priftin (rifapentine) tablet, film coated
[sanofi-aventis U.S. LLC]
DESCRIPTION
PRIFTIN® (rifapentine) for oral administration contains 150 mg of the active ingredient rifapentine per tablet.
The 150 mg tablets also contain, as inactive ingredients: calcium stearate, disodium EDTA, FD&C Blue No. 2 aluminum lake, hydroxypropyl cellulose, hypromellose USP, microcrystalline cellulose, polyethylene glycol, pregelatinized starch, propylene glycol, sodium ascorbate, sodium lauryl sulfate, sodium starch glycolate, synthetic red iron oxide, and titanium dioxide.
Rifapentine is a rifamycin derivative antibiotic and has a similar profile of microbiological activity to rifampin (rifampicin). The molecular weight is 877.04.
The molecular formula is C47H64N4O12.
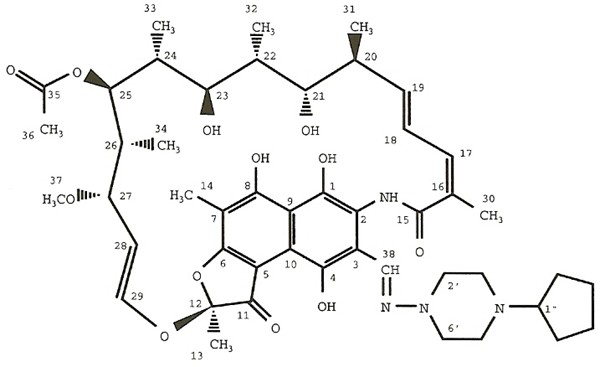
The chemical name for rifapentine is rifamycin, 3-[[(4-cyclopentyl-1-piperazinyl)imino]methyl]-
or
3-[N-(4-Cyclopentyl-1-piperazinyl)formimidoyl] rifamycin or 5,6,9,17,19,21-hexahydroxy-23-methoxy-2,4,12,16,18,20,22-heptamethyl-8-[N-(4-cyclopentyl-l-piperazinyl)-formimidoyl]-2,7-(epoxypentadeca[1,11,13]trienimino)naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate. It has the following structure:
ACTIONS/CLINICAL PHARMACOLOGY
Pharmacokinetics
Absorption
The absolute bioavailability of rifapentine has not been determined. The relative bioavailability (with an oral solution as a reference) of rifapentine after a single 600 mg dose to healthy adult volunteers was 70%. The maximum concentrations were achieved from 5 to 6 hours after administration of the 600 mg rifapentine dose. Food (850 total calories: 33 g protein, 55 g fat and 58 g carbohydrate) increased AUC(0–∞) and Cmax by 43% and 44%, respectively over that observed when administered under fasting conditions. When oral doses of rifapentine were administered once daily or once every 72 hours to healthy volunteers for 10 days, single dose AUC(0–∞) value of rifapentine was similar to its steady-state AUCss (0–24h) or AUCss (0–72h) values, suggesting no significant auto-induction effect on steady-state pharmacokinetics of rifapentine. Steady-state conditions were achieved by day 10 following daily administration of rifapentine 600 mg. The pharmacokinetic characteristics of rifapentine and 25-desacetyl rifapentine (active metabolite) on day 10 following oral administration of 600 mg rifapentine every 72 hours to healthy volunteers are contained in the following table.
| Parameter | Rifapentine | 25-desacetyl Rifapentine |
|---|---|---|
| Mean ± SD (n=12) | ||
| Cmax (µg/mL) | 15.05 ± 4.62 | 6.26 ± 2.06 |
| AUC (0–72h)(µg*h/mL) | 319.54 ± 91.52 | 215.88 ± 85.96 |
| T1/2(h) | 13.19 ± 1.38 | 13.35 ± 2.67 |
| Tmax (h) | 4.83 ± 1.80 | 11.25 ± 2.73 |
| Clpo (L/h) | 2.03 ± 0.60 | -- |
Distribution
In a population pharmacokinetic analysis in 351 tuberculosis patients who received 600 mg rifapentine in combination with isoniazid, pyrazinamide and ethambutol, the estimated apparent volume of distribution was 70.2 ± 9.1 L. In healthy volunteers, rifapentine and 25-desacetyl rifapentine were 97.7% and 93.2% bound to plasma proteins, respectively. Rifapentine was mainly bound to albumin. Similar extent of protein binding was observed in healthy volunteers, asymptomatic HIV-infected subjects and hepatically impaired subjects.
Metabolism/Excretion
Following a single 600 mg oral dose of radiolabelled rifapentine to healthy volunteers (n=4), 87% of the total 14C rifapentine was recovered in the urine (17%) and feces (70%). Greater than 80% of the total 14C rifapentine dose was excreted from the body within 7 days. Rifapentine was hydrolyzed by an esterase enzyme to form a microbiologically active 25-desacetyl rifapentine. Rifapentine and 25-desacetyl rifapentine accounted for 99% of the total radioactivity in plasma. Plasma AUC(0–∞) and Cmax values of the 25-desacetyl rifapentine metabolite were one-half and one-third those of the rifapentine, respectively. Based upon relative in vitro activities and AUC(0–∞) values, rifapentine and 25-desacetyl rifapentine potentially contributes 62% and 38% to the clinical activities against M. tuberculosis, respectively.
