HUMATROPE
-
somatropin
Eli Lilly and Company
----------
|
|||||||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Pediatric Patients
Growth Hormone Deficiency — Humatrope is indicated for the treatment of pediatric patients who have growth failure due to inadequate secretion of endogenous growth hormone (GH).
Short Stature Associated with Turner Syndrome — Humatrope is indicated for the treatment of short stature associated with Turner syndrome [see Clinical Studies (14.2)].
Idiopathic Short Stature — Humatrope is indicated for the treatment of idiopathic short stature, also called non-GH-deficient short stature, defined by height SDS ≤-2.25 and associated with growth rates unlikely to permit attainment of adult height in the normal range, in pediatric patients for whom diagnostic evaluation excludes other causes of short stature that should be observed or treated by other means [see Clinical Studies (14.3)]; SDS = standard deviation scores.
SHOX Deficiency — Humatrope is indicated for the treatment of short stature or growth failure in children with short stature homeobox-containing gene (SHOX) deficiency [see Clinical Studies (14.4)].
Small for Gestational Age — Humatrope is indicated for the treatment of growth failure in children born small for gestational age (SGA) who fail to demonstrate catch-up growth by age two to four years [see Clinical Studies (14.5)].
1.2 Adult Patients
Humatrope is indicated for the replacement of endogenous GH in adults with GH deficiency who meet either of the following two criteria [see Clinical Studies (14.1)]:
Adult-Onset (AO): Patients who have GH deficiency, either alone or associated with multiple hormone deficiencies (hypopituitarism), as a result of pituitary disease, hypothalamic disease, surgery, radiation therapy, or trauma; or
Childhood-Onset (CO): Patients who were GH deficient during childhood as a result of congenital, genetic, acquired, or idiopathic causes.
Patients who were treated with somatropin for GH deficiency in childhood and whose epiphyses are closed should be reevaluated before continuation of somatropin therapy at the reduced dose level recommended for GH deficient adults. According to current standards, confirmation of the diagnosis of adult GH deficiency in both groups involves an appropriate GH provocative test with two exceptions: (1) patients with multiple other pituitary hormone deficiencies due to organic disease; and (2) patients with congenital/genetic GH deficiency.
2 DOSAGE AND ADMINISTRATION
For subcutaneous injection.
Therapy with Humatrope should be supervised by a physician who is experienced in the diagnosis and management of pediatric patients with short stature associated with GH deficiency, Turner syndrome, idiopathic short stature, SHOX deficiency, small for gestational age birth, or adult patients with either childhood-onset or adult-onset GH deficiency.
2.1 Reconstitution
Vial — Each 5-mg vial of Humatrope should be reconstituted with 1.5 to 5 mL of Diluent for Humatrope. The diluent should be injected into the vial of Humatrope by aiming the stream of liquid gently against the vial wall. Following reconstitution, the vial should be swirled with a GENTLE rotary motion until the contents are completely dissolved. DO NOT SHAKE. The resulting solution should be clear. If the solution is cloudy or contains particulate matter, the contents MUST NOT be injected.
If sensitivity to the diluent should occur, the vials may be reconstituted with Bacteriostatic Water for Injection (Benzyl Alcohol preserved), USP or Sterile Water for Injection, USP. When Humatrope is reconstituted with Bacteriostatic Water for Injection, USP, the solution should be kept refrigerated at 36° to 46°F (2° to 8°C) and used within 14 days. It is important to note that benzyl alcohol used as a preservative in Bacteriostatic Water has been associated with toxicity in newborns. Therefore, Bacteriostatic Water for Injection must not be used to reconstitute Humatrope for use in a newborn infant. When Humatrope is to be administered to a newborn infant it should be reconstituted with the diluent provided or, if the infant is sensitive to the diluent, Sterile Water for Injection, USP. When reconstituted with Sterile Water for Injection the solution should be kept refrigerated at 36° to 46°F (2° to 8°C) and used within 24 hours.
Cartridge — The Humatrope cartridge has been designed for use only with the Humatrope injection device. Each cartridge of Humatrope should be reconstituted using only the diluent syringe that accompanies the cartridge and should not be reconstituted with the Diluent for Humatrope provided with Humatrope vials. The reconstituted solution should be clear. If the solution is cloudy or contains particulate matter, the contents MUST NOT be injected. Humatrope cartridges should not be used if the patient is allergic to metacresol or glycerin.
The somatropin concentrations for the reconstituted Humatrope cartridges are as follows:
| 6 mg cartridge (gold) | 2.08 mg/mL |
| 12 mg cartridge (teal) | 4.17 mg/mL |
| 24 mg cartridge (purple) | 8.33 mg/mL |
[See How Supplied (16.2) and Information for the Patient for comprehensive directions on Humatrope cartridge reconstitution].
2.2 General Administration Guidelines
For all indications, the following general principles for administration should be followed:
- When using the Humatrope vial the septum of the vial should be wiped with an alcoholic antiseptic solution before and after each injection to prevent contamination of the contents by repeated needle insertions. Sterile disposable syringes and needles should be used. The volume of the syringe should be small enough so that the prescribed dose can be withdrawn from the vial with reasonable accuracy.
- When using the Humatrope cartridge a sterile disposable needle should be used for each injection.
- Humatrope should be administered by subcutaneous injection with regular rotation of injection sites to avoid lipoatrophy.
- For pediatric patients the calculated weekly Humatrope dosage should be divided into equal doses given either 6 or 7 days per week.
- For adult patients the prescribed dose should be administered daily.
2.3 Dosing for Pediatric Patients
The Humatrope dosage and administration schedule should be individualized for each patient based on the growth response. Failure to increase height velocity, particularly during the first year of treatment, should prompt close assessment of compliance and evaluation of other causes of poor growth, such as hypothyroidism, under–nutrition, advanced bone age and antibodies to recombinant human growth hormone. Response to somatropin treatment tends to decrease with time. Somatropin treatment for stimulation of linear growth should be discontinued once epiphyseal fusion has occurred.
The recommended weekly dosages in milligrams (mg) per kilogram (kg) of body weight for pediatric patients are:
|
a Recent literature has recommended initial treatment with larger doses of somatropin (e.g., 0.067 mg/kg/day), especially in very short children (i.e., height SDS <–3), and/or older pubertal children, and that a reduction in dosage (e.g., gradually towards 0.033 mg/kg/day) should be considered if substantial catch-up growth is observed during the first few years of therapy. On the other hand, in younger SGA children (e.g., approximately <4 years) (who respond the best in general) with less severe short stature (i.e., baseline height SDS values between -2 and -3), consideration should be given to initiating treatment at a lower dose (e.g., 0.033 mg/kg/day), and titrating the dose as needed over time. In all children, clinicians should carefully monitor the growth response, and adjust the somatropin dose as necessary. |
|
| Growth hormone deficiency | 0.026 to 0.043 mg/kg/day (0.18 to 0.30 mg/kg/week) |
| Turner syndrome | up to 0.054 mg/kg/day (0.375 mg/kg/week) |
| Idiopathic short stature | up to 0.053 mg/kg/day (0.37 mg/kg/week) |
| SHOX deficiency | 0.050 mg/kg/day (0.35 mg/kg/week) |
| Small for gestational age | up to 0.067 mg/kg/day (0.47 mg/kg/week)a |
2.4 Dosing for Patients with Adult Growth Hormone Deficiency
Either of two approaches to Humatrope dosing may be followed: a non-weight-based regimen or a weight-based regimen.
Non-weight based — based on published consensus guidelines, a starting dose of approximately 0.2 mg/day (range, 0.15-0.30 mg/day) may be used without consideration of body weight. This dose can be increased gradually every 1-2 months by increments of approximately 0.1-0.2 mg/day, according to individual patient requirements based on the clinical response and serum insulin-like growth factor I (IGF-I) concentrations. The dose should be decreased as necessary on the basis of adverse events and/or serum IGF-I concentrations above the age- and gender-specific normal range. Maintenance dosages vary considerably from person to person, and between male and female patients.
Weight-based — based on the dosing regimen used in the original adult GH deficiency registration trials, the recommended dosage at the start of treatment is not more than 0.006 mg/kg (6 μg/kg) daily. The dose may be increased according to individual patient requirements to a maximum of 0.0125 mg/kg (12.5 μg/kg) daily. Clinical response, side effects, and determination of age- and gender-adjusted serum IGF-I concentrations should be used as guidance in dose titration.
A lower starting dose and smaller dose increments should be considered for older patients, who are more prone to the adverse effects of somatropin than younger individuals. In addition, obese individuals are more likely to manifest adverse effects when treated with a weight-based regimen. Estrogen-replete women may need higher doses than men. Oral estrogen administration may increase the dose requirements in women.
3 DOSAGE FORMS AND STRENGTHS
Humatrope is a sterile, white lyophilized powder available in the following vial and cartridge sizes:
- 5 mg vial and a 5-mL vial of Diluent for Humatrope
- 6 mg cartridge (gold) and a prefilled syringe of Diluent for Humatrope
- 12 mg cartridge (teal) and a prefilled syringe of Diluent for Humatrope
- 24 mg cartridge (purple) and a prefilled syringe of Diluent for Humatrope
Humatrope cartridges should be used only with the appropriate corresponding pen device.
4 CONTRAINDICATIONS
4.1 Acute Critical Illness
Treatment with pharmacologic amounts of somatropin is contraindicated in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure. Two placebo-controlled clinical trials in non-GH deficient adult patients (n=522) with these conditions in intensive care units revealed a significant increase in mortality (41.9% vs. 19.3%) among somatropin-treated patients (doses 5.3-8.0 mg/day) compared to those receiving placebo [see Warnings and Precautions (5.1)].
4.2 Prader-Willi Syndrome in Children
Somatropin is contraindicated in patients with Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea, or have severe respiratory impairment. There have been reports of sudden death when somatropin was used in such patients. Humatrope is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome. [See Warnings and Precautions (5.2)].
4.3 Active Malignancy
In general, somatropin is contraindicated in the presence of active malignancy. Any preexisting malignancy should be inactive and its treatment complete prior to instituting therapy with somatropin. Somatropin should be discontinued if there is evidence of recurrent activity. Since GH deficiency may be an early sign of the presence of a pituitary tumor (or, rarely, other brain tumors), the presence of such tumors should be ruled out prior to initiation of treatment. Somatropin should not be used in patients with any evidence of progression or recurrence of an underlying intracranial tumor.
4.4 Diabetic Retinopathy
Somatropin is contraindicated in patients with active proliferative or severe non-proliferative diabetic retinopathy.
4.5 Closed Epiphyses
Somatropin should not be used for growth promotion in pediatric patients with closed epiphyses.
4.6 Hypersensitivity
Humatrope is contraindicated in patients with a known hypersensitivity to somatropin or diluent. Localized reactions are the most common hypersensitivity reactions.
5 WARNINGS AND PRECAUTIONS
5.1 Acute Critical Illness
Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin [see Contraindications (4.1)]. The safety of continuing somatropin treatment in patients receiving replacement doses for approved indications who concurrently develop these illnesses has not been established. Therefore, the potential benefit of treatment continuation with somatropin in patients experiencing acute critical illnesses should be weighed against the potential risk.
5.2 Prader-Willi Syndrome in Children
There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. Patients with Prader-Willi syndrome should be evaluated for signs of upper airway obstruction and sleep apnea before initiation of treatment with somatropin. If, during treatment with somatropin, patients show signs of upper airway obstruction (including onset of, or increased, snoring) and/or new onset sleep apnea, treatment should be interrupted. All patients with Prader-Willi syndrome treated with somatropin should also have effective weight control and be monitored for signs of respiratory infection, which should be diagnosed as early as possible and treated aggressively [see Contraindications (4.2)]. Humatrope is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
5.3 Neoplasms
Patients with preexisting tumors or GH deficiency secondary to an intracranial lesion should be examined routinely for progression or recurrence of the underlying disease process. In pediatric patients, clinical literature has revealed no relationship between somatropin replacement therapy and central nervous system (CNS) tumor recurrence or new extracranial tumors. However, in childhood cancer survivors, an increased risk of a second neoplasm has been reported in patients treated with somatropin after their first neoplasm. Intracranial tumors, in particular meningiomas, in patients treated with radiation to the head for their first neoplasm, were the most common of these second neoplasms. In adults, it is unknown whether there is any relationship between somatropin replacement therapy and CNS tumor recurrence.
Patients should be monitored carefully for any malignant transformation of skin lesions (e.g., changes in pre-existing cutaneous nevi).
5.4 Glucose Intolerance and Diabetes Mellitus
Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses in susceptible patients. As a result, previously undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked, and new onset type 2 diabetes mellitus has been reported in patients taking somatropin. Therefore, glucose levels should be monitored periodically in all patients treated with somatropin, especially in those with risk factors for diabetes mellitus, such as obesity, Turner syndrome, or a family history of diabetes mellitus. Patients with preexisting type 1 or type 2 diabetes mellitus or impaired glucose tolerance should be monitored closely during somatropin therapy. The doses of antihyperglycemic drugs (e.g., insulin or oral agents) may require adjustment when somatropin therapy is instituted in these patients.
5.5 Intracranial Hypertension
Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin products. Symptoms usually occurred within the first eight (8) weeks after the initiation of somatropin therapy. In all reported cases, IH-associated signs and symptoms rapidly resolved after cessation of therapy or a reduction of the somatropin dose. Funduscopic examination should be performed routinely before initiating treatment with somatropin to exclude preexisting papilledema, and periodically during the course of somatropin therapy. If papilledema is observed by funduscopy during somatropin treatment, treatment should be stopped. If somatropin-induced IH is diagnosed, treatment with somatropin can be restarted at a lower dose after IH-associated signs and symptoms have resolved. Patients with Turner syndrome may be at increased risk for the development of IH.
5.6 Fluid Retention
Fluid retention during somatropin replacement therapy in adults may frequently occur. Clinical manifestations of fluid retention are usually transient and dose dependent.
5.7 Hypopituitarism
Patients with hypopituitarism (multiple pituitary hormone deficiencies) should have their other hormonal replacement treatments closely monitored during somatropin treatment.
5.8 Hypothyroidism
Undiagnosed/untreated hypothyroidism may prevent an optimal response to somatropin, in particular, the growth response in children. Patients with Turner syndrome have an inherently increased risk of developing autoimmune thyroid disease and primary hypothyroidism. In patients with GH deficiency, central (secondary) hypothyroidism may first become evident or worsen during somatropin treatment. Therefore, patients treated with somatropin should have periodic thyroid function tests performed, and thyroid hormone replacement therapy should be initiated or appropriately adjusted when indicated.
5.9 Slipped Capital Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis may occur more frequently in patients with endocrine disorders (including pediatric GH deficiency and Turner syndrome) or in patients undergoing rapid growth. Any pediatric patient with the onset of a limp or complaints of hip or knee pain during somatropin therapy should be carefully evaluated.
5.10 Progression of Preexisting Scoliosis in Pediatric Patients
Progression of scoliosis can occur in patients who experience rapid growth. Because somatropin increases growth rate, patients with a history of scoliosis who are treated with somatropin should be monitored for progression of scoliosis. However, somatropin has not been shown to increase the occurrence of scoliosis. Skeletal abnormalities including scoliosis are commonly seen in untreated patients with Turner syndrome. Scoliosis is also commonly seen in untreated patients with Prader-Willi syndrome. Physicians should be alert to these abnormalities, which may manifest during somatropin therapy.
5.11 Otitis Media and Cardiovascular Disorders in Patients with Turner Syndrome
Patients with Turner syndrome should be evaluated carefully for otitis media and other ear disorders, as these patients have an increased risk of ear and hearing disorders. Somatropin treatment may increase the occurrence of otitis media in patients with Turner syndrome. In addition, patients with Turner syndrome should be monitored closely for cardiovascular disorders (e.g., hypertension, aortic aneurysm or dissection, stroke) as patients with Turner syndrome are also at increased risk for these conditions.
5.12 Pancreatitis
Cases of pancreatitis have been reported rarely in children and adults receiving somatropin treatment, with some evidence supporting a greater risk in children compared with adults. Published literature indicates that girls who have Turner syndrome may be at greater risk than other somatropin-treated children. Pancreatitis should be considered in any somatropin-treated patient, especially a child, who develops abdominal pain.
5.13 Local and Systemic Reactions
When somatropin is administered subcutaneously at the same site over a long period of time, tissue atrophy may result. This can be avoided by rotating the injection site [see Dosage and Administration (2.2)]. As with any protein, local or systemic allergic reactions may occur. Parents/patients should be informed that such reactions are possible and that prompt medical attention should be sought if allergic reactions occur.
5.14 Laboratory Tests
Serum levels of inorganic phosphorus, alkaline phosphatase, parathyroid hormone and IGF-I may increase after somatropin therapy.
6 ADVERSE REACTIONS
6.1 Most Serious and/or Most Frequently Observed Adverse Reactions
This list presents the most seriousa and/or most frequently observedb adverse reactions during treatment with somatropin (including events observed in patients who received brands of somatropin other than Humatrope):
- aSudden death in pediatric patients with Prader-Willi syndrome who had risk factors including severe obesity, history of upper airway obstruction or sleep apnea and unidentified respiratory infection [see Contraindications (4.2) and Warnings and Precautions (5.2)]
- aIntracranial tumors, in particular meningiomas, in teenagers/young adults treated with radiation to the head for a first neoplasm who subsequently receive somatropin [see Contraindications (4.3) and Warnings and Precautions (5.3)]
- aPancreatitis [see Warnings and Precautions (5.12)]
- a,bGlucose intolerance including impaired glucose tolerance/impaired fasting glucose as well as overt diabetes mellitus [see Warnings and Precautions (5.4)]
- aIntracranial hypertension [see Warnings and Precautions (5.5)]
- aSignificant diabetic retinopathy [see Contraindications (4.4)]
- aSlipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.9)]
- aProgression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.10)]
- bFluid retention manifested by edema, arthralgia, myalgia, nerve compression syndromes including carpal tunnel syndrome/paraesthesias [see Warnings and Precautions (5.6)]
- aUnmasking of latent central hypothyroidism [see Warnings and Precautions (5.8)]
- aInjection site reactions/rashes and lipoatrophy (as well as rare generalized hypersensitivity reactions) [see Warnings and Precautions (5.13)]
6.2 Clinical Trials Experience
Because clinical trials are conducted under varying conditions, adverse reaction rates observed during the clinical trials performed with one somatropin formulation cannot always be directly compared to the rates observed during the clinical trials performed with a second somatropin formulation, and may not reflect the adverse reaction rates observed in practice.
Pediatric Patients
GH Deficiency
As with all protein pharmaceuticals, a small percentage of patients may develop antibodies to the protein. During the first 6 months of Humatrope therapy in 314 naive patients, only 1.6% developed specific antibodies to Humatrope (binding capacity ≥0.02 mg/L). None had antibody concentrations which exceeded 2 mg/L. Throughout 8 years of this same study, two patients (0.6%) had binding capacity >2 mg/L. Neither patient demonstrated a decrease in growth velocity at or near the time of increased antibody production. It has been reported that growth attenuation from pituitary-derived GH may occur when antibody concentrations are >1.5 mg/L.
In addition to an evaluation of compliance with the treatment program and of thyroid status, testing for antibodies to somatropin should be carried out in any patient who fails to respond to therapy.
In studies with GH deficient pediatric patients, injection site pain was reported infrequently. A mild and transient edema, which appeared in 2.5% of patients, was observed early during the course of treatment.
Turner Syndrome
In a randomized, concurrent-controlled, open-label trial, there was a statistically significant increase in the occurrence of otitis media (43% vs. 26%), ear disorders (18% vs. 5%) and surgical procedures (45% vs. 27%) in patients receiving Humatrope compared with untreated control patients (Table 1). A similar increase in otitis media was observed in an 18-month placebo-controlled trial.
|
a Open-label study. |
|||
|
b Dose=0.3 mg/kg/wk. |
|||
| Treatment Groupa | |||
| Adverse Reaction | Untreated | Humatropeb | Significance |
| Total Number of Patients | 62 | 74 | |
| Surgical procedure | 17 (27.4%) | 33 (44.6%) | p≤0.05 |
| Otitis media | 16 (25.8%) | 32 (43.2%) | p≤0.05 |
| Ear disorders | 3 (4.8%) | 13 (17.6%) | p≤0.05 |
Idiopathic Short Stature
In a randomized, placebo-controlled study of Humatrope treatment (0.22 mg/kg/week) to adult height in patients with idiopathic short stature, the adverse events reported in Humatrope-treated patients (Table 2) were similar to those observed in other pediatric populations treated with Humatrope. Mean serum glucose concentration did not change during Humatrope treatment. Mean fasting serum insulin concentration increased 10% in the Humatrope treatment group at the end of treatment relative to baseline, but remained within the normal reference range. For the same duration of treatment, the mean fasting serum insulin concentration decreased by 2% in the placebo group. The occurrence rates of above-range values for glucose, insulin, and HbA1c were similar in the Humatrope (somatropin)- and placebo-treated groups. No patient developed diabetes mellitus. Consistent with the known mechanism of growth hormone action, Humatrope-treated patients had greater mean increases, relative to baseline, in serum insulin-like growth factor-I (IGF-I) than placebo-treated patients at each study observation. However, there was no significant difference between the Humatrope and placebo treatment groups in the proportion of patients who had at least one serum IGF-I concentration more than 2.0 SD above the age- and gender-appropriate mean (Humatrope: 9 of 35 patients [26%]; placebo: 7 of 28 patients [25%]).
| Treatment Group | ||
| Adverse Reaction | Placebo | Humatrope |
| Total Number of Patients | 31 | 37 |
| Scoliosis | 4 (12.9%) | 7 (18.9%) |
| Otitis media | 2 (6.5%) | 6 (16.2%) |
| Hyperlipidemia | 1 (3.2%) | 3 (8.1%) |
| Gynecomastia | 1 (3.2%) | 2 (5.4%) |
| Hip pain | 0 | 1 (2.7%) |
| Arthralgia | 1 (3.2%) | 4 (10.8%) |
| Arthrosis | 2 (6.5%) | 4 (10.8%) |
| Myalgia | 4 (12.9%) | 9 (24.3%) |
| Hypertension | 0 | 1 (2.7%) |
The adverse events observed in the dose-response study (239 patients treated for 2 years) did not indicate a pattern suggestive of a somatropin dose effect. Among Humatrope dose groups, mean fasting blood glucose, mean glycosylated hemoglobin, and the incidence of elevated fasting blood glucose concentrations were similar. One patient developed abnormalities of carbohydrate metabolism (glucose intolerance and high serum HbA1c) on treatment.
SHOX Deficiency
Clinically significant adverse events (adverse events previously observed in association with growth hormone treatment in general) were assessed prospectively during the 2-year randomized, open-label study; those observed are presented in Table 3. In both treatment groups, the mean fasting plasma glucose concentration at the end of the first year was similar to the baseline value and remained in the normal range. No patient developed diabetes mellitus or had an above normal value for fasting plasma glucose at the end of one-year of treatment. During the 2 year study period, the proportion of patients who had at least one IGF-I concentration greater than 2.0 SD above the age- and gender-appropriate mean was 10 of 27 [37.0%] for the Humatrope-treated group vs. 0 of 24 patients [0.0%] for the untreated group. The proportion of patients who had at least one IGFBP-3 concentration greater than 2.0 SD above the age and gender appropriate mean was 16 of 27 [59.3%] for the Humatrope treated group vs. 7 of 24 [29.2%] for the untreated group.
|
a All events were non-serious. |
||
|
b Events are included only if reported for a greater number of Humatrope-treated than Untreated patients. |
||
|
c Percentage calculated for males only (1/12). |
||
| Adverse Reaction | Treatment Group | |
| Untreated | Humatrope | |
| Total Number of Patients | 25 | 27 |
| Patients with at least one event | 2 | 5 |
| Arthralgia | 2 (8.0%) | 3 (11.1%) |
| Gynecomastiac | 0 (0.0%) | 1 (8.3%) |
| Excessive number of cutaneous nevi | 0 (0.0%) | 2 (7.4%) |
| Scoliosis | 0 (0.0%) | 1 (3.7%) |
Small for Gestational Age
Study 1 — In a 2-year, multicenter, randomized study, 193 non-GH deficient children with short stature born SGA who failed to demonstrate catch-up growth were treated with 2 different Humatrope treatment regimens: a fixed dose of 0.067 mg/kg/day (FHD group) or an individually adjusted dose regimen (IAD group; starting dose 0.035 mg/kg/day which could be increased as early as Month 3 to 0.067 mg/kg/day based on a validated growth prediction model). The most frequently reported adverse events were common childhood infectious diseases. Adverse events possibly/probably related to Humatrope were otitis media and headaches (where there was a suggestion of a modest dose response), and slipped capital femoral epiphysis (1 child) [see Warnings and Precautions (5.9) and Adverse Reactions (6.1)]. There were no clear cut cases of new-onset diabetes mellitus, no children treated for hyperglycemia, and no children whose fasting blood glucose exceeded 126 mg/dL at any time during the study. However, 6 children (4 in the FHD group and 2 in the IAD group whose dose was increased from 0.035 mg/kg/day to 0.067 mg/kg/day [one at Month 3 and one at Year 1]) manifested impaired fasting glucose at Year 2. Two of these six children displayed impaired fasting glucose during the study as well, and one of them was required to discontinue Humatrope at Month 15 as a consequence [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)]. A modestly dose-dependent increase in mean serum IGF-I SDS concentrations within the reference range was observed; of note, at study completion, 20-25% of these children had serum IGF-I SDS values > +2.
Study 2 — A 2-year, open-label, single-arm study of Humatrope at a dosage of 0.067 mg/kg/day in 35 non-GH deficient children with short stature born SGA who failed to demonstrate catch-up growth did not reveal further safety data of note.
Study 3 — Additional safety information was obtained from 340 short children born SGA followed in an observational study who received an average Humatrope dosage of 0.041 mg/kg/day (maximum dose: 0.084 mg/kg/day) for an average of 3.0 years. Type 2 diabetes mellitus apparently precipitated by Humatrope therapy was reported in a single patient, but appeared to resolve after discontinuation of Humatrope treatment, as the child had a normal oral glucose tolerance test and was receiving no antihyperglycemic medications 9 months after the drug was discontinued. One patient manifested carpal tunnel syndrome [see Adverse Reactions (6.1)] and another developed an exacerbation of preexisting scoliosis [see Warnings and Precautions (5.10) and Adverse Reactions (6.1)] which may have been related to Humatrope treatment.
In both Study 1 and Study 2, after treatment with Humatrope, bone maturation did not accelerate excessively, and the timing of puberty was age-appropriate in boys and girls.
Therefore, it can be concluded that no novel adverse events potentially related to treatment with Humatrope were reported in either short-term study or were apparent after a review of the post-marketing, observational, safety database.
Adult Patients
In clinical studies in which high doses of Humatrope were administered to healthy adult volunteers, the following events occurred infrequently: headache, localized muscle pain, weakness, mild hyperglycemia, and glucosuria.
Adult-Onset GH Deficiency
In the first 6 months of controlled blinded trials during which patients received either Humatrope or placebo, adult-onset GH deficient adults who received Humatrope experienced a statistically significant increase in edema (Humatrope 17.3% vs. placebo 4.4%, p=0.043) and peripheral edema (11.5% vs. 0%, respectively, p=0.017). In patients with adult-onset GH deficiency, edema, muscle pain, joint pain, and joint disorder were reported early in therapy and tended to be transient or responsive to dosage titration.
Two of 113 adult-onset patients developed carpal tunnel syndrome after beginning maintenance therapy without a low dose (0.00625 mg/kg/day) lead-in phase. Symptoms abated in these patients after dosage reduction.
All treatment-emergent adverse events with ≥5% overall occurrence rate during 12 or 18 months of replacement therapy with Humatrope are shown in Table 4 (adult-onset patients) and in Table 5 (childhood-onset patients).
Adult patients treated with Humatrope who had been diagnosed with GH deficiency in childhood reported side effects less frequently than those with adult-onset GH deficiency.
|
a Abbreviations: GH=Humatrope; N=number of patients receiving treatment in the period stated; n=number of patients reporting each treatment-emergent adverse event. |
||||
|
b p=0.04 as compared to placebo (6 months). |
||||
|
c p=0.02 as compared to placebo (6 months). |
||||
| Adverse Reaction | 18 Months Exposure [Placebo (6 Months)/GH (12 Months)] (N=46) |
18 Months GH Exposure (N=52) |
||
| n | % | n | % | |
| Edemab | 7 | 15.2 | 11 | 21.2 |
| Arthralgia | 7 | 15.2 | 9 | 17.3 |
| Paresthesia | 6 | 13.0 | 9 | 17.3 |
| Myalgia | 6 | 13.0 | 7 | 13.5 |
| Pain | 6 | 13.0 | 7 | 13.5 |
| Rhinitis | 5 | 10.9 | 7 | 13.5 |
| Peripheral edemac | 8 | 17.4 | 6 | 11.5 |
| Back pain | 5 | 10.9 | 5 | 9.6 |
| Headache | 5 | 10.9 | 4 | 7.7 |
| Hypertension | 2 | 4.3 | 4 | 7.7 |
| Acne | 0 | 0 | 3 | 5.8 |
| Joint disorder | 1 | 2.2 | 3 | 5.8 |
| Surgical procedure | 1 | 2.2 | 3 | 5.8 |
| Flu syndrome | 3 | 6.5 | 2 | 3.9 |
Childhood-Onset GH Deficiency
Two double-blind, placebo-controlled trials were conducted in 67 adult patients with childhood-onset GH deficiency who had received previous somatropin treatment during childhood. Patients were randomized to receive either placebo injections or Humatrope (0.00625 mg/kg/day [6.25 μg/kg/day] for the first 4 weeks, then 0.0125 mg/kg/day [12.5 μg/kg/day] thereafter) for the first 6 months, followed by open-label Humatrope for the next 12 months for all patients. The patients in these studies reported side effects less frequently than those with adult-onset GH deficiency. During the placebo-controlled phase (first 6 months) of the study, elevations of serum glutamic oxaloacetic transferase were reported significantly more often for Humatrope-treated (12.5%) than placebo-treated patients (0.0%, p=0.031). No other events were reported significantly more often for Humatrope-treated patients during the placebo-controlled phase. The following events were reported for at least 5% of patients in either of the 2 treatment groups over the 18-month duration of the study, listed in descending order of maximum frequency for either group: aspartate aminotransferase increased 13%, headache 11%, edema 9%, pain 9%, alanine aminotransferase increased 6%, asthenia 6%, myalgia 6%, respiratory disorder 6%.
|
a Abbreviations: GH=Humatrope; N=number of patients receiving treatment in the period stated; n=number of patients reporting each treatment-emergent adverse event; ALT=alanine aminotransferase, formerly SGPT; AST=aspartate aminotransferase, formerly SGOT. |
||||
|
b p=0.03 as compared to placebo (6 months). |
||||
| Adverse Reaction | 18 Months Exposure [Placebo (6 Months)/GH (12 Months)] (N=35) | 18 Months GH Exposure
(N=32) |
||
| n | % | n | % | |
| Flu syndrome | 8 | 22.9 | 5 | 15.6 |
| AST increasedb | 2 | 5.7 | 4 | 12.5 |
| Headache | 4 | 11.4 | 3 | 9.4 |
| Asthenia | 1 | 2.9 | 2 | 6.3 |
| Cough increased | 0 | 0 | 2 | 6.3 |
| Edema | 3 | 8.6 | 2 | 6.3 |
| Hypesthesia | 0 | 0 | 2 | 6.3 |
| Myalgia | 2 | 5.7 | 2 | 6.3 |
| Pain | 3 | 8.6 | 2 | 6.3 |
| Rhinitis | 2 | 5.7 | 2 | 6.3 |
| ALT increased | 2 | 5.7 | 2 | 6.3 |
| Respiratory disorder | 2 | 5.7 | 1 | 3.1 |
| Gastritis | 2 | 5.7 | 0 | 0 |
| Pharyngitis | 5 | 14.3 | 1 | 3.1 |
6.3 Post-Marketing Experience
Because these adverse events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The adverse events reported during post-marketing surveillance do not differ from those listed/discussed above in Sections 6.1 and 6.2 in children and adults.
Other adverse events that have been reported in somatropin-treated patients include the following:
- Neurologic — Headaches (common in children and occasional in adults).
- Skin — Increase in size or number of cutaneous nevi, especially in patients with Turner syndrome and those with SHOX deficiency [see Warnings and Precautions (5.3)].
- Endocrine — Gynecomastia.
- Gastrointestinal — Pancreatitis. Cases of pancreatitis have been reported rarely in children and adults receiving somatropin treatment, with some evidence supporting a greater risk in children compared with adults. Published literature indicates that girls who have Turner syndrome may be at greater risk than other somatropin-treated children. Pancreatitis should be considered in any somatropin-treated patient, especially a child, who develops abdominal pain [see Warnings and Precautions (5.12)].
- Metabolic — New-onset type 2 diabetes mellitus in patients.
- Neoplasia — Leukemia has been reported in a small number of GH deficient children treated with somatropin, somatrem (methionylated rhGH), and GH of pituitary origin. It is uncertain whether these cases of leukemia are related to GH therapy, the pathology of GH deficiency itself, or other associated treatments such as radiation therapy. On the basis of current evidence, experts have not been able to conclude that GH therapy per se was responsible for these cases of leukemia. The risk for children with GH deficiency, if any, remains to be established [see Contraindications (4.3) and Warnings and Precautions (5.3)].
In an ongoing post-marketing observational study of somatropin treatment in 3,102 GH-deficient adults, hypertension, dyspnea, and sleep apnea were reported by 1% to less than 10% of patients after various durations of treatment.
7 DRUG INTERACTIONS
7.1 11β-Hydroxysteroid Dehydrogenase Type 1
The microsomal enzyme 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) is required for conversion of cortisone to its active metabolite, cortisol, in hepatic and adipose tissue. GH and somatropin inhibit 11βHSD-1. Consequently, individuals with untreated GH deficiency have relative increases in 11βHSD-1 and serum cortisol. Introduction of somatropin treatment may result in inhibition of 11βHSD-1 and reduced serum cortisol concentrations. As a consequence, previously undiagnosed central (secondary) hypoadrenalism may be unmasked and glucocorticoid replacement may be required in patients treated with somatropin. In addition, patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of somatropin treatment; this may be especially true for patients treated with cortisone acetate and prednisone since conversion of these drugs to their biologically active metabolites is dependent on the activity of 11βHSD-1.
7.2 Pharmacologic Glucocorticoid Therapy and Supraphysiologic Glucocorticoid Treatment
Pharmacologic glucocorticoid therapy and supraphysiologic glucocorticoid treatment may attenuate the growth promoting effects of somatropin in children. Therefore, glucocorticoid replacement dosing should be carefully adjusted in children receiving concomitant somatropin and glucocorticoid treatments to avoid both hypoadrenalism and an inhibitory effect on growth.
7.3 Cytochrome P450-Metabolized Drugs
Limited published data indicate that somatropin treatment increases cytochrome P450 (CP450)-mediated antipyrine clearance in man. These data suggest that somatropin administration may alter the clearance of compounds metabolized by CP450 liver enzymes (e.g., corticosteroids, sex steroids, anticonvulsants, cyclosporine). Therefore, careful monitoring is advised when somatropin is administered in combination with drugs metabolized by CP450 liver enzymes. However, formal drug interaction studies have not been conducted.
7.4 Oral Estrogen
Because oral estrogens may reduce the serum IGF-I response to somatropin treatment, girls and women receiving oral estrogen replacement may require greater somatropin dosages [see Dosage and Administration (2.4)].
7.5 Insulin and/or Other Hypoglycemic Agents
Patients with diabetes mellitus who receive concomitant treatment with somatropin may require adjustment of their doses of insulin and/or other hypoglycemic agents [see Warnings and Precautions (5.4)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C — Animal reproduction studies have not been conducted with Humatrope. It is not known whether Humatrope can cause fetal harm when administered to a pregnant woman or can affect reproductive capacity. Humatrope should be given to a pregnant woman only if clearly needed.
8.3 Nursing Mothers
There have been no studies conducted with Humatrope in nursing mothers. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Humatrope is administered to a nursing woman.
8.5 Geriatric Use
The safety and effectiveness of Humatrope in patients aged 65 years and over has not been evaluated in clinical studies. Elderly patients may be more sensitive to the action of somatropin, and therefore may be more prone to development of adverse reactions. A lower starting dose and smaller dose increments should be considered for older patients [see Dosage and Administration (2.4)].
9 DRUG ABUSE AND DEPENDENCE
Inappropriate use of somatropin by individuals who do not have indications for which somatropin is approved, may result in significant negative health consequences. Somatropin is not a drug of dependence.
10 OVERDOSAGE
Short-term — Acute overdosage could lead initially to hypoglycemia and subsequently to hyperglycemia.
Long-term — Long-term overdosage could result in signs and symptoms of gigantism or acromegaly consistent with the known effects of excess endogenous human GH.
11 DESCRIPTION
Humatrope (somatropin, rDNA origin, for injection) is a polypeptide hormone of recombinant DNA origin. Humatrope is synthesized in a strain of Escherichia coli that has been modified by the addition of the gene for human GH. The peptide is comprised of 191 amino acid residues and has a molecular weight of about 22,125 daltons. The amino acid sequence of the peptide is identical to that of human GH of pituitary origin.
Humatrope is a sterile, white, lyophilized powder intended for subcutaneous or intramuscular administration after reconstitution to its liquid form. Humatrope is a highly purified preparation. Phosphoric acid and/or sodium hydroxide may have been added to adjust the pH. Reconstituted solutions have a pH of approximately 7.5. This product is oxygen sensitive.
Vial — Each vial of Humatrope contains 5 mg somatropin (15 IU or 225 nanomoles); 25 mg mannitol; 5 mg glycine; and 1.13 mg dibasic sodium phosphate. Each vial is supplied in a combination package with an accompanying 5-mL vial of diluting solution (diluent). The diluent contains Water for Injection with 0.3% metacresol as a preservative and 1.7% glycerin.
Cartridge — Cartridges of Humatrope contain either 6 mg (18 IU), 12 mg (36 IU), or 24 mg (72 IU) of somatropin. Each Humatrope cartridge contains the following:
| Cartridge | |||
| 6 mg
(gold) | 12 mg
(teal) | 24 mg
(purple) |
|
| Component | |||
| Somatropin | 6 mg | 12 mg | 24 mg |
| Mannitol | 18 mg | 36 mg | 72 mg |
| Glycine | 6 mg | 12 mg | 24 mg |
| Dibasic sodium phosphate | 1.36 mg | 2.72 mg | 5.43 mg |
Each cartridge is supplied in a combination package with an accompanying syringe containing approximately 3 mL of diluting solution (diluent). The diluent contains Water for Injection; 0.3% metacresol as a preservative; and 1.7%, 0.29%, and 0.29% glycerin in the 6, 12, and 24 mg cartridges, respectively.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
GH binds to dimeric GH receptors located within the cell membranes of target tissue cells. This interaction results in intracellular signal transduction and subsequent induction of transcription and translation of GH-dependent proteins including IGF-I, IGF BP-3 and acid-labile subunit. GH has direct tissue and metabolic effects, including stimulation of chondrocyte differentiation, stimulation of lipolysis and stimulation of hepatic glucose output. In addition, some effects of somatropin are mediated indirectly by IGF-I, including stimulation of protein synthesis and chondrocyte proliferation.
12.2 Pharmacodynamics
In vitro, preclinical, and clinical testing have demonstrated that Humatrope is therapeutically equivalent to human GH of pituitary origin and achieves equivalent pharmacokinetic profiles in healthy adults. The following effects have been reported for human GH of pituitary origin, and/or somatropin.
Cell Growth — Total numbers of muscle cells are reduced in GH deficient children. Somatropin increases the number and size of muscle cells in such children.
Skeletal Growth — Somatropin stimulates skeletal growth in children with GH deficiency as a result of effects on the growth plates (epiphyses) of long bones. Concentrations of IGF-I, which play a role in skeletal growth, are low in the serum of GH deficient children but increase during somatropin treatment in most patients. The stimulation of skeletal growth increases linear growth rate (height velocity) in most somatropin-treated children.
Protein Metabolism — Linear growth is facilitated in part by increased cellular protein synthesis as reflected by nitrogen retention, which can be demonstrated by decreased urinary nitrogen excretion and serum urea nitrogen.
Connective Tissue Metabolism — Somatropin stimulates the synthesis of chondroitin sulfate and collagen, and increases the urinary excretion of hydroxyproline.
Carbohydrate Metabolism — GH has a physiological role in the maintenance of normoglycemia during times of substrate restriction (e.g., fasting), via mechanisms such as stimulation of hepatic gluconeogenesis and suppression of insulin-stimulated glucose uptake by peripheral tissues. Because of these actions GH is considered an insulin antagonist with respect to carbohydrate metabolism. Consequently, the fasting hypoglycemia that may occur in some children with hypopituitarism may be improved by somatropin treatment. As an extension of its physiological actions, supraphysiological GH concentrations may increase glucose production sufficiently to stimulate insulin secretion to maintain normoglycemia. Large doses of somatropin may impair glucose tolerance if compensatory insulin secretion is inadequate. Administration of somatropin to healthy adults and patients with Turner syndrome resulted in increases in mean serum fasting and postprandial insulin concentrations, although mean values remained in the normal range. In addition, mean HbA1c concentrations and mean fasting and postprandial glucose concentrations remained in the normal range.
Lipid Metabolism — Somatropin stimulates intracellular lipolysis, and administration of somatropin leads to an increase in plasma free fatty acids and triglycerides. Untreated GH deficiency is associated with increased body fat stores, including increased abdominal visceral and subcutaneous adipose tissue. Treatment of GH deficient patients with somatropin results in a general reduction of fat stores, and decreased serum concentrations of low density lipoprotein (LDL) cholesterol.
Mineral Metabolism — Administration of somatropin results in an increase in total body potassium and phosphorus and to a lesser extent sodium, probably as the result of cell growth. Serum concentrations of inorganic phosphate increase in somatropin-treated GH deficient children because of the metabolic activities associated with bone growth. Although urinary calcium excretion is increased, there is a simultaneous increase in calcium absorption from the intestine. Consequently, serum calcium concentrations generally are not altered, although negative calcium balance may occur occasionally during somatropin treatment. Associated with the changes in mineral metabolism, parathyroid hormone may increase during somatropin treatment.
12.3 Pharmacokinetics
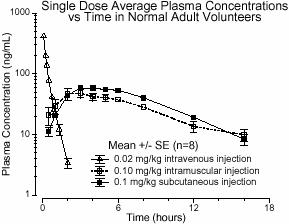
Absorption — Humatrope has been studied following intramuscular, subcutaneous, and intravenous administration in adult volunteers (see Figure 1). The absolute bioavailability of somatropin is 75% and 63% after subcutaneous and intramuscular administration, respectively.
Distribution — The volume of distribution of somatropin after intravenous injection is about 0.07 L/kg (Table 6).
Metabolism — Extensive metabolism studies have not been conducted. The metabolic fate of somatropin involves classical protein catabolism in both the liver and kidneys. In renal cells, at least a portion of the breakdown products of somatropin is returned to the systemic circulation. In healthy volunteers, mean somatropin clearance is 0.14 L/hr/kg. The mean half-life of intravenous somatropin is 0.36 hours, whereas subcutaneously and intramuscularly administered somatropin have mean half-lives of 3.8 and 4.9 hours, respectively. The longer half-life observed after subcutaneous or intramuscular administration is due to slow absorption from the injection site.
Excretion — Urinary excretion of intact Humatrope has not been measured. Small amounts of somatropin have been detected in the urine of pediatric patients following replacement therapy.
Geriatric patients — The pharmacokinetics of Humatrope have not been studied in patients greater than 65 years of age.
Pediatric patients — The pharmacokinetics of Humatrope in pediatric patients are similar to those of adults.
Gender — No gender-specific pharmacokinetic studies have been performed with Humatrope. The available literature indicates that the pharmacokinetics of somatropin are similar in men and women.
Race — No data are available.
Renal, hepatic insufficiency — No studies have been performed with Humatrope.
|
a Abbreviations: Cmax=maximum concentration; t1/2=half-life; AUC0-∞=area under the curve; Cls=systemic clearance; Vβ=volume distribution; iv=intravenous; SD=standard deviation; im=intramuscular; sc=subcutaneous. |
|||||
|
b Based on previous International Standard of 2.7 IU=1 mg. |
|||||
| Cmax
(ng/mL) | t1/2
(hr) | AUC0-∞
(ng•hr/mL) | Cls
(L/kg•hr) | Vβ
(L/kg) |
|
| 0.02 mg (0.05 IUb)/kg, iv
Mean (SD) | 415 (75) | 0.363 (0.053) | 156 (33) | 0.135 (0.029) | 0.0703 (0.0173) |
| 0.1 mg (0.27 IUb)/kg, im
Mean (SD) | 53.2 (25.9) | 4.93 (2.66) | 495 (106) | 0.215 (0.047) | 1.55 (0.91) |
| 0.1 mg (0.27 IUb)/kg, sc
Mean (SD) | 63.3 (18.2) | 3.81 (1.40) | 585 (90) | 0.179 (0.028) | 0.957 (0.301) |
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
There has been no evidence to date of Humatrope-induced mutagenicity. No long-term animal studies for carcinogenicity or impairment of fertility with somatropin have been performed.
14 CLINICAL STUDIES
14.1 Adult Patients with Growth Hormone Deficiency
Two multicenter trials in patients with adult-onset GH deficiency (n=98) and two studies in patients with childhood-onset GH deficiency (n=67) were designed to assess the effects of replacement therapy with Humatrope. These four studies each included a 6-month randomized, blinded, placebo-controlled phase, during which approximately half of the patients received placebo injections, while the other half received Humatrope injections. The Humatrope dosages for all studies were identical: 1 month of treatment at 0.00625 mg/kg/day (6.25μg/kg/day) followed by 0.0125 mg/kg/day (12.5 μg/kg/day) for the next 5 months. The 6-month, double-blind phase was followed by 12 months of open-label Humatrope treatment for all patients. The primary efficacy measures were body composition (lean body mass and fat mass), lipid parameters, and quality of life, as measured by the Nottingham Health Profile (a general health-related quality of life questionnaire). Lean body mass was determined by bioelectrical impedance analysis (BIA), validated with potassium 40. Body fat was assessed by BIA and sum of skinfold thickness. Lipid subfractions were analyzed by standard assay methods in a central laboratory. Adult-onset patients and childhood-onset patients differed by diagnosis (organic vs. idiopathic pituitary disease), body size (average vs. small [mean height and weight]), and age (mean 44 vs. 29 years).
In patients with adult-onset GH deficiency, Humatrope treatment (vs. placebo) resulted in an increase in mean lean body mass (2.59 vs. -0.22 kg, p<0.001) and a decrease in body fat (-3.27 vs. 0.56 kg, p<0.001). Similar changes were seen in childhood-onset GH deficient patients. These significant changes in lean body mass persisted throughout the 18-month period for both the adult-onset and childhood-onset groups; the changes in fat mass persisted in the childhood-onset group. Serum concentrations of high-density lipoprotein (HDL) cholesterol which were low at baseline (mean, 30.1 mg/mL and 33.9 mg/mL in adult-onset and childhood-onset patients, respectively) had normalized by the end of 18 months of Humatrope treatment (mean change of 13.7 and 11.1 mg/dL for the adult-onset and childhood-onset groups, respectively p<0.001). After 6 months, the physical mobility and social isolation domains on the Nottingham Health Profile were significantly improved in Humatrope-treated vs. placebo-treated patients with adult-onset GH deficiency (p<0.01) (Table 7). There were no significant between-group differences (Humatrope vs. placebo) for the other Nottingham Health Profile domains (energy level, emotional reactions, sleep, pain) in patients with adult-onset GH deficiency, and no significant between-group differences in any of the domains were demonstrated for patients with childhood-onset GH deficiency.
Two additional studies on the effect of Humatrope on exercise capacity were conducted. Improved physical function was documented by increased exercise capacity (VO2 max, p<0.005) and work performance (Watts, p<0.01).
|
a An improvement in score is indicated by a more negative change in the score. |
|||
|
b To account for multiple analyses, appropriate statistical methods were applied and the required level of significance is 0.01. |
|||
|
c NS=not significant. |
|||
| Outcome Measure | Placebo (6 Months) | Humatrope Therapy (6 Months) | Significance |
| Energy level | -11.4 | -15.5 | NSc |
| Physical mobility | -3.1 | -10.5 | p<0.01 |
| Social isolation | 0.5 | -4.7 | p<0.01 |
| Emotional reactions | -4.5 | -5.4 | NSc |
| Sleep | -6.4 | -3.7 | NSc |
| Pain | -2.8 | -2.9 | NSc |
Two studies evaluating the effect of Humatrope on bone mineralization were conducted subsequently. In a 2-year, randomized, double-blind, placebo-controlled trial, 67 patients with previously untreated adult-onset GH deficiency received placebo or Humatrope injections titrated to maintain serum IGF-I within the age-adjusted normal range. In men, but not women, lumbar spine bone mineral density (BMD) increased with Humatrope treatment compared to placebo, with a treatment difference of approximately 4% (p=0.001). There was no significant change in hip BMD with Humatrope treatment in men or women, when compared to placebo.
In a 2-year, open-label, randomized trial, 149 patients with childhood-onset GH deficiency who had completed pediatric somatropin therapy, had attained final height (height velocity <1 cm/yr) and were confirmed to be GH-deficient as young adults (commonly referred to as transition patients), were randomized to receive Humatrope 0.0125 mg/kg/day (12.5 μg/kg/day), Humatrope 0.025 mg/kg/day (25 μg/kg/day), or no injections (control). Patients who were randomized to treatment with Humatrope at 12.5 μg/kg/day achieved a 2.9% greater increase from baseline than control patients in total body bone mineral content (BMC) (8.1 ± 9.0% vs. 5.2 ± 8.2%, p=0.02), whereas patients treated with Humatrope at 25 μg/kg/day had no significant change in BMC. These results include data from patients who received less than 2 years of treatment. A greater treatment effect was observed for patients who completed 2 years of treatment. Increases in lumbar spine BMD and BMC were also statistically significant compared to control with the 12.5 μg/kg/day dose but not the 25 μg/kg/day dose. Hip BMD and BMC did not change significantly compared to control with either dose. The effect of GH treatment on BMC and BMD in transition patients at doses lower than12.5 μg/kg/day was not studied. The effect of Humatrope on the occurrence of osteoporotic fractures has not been studied.
14.2 Pediatric Patients with Turner Syndrome
One long-term, randomized, open-label, Canadian multicenter, concurrently controlled study, two long-term, open-label multicenter, historically controlled US studies and one long-term, randomized, US dose-response study were conducted to evaluate the efficacy of somatropin treatment of short stature due to Turner syndrome.
The Canadian randomized study compared near-adult height outcomes for Humatrope-treated patients to those of a concurrent control group who received no injections. The Humatrope-treated patients received a dosage of 0.3 mg/kg/week given in divided doses 6 times per week from a mean age of 11.7 years for a mean duration of 4.7 years. Puberty was induced with a standardized estrogen regimen initiated at 13 years of age for both treatment groups. The Humatrope-treated group (n=27) attained a mean (± SD) near-final height of 146.0 ± 6.2 cm; the untreated control group (n=19) attained a near-final height of 142.1 ± 4.8 cm. By analysis of covariance (with adjustments for baseline height and mid-parental height), the effect of somatropin treatment was a mean height increase of 5.4 cm (p=0.001).
In two of the US studies, the effect of long-term somatropin treatment (0.375 mg/kg/week given in divided doses either 3 times per week or daily) on adult height was determined by comparing adult heights in the treated patients with those of age-matched historical controls with Turner syndrome who received no growth-promoting therapy. Puberty was induced with a standardized estrogen regimen initiated after 14 years of age in one study; in the second study patients treated with early somatropin (before 11 years of age) were randomized to begin pubertal induction at either age 12 (n=26) or 15 (n=29) years (conjugated estrogens, 0.3 mg escalating to 0.625 mg daily); those whose somatropin was initiated after 11 years of age began estrogen replacement after 1 year of somatropin. Mean height gains from baseline to adult (or near-adult) height ranged from 5.0 to 8.3 cm, depending on age at initiation of somatropin treatment and estrogen replacement (Table 8).
In the third US study, a randomized, blinded dose-response study, patients were treated from a mean age of 11.1 years for a mean duration of 5.3 years with a weekly Humatrope dosage of either 0.27 mg/kg or 0.36 mg/kg administered in divided doses 3 or 6 times weekly. The mean near-final height of Humatrope-treated patients was 148.7 ± 6.5 cm (n=31). When compared to historical control data, the mean gain in adult height was approximately 5 cm.
In summary, patients with Turner syndrome (total n=181 from the 4 studies above) treated to adult height achieved statistically significant average height gains ranging from 5.0 to 8.3 cm.
|
a Data shown are mean values. |
|||||||
|
b RCT: randomized controlled trial; MHT: matched historical controlled trial; RDT: randomized dose-response trial. |
|||||||
|
c Analysis of covariance vs. controls. |
|||||||
|
d Compared with historical data. |
|||||||
|
e GH age <11 yr, estrogen age 15 yr. |
|||||||
|
f GH age <11 yr, estrogen age 12 yr. |
|||||||
|
g GH age >11 yr, estrogen at month 12. |
|||||||
| Study | Group | Study Designb | Number at Adult Height | GH Age (yr) | Estrogen Age (yr) | GH Duration (yr) | Adult Height Gain (cm)c |
| Canadian | RCT | 27 | 11.7 | 13 | 4.7 | 5.4 | |
| US 1 | MHT | 17 | 9.1 | 15.2 | 7.6 | 7.4 | |
| US 2 | Ae | MHT | 29 | 9.4 | 15 | 6.1 | 8.3 |
| Bf | 26 | 9.6 | 12.3 | 5.6 | 5.9 | ||
| Cg | 51 | 12.7 | 13.7 | 3.8 | 5 | ||
| US 3 | RDT | 31 | 11.1 | 8-13.5 | 5.3 | ~5d | |
14.3 Pediatric Patients with Idiopathic Short Stature
Two randomized, multicenter trials, 1 placebo-controlled and 1 dose-response, were conducted in pediatric patients with idiopathic short stature, also called non-GH-deficient short stature. The diagnosis of idiopathic short stature was made after excluding other known causes of short stature, as well as GH deficiency. Limited safety and efficacy data are available below the age of 7 years. No specific studies have been conducted in pediatric patients with familial short stature. The placebo-controlled study enrolled 71 pediatric patients (55 males, 16 females) 9 to 15 years old (mean age 12.4 ± 1.5 years), with short stature, 68 of whom received Humatrope. Patients were predominately prepubertal (Tanner I, 45%) or in early puberty (Tanner II, 47%) at baseline. In this double-blind trial, patients received subcutaneous injections of either Humatrope 0.222 mg/kg/week (equivalent to 32 μg/kg/day), or placebo given in divided doses 3 times per week until height velocity decreased to ≤1.5 cm/year (“final height”). Final height measurements were available for 33 subjects (22 Humatrope, 11 placebo) after a mean treatment duration of 4.4 years (range 0.1-9.1 years).
The Humatrope-treated group achieved a mean final height SDS of -1.8 (Table 9), whereas placebo-treated patients had a mean final height SDS of -2.3 (mean treatment difference, 0.51 SDS, p=0.017). Height gain across the duration of the study and final height SDS minus baseline predicted height SDS were also significantly greater in Humatrope-treated patients than in placebo-treated patients (Tables 9 and 10). In addition, the number of patients whose final height was above the 5th percentile of the general population height standard for age and sex was significantly greater in the Humatrope group than the placebo group (41% vs. 0%, p<0.05), as was the number of patients who gained at least 1 SDS unit in height across the duration of the study (50% vs. 0%, p<0.05).
|
a Abbreviations: BPH=baseline predicted height; CI=confidence interval; FH=final height; NA=not applicable; SDS=standard deviation score. |
||||
|
b For final height population. |
||||
|
c Between-group comparison was performed using analysis of covariance with baseline predicted height SDS as the covariate. Treatment effect is expressed as least squares mean (95% CI). |
||||
| Placebo (n=11) Mean (SD) | Humatrope (n=22) Mean (SD) | Treatment Effect Mean (95%CI) | p-value | |
| Baseline height SDS | -2.75 (0.6) | -2.7 (0.6) | NA | 0.77 |
| BPH SDS | -2.3 (0.8) | -2.1 (0.7) | NA | 0.53 |
| Final height SDSc | -2.3 (0.6) | -1.8 (0.8) | 0.51 (0.10, 0.92) | 0.017 |
| FH SDS - baseline height SDS | 0.4 (0.2) | 0.9 (0.7) | 0.51 (0.04, 0.97) | 0.034 |
| FH SDS - BPH SDS | -0.1 (0.6) | 0.3 (0.6) | 0.46 (0.02, 0.89) | 0.043 |
The dose-response study included 239 pediatric patients (158 males, 81 females), 5 to 15 years old, (mean age 9.8 ± 2.3 years). Mean ± SD baseline characteristics included: height SDS -3.21 ± 0.70, predicted adult height SDS -2.63 ± 1.08, and height velocity SDS -1.09 ± 1.15. All but 3 patients were prepubertal. Patients were randomized to one of three Humatrope treatment groups: 0.24 mg/kg/week (equivalent to 34 μg/kg/day); 0.24 mg/kg/week for 1 year, followed by 0.37 mg/kg/week (equivalent to 53 μg/kg/day); and 0.37 mg/kg/week. The primary hypothesis of this study was that treatment with Humatrope would increase height velocity during the first 2 years of therapy in a dose-dependent manner. Additionally, after completing the initial 2-year dose-response phase of the study, 50 patients were followed to final height.
Patients who received the Humatrope dosage of 0.37 mg/kg/week had a significantly greater increase in mean height velocity after 2 years of treatment than patients who received 0.24 mg/kg/week (4.04 vs. 3.27 cm/year, p=0.003). The mean difference between final height and baseline predicted height was 7.2 cm for patients who received Humatrope 0.37 mg/kg/week and 5.4 cm for patients who received 0.24 mg/kg/week (Table 10). While no patient had height above the 5th percentile in any dosage group at baseline, 82% of the patients who received 0.37 mg/kg/week and 47% of the patients who received 0.24 mg/kg/week achieved final heights above the 5th percentile of the general population height standards (p=NS).
|
a Abbreviations: FH=final height; PH=predicted height; CI=confidence interval; cm=centimeters. |
|||||
| Placebo-controlled Trial 3x per week dosing | Dose Response Trial 6x per week dosing |
||||
| Placebo (n=10) | Humatrope 0.22 mg/kg (n=22) | Humatrope 0.24 mg/kg (n=13) | Humatrope 0.24/0.37 mg/kg (n=13) | Humatrope 0.37 mg/kg (n=13) |
|
| FH - Baseline PH Mean (95% CI), cm | -0.7 (-3.6, 2.3) | +2.2 (0.4, 3.9) | +5.4 (2.8, 7.9) | +6.7 (4.1, 9.2) | +7.2 (4.6, 9.8) |
14.4 Pediatric Patients with SHOX Deficiency
SHOX deficiency may result either from a deletion of one copy of the short stature homeobox-containing (SHOX) gene or from a mutation within or outside one copy of the SHOX gene that impairs the production or function of SHOX protein.
A randomized, controlled, two-year, three-arm, open-label study was conducted to evaluate the efficacy of Humatrope treatment of short stature in pediatric patients with SHOX deficiency who were not GH–deficient. 52 patients (24 male, 28 female) with SHOX deficiency, 3.0 to 12.3 years of age, were randomized to either a Humatrope-treated arm (27 patients; mean age 7.3 ± 2.1 years) or an untreated control arm (25 patients; mean age 7.5 ± 2.7 years). To determine the comparability of treatment effect between patients with SHOX deficiency and patients with Turner syndrome, the third study arm enrolled 26 patients with Turner syndrome, 4.5 to 11.8 years of age (mean age 7.5 ± 1.9 years), to Humatrope treatment. All patients were prepubertal at study entry. Patients in the Humatrope-treated group(s) received daily subcutaneous injections of 0.05 mg/kg (50 μg/kg) of Humatrope, equivalent to 0.35 mg/kg/week. Patients in the untreated group received no injections.
Patients with SHOX deficiency who received Humatrope had significantly greater first-year height velocity than untreated patients (8.7 cm/year vs. 5.2 cm/year, p<0.001, primary efficacy analysis) and similar first-year height velocity to Humatrope-treated patients with Turner syndrome (8.7 cm/year vs. 8.9 cm/year). In addition, patients who received Humatrope had significantly greater second year height velocity, and first- and second-year height gain (cm and SDS) than untreated patients (Table 11).
|
a Positive values favor Humatrope |
||||
|
b Statistically significantly different from untreated, p<0.001. |
||||
|
c Statistically significantly different from untreated, p<0.05. |
||||
| SHOX Deficiency | Turner Syndrome | |||
| Untreated (n=24) Mean (SD) | Humatrope (n=27) Mean (SD) | Treatment Differencea Mean (95% CI) | Humatrope (n=26) Mean (SD) |
|
| Height Velocity (cm/yr) | ||||
| 1st Year | 5.2 (1.1) | 8.7 (1.6)b | +3.5 (2.8, 4.2) | 8.9 (2.0) |
| 2nd Year | 5.4 (1.2) | 7.3 (1.1)b | +2.0 (1.3, 2.6) | 7.0 (1.1) |
| Height Gain (cm) | ||||
| Baseline to 1st Year | +5.4 (1.2) | +9.1 (1.5)b | +3.7 (2.9, 4.5) | +8.9 (1.9) |
| Baseline to 2nd Year | +10.5 (1.9) | +16.4 (2.0)b | +5.8 (4.6, 7.1) | +15.7 (2.7) |
| Height SDS Gain | ||||
| Baseline to 1st Year | +0.1 (0.5) | +0.7 (0.5)b | +0.5 (0.3, 0.8) | +0.8 (0.5) |
| Baseline to 2nd Year | +0.2 (0.5) | +1.2 (0.7)b | +1.0 (0.7, 1.3) | +1.2 (0.7) |
| Patients with height SDS > -2.0 at 2 years | 1 (4%) | 11 (41%)c | -- | 8 (31%) |
14.5 Pediatric Patients Born Small for Gestational Age (SGA) Who Fail to Demonstrate Catch-up Growth by Age 2 - 4 Years
Data from 2 clinical trials demonstrate the effectiveness of Humatrope in promoting linear growth in short children born SGA who fail to demonstrate catch-up growth.
The primary objective of Study 1 was to demonstrate that the increase from baseline in height SDS after 1 year of treatment would be similar when Humatrope is administered according to an individually adjusted dose (IAD) regimen or a fixed high dose (FHD) regimen. The height increases would be considered similar if the lower bound of the 95% confidence interval (CI) for the mean difference between the groups (IAD – FHD) was greater than -0.5 height SDS. This 2-year, open-label, multicenter, European study enrolled 193 prepubertal, non-GH deficient children with mean chronological age 6.8 ± 2.4 years (range: 3.0 to 12.3). Additional study entry criteria included birth weight <10th percentile and/or birth length SDS <-2 for gestational age, and height SDS for chronological age ≤-3. Exclusion criteria included syndromal conditions (e.g., Turner syndrome), chronic disease (e.g., diabetes mellitus), and tumor activity. Children were randomized to either a FHD (0.067 mg/kg/day [0.47 mg/kg/week]; n=99) or an IAD treatment group (n=94). The initial Humatrope dosage in the IAD treatment group was 0.035 mg/kg/day (0.25 mg/kg/week). The dosage was increased to 0.067 mg/kg/day in those patients in the IAD group whose 1-year height gain predicted at Month 3 was <0.75 height SDS (n=40) or whose actual height gain measured at Year 1 was <0.75 height SDS (n=11). Approximately 85% of the randomized patients completed 2 years of therapy.
At baseline, the FHD and IAD treatment groups had comparable height SDS (mean -3.9; Table 12). Although the mean 1-year height increase in the IAD group was statistically significantly lower than that observed in the FHD group, the study achieved its primary objective by demonstrating that the increase from baseline in height SDS in the IAD group was clinically similar (non-inferior) to that in the FHD group (mean between-group difference = -0.3 SDS [95% CI: -0.4, -0.2 SDS]). The mean changes from baseline in height SDS at the end of the 2-year study were 1.4 and 1.6 SDS in the IAD and FHD groups, respectively. The results were similar when children who entered puberty during the study were removed from the analysis.
|
a Abbreviations: IAD=individually adjusted dose; FHD=fixed high dose; SD=standard deviation; SDS=standard deviation score |
|||
|
b Least squares mean difference ± standard error and 95% confidence interval based on ANCOVA model with treatment and gender as fixed effects, and baseline height SDS, baseline chronological age, baseline bone age, and mid-parental target height SDS as covariates. |
|||
|
c Only children with actual height measurements were included in the Year 1 and Year 2 analyses. |
|||
| IAD Group 0.035 to 0.067 mg/kg/day Mean (SD) | FHD Group 0.067 mg/kg/day Mean (SD) | Between-Group Difference IAD – FHDb |
|
| Baseline | (n=86) -3.9 (0.6) | (n=93) -3.9 (0.7) | -0.0 ± 0.1 (-0.2, 0.2) p-value = 0.95 |
| Year 1 Height SDS Change from baseline | (n=86)c
-3.0 (0.7) 0.9 (0.4) | (n=93)c
-2.7 (0.7) 1.1 (0.4) | -0.3 ± 0.1 (-0.4, -0.2) p-value <0.001 |
| Year 2 Height SDS Change from baseline | (n=82)c
-2.5 (0.8) 1.4 (0.5) | (n=88)c
-2.2 (0.7) 1.6 (0.5) | -0.3 ± 0.1 (-0.4, -0.1) p-value = 0.003 |
Study 2 was an open-label, multicenter, single arm study conducted in France, during which 35 prepubertal, non-GH deficient children were treated for 2 years with Humatrope 0.067 mg/kg/day (0.47 mg/kg/week). Mean chronological age at baseline was 9.3 ± 0.9 years (range: 6.7 to 10.8). Additional study entry criteria included birth length SDS <-2 or <3rd percentile for gestational age, and height SDS for chronological age <-2. Exclusion criteria included syndromal conditions (e.g., Turner syndrome), chronic disease (e.g., diabetes mellitus), and any active disease. All 35 patients completed the study. Mean height SDS increased from a baseline value of -2.7 (SD 0.5) to -1.5 (SD 0.6) after 2 years of Humatrope treatment.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Vials
Vial Kit — (6s) NDC 0002-7335-16
5 mg vial (no. 7335) and 5-mL vial of Diluent for Humatrope (No. 7336)
Cartridges
Cartridge Kit (MS8147) NDC 0002-8147-01 (gold)
6 mg cartridge (gold) (VL7554), and prefilled syringe of Diluent for Humatrope (VL7616)
Cartridge Kit (MS8148) NDC 0002-8148-01 (teal)
12 mg cartridge (teal) (VL7555), and prefilled syringe of Diluent for Humatrope (VL7617)
Cartridge Kit (MS8149) NDC 0002-8149-01 (purple)
24 mg cartridge (purple) (VL7556), and prefilled syringe of Diluent for Humatrope (VL7617)
16.2 Storage and Handling
Vials
Before Reconstitution — Vials of Humatrope and Diluent for Humatrope are stable when refrigerated at 2° to 8°C (36° to 46°F). Avoid freezing Diluent for Humatrope. Expiration dates are stated on the labels.
After Reconstitution — Vials of Humatrope are stable for up to 14 days when reconstituted with Diluent for Humatrope or Bacteriostatic Water for Injection, USP and refrigerated at 2° to 8°C (36° to 46°F). Avoid freezing the reconstituted vial of Humatrope.
After Reconstitution with Sterile Water, USP — Use only one dose per Humatrope vial and discard the unused portion. If the solution is not used immediately, it must be refrigerated at 2° to 8°C (36° to 46°F) and used within 24 hours.
Cartridges
Before Reconstitution — Cartridges of Humatrope and Diluent for Humatrope are stable when refrigerated at 2° to 8°C (36° to 46°F). Avoid freezing Diluent for Humatrope. Expiration dates are stated on the labels.
After Reconstitution — Cartridges of Humatrope are stable for up to 28 days when reconstituted with Diluent for Humatrope and refrigerated at 2° to 8°C (36° to 46°F). Store the Humatrope injection device without the needle attached. Avoid freezing the reconstituted cartridge of Humatrope. Cartridges should be reconstituted only with the supplied diluent. Cartridges should not be reconstituted with the Diluent for Humatrope provided with Humatrope vials, or with any other solution.
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling.
Patients being treated with Humatrope (and/or their parents) should be informed about the potential benefits and risks associated with Humatrope treatment, and the contents of the Patient Information Insert should be reviewed. This information is intended to educate patients (and caregivers); it is not a disclosure of all possible intended or adverse effects.
Patients and caregivers who will administer Humatrope should receive appropriate training and instruction on the proper use of Humatrope from the physician or other suitably qualified health care professional. A puncture-resistant container for the disposal of used needles and syringes should be strongly recommended. Patients and/or parents should be thoroughly instructed in the importance of proper disposal, and cautioned against any reuse of needles and syringes. This information is intended to aid in the safe and effective administration of the medication.
Literature revised February 23, 2011
Manufactured by Lilly France
F-67640 Fegersheim, France
for Eli Lilly and Company
Indianapolis, IN 46285, USA
Copyright © 1987, 2011, Eli Lilly and Company. All rights reserved.
PA 286 FSAM 02
HUMATROPE®
Somatropin (rDNA origin) for Injection
INFORMATION FOR THE PATIENT
Do not mix (reconstitute) the drug or inject it until you have been thoroughly trained in the proper techniques by your doctor. Use sterile techniques as instructed by your doctor. Destroy and discard syringes and/or needles after each use.
Humatrope should be kept refrigerated (36° to 46°F [2°to 8°C]) before and after reconstitution. Do not freeze. Reconstituted Humatrope should be used within 14 days.
Reconstituting the Vial of Humatrope
Reconstitute Humatrope only with Diluent for Humatrope. Do not use other solutions for reconstitution unless instructed to do so by your doctor. Your doctor will also tell you what size syringe and needle to use and how much diluent to add to the vial of Humatrope.
Always start by washing your hands.
1. Remove and discard plastic caps from tops of vials of diluent and Humatrope. Wipe tops of both vials with an alcohol swab (Figure 1). Remove needle cover and save. Pull back on syringe plunger to draw up an amount of air equal to the amount of diluent your doctor has prescribed. Insert needle in stopper of diluent vial, and inject air into vial.
2. Hold vial upside down and, making sure needle tip remains in solution, withdraw the amount of diluent your doctor has prescribed (Figure 2). After making sure that no air bubbles are in the syringe, turn vial upright and, holding barrel, remove syringe.
3. Insert same needle into vial of Humatrope and gently aim needle tip toward wall of vial. Slowly inject the diluent by aiming the stream of liquid against the wall of vial (Figure 3). Do not aim it at the white powder at the bottom of the vial. To equalize the pressure, withdraw a volume of air equal to the amount of diluent added before removing the syringe from the vial. If the needle can be removed from the barrel of the syringe, remove, destroy, and discard the needle. If the needle and syringe are made as 1 unit, destroy and discard the entire unit.
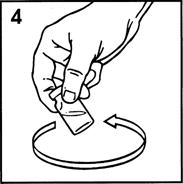
4. Swirl the vial with a gentle rotary motion until contents are completely dissolved (Figure 4). Do not shake.
Preparing the Injection
- Do not use reconstituted Humatrope if it is cloudy or contains particles.
- If the needle can be removed from the type of syringe you are using, a new needle should be placed on the syringe before the injection. If the syringe and needle are made as 1 unit, another unit should be used for the injection.
- Before and after injection, the rubber stopper of the vial should be wiped with rubbing alcohol or an alcoholic antiseptic solution to prevent contamination of the contents by repeated needle insertions.
- Remove the needle cover and draw an amount of air into the syringe equal to your dose of Humatrope.
- Insert needle into vial of reconstituted Humatrope and inject the air into the vial. Turn the vial upside down, and, making sure needle tip is in solution, withdraw your correct dose (see Figure 2). Make sure that no air bubbles are in the syringe.
- Remove syringe and replace needle cover. Write date of reconstitution on vial label, and discard unused diluent.
- Return unused portion of reconstituted Humatrope to refrigerator and use within 14 days.
- Destroy needle or the needle and syringe after use.
Injecting Humatrope
- Gently tap injection site several times with fingers.
- Wipe the area thoroughly with an alcohol swab. Use a circular motion and work outward from the inside of the circle.
-
Subcutaneous Injection: With the thumb and forefinger, stabilize the skin by spreading or pinching up a large area of skin.
- Holding the syringe at a 90-degree angle to injection site, quickly insert the needle all the way into the skin.
- Slowly inject the solution.
- Remove the needle quickly, and apply pressure over the injection site with a dry gauze pad or cotton ball. Rub for several seconds.
- Destroy needle or needle and syringe after use.
-
Intramuscular Injection: With the thumb and first 2 fingers, press the skin down firmly against a large muscle mass, such as the thigh.
- Holding the syringe at a 90-degree angle to injection site, quickly insert the needle all the way into the skin.
- When the needle is in place, slowly pull back on the plunger. If blood enters the syringe, remove needle, discard syringe and drug, and prepare another injection.
- If no blood enters the syringe, slowly inject the solution.
- Remove the needle quickly, and apply pressure over the injection site with a dry gauze pad or cotton ball. Rub for several seconds.
- Destroy needle or needle and syringe after use.
If you have any questions, consult your doctor.
Text revised July 6, 1990
Literature revised March 1, 2002
ELI LILLY AND COMPANY, INDIANAPOLIS, IN 46285, USA
PA 1685 AMP
INFORMATION AND PATIENT INSTRUCTIONS
HUMATROPE®
Somatropin (rDNA origin) for Injection
CARTRIDGES
HUMATROPE CARTRIDGES ARE ONLY TO BE USED WITH HUMATROPEN®OR HUMATROPEN®3 INJECTION DEVICES.
Important Things to Know
It is important to learn the names of the parts of the Humatrope Cartridge Kit and how these parts work before injecting yourself or your child. Make sure you have been properly trained by your nurse, pharmacist or doctor before you mix the drug (add the diluent liquid to the dry Humatrope powder) or inject it. Wash your hands and be careful to follow the instructions given to you by your nurse, pharmacist or doctor. After mixing, throw away the diluent syringe in a puncture-resistant container such as the type your nurse, pharmacist or doctor has told you to use.
Storage
Humatrope must be kept refrigerated (36° to 46°F [2° to 8°C]) before and after it is mixed. Do not freeze. Once Humatrope has been mixed and is in liquid form, it must be used within 28 days. Throw away any mixed Humatrope left over after 28 days. Before giving an injection, check the date on the cartridge. Do not use the cartridge if it has expired.
WARNING
HUMATROPE CARTRIDGES SHOULD NOT BE USED IF THE PATIENT IS ALLERGIC TO METACRESOL OR GLYCERIN.
Contents
- one cartridge with 6, 12, or 24 mg of dried Humatrope
- one prefilled syringe with diluent (the liquid used to mix the dried Humatrope)
NOTE: There are three kinds of Humatrope cartridges that have different amounts of Humatrope (6, 12, or 24 mg). Make sure that you have the cartridge that your doctor prescribed.
Mixing the Humatrope in the Cartridge
Use only the prefilled diluent syringe to mix the Humatrope in the cartridge. DO NOT use the diluent that comes in the Humatrope vial box, or any other liquid.
Reconstitution Instructions
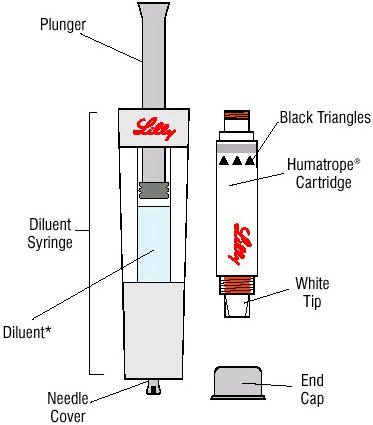
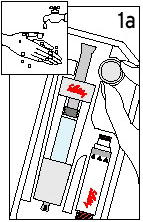
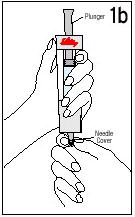
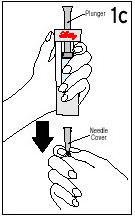
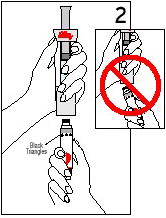
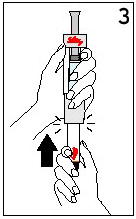
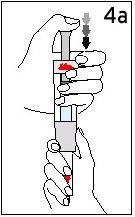
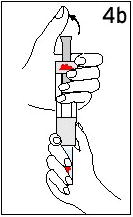
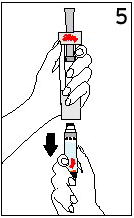
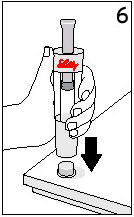
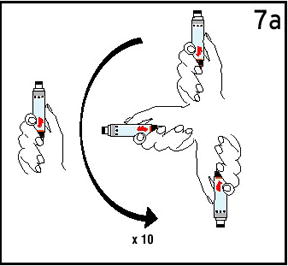
Injections can be given in the following areas:
- Abdomen (above, below, or either side of the navel)
- Front of the upper thighs
- Upper, outer buttocks
- Back of the arms above the elbow and below the shoulder
Discuss use of the pen injection device, the right places to inject, and site rotation with your nurse or doctor.
Literature revised August 1, 2005
Manufactured by Lilly France S.A.S.
F-67640 Fegersheim, France
for Eli Lilly and Company
Indianapolis, IN 46285, USA
www.humatrope.com
PA 9330 FSAMP
PACKAGE LABEL – Humatrope 5 mg Vial Kit
NDC 0002-7335-16
6 of VIAL No. 7335
6 of VIAL No. 7336
1—6
6 COMBINATION PACKAGES
Humatrope®
somatropin (rDNA origin) for injection
Includes:
Six Vials of Humatrope® 5 mg
(somatropin [rDNA origin] for injection)
Six Vials of Sterile Diluent 5 mL
RX only
Refrigerate
Do Not Freeze
Do Not Shake
Lilly


PACKAGE LABEL – Humatrope 6 mg Cartridge Kit
NDC 0002-8147-01
MS 8147
6 mg Combination Package
Humatrope®
somatropin (rDNA origin) for injection
6 mg
Cartridge Kit
for use only with the Humatrope® (somatropin [rDNA origin] for injection) pen injection device
Rx only
Refrigerate
Do Not Freeze
Do Not Shake
Kit contains:
One Humatrope Cartridge 6 mg
One Prefilled Diluent Syringe
www.humatrope.com
Lilly
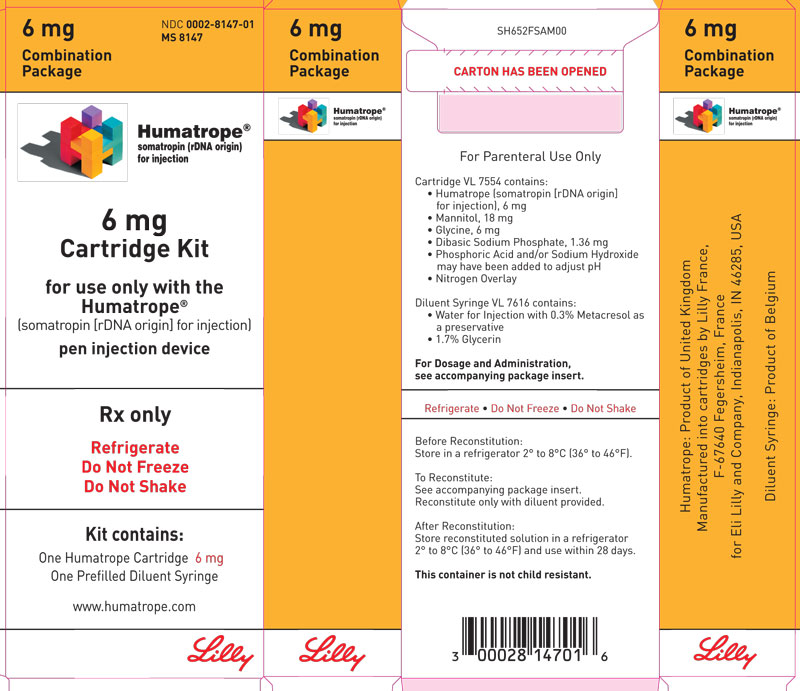
PACKAGE LABEL – Humatrope 12 mg Cartridge Kit
NDC 0002-8148-01
MS 8148
12 mg Combination Package
Humatrope®
somatropin (rDNA origin) for injection
12 mg
Cartridge Kit
for use only with the Humatrope® (somatropin [rDNA origin] for injection) pen injection device
Rx only
Refrigerate
Do Not Freeze
Do Not Shake
Kit contains:
One Humatrope Cartridge 12 mg
One Prefilled Diluent Syringe
www.humatrope.com
Lilly

PACKAGE LABEL – Humatrope 24 mg Cartridge Kit
NDC 0002-8149-01
MS 8149
24 mg Combination Package
Humatrope®
somatropin (rDNA origin) for injection
24 mg
Cartridge Kit
for use only with the Humatrope® (somatropin [rDNA origin] for injection) pen injection device
Rx only
Refrigerate
Do Not Freeze
Do Not Shake
Kit contains:
One Humatrope Cartridge 24 mg
One Prefilled Diluent Syringe
www.humatrope.com
Lilly
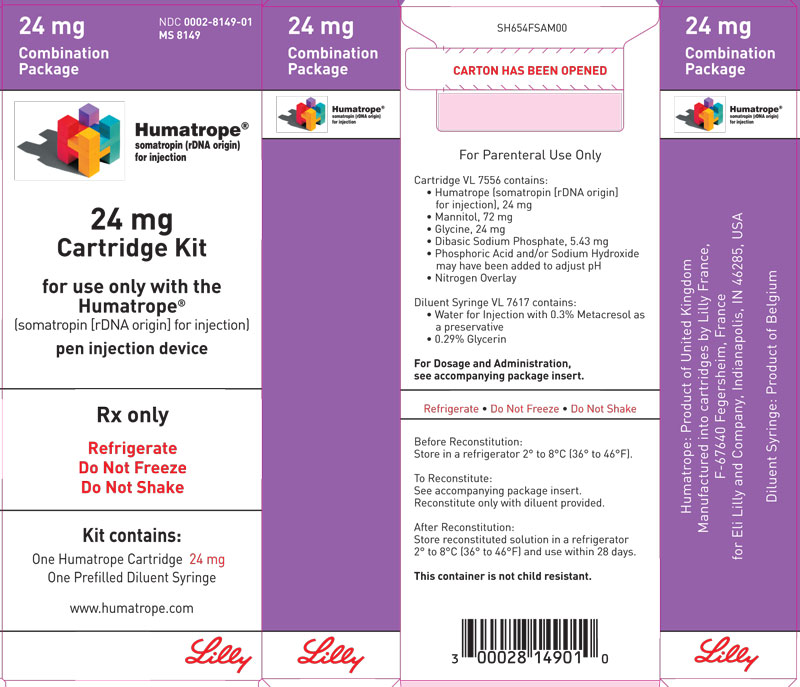
| HUMATROPE
somatropin kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019640 | 04/01/1987 | |
| HUMATROPE
somatropin kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019640 | 01/27/2006 | |
| HUMATROPE
somatropin kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019640 | 01/27/2006 | |
| HUMATROPE
somatropin kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019640 | 01/27/2006 | |
| Labeler - Eli Lilly and Company (006421325) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Eli Lilly and Company (Indianapolis) | 006421325 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Lilly France S.A.S. | 395346919 | MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Eli Lilly and Company Limited (Speke) | 230761368 | API MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Baxter Pharmaceutical Solutions LCC | 604719430 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Catalent Belgium S.A. | 370696762 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Covance Laboratories, Inc. | 006453670 | ANALYSIS | |