COMTAN
-
entacapone tablet, film coated
Novartis Pharmaceuticals Corporation
----------
COMTAN®(entacapone) Tablets
Rx only
Prescribing Information
DESCRIPTION
Comtan® (entacapone) is available as tablets containing 200-mg entacapone.
Entacapone is an inhibitor of catechol-O-methyltransferase (COMT), used in the treatment of Parkinson's Disease as an adjunct to levodopa/carbidopa therapy. It is a nitrocatechol-structured compound with a relative molecular mass of 305.29. The chemical name of entacapone is (E)-2-cyano-3-(3,4-dihydroxy-5-nitrophenyl)-N,N-diethyl-2-propenamide. Its empirical formula is C14H15N3O5 and its structural formula is:
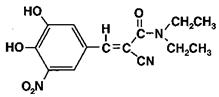
The inactive ingredients of the Comtan tablet are microcrystalline cellulose, mannitol, croscarmellose sodium, hydrogenated vegetable oil, hydroxypropyl methylcellulose, polysorbate 80, glycerol 85%, sucrose, magnesium stearate, yellow iron oxide, red oxide, and titanium dioxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Entacapone is a selective and reversible inhibitor of catechol-O-methyltransferase (COMT).
In mammals, COMT is distributed throughout various organs with the highest activities in the liver and kidney. COMT also occurs in the heart, lung, smooth and skeletal muscles, intestinal tract, reproductive organs, various glands, adipose tissue, skin, blood cells, and neuronal tissues, especially in glial cells. COMT catalyzes the transfer of the methyl group of S-adenosyl-L-methionine to the phenolic group of substrates that contain a catechol structure. Physiological substrates of COMT include dopa, catecholamines (dopamine, norepinephrine, and epinephrine) and their hydroxylated metabolites. The function of COMT is the elimination of biologically active catechols and some other hydroxylated metabolites. In the presence of a decarboxylase inhibitor, COMT becomes the major metabolizing enzyme for levodopa, catalyzing the metabolism to 3-methoxy-4-hydroxy-L-phenylalanine (3-OMD) in the brain and periphery.
The mechanism of action of entacapone is believed to be through its ability to inhibit COMT and alter the plasma pharmacokinetics of levodopa. When entacapone is given in conjunction with levodopa and an aromatic amino acid decarboxylase inhibitor, such as carbidopa, plasma levels of levodopa are greater and more sustained than after administration of levodopa and an aromatic amino acid decarboxylase inhibitor alone. It is believed that at a given frequency of levodopa administration, these more sustained plasma levels of levodopa result in more constant dopaminergic stimulation in the brain, leading to greater effects on the signs and symptoms of Parkinson's Disease. The higher levodopa levels also lead to increased levodopa adverse effects, sometimes requiring a decrease in the dose of levodopa.
In animals, while entacapone enters the CNS to a minimal extent, it has been shown to inhibit central COMT activity. In humans, entacapone inhibits the COMT enzyme in peripheral tissues. The effects of entacapone on central COMT activity in humans have not been studied.
Pharmacodynamics
COMT Activity in Erythrocytes
Studies in healthy volunteers have shown that entacapone reversibly inhibits human erythrocyte catechol-O-methyltransferase (COMT) activity after oral administration. There was a linear correlation between entacapone dose and erythrocyte COMT inhibition, the maximum inhibition being 82% following an 800-mg single dose. With a 200-mg single dose of entacapone, maximum inhibition of erythrocyte COMT activity is on average 65% with a return to baseline level within 8 hours.
Effect on the Pharmacokinetics of Levodopa and its Metabolites
When 200 mg entacapone is administered together with levodopa/carbidopa, it increases the area under the curve (AUC) of levodopa by approximately 35% and the elimination half-life of levodopa is prolonged from 1.3 h - 2.4 h. In general, the average peak levodopa plasma concentration and the time of its occurrence (Tmax of 1 hour) are unaffected. The onset of effect occurs after the first administration and is maintained during long-term treatment. Studies in Parkinson's Disease patients suggest that the maximal effect occurs with 200-mg entacapone. Plasma levels of 3-OMD are markedly and dose-dependently decreased by entacapone when given with levodopa/carbidopa.