Special Populations
Gender
In a population pharmacokinetics analysis of sparse blood samples obtained from 351 tuberculosis patients who received 600 mg rifapentine in combination with isoniazid, pyrazinamide and ethambutol, the estimated apparent oral clearance of rifapentine for males and females was 2.51 ± 0.14 L/h and 1.69 ± 0.41 L/h, respectively. The clinical significance of the difference in the estimated apparent oral clearance is not known.
Elderly
Following oral administration of a single 600 mg dose of rifapentine to elderly (≥65 years) male healthy volunteers (n=14), the pharmacokinetics of rifapentine and 25-desacetyl metabolite were similar to that observed for young (18 to 45 years) healthy male volunteers (n=20).
Pediatric (Adolescents)
In a pharmacokinetics study of rifapentine in healthy adolescents (age 12 to 15), 600 mg rifapentine was administered to those weighing ≥45 kg (n=10) and 450 mg was administered to those weighing <45 kg (n=2). The pharmacokinetics of rifapentine were similar to those observed in healthy adults.
Renal Impaired Patients
The pharmacokinetics of rifapentine have not been evaluated in renal impaired patients. Although only about 17% of an administered dose is excreted via the kidneys, the clinical significance of impaired renal function on the disposition of rifapentine and its 25-desacetyl metabolite is not known.
Hepatic Impaired Patients
Following oral administration of a single 600 mg dose of rifapentine to mild to severe hepatic impaired patients (n=15), the pharmacokinetics of rifapentine and 25-desacetyl metabolite were similar in patients with various degrees of hepatic impairment and to that observed in another study for healthy volunteers (n=12). Since the elimination of these agents are primarily via the liver, the clinical significance of impaired hepatic function on the disposition of rifapentine and its 25-desacetyl metabolite is not known.
Asymptomatic HIV-Infected Volunteers
Following oral administration of a single 600 mg dose of rifapentine to asymptomatic HIV-infected volunteers (n=15) under fasting conditions, mean Cmax and AUC(0–∞) of rifapentine were lower (20–32%) than that observed in other studies in healthy volunteers (n=55). In a cross-study comparison, mean Cmax and AUC values of the 25-desacetyl metabolite of rifapentine, when compared to healthy volunteers were higher (6–21%) in one study (n=20), but lower (15–16%) in a different study (n=40). The clinical significance of this observation is not known. Food (850 total calories: 33 g protein, 55 g fat, and 58 g carbohydrate) increases the mean AUC and Cmax of rifapentine observed under fasting conditions in asymptomatic HIV-infected volunteers by about 51% and 53%, respectively.
Microbiology
Mechanism of Action
Rifapentine, a cyclopentyl rifamycin, inhibits DNA-dependent RNA polymerase in susceptible strains of Mycobacterium tuberculosis but not in mammalian cells. At therapeutic levels, rifapentine exhibits bactericidal activity against both intracellular and extracellular M. tuberculosis organisms. Both rifapentine and the 25-desacetyl metabolite accumulate in human monocyte-derived macrophages with intracellular/extracellular ratios of approximately 24:1 and 7:1, respectively.
Resistance Development
In the treatment of tuberculosis (see INDICATIONS AND USAGE), a small number of resistant cells present within large populations of susceptible cells can rapidly become predominant. Rifapentine resistance development in M. tuberculosis strains is principally due to one of several single point mutations that occur in the rpoB portion of the gene coding for the beta subunit of the DNA-dependent RNA polymerase. The incidence of rifapentine resistant mutants in an otherwise susceptible population of M. tuberculosis strains is approximately one in 107 to 108 bacilli. Due to the potential for resistance development to rifapentine, appropriate susceptibility tests should be performed in the event of persistently positive cultures.
M. tuberculosis organisms resistant to other rifamycins are likely to be resistant to rifapentine. A high level of cross-resistance between rifampin and rifapentine has been demonstrated with M. tuberculosis strains. Cross-resistance does not appear between rifapentine and non-rifamycin antimycobacterial agents such as isoniazid and streptomycin.
In Vitro Activity of Rifapentine against M. tuberculosis
Rifapentine and its 25-desacetyl metabolite have demonstrated in vitro activity against rifamycin-susceptible strains of Mycobacterium tuberculosis including cidal activity against phagocytized M. tuberculosis organisms grown in activated human macrophages.
In vitro results indicate that rifapentine MIC values for M. tuberculosis organisms are influenced by study conditions. Rifapentine MIC values were substantially increased employing egg-based medium compared to liquid or agar-based solid media. The addition of Tween 80 in these assays has been shown to lower MIC values for rifamycin compounds.
In mouse infection studies a therapeutic effect, in terms of enhanced survival time or reduction of organ bioburden, has been observed in M. tuberculosis-infected animals treated with various intermittent rifapentine-containing regimens. Animal studies have shown that the activity of rifapentine is influenced by dose and frequency of administration.
Susceptibility testing for Mycobacterium tuberculosis
Breakpoints to determine whether clinical isolates of M. tuberculosis are susceptible or resistant to rifapentine have not been established. The clinical relevance of rifapentine in vitro susceptibility test results for other mycobacterial species has not been determined.
CLINICAL TRIALS
A total of 722 patients were enrolled in Clinical Study 008, an open label, prospective, randomized, parallel group, active controlled trial, for the treatment of pulmonary tuberculosis. This population was mostly comprised of Black (>60%) or Multiracial (>31%) patients and the mean ± standard deviation age was 37 ± 11 years. Treatment groups were comparable with respect to age and race. The percentage of male patients was higher in the rifapentine combination group (80%) than in the rifampin combination group (73%). The study was divided into two phases on the basis of dosing frequency. For the first phase, designated as the Intensive Phase, 361 patients were randomized to receive rifapentine, isoniazid, pyrazinamide, and ethambutol for 60 days and 361 patients were randomized to receive rifampin, isoniazid, pyrazinamide, and ethambutol for 60 days. (Ethambutol was to be discontinued once baseline susceptibility test results were available.) Rifapentine and isoniazid were each administered at a fixed dose regardless of body weight. Rifampin, pyrazinamide, and ethambutol were administered based on body weight according to Table 2-1. Note: All drugs were administered daily in the Intensive Phase except for rifapentine which was administered twice weekly.
During the second phase, designated as the Continuation Phase, 321 patients who had received rifapentine in the Intensive Phase continued to receive rifapentine and isoniazid once weekly for up to 120 days. Three hundred seven patients who had received rifampin in the Intensive Phase continued to receive rifampin and isoniazid during the Continuation Phase twice weekly for up to 120 days. Rifampin and isoniazid were administered based on body weight according to Table 2-1.
Patients in either treatment group were scheduled to receive study drug over a 180-day period with a subsequent 24-month follow-up. Additionally, both treatment groups received pyridoxine (Vitamin B6) over the 180-day treatment period.
|
||||
| Rifapentine Combination Treatment | ||||
| Intensive | Rifapentine | Isoniazid | Pyrazinamide | Ethambutol* |
| Phase | (mg) | (mg) | (mg) | (mg) |
| Twice Weekly | Daily | Daily | Daily | |
| Patient Weight | ||||
| <50 kg | 600 | 300 | 1500 | 800 |
| ≥50 kg | 600 | 300 | 2000 | 1200 |
| Continuation Phase | Rifapentine (mg) | Isoniazid (mg) | ||
| Once Weekly | Once Weekly | |||
| Patient Weight | ||||
| <50 kg | 600 | 600 | ||
| ≥50 kg | 600 | 900 | ||
| Rifampin Combination Treatment | ||||
| Intensive | Rifampin | Isoniazid | Pyrazinamide | Ethambutol |
| Phase | (mg) | (mg) | (mg) | (mg) |
| Daily | Daily | Daily | Daily | |
| Patient Weight | ||||
| <50 kg | 450 | 300 | 1500 | 800 |
| ≥50 kg | 600 | 300 | 2000 | 1200 |
| Continuation | Rifampin | Isoniazid | ||
| Phase | (mg) | (mg) | ||
| Twice Weekly | Twice Weekly | |||
| Patient Weight | ||||
| <50 kg | 450 | 600 | ||
| ≥50 kg | 600 | 900 | ||
Table 2-2 presents clinical outcome in Study 008.
| Rifapentine Combination | Rifampin Combination | ||
|---|---|---|---|
|
|||
| Status of End of Treatment | |||
| Converted | 87% (248/286) | 80% (226/283) | |
| Not Converted | 1% (4/286) | 3% (8/283) | |
| Lost to Follow-up | 12% (34/286) | 17% (49/283) | |
| Status Through 24 Month Follow-up: | |||
| Relapsed | 12% (29/248) | 7% (15/226) | |
| Sputum Negative | 57% (142/248) | 64% (145/226) | |
| Lost to Follow-up | 31% (77/248) | 29% (66/226) | |
Risk of relapse was higher in the rifapentine regimen. During the Intensive Phase of treatment the rate of noncompliance with companion medications was somewhat higher for the rifapentine regimen than for the rifampin regimen. Most of the relapses occurred among those with poor compliance with these companion medications and this group also had the largest risk of relapse for the rifapentine regimen relative to the rifampin regimen. This factor appears to explain most, but not all, of the higher relapse rate observed in the rifapentine arm. Failure to convert sputum after two months of treatment (ie, end of Intensive Phase) was associated with a greater risk of relapse for both treatment regimens. Relapse rates were also higher for males in both regimens. Relapse in the rifapentine group was not associated with development of mono‑resistance to rifampin.
In vitro susceptibility testing was conducted against M. tuberculosis isolates recovered from 620 patients enrolled in the study. Rifapentine and rifampin MIC values were determined employing the radiometric susceptibility testing method utilizing 7H12 broth at pH 6.8 (NCCLS procedure M24-T). Six hundred and twelve patients had M. tuberculosis isolates that were susceptible to rifampin (MIC < 0.5 μg/ml). Of these patients, six hundred and ten had M. tuberculosis isolates (99.7%) with rifapentine MICs of < 0.125 μg/ml. The other two patients that had rifampin susceptible M. tuberculosis isolates had rifapentine MICs of 0.25 µg/ml. The remaining eight patients had M. tuberculosis isolates that were resistant to rifampin (MIC > 8.0 μg/ml). These M. tuberculosis isolates had rifapentine MICs of > 8.0 μg/ml. In this study high rifampin and rifapentine MICs were associated with multi-drug resistant M. tuberculosis (MDRTB) isolates. Rifamycin mono-resistance was not observed in either treatment arm. This information is provided for comparative purposes only as rifapentine breakpoints have not been established.
INDICATIONS AND USAGE
PRIFTIN is indicated for the treatment of pulmonary tuberculosis. PRIFTIN must always be used in conjunction with at least one other antituberculosis drug to which the isolate is susceptible. In the intensive phase of the short-course treatment of pulmonary tuberculosis, PRIFTIN should be administered twice weekly for two months, with an interval of no less than 3 days (72 hours) between doses, as part of an appropriate regimen which includes daily companion drugs (Table 2-1). It may also be necessary to add either streptomycin or ethambutol until the results of susceptibility testing are known. Compliance with all drugs in the Intensive Phase (ie, PRIFTIN, isoniazid, pyrazinamide, ethambutol or streptomycin) is imperative to assure early sputum conversion and protection against relapse. Following the intensive phase, Continuation Phase treatment should be continued with PRIFTIN for 4 months. During this phase, PRIFTIN should be administered on a once-weekly basis in combination with an appropriate antituberculous agent for susceptible organisms (Table 2-1) (see DOSAGE AND ADMINISTRATION section).
In the treatment of tuberculosis, the small number of resistant cells present within large populations of susceptible cells can rapidly become the predominant type. Consequently, clinical samples for mycobacterial culture and susceptibility testing should be obtained prior to the initiation of therapy, as well as during treatment to monitor therapeutic response. The susceptibility of M. tuberculosis organisms to isoniazid, rifampin, pyrazinamide, ethambutol, rifapentine and other appropriate agents should be measured. If test results show resistance to any of these drugs and the patient is not responding to therapy, the drug regimen should be modified.
CONTRAINDICATIONS
This product is contraindicated in patients with a history of hypersensitivity to any of the rifamycins (eg, rifampin and rifabutin).
WARNINGS
Poor compliance with the dosage regimen, particularly the daily administered non-rifamycin drugs in the Intensive Phase, was associated with late sputum conversion and a high relapse rate in the rifapentine arm of Clinical Study 008. Therefore, compliance with the full course of therapy must be emphasized, and the importance of not missing any doses must be stressed. (See PRECAUTIONS and DOSAGE AND ADMINISTRATION.)
Since antituberculous multidrug treatments, including the rifamycin class, are associated with serious hepatic events, patients with abnormal liver tests and/or liver disease should only be given rifapentine in cases of necessity and then with caution and under strict medical supervision. In these patients, careful monitoring of liver tests (especially serum transaminases) should be carried out prior to therapy and then every 2 to 4 weeks during therapy. If signs of liver disease occur or worsen, rifapentine should be discontinued. Hepatotoxicity of other antituberculosis drugs (eg, isoniazid, pyrazinamide) used in combination with rifapentine should also be taken into account.
Hyperbilirubinemia resulting from competition for excretory pathways between rifapentine and bilirubin cannot be excluded since competition between the related drug rifampin and bilirubin can occur. An isolated report showing a moderate rise in bilirubin and/or transaminase level is not in itself an indication for interrupting treatment; rather, the decision should be made after repeating the tests, noting trends in the levels and considering them in conjunction with the patient's clinical condition.
Pseudomembranous colitis has been reported to occur with various antibiotics, including other rifamycins. Diarrhea, particularly if severe and/or persistent, occurring during treatment or in the initial weeks following treatment may be symptomatic of Clostridium difficile-associated disease, the most severe form of which is pseudomembranous colitis. If pseudomembranous colitis is suspected, rifapentine should be stopped immediately and the patient should be treated with supportive and specific treatment without delay (eg, oral vancomycin). Products inhibiting peristalsis are contraindicated in this clinical situation.
Experience in HIV-infected patients is limited. In an ongoing CDC TB trial, five out of 30 HIV‑infected patients randomized to once weekly rifapentine (plus INH) in the Continuation Phase who completed treatment, relapsed. Four of these patients developed rifampin mono-resistant (RMR) TB. Each RMR patient had late-stage HIV infection, low CD4 counts and extrapulmonary disease, and documented co-administration of antifungal azoles (See Reference 1). These findings are consistent with the literature in which an emergence of RMR TB in HIV-infected TB patients has been reported in recent years. Further study in this sub-population is warranted. As with other antituberculous treatments, when rifapentine is used in HIV‑infected patients, a more aggressive regimen should be employed (eg, more frequent dosing). Based on results to date of the CDC trial (see above), once weekly dosing during the Continuation Phase of treatment is not recommended at this time.
Because rifapentine has been shown to increase indinavir metabolism (see DRUG INTERACTIONS), it should be used with extreme caution, if at all, in patients who are also taking protease inhibitors.
PRECAUTIONS
General
Rifapentine may produce a predominately red-orange discoloration of body tissues and/or fluids (eg, skin, teeth, tongue, urine, feces, saliva, sputum, tears, sweat, and cerebrospinal fluid).
Contact lenses or dentures may become permanently stained.
Rifapentine should not be used in patients with porphyria. Rifampin has enzyme-inducing properties, including induction of delta amino levulinic acid synthetase. Isolated reports have associated porphyria exacerbation with rifampin administration. Based on these isolated reports with rifampin, it may be assumed that rifapentine has a similar effect.
Information for Patients
The patient should be told that PRIFTIN may produce a reddish coloration of the urine, sweat, sputum, tears, and breast milk and the patient should be forewarned that contact lenses or dentures may be permanently stained. The patient should be advised that the reliability of oral or other systemic hormonal contraceptives may be affected; consideration should be given to using alternative contraceptive measures. For those patients with a propensity to nausea, vomiting, or gastrointestinal upset, administration of PRIFTIN with food may be useful. Patients should be instructed to notify their physician promptly if they experience any of the following: fever, loss of appetite, malaise, nausea and vomiting, darkened urine, yellowish discoloration of the skin and eyes, and pain or swelling of the joints.
Compliance with the full course of therapy must be emphasized, and the importance of not missing any doses of the daily administered companion medications in the Intensive Phase must be stressed. (See DOSAGE AND ADMINISTRATION and WARNINGS).
Laboratory Tests
Adults treated for tuberculosis with rifapentine should have baseline measurements of hepatic enzymes, bilirubin, a complete blood count, and a platelet count (or estimate).
Patients should be seen at least monthly during therapy and should be specifically questioned concerning symptoms associated with adverse reactions. All patients with abnormalities should have follow-up, including laboratory testing, if necessary. Routine laboratory monitoring for toxicity in people with normal baseline measurements is generally not necessary.
Therapeutic concentrations of rifampin have been shown to inhibit standard microbiological assays for serum folate and Vitamin B12. Similar drug-laboratory interactions should be considered for rifapentine; thus, alternative assay methods should be considered.
Drug Interaction
Rifapentine-Indinavir Interaction
In a study in which 600 mg rifapentine was administered twice weekly for 14 days followed by rifapentine twice weekly plus 800 mg indinavir 3 times a day for an additional 14 days, indinavir Cmax decreased by 55% while AUC reduced by 70%. Clearance of indinavir increased by 3-fold in the presence of rifapentine while half-life did not change. But when indinavir was administered for 14 days followed by coadministration with rifapentine for an additional 14 days, indinavir did not affect the pharmacokinetics of rifapentine. Rifapentine should be used with extreme caution, if at all, in patients who are also taking protease inhibitors. (See WARNINGS and DOSAGE AND ADMINISTRATION.)
Rifapentine is an inducer of cytochromes P4503A4 and P4502C8/9. Therefore, rifapentine may increase the metabolism of other coadministered drugs that are metabolized by these enzymes. Induction of enzyme activities by rifapentine occurred within 4 days after the first dose. Enzyme activities returned to baseline levels 14 days after discontinuing rifapentine. In addition, the magnitude of enzyme induction by rifapentine was dose and dosing frequency dependent; less enzyme induction occurred when 600 mg oral doses of rifapentine were given once every 72 hours versus daily. In vitro and in vivo enzyme induction studies have suggested rifapentine induction potential may be less than rifampin but more potent than rifabutin. Rifampin has been reported to accelerate the metabolism and may reduce the activity of the following drugs; hence, rifapentine may also increase the metabolism and decrease the activity of these drugs. Dosage adjustments of the following drugs or of drugs metabolized by cytochrome P4503A4 or P4502C8/9 may be necessary if they are given concurrently with rifapentine. Patients using oral or other systemic hormonal contraceptives should be advised to change to nonhormonal methods of birth control.
Anticonvulsants: eg, phenytoin
Antiarrhythmics: eg, disopyramide, mexiletine, quinidine, tocainide
Antibiotics: eg, chloramphenicol, clarithromycin, dapsone, doxycycline, fluoroquinolones (such as ciprofloxacin)
Oral anticoagulants: eg, warfarin
Antifungals: eg, fluconazole, itraconazole, ketoconazole
Barbiturates
Benzodiazepines: eg, diazepam
Beta-blockers, calcium channel blockers: eg, diltiazem, nifedipine, verapamil
Corticosteroids
Cardiac glycoside preparations
Clofibrate
Oral or other systemic hormonal contraceptives
Haloperidol
HIV protease inhibitors: eg, indinavir, ritonavir, nelfinavir, saquinavir (see Rifapentine-Indinavir Interaction above)
Oral hypoglycemic agents: eg, sulfonylureas
Immunosuppressants: eg, cyclosporine, tacrolimus
Levothyroxine
Narcotic analgesics: eg, methadone
Progestins
Quinine
Reverse transcriptase inhibitors: eg, delavirdine, zidovudine
Sildenafil
Theophylline
Tricyclic antidepressants: eg, amitriptyline, nortriptyline
The conversion of rifapentine to 25-desacetyl rifapentine is mediated by an esterase enzyme. There is minimal potential for rifapentine metabolism to be inhibited or induced by another drug, or for rifapentine to inhibit the metabolism of another drug based upon the characteristics of the esterase enzymes. Rifapentine does not induce its own metabolism. Since rifapentine is highly bound to albumin, drug displacement interactions may also occur.
In Clinical Study 008 patients were advised to take rifapentine at least 1 hour before or 2 hours after ingestion of antacids.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with rifapentine have not been completed. Rifapentine was negative in the following genotoxicity tests: in vitro gene mutation assay in bacteria (Ames test); in vitro point mutation test in Aspergillus nidulans; in vitro gene conversion assay in Saccharomyces cerevisiae; host‑mediated (mouse) gene conversion assay with Saccharomyces cerevisiae; in vitro Chinese hamster ovary cell/hypoxanthine-guanine-phosphoribosyl transferase (CHO/HGPRT) forward mutation assay; in vitro chromosomal aberration assay utilizing rat lymphocytes; and in vivo mouse bone marrow micronucleus assay. The 25-desacetyl metabolite of rifapentine was also negative in the in vitro gene mutation assay in bacteria (Ames test), the in vitro Chinese hamster ovary cell/hypoxanthine-guanine-phosphoribosyl transferase (CHO/HGPRT) forward mutation assay, and the in vivo mouse bone marrow micronucleus assay. This metabolite did induce chromosomal aberrations in an in vitro chromosomal aberration assay. Fertility and reproductive performance were not affected by oral administration of rifapentine to male and female rats at doses of up to one-third of the human dose (based on body surface area conversions).
Pregnancy Category C
Teratogenic Effects
Rifapentine has been shown to be teratogenic in rats and rabbits. In rats, when given in doses 0.6 times the human dose (based on body surface area comparisons) during the period of organogenesis, pups showed cleft palates, right aortic arch and increased incidence of delayed ossification and increased number of ribs. Rabbits treated with drug at doses between 0.3 and 1.3 times the human dose (based on body surface area comparison) displayed major malformations including ovarian agenesis, pes varus, arhinia, microphthalmia and irregularities of the ossified facial tissues (4 of 321 examined fetuses).
Nonteratogenic Effects
In rats, rifapentine administration was associated with increased resorption rate and post implantation loss, decreased mean fetus weight, increased number of stillborn pups and slightly increased mortality during lactation. Rabbits given 1.3 times the human dose (based on body surface area comparisons) showed higher post-implantation losses and an increased incidence of stillborn pups.
When rifapentine was administered at 0.3 times the human dose (based on body surface area comparisons) to mated female rats late in gestation (from day 15 of gestation to day 21 postpartum), pup weights and gestational survival (live pups born/pups born) were reduced compared to controls.
Pregnancy–Human Experience
There are no adequate and well-controlled studies in pregnant women. In Clinical Study 008, six patients randomized to rifapentine became pregnant; two had normal deliveries; two had first trimester spontaneous abortions, one had an elective abortion and one patient was lost to follow-up. Of the two patients who spontaneously aborted, co-morbid conditions of ethanol abuse in one and HIV infection in the other were noted.
When administered during the last few weeks of pregnancy, rifampin can cause postnatal hemorrhages in the mother and infant for which treatment with Vitamin K may be indicated. Thus, patients and infants who receive rifapentine during the last few weeks of pregnancy should have appropriate clotting parameters evaluated.
Rifapentine should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether rifapentine is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Since rifapentine may produce a red-orange discoloration of body fluids, there is a potential for discoloration of breast milk.
Pediatric Use
The safety and effectiveness of rifapentine in pediatric patients under the age of 12 have not been established. A pharmacokinetic study was conducted in 12- to 15-year-old healthy volunteers. (See ACTIONS/CLINICAL PHARMACOLOGY Special Populations for pharmacokinetic information).
Geriatric Use
Clinical studies of PRIFTIN did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy. (See ACTIONS/CLINICAL PHARMACOLOGY, Pharmacokinetics, Special Populations-Elderly).
ADVERSE REACTIONS
The investigators in the tuberculosis treatment clinical trial (Study 008) assessed the causality of adverse events as definitely, probably, possibly, unlikely or not related to one of the two drug regimens tested. The following table (Table 2-3) presents treatment-related adverse events deemed by the investigators to be at least possibly related to any of the four drugs in the regimens (rifapentine/rifampin, isoniazid, pyrazinamide, or ethambutol) which occurred in ≥1% of patients. Hyperuricemia was the most frequently reported event that was assessed as treatment related and was most likely related to the pyrazinamide since no cases were reported in the Continuation Phase when this drug was no longer included in the treatment regimen.
| Intensive Phase* | Continuation Phase† | Total | ||||
|---|---|---|---|---|---|---|
| Preferred Term | Rifapentine Combination (N=361) N (%) | Rifampin Combination (N=361) N (%) | Rifapentine Combination (N=321) N (%) | Rifampin Combination (N=307) N (%) | Rifapentine Combination (N=361) N(%) | Rifampin Combination (N=361) N (%) |
| Note: ≥1% refers to rifapentine in the TOTAL column. | ||||||
| Note: A patient may have experienced the same adverse event more than once during the course of the study, therefore, patient counts across the columns may not equal the patient counts in the TOTAL column. |
||||||
|
||||||
| Hyperuricemia | 78 (21.6) | 55 (15.2) | 0 | 0 | 78 (21.6) | 55 (15.2) |
| ALT increased | 12 (3.3) | 17 (4.7) | 6 (1.9) | 7 (2.3) | 18 (5.0) | 24 (6.6) |
| AST increased | 11 (3.0) | 16 (4.4) | 5 (1.6) | 7 (2.3) | 15 (4.2) | 23 (6.4) |
| Neutropenia | 7 (1.9) | 9 (2.5) | 12 (3.7) | 9 (2.9) | 18 (5.0) | 18 (5.0) |
| Pyuria | 11 (3.0) | 10 (2.8) | 6 (1.9) | 3 (1.0) | 14 (3.9) | 12 (3.3) |
| Proteinuria | 15 (4.2) | 10 (2.8) | 2 (0.6) | 1 (0.3) | 17 (4.7) | 11 (3.0) |
| Hematuria | 10 (2.8) | 12 (3.3) | 4 (1.2) | 4 (1.3) | 13 (3.6) | 15 (4.2) |
| Lymphopenia | 14 (3.9) | 13 (3.6) | 3 (0.9) | 1 (0.3) | 16 (4.4) | 14 (3.9) |
| Urinary casts | 11 (3.0) | 3 (0.8) | 4 (1.2) | 0 | 14 (3.9) | 3 (0.8) |
| Rash | 9 (2.5) | 19 (5.3) | 4 (1.2) | 3 (1.0) | 13 (3.6) | 21 (5.8) |
| Pruritus | 8 (2.2) | 15 (4.2) | 1 (0.3) | 1 (0.3) | 9 (2.5) | 16 (4.4) |
| Acne | 5 (1.4) | 3 (0.8) | 2 (0.6) | 1 (0.3) | 7 (1.9) | 4 (1.1) |
| Anorexia | 6 (1.7) | 8 (2.2) | 3 (0.9) | 4 (1.3) | 8 (2.2) | 10 (2.8) |
| Anemia | 7 (1.9) | 9 (2.5) | 2 (0.6) | 1 (0.3) | 9 (2.5) | 10 (2.8) |
| Leukopenia | 4 (1.1) | 4 (1.1) | 3 (0.9) | 5 (1.6) | 7 (1.9) | 8 (2.2) |
| Arthralgia | 9 (2.5) | 7 (1.9) | 0 | 0 | 9 (2.5) | 7 (1.9) |
| Pain | 7 (1.9) | 5 (1.4) | 0 | 1 (0.3) | 7 (1.9) | 6 (1.7) |
| Nausea | 7 (1.9) | 2 (0.6) | 0 | 1 (0.3) | 7 (1.9) | 3 (0.8) |
| Vomiting | 4 (1.1) | 6 (1.7) | 1 (0.3) | 1 (0.3) | 5 (1.4) | 7 (1.9) |
| Headache | 3 (0.8) | 4 (1.1) | 1 (0.3) | 3 (1.0) | 4 (1.1) | 7 (1.9) |
| Dyspepsia | 3 (0.8) | 5 (1.4) | 2 (0.6) | 3 (1.0) | 4 (1.1) | 8 (2.2) |
| Hypertension | 3 (0.8) | 0 (0.0) | 1 (0.3) | 1 (0.3) | 4 (1.1) | 1 (0.3) |
| Dizziness | 4 (1.1) | 0 | 0 | 1 (0.3) | 4 (1.1) | 1 (0.3) |
| Thrombocytosis | 4 (1.1) | 2 (0.6) | 0 | 0 | 4 (1.1) | 2 (0.6) |
| Diarrhea | 4 (1.1) | 0 | 0 | 0 | 4 (1.1) | 0 |
| Rash maculopapular | 4 (1.1) | 3 (0.8) | 0 | 0 | 4 (1.1) | 3 (0.8) |
| Hemoptysis | 2 (0.6) | 0 | 2 (0.6) | 0 | 4 (1.1) | 0 |
Treatment-related adverse events of moderate or severe intensity in <1% of the rifapentine combination therapy patients in Study 008 are presented below by body system.
Hepatic & Biliary: bilirubinemia, hepatitis
Dermatologic: urticaria, skin discoloration
Hematologic: thrombocytopenia, neutrophilia, leukocytosis, purpura, hematoma
Metabolic & Nutritional: hyperkalemia, hypovolemia, alkaline phosphatase increased, LDH increased
Body as a Whole - General: peripheral edema, fatigue
Gastrointestinal: constipation, esophagitis, gastritis, pancreatitis
Musculoskeletal: gout, arthrosis
Psychiatric: aggressive reaction
Three patients (two rifampin combination therapy patients and one rifapentine combination therapy patient) were discontinued in the Intensive Phase as a result of hepatitis with increased liver function tests (ALT, AST, LDH, and bilirubin). Concomitant medications for all three patients included isoniazid, pyrazinamide, ethambutol, and pyridoxine. The two rifampin patients and one rifapentine patient recovered without sequelae.
Twenty-two deaths occurred in Study 008 (eleven in the rifampin combination therapy group and eleven in the rifapentine combination therapy group). None of the deaths were attributed to study medication. In the study, 18/361 (5.0%) rifampin combination therapy patients discontinued the study due to an adverse event compared to 11/361 (3.0%) rifapentine combination therapy patients.
The overall occurrence rate of treatment-related adverse events was higher in males with the rifapentine combination regimen (50%) versus the rifampin combination regimen (43%), while in females the overall rate was greater in the rifampin combination group (68%) compared to the rifapentine combination group (59%). However, there were higher frequencies of treatment-related hematuria and ALT increases for female patients in both treatment groups compared to those for male patients.
Adverse events associated with rifampin may occur with rifapentine: effects of enzyme induction to increase metabolism resulting in decreased concentration of endogenous substrates, including adrenal hormones, thyroid hormones, and vitamin D.
OVERDOSAGE
There is no experience with the treatment of acute overdose with rifapentine at doses exceeding 1200 mg per dose.
In a pharmacokinetic study involving healthy volunteers (n=9), single oral doses up to 1200 mg have been administered without serious adverse events. The only adverse events reported with the 1200 mg dose were heartburn (3/8), headache (2/8) and increased urinary frequency (1/8). In clinical trials, tuberculosis patients ranging in age from 20 to 74 years accidentally received continuous daily doses of rifapentine 600 mg. Some patients received continuous daily dosing for up to 20 days without evidence of serious adverse effects. One patient experienced a transient elevation in SGPT and glucose (the latter attributed to pre-existing diabetes); a second patient experienced slight pruritus. While there is no experience with the treatment of acute overdose with rifapentine, clinical experience with rifamycins suggests that gastric lavage to evacuate gastric contents (within a few hours of overdose), followed by instillation of an activated charcoal slurry into the stomach, may help adsorb any remaining drug from the gastrointestinal tract.
Rifapentine and 25-desacetyl rifapentine are 97.7% and 93.2% plasma protein bound, respectively. Rifapentine and related compounds excreted in urine account for only 17% of the administered dose, therefore, neither hemodialysis nor forced diuresis is expected to enhance the systemic elimination of unchanged rifapentine from the body of a patient with PRIFTIN overdose.
DOSAGE AND ADMINISTRATION
PRIFTIN should not be used alone, in initial treatment or in retreatment of pulmonary tuberculosis. In the intensive phase of short-course therapy which is to continue for 2 months, 600 mg (four 150 mg tablets) of PRIFTIN should be given twice weekly with an interval of not less than 3 days (72 hours) between doses. For those patients with propensity to nausea, vomiting or gastrointestinal upset, administration of PRIFTIN with food may be useful. In the Intensive Phase, PRIFTIN must be administered in combination as part of an appropriate regimen which includes daily companion drugs. Compliance with all drugs in the Intensive Phase (ie, PRIFTIN, isoniazid, pyrazinamide, ethambutol, or streptomycin), especially on days when rifapentine is not administered, is imperative to assure early sputum conversion and protection against relapse. The Advisory Council for the Elimination of Tuberculosis, the American Thoracic Society and the Centers for Disease Control and Prevention also recommend that either streptomycin or ethambutol be added to the regimen unless the likelihood of isoniazid resistance is very low. The need for streptomycin or ethambutol should be reassessed when the results of susceptibility testing are known. An initial treatment regimen with less than four drugs may be considered if there is little possibility of drug resistance (that is, less than 4% primary resistance to isoniazid in the community, and the patient has had no previous treatment with antituberculosis medications, is not from a country with a high prevalence of drug resistance, and has no known exposure to a drug-resistant case) (see Reference 2).
Following the intensive phase, treatment should be continued with PRIFTIN once weekly for 4 months in combination with isoniazid or an appropriate agent for susceptible organisms. If the patient is still sputum smear or culture positive, if resistant organisms are present, or if the patient is HIV positive, follow the ATS/CDC treatment guidelines (see Reference 2).
Concomitant administration of pyridoxine (Vitamin B6) is recommended in the malnourished, in those predisposed to neuropathy (eg, alcoholics and diabetics), and in adolescents.
The above recommendations apply to patients with drug-susceptible organisms. Patients with drug-resistant organisms may require longer duration treatment with other drug regimens.
HOW SUPPLIED
PRIFTIN (rifapentine) 150 mg round normal convex dark-pink film-coated tablets debossed "Priftin" on top and "150" on the bottom, are packaged in aluminum formable foil blister strips placed in cartons of 32 tablets (4 strips of 8). Each strip of 8 tablets is inserted into an aluminum foil laminated pouch. (NDC 0088-2100-03).
Store at 25°C (77°F); excursions permitted 15–30°C (59–86°F) (see USP Controlled Room Temperature). Protect from excessive heat and humidity.
Revised December 2006
sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
References
1. Vernon A, et al. Acquired rifamycin monoresistance in patients with HIV-related tuberculosis treated with once-weekly rifapentine and isoniazid. The Lancet 1999; 353: 1843–1847.
2. American Thoracic Society, CDC. Treatment of tuberculosis and tuberculosis infection in adults and children. Am J Respir Crit Care Med. 149:1359–1374, 1994.
| Priftin (rifapentine) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
Revised: 08/2007sanofi-aventis U.S. LLC