient Information before you receive your first treatment with DOCEFREZ and each time before you are treated. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about DOCEFREZ?
DOCEFREZ can cause serious side effects, including death.
-
The chance of death in people who receive DOCEFREZ is higher if you:
- have liver problems
- receive high doses of DOCEFREZ
- have non-small cell lung cancer and have been treated with chemotherapy medicines that contain platinum
- DOCEFREZ can affect your blood cells. Your doctor should do routine blood tests during treatment with DOCEFREZ. This will include regular checks of your white blood cell counts. If your white blood cells are too low, your doctor may not treat you with DOCEFREZ until you have enough white blood cells. People with low white blood counts can develop life-threatening infections. The earliest sign of infection may be fever. Follow your doctor’s instructions for how often to take your temperature while taking DOCEFREZ. Call your doctor right away if you have a fever.
-
Serious allergic reactions can happen in people who take DOCEFREZ. Serious allergic reactions are medical emergencies that can lead to death and must be treated right away. Tell your doctor right away if you have any of these signs of a serious allergic reaction:
- trouble breathing
- sudden swelling of your face, lips, tongue, throat, or trouble swallowing
- hives (raised bumps), rash, or redness all over your body
- Your body may hold too much fluid (severe fluid retention) during treatment with DOCEFREZ. This can be life threatening. To decrease the chance of this happening, you must take another medicine, a corticosteroid, before each DOCEFREZ treatment. You must take the corticosteroid exactly as your doctor tells you. Tell your doctor or nurse before your DOCEFREZ treatment if you forget to take corticosteroid dose or do not take it as your doctor tells you.
DOCEFREZ is a prescription anti-cancer medicine used to treat certain people with:
- breast cancer
- non-small cell lung cancer
- prostate cancer
Who should not take DOCEFREZ?
Do not take DOCEFREZ if you:
- have had a severe allergic reaction to:
- docetaxel, the active ingredient in DOCEFREZ, or
- any other medicines that contain polysorbate 80. Ask your doctor or pharmacist if you are not sure.
See “What is the most important information I should know about DOCEFREZ?” for the signs and symptoms of a severe allergic reaction.
- have a low white blood cell count.
Before you receive DOCEFREZ, tell your doctor if you:
- are allergic to any medicines. See “Who should not take DOCEFREZ?” Also, see the end of this leaflet for a list of the ingredients in DOCEFREZ.
- have liver problems
- have any other medical conditions
- are pregnant or plan to become pregnant. DOCEFREZ can harm your unborn baby.
- are breast-feeding or plan to breast-feed. It is not known if DOCEFREZ passes into your breast milk. You and your doctor should decide if you will take DOCEFREZ or breast-feed.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How will I receive DOCEFREZ?
- DOCEFREZ will be given to you as an intravenous (IV) injection into your vein, usually over 1 hour.
- DOCEFREZ is usually given every 3 weeks.
- Your doctor will decide how long you will receive treatment with DOCEFREZ
- Your doctor will check your blood cell counts and other blood tests during your treatment with DOCEFREZ to check for side effects of DOCEFREZ.
- Your doctor may stop your treatment, change the timing of your treatment, or change the dose of your treatment if you have certain side effects while taking DOCEFREZ.
DOCEFREZ may cause serious side effects including death.
- See “What is the most important information I should know about DOCEFREZ?”
- Acute Myeloid Leukemia (AML), a type of blood cancer, can happen in people who take DOCEFREZ along with certain other medicines. Tell your doctor about all the medicines you take.
- Other Blood Disorders – Changes in blood counts due to leukemia and other blood disorders may occur years after treatment with Docefrez.
- Skin Reactions including redness and swelling of your arms and legs with peeling of your skin.
- Neurologic Symptoms including numbness, tingling, or burning in your hands and feet.
- changes in your sense of taste
- feeling short of breath
- constipation
- decreased appetite
- changes in your fingernails or toenails
- swelling of your hands, face or feet
- feeling weak or tired
- joint and muscle pain
- nausea and vomiting
- diarrhea
- mouth or lips sores
- hair loss
- rash
- redness of the eye, excess tearing
- skin reactions at the site of DOCEFREZ administration such as increased skin pigmentation, redness, tenderness, swelling, warmth or dryness of the skin.
- tissue damage if DOCEFREZ leaks out of the vein into the tissues
These are not all the possible side effects of DOCEFREZ. For more information ask your doctor or pharmacist.
Call your doctor or for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about DOCEFREZ
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. This Patient Information leaflet summarizes the most important information about DOCEFREZ. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about DOCEFREZ that is written for healthcare professionals.
For more information contact Caraco Pharmaceutical Laboratories, Ltd. at 1-800-818-4555.
What are the ingredients in DOCEFREZ?
Active ingredient: docetaxel
Inactive ingredients include: ethanol in polysorbate 80 (Diluent)
| Every three-week injection of DOCEFREZ for breast, and non-small cell lung cancers
Take your oral corticosteroid medicine as your doctor tells you. Oral corticosteroid dosing: Day 1 Date:_________ Time:______AM _______PM Day 2 Date:_________ Time:______AM _______PM (DOCEFREZ Treatment Day) Day 3 Date:_________ Time:______AM _______PM |
|
Every three-week injection of DOCEFREZ for prostate cancer Take your oral corticosteroid medicine as your doctor tells you. Oral corticosteroid dosing: Date:___________ Time:___________ Date:___________ Time:___________ (DOCEFREZ Treatment Day) Time:___________ |
Manufactured by:
Sun Pharmaceutical Ind. Ltd.
Halol-Baroda Highway,
Halol-389 350, Gujarat, India.
Distributed by:
Caraco Pharmaceutical Laboratories, Ltd.
1150 Elijah McCoy Drive
Detroit, MI 48202
PJPI0307C
ISS. 05/2011
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DOCEFREZ-20MG-LABEL
NDC 47335-285-40
DocefrezTM
(Docetaxel) for Injection
20 mg
For Intravenous Infusion Only
Each Docefrez for Injection vial contains a slight overfill to deliver 20 mg of Docetaxel per 0.8 mL after reconstitution
Rx ONLY

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DILUENT-20MG-LABEL
NDC 47335-287-40
DILUENT for DocefrezTM 20 mg
1.13 mL
Single Use Vial-Discard Unused Portion.
Rx ONLY

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DOCEFREZ-20MG-CARTON
New Concentration and Preparation
NDC 47335-285-41
DocefrezTM
(Docetaxel) for Injection
20 mg
For Intravenous Infusion Only
Each Docefrez for Injection vial contains a slight overfill to deliver 20 mg of Docetaxel per 0.8 mL after reconstitution
Each carton contains:
One vial of Docefrez (docetaxel) for Injection 20 mg
One vial of DILUENT for Docefrez 20 mg
Rx ONLY

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DOCEFREZ-80MG-LABEL
NDC 47335-286-40
DocefrezTM
(Docetaxel) for Injection
80 mg
For Intravenous Infusion Only
Each Docefrez for Injection vial contains a slight overfill to deliver 80 mg of Docetaxel
(24 mg/mL after reconstitution)

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DILUENT-80MG-LABEL
NDC 47335-288-40
DILUENT for DocefrezTM 80 mg
4.21 mL
Rx ONLY

PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-DOCEFREZ-80MG-CARTON
New Concentration and Preparation
NDC 47335-286-41
DocefrezTM
(Docetaxel) for Injection
80 mg
For Intravenous Infusion Only
Each Docefrez for Injection vial contains a slight overfill to deliver 80 mg of Docetaxel (24 mg/mL after reconstitution)
Each carton contains:
One vial of Docefrez (docetaxel) for Injection 80 mg
One vial of DILUENT for Docefrez 80 mg
Rx ONLY

| DOCEFREZ
docetaxel kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA022534 | 05/03/2011 | |
| DOCEFREZ
docetaxel kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA022534 | 05/03/2011 | |
| Labeler - Sun Pharma Global FZE (864347344) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Sun Pharmaceutical Industries Limited | 725959238 | MANUFACTURE, ANALYSIS | |