Pharmacokinetics of Entacapone
Entacapone pharmacokinetics are linear over the dose range of 5 mg - 800 mg, and are independent of levodopa/carbidopa coadministration. The elimination of entacapone is biphasic, with an elimination half-life of 0.4 h - 0.7 h based on the β-phase and 2.4 h based on the γ-phase. The γ-phase accounts for approximately 10% of the total AUC. The total body clearance after i.v. administration is 850 mL/min. After a single 200-mg dose of Comtan (entacapone), the Cmax is approximately 1.2 µg/mL.
Absorption
Entacapone is rapidly absorbed, with a Tmax of approximately 1 hour. The absolute bioavailability following oral administration is 35%. Food does not affect the pharmacokinetics of entacapone.
Distribution
The volume of distribution of entacapone at steady state after i.v. injection is small (20 L). Entacapone does not distribute widely into tissues due to its high plasma protein binding. Based on in vitro studies, the plasma protein binding of entacapone is 98% over the concentration range of 0.4 - 50 µg/mL. Entacapone binds mainly to serum albumin.
Metabolism and Elimination
Entacapone is almost completely metabolized prior to excretion, with only a very small amount (0.2% of dose) found unchanged in urine. The main metabolic pathway is isomerization to the cis-isomer, followed by direct glucuronidation of the parent and cis-isomer; the glucuronide conjugate is inactive. After oral administration of a 14C-labeled dose of entacapone, 10% of labeled parent and metabolite is excreted in urine and 90% in feces.
Special Populations
Entacapone pharmacokinetics are independent of age. No formal gender studies have been conducted. Racial representation in clinical trials was largely limited to Caucasians (there were only 4 blacks in one US trial and no Asians in any of the clinical trials); no conclusions can therefore be reached about the effect of Comtan on groups other than Caucasian.
Hepatic Impairment
A single 200-mg dose of entacapone, without levodopa/dopa decarboxylase inhibitor coadministration, showed approximately twofold higher AUC and Cmax values in patients with a history of alcoholism and hepatic impairment (n=10) compared to normal subjects (n=10). All patients had biopsy-proven liver cirrhosis caused by alcohol. According to Child-Pugh grading 7 patients with liver disease had mild hepatic impairment and 3 patients had moderate hepatic impairment. As only about 10% of the entacapone dose is excreted in urine as parent compound and conjugated glucuronide, biliary excretion appears to be the major route of excretion of this drug. Consequently, entacapone should be administered with care to patients with biliary obstruction.
Renal Impairment
The pharmacokinetics of entacapone have been investigated after a single 200-mg entacapone dose, without levodopa/dopa decarboxylase inhibitor coadministration, in a specific renal impairment study. There were three groups: normal subjects (n=7; creatinine clearance >1.12 mL/sec/1.73 m2), moderate impairment (n=10; creatinine clearance ranging from 0.60 - 0.89 mL/sec/1.73 m2), and severe impairment (n=7; creatinine clearance ranging from 0.20 - 0.44 mL/sec/1.73 m2). No important effects of renal function on the pharmacokinetics of entacapone were found.
Drug Interactions
See PRECAUTIONS, Drug Interactions.
Clinical Studies
The effectiveness of Comtan (entacapone) as an adjunct to levodopa in the treatment of Parkinson's Disease was established in three 24-week multicenter, randomized, double-blind placebo-controlled trials in patients with Parkinson's Disease. In two of these trials, the patients' disease was "fluctuating", i.e., was characterized by documented periods of "On" (periods of relatively good functioning) and "Off" (periods of relatively poor functioning), despite optimum levodopa therapy. There was also a withdrawal period following 6 months of treatment. In the third trial patients were not required to have been experiencing fluctuations. Prior to the controlled part of the trials, patients were stabilized on levodopa for 2 - 4 weeks. Comtan has not been systematically evaluated in patients who do not experience fluctuations.
In the first two studies to be described, patients were randomized to receive placebo or entacapone 200 mg administered concomitantly with each dose of levodopa/carbidopa (up to 10 times daily, but averaging 4-6 doses per day). The formal double-blind portion of both trials was 6 months long. Patients recorded the time spent in the "On" and "Off" states in home diaries periodically throughout the duration of the trial. In one study, conducted in the Nordic countries, the primary outcome measure was the total mean time spent in the "On" state during an 18-hour diary recorded day (6 AM to midnight). In the other study, the primary outcome measure was the proportion of awake time spent over 24 hours in the "On" state.
In addition to the primary outcome measure, the amount of time spent in the "Off" state was evaluated, and patients were also evaluated by subparts of the Unified Parkinson's Disease Rating Scale (UPDRS), a frequently used multi-item rating scale intended to assess mentation (Part I), activities of daily living (Part II), motor function (Part III), complications of therapy (Part IV), and disease staging (Part V & VI); an investigator's and patient's global assessment of clinical condition, a 7-point subjective scale designed to assess global functioning in Parkinson's Disease; and the change in daily levodopa/carbidopa dose.
In one of the studies, 171 patients were randomized in 16 centers in Finland, Norway, Sweden, and Denmark (Nordic study), all of whom received concomitant levodopa plus dopa-decarboxylase inhibitor (either levodopa/carbidopa or levodopa/benserazide). In the second trial, 205 patients were randomized in 17 centers in North America (US and Canada); all patients received concomitant levodopa/carbidopa.
The following tables display the results of these two trials:
| Primary Measure from Home Diary (from an 18-hour Diary Day) | |||
|---|---|---|---|
| Baseline | Change from Baseline at Month 6* | p-value vs. placebo |
|
| Hours of Awake Time "On" | |||
| Placebo | 9.2 | +0.1 | – |
| Comtan | 9.3 | +1.5 | <0.001 |
| Duration of "On" time after first AM dose (hrs) | |||
| Placebo | 2.2 | 0.0 | – |
| Comtan | 2.1 | +0.2 | <0.05 |
| Secondary Measures from Home Diary (from an 18-hour Diary Day) | |||
| Hours of Awake Time "Off" | |||
| Placebo | 5.3 | 0.0 | – |
| Comtan | 5.5 | - 1.3 | <0.001 |
| Proportion of Awake Time "On" † (%) | |||
| Placebo | 63.8 | +0.6 | – |
| Comtan | 62.7 | +9.3 | <0.001 |
| Levodopa Total Daily Dose (mg) | |||
| Placebo | 705 | +14 | – |
| Comtan | 701 | - 87 | <0.001 |
| Frequency of Levodopa Daily Intakes | |||
| Placebo | 6.1 | +0.1 | – |
| Comtan | 6.2 | - 0.4 | <0.001 |
| Other Secondary Measures | |||
| Baseline | Change from Baseline at Month 6 | p-value vs. placebo |
|
| Investigator's Global (overall) % Improved‡ | |||
| Placebo | – | 28 | – |
| Comtan | – | 56 | <0.01 |
| Patient's Global (overall) % Improved‡ | |||
| Placebo | – | 22 | – |
| Comtan | – | 39 | N.S.§ |
| UPDRS Total | |||
| Placebo | 37.4 | -1.1 | – |
| Comtan | 38.5 | -4.8 | <0.01 |
| UPDRS Motor | |||
| Placebo | 24.6 | -0.7 | – |
| Comtan | 25.5 | -3.3 | <0.05 |
| UPDRS ADL | |||
| Placebo | 11.0 | -0.4 | – |
| Comtan | 11.2 | -1.8 | <0.05 |
| Primary Measure from Home Diary (for a 24-hour Diary Day) | |||
|---|---|---|---|
| Mean; the month 6 values represent the average of weeks 8, 16, and 24, by protocol-defined outcome measure. | |||
| Baseline | Change from Baseline at Month 6 | p-value vs. placebo |
|
| Percent of Awake Time "On" | |||
| Placebo | 60.8 | +2.0 | – |
| Comtan | 60.0 | +6.7 | <0.05 |
| Secondary Measures from Home Diary (for a 24-hour Diary Day) | |||
| Hours of Awake Time "Off" | |||
| Placebo | 6.6 | - 0.3 | – |
| Comtan | 6.8 | - 1.2 | <0.01 |
| Hours of Awake Time "On" | |||
| Placebo | 10.3 | + 0.4 | – |
| Comtan | 10.2 | + 1.0 | N.S.* |
| Levodopa Total Daily Dose (mg) | |||
| Placebo | 758 | + 19 | – |
| Comtan | 804 | - 93 | <0.001 |
| Frequency of Levodopa Daily Intakes | |||
| Placebo | 6.0 | + 0.2 | – |
| Comtan | 6.2 | 0.0 | N.S.* |
| Other Secondary Measures | |||
| Baseline | Change from Baseline at Month 6 | p-value vs. placebo |
|
| Investigator's Global (overall) % Improved† | |||
| Placebo | – | 21 | – |
| Comtan | – | 34 | <0.05 |
| Patient's Global (overall) % Improved† | |||
| Placebo | – | 20 | – |
| Comtan | – | 31 | <0.05 |
| UPDRS Total‡ | |||
| Placebo | 35.6 | +2.8 | – |
| Comtan | 35.1 | -0.6 | <0.05 |
| UPDRS Motor‡ | |||
| Placebo | 22.6 | +1.2 | – |
| Comtan | 22.0 | -0.9 | <0.05 |
| UPDRS ADL‡ | |||
| Placebo | 11.7 | +1.1 | – |
| Comtan | 11.9 | 0.0 | <0.05 |
Effects on "On" time did not differ by age, sex, weight, disease severity at baseline, levodopa dose and concurrent treatment with dopamine agonists or selegiline.
Withdrawal of entacapone
In the North American study, abrupt withdrawal of entacapone, without alteration of the dose of levodopa/carbidopa, resulted in a significant worsening of fluctuations, compared to placebo. In some cases, symptoms were slightly worse than at baseline, but returned to approximately baseline severity within two weeks following levodopa dose increase on average by 80 mg. In the Nordic study, similarly, a significant worsening of parkinsonian symptoms was observed after entacapone withdrawal, as assessed two weeks after drug withdrawal. At this phase, the symptoms were approximately at baseline severity following levodopa dose increase by about 50 mg.
In the third placebo controlled trial, a total of 301 patients were randomized in 32 centers in Germany and Austria. In this trial, as in the other two trials, entacapone 200 mg was administered with each dose of levodopa/dopa decarboxylase inhibitor (up to 10 times daily) and UPDRS Parts II and III and total daily "On" time were the primary measures of effectiveness. The following results were seen for the primary measures, as well as for some secondary measures:
| Primary Measures | |||
|---|---|---|---|
| Baseline | Change from Baseline at Month 6 | p-value vs. placebo (LOCF) |
|
| UPDRS ADL* | |||
| Placebo | 12.0 | +0.5 | – |
| Comtan | 12.4 | -0.4 | <0.05 |
| UPDRS Motor* | |||
| Placebo | 24.1 | +0.1 | – |
| Comtan | 24.9 | -2.5 | <0.05 |
| Hours of Awake Time "On" (Home diary)† | |||
| Placebo | 10.1 | +0.5 | – |
| Comtan | 10.2 | +1.1 | N.S.‡ |
| Secondary Measures | |||
| Baseline | Change from Baseline at Month 6 | p-value vs. placebo |
|
| UPDRS Total* | |||
| Placebo | 37.7 | +0.6 | – |
| Comtan | 39.0 | -3.4 | <0.05 |
| Percent of Awake Time "On" (Home diary)† | |||
| Placebo | 59.8 | +3.5 | – |
| Comtan | 62.0 | +6.5 | N.S.‡ |
| Hours of Awake Time "Off" (Home diary)† | |||
| Placebo | 6.8 | -0.6 | – |
| Comtan | 6.3 | -1.2 | 0.07 |
| Levodopa Total Daily Dose (mg)* | |||
| Placebo | 572 | +4 | – |
| Comtan | 566 | -35 | N.S.‡ |
| Frequency of Levodopa Daily Intake* | |||
| Placebo | 5.6 | +0.2 | – |
| Comtan | 5.4 | 0.0 | <0.01 |
| Global (overall) % Improved§ | |||
| Placebo | – | 34 | – |
| Comtan | – | 38 | N.S.‡ |
INDICATIONS
Comtan (entacapone) is indicated as an adjunct to levodopa/carbidopa to treat patients with idiopathic Parkinson's Disease who experience the signs and symptoms of end-of-dose "wearing-off" (see CLINICAL PHARMACOLOGY, Clinical Studies).
Comtan's effectiveness has not been systematically evaluated in patients with idiopathic Parkinson's Disease who do not experience end-of-dose "wearing-off".
CONTRAINDICATIONS
Comtan (entacapone) tablets are contraindicated in patients who have demonstrated hypersensitivity to the drug or its ingredients.
WARNINGS
Comtan should be administered with caution to patients with ischemic heart disease.
Monoamine oxidase (MAO) and COMT are the two major enzyme systems involved in the metabolism of catecholamines. It is theoretically possible, therefore, that the combination of Comtan (entacapone) and a non-selective MAO inhibitor (e.g., phenelzine and tranylcypromine) would result in inhibition of the majority of the pathways responsible for normal catecholamine metabolism. For this reason, patients should ordinarily not be treated concomitantly with Comtan and a non-selective MAO inhibitor.
Entacapone can be taken concomitantly with a selective MAO-B inhibitor (e.g., selegiline).
Drugs Metabolized By Catechol-O-Methyltransferase (COMT)
When a single 400-mg dose of entacapone was given together with intravenous isoprenaline (isoproterenol) and epinephrine without coadministered levodopa/dopa decarboxylase inhibitor, the overall mean maximal changes in heart rate during infusion were about 50% and 80% higher than with placebo, for isoprenaline and epinephrine, respectively.
Therefore, drugs known to be metabolized by COMT, such as isoproterenol, epinephrine, norepinephrine, dopamine, dobutamine, alpha-methyldopa, apomorphine, isoetherine, and bitolterol should be administered with caution in patients receiving entacapone regardless of the route of administration (including inhalation), as their interaction may result in increased heart rates, possibly arrhythmias, and excessive changes in blood pressure.
Ventricular tachycardia was noted in one 32-year-old healthy male volunteer in an interaction study after epinephrine infusion and oral entacapone administration. Treatment with propranolol was required. A causal relationship to entacapone administration appears probable but cannot be attributed with certainty.
PRECAUTIONS
Hypotension/Syncope
Dopaminergic therapy in Parkinson's Disease patients has been associated with orthostatic hypotension. Entacapone enhances levodopa bioavailability and, therefore, might be expected to increase the occurrence of orthostatic hypotension. In Comtan (entacapone) clinical trials, however, no differences from placebo were seen for measured orthostasis or symptoms of orthostasis. Orthostatic hypotension was documented at least once in 2.7% and 3.0% of the patients treated with 200 mg Comtan and placebo, respectively. A total of 4.3% and 4.0% of the patients treated with 200 mg Comtan and placebo, respectively, reported orthostatic symptoms at some time during their treatment and also had at least one episode of orthostatic hypotension documented (however, the episode of orthostatic symptoms itself was not accompanied by vital sign measurements). Neither baseline treatment with dopamine agonists or selegiline, nor the presence of orthostasis at baseline, increased the risk of orthostatic hypotension in patients treated with Comtan compared to patients on placebo.
In the large controlled trials, approximately 1.2% and 0.8% of 200 mg entacapone and placebo patients, respectively, reported at least one episode of syncope. Reports of syncope were generally more frequent in patients in both treatment groups who had an episode of documented hypotension (although the episodes of syncope, obtained by history, were themselves not documented with vital sign measurement).
Diarrhea and Colitis
In clinical trials, diarrhea developed in 60 of 603 (10.0%) and 16 of 400 (4.0%) of patients treated with 200 mg Comtan and placebo, respectively. In patients treated with Comtan, diarrhea was generally mild to moderate in severity (8.6%) but was regarded as severe in 1.3%. Diarrhea resulted in withdrawal in 10 of 603 (1.7%) patients, 7 (1.2%) with mild and moderate diarrhea and 3 (0.5%) with severe diarrhea. Diarrhea generally resolved after discontinuation of Comtan. Two patients with diarrhea were hospitalized. Typically, diarrhea presents within 4 - 12 weeks after entacapone is started, but it may appear as early as the first week and as late as many months after the initiation of treatment. Diarrhea may be associated with weight loss, dehydration, and hypokalemia.
Post-marketing experience has shown that diarrhea may be a sign of drug-induced microscopic colitis, primarily lymphocytic colitis. In these cases diarrhea has usually been moderate to severe, watery, and non-bloody, at times associated with dehydration, abdominal pain, weight loss, and hypokalemia. In the majority of cases, diarrhea and other colitis-related symptoms resolved or significantly improved when Comtan treatment was stopped. In some patients with biopsy confirmed colitis, diarrhea had resolved or significantly improved after discontinuation of Comtan but recurred after retreatment with Comtan.
If prolonged diarrhea is suspected to be related to Comtan, the drug should be discontinued and appropriate medical therapy considered. If the cause of prolonged diarrhea remains unclear or continues after stopping entacapone, then further diagnostic investigations including colonoscopy and biopsies should be considered.
Hallucinations
Dopaminergic therapy in Parkinson's Disease patients has been associated with hallucinations. In clinical trials, hallucinations developed in approximately 4.0% of patients treated with 200 mg Comtan or placebo. Hallucinations led to drug discontinuation and premature withdrawal from clinical trials in 0.8% and 0% of patients treated with 200 mg Comtan and placebo, respectively. Hallucinations led to hospitalization in 1.0% and 0.3% of patients in the 200 mg Comtan and placebo groups, respectively.
Dyskinesia
Comtan may potentiate the dopaminergic side effects of levodopa and may cause and/or exacerbate preexisting dyskinesia. Although decreasing the dose of levodopa may ameliorate this side effect, many patients in controlled trials continued to experience frequent dyskinesias despite a reduction in their dose of levodopa. The rates of withdrawal for dyskinesia were 1.5% and 0.8% for 200 mg Comtan and placebo, respectively.
Other Events Reported With Dopaminergic Therapy
The events listed below are rare events known to be associated with the use of drugs that increase dopaminergic activity, although they are most often associated with the use of direct dopamine agonists.
Rhabdomyolysis
Cases of severe rhabdomyolysis have been reported with Comtan use. The complicated nature of these cases makes it impossible to determine what role, if any, Comtan played in their pathogenesis. Severe prolonged motor activity including dyskinesia may account for rhabdomyolysis. One case, however, included fever and alteration of consciousness. It is therefore possible that the rhabdomyolysis may be a result of the syndrome described in Hyperpyrexia and Confusion (see PRECAUTIONS, Other Events Reported With Dopaminergic Therapy).
Hyperpyrexia and Confusion
Cases of a symptom complex resembling the neuroleptic malignant syndrome characterized by elevated temperature, muscular rigidity, altered consciousness, and elevated CPK have been reported in association with the rapid dose reduction or withdrawal of other dopaminergic drugs. Several cases with similar signs and symptoms have been reported in association with Comtan therapy, although no information about dose manipulation is available. The complicated nature of these cases makes it difficult to determine what role, if any, Comtan may have played in their pathogenesis. No cases have been reported following the abrupt withdrawal or dose reduction of entacapone treatment during clinical studies.
Prescribers should exercise caution when discontinuing entacapone treatment. When considered necessary, withdrawal should proceed slowly. If a decision is made to discontinue treatment with Comtan, recommendations include monitoring the patient closely and adjusting other dopaminergic treatments as needed. This syndrome should be considered in the differential diagnosis for any patient who develops a high fever or severe rigidity. Tapering Comtan has not been systematically evaluated.
Fibrotic Complications
Cases of retroperitoneal fibrosis, pulmonary infiltrates, pleural effusion, and pleural thickening have been reported in some patients treated with ergot derived dopaminergic agents. These complications may resolve when the drug is discontinued, but complete resolution does not always occur. Although these adverse events are believed to be related to the ergoline structure of these compounds, whether other, nonergot derived drugs (e.g., entacapone) that increase dopaminergic activity can cause them is unknown. It should be noted that the expected incidence of fibrotic complications is so low that even if entacapone caused these complications at rates similar to those attributable to other dopaminergic therapies, it is unlikely that it would have been detected in a cohort of the size exposed to entacapone. Four cases of pulmonary fibrosis were reported during clinical development of entacapone; three of these patients were also treated with pergolide and one with bromocriptine. The duration of treatment with entacapone ranged from 7 - 17 months.
Melanoma
Epidemiological studies have shown that patients with Parkinson's disease have a higher risk (2- to approximately 6-fold higher) of developing melanoma than the general population. Whether the increased risk observed was due to Parkinson's disease or other factors, such as drugs used to treat Parkinson's disease, is unclear.
For the reasons stated above, patients and providers are advised to monitor for melanomas frequently and on a regular basis when using Comtan for any indication. Ideally, periodic skin examination should be performed by appropriately qualified individuals (e.g., dermatologists).
Renal Toxicity
In a 1 year toxicity study, entacapone (plasma exposure 20 times that in humans receiving the maximum recommended daily dose of 1600 mg) caused an increased incidence in male rats of nephrotoxicity that was characterized by regenerative tubules, thickening of basement membranes, infiltration of mononuclear cells and tubular protein casts. These effects were not associated with changes in clinical chemistry parameters, and there is no established method for monitoring for the possible occurrence of these lesions in humans. Although this toxicity could represent a species-specific effect, there is not yet evidence that this is so.
Hepatic Impairment
Patients with hepatic impairment should be treated with caution. The AUC and Cmax of entacapone approximately doubled in patients with documented liver disease compared to controls. (See CLINICAL PHARMACOLOGY, Pharmacokinetics of Entacapone and DOSAGE AND ADMINISTRATION).
Information for Patients
Patients should be instructed to take Comtan only as prescribed.
Patients should be informed that hallucinations can occur.
Patients should be advised that they may develop postural (orthostatic) hypotension with or without symptoms such as dizziness, nausea, syncope, and sweating. Hypotension may occur more frequently during initial therapy. Accordingly, patients should be cautioned against rising rapidly after sitting or lying down, especially if they have been doing so for prolonged periods, and especially at the initiation of treatment with Comtan.
Patients should be advised that they should neither drive a car nor operate other complex machinery until they have gained sufficient experience on Comtan to gauge whether or not it affects their mental and/or motor performance adversely. Because of the possible additive sedative effects, caution should be used when patients are taking other CNS depressants in combination with Comtan.
Patients should be informed that nausea may occur, especially at the initiation of treatment with Comtan.
Patients should be informed that diarrhea may occur with Comtan and it may have a delayed onset. Sometimes prolonged diarrhea may be caused by colitis (inflammation of the large intestine). Patients with diarrhea should drink fluids to maintain adequate hydration and monitor for weight loss. If diarrhea associated with Comtan is prolonged, discontinuing the drug is expected to lead to resolution, if diarrhea continues after stopping entacapone, further diagnostic investigations may be needed.
Patients should be advised of the possibility of an increase in dyskinesia.
Patients should be advised that treatment with entacapone may cause a change in the color of their urine (a brownish orange discoloration) that is not clinically relevant. In controlled trials, 10% of patients treated with Comtan reported urine discoloration compared to 0% of placebo patients.
Although Comtan has not been shown to be teratogenic in animals, it is always given in conjunction with levodopa/carbidopa, which is known to cause visceral and skeletal malformations in the rabbit. Accordingly, patients should be advised to notify their physicians if they become pregnant or intend to become pregnant during therapy (see PRECAUTIONS, Pregnancy).
Entacapone is excreted into maternal milk in rats. Because of the possibility that entacapone may be excreted into human maternal milk, patients should be advised to notify their physicians if they intend to breastfeed or are breastfeeding an infant.
There have been reports of patients experiencing intense urges to gamble, increased sexual urges, and other intense urges and the inability to control these urges while taking one or more of the medications that increase central dopaminergic tone, that are generally used for the treatment of Parkinson's disease, including Comtan. Although it is not proven that the medications caused these events, these urges were reported to have stopped in some cases when the dose was reduced or the medication was stopped. Prescribers should ask patients about the development of new or increased gambling urges, sexual urges or other urges while being treated with Comtan. Patients should inform their physician if they experience new or increased gambling urges, increased sexual urges or other intense urges while taking Comtan. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking Comtan.
Laboratory Tests
Comtan is a chelator of iron. The impact of entacapone on the body's iron stores is unknown; however, a tendency towards decreasing serum iron concentrations was noted in clinical trials. In a controlled clinical study serum ferritin levels (as marker of iron deficiency and subclinical anemia) were not changed with entacapone compared to placebo after one year of treatment and there was no difference in rates of anemia or decreased hemoglobin levels.
Special Populations
Patients with hepatic impairment should be treated with caution (see INDICATIONS, DOSAGE AND ADMINISTRATION).
Drug Interactions
In vitro studies of human CYP enzymes showed that entacapone inhibited the CYP enzymes 1A2, 2A6, 2C9, 2C19, 2D6, 2E1 and 3A only at very high concentrations (IC50 from 200 to over 1000 µM; an oral 200 mg dose achieves a highest level of approximately 5 µM in people); these enzymes would therefore not be expected to be inhibited in clinical use.
Protein Binding
Entacapone is highly protein bound (98%). In vitro studies have shown no binding displacement between entacapone and other highly bound drugs, such as warfarin, salicylic acid, phenylbutazone, and diazepam.
Drugs Metabolized by Catechol-O-methyltransferase (COMT)
See WARNINGS.
Hormone levels
Levodopa is known to depress prolactin secretion and increase growth hormone levels. Treatment with entacapone coadministered with levodopa/dopa decarboxylase inhibitor does not change these effects.
Effect of Entacapone on the Metabolism of Other Drugs
See WARNINGS regarding concomitant use of Comtan and non-selective MAO inhibitors.
No interaction was noted with the MAO-B inhibitor selegiline in two multiple-dose interaction studies when entacapone was coadministered with a levodopa/dopa decarboxylase inhibitor (n=29). More than 600 Parkinson's Disease patients in clinical trials have used selegiline in combination with entacapone and levodopa/dopa decarboxylase inhibitor.
As most entacapone excretion is via the bile, caution should be exercised when drugs known to interfere with biliary excretion, glucuronidation, and intestinal beta-glucuronidase are given concurrently with entacapone. These include probenecid, cholestyramine, and some antibiotics (e.g., erythromycin, rifampicin, ampicillin and chloramphenicol).
No interaction with the tricyclic antidepressant imipramine was shown in a single-dose study with entacapone without coadministered levodopa/dopa-decarboxylase inhibitor.
Carcinogenesis
Two-year carcinogenicity studies of entacapone were conducted in mice and rats. Rats were treated once daily by oral gavage with entacapone doses of 20, 90, or 400 mg/kg. An increased incidence of renal tubular adenomas and carcinomas was found in male rats treated with the highest dose of entacapone. Plasma exposures (AUC) associated with this dose were approximately 20 times higher than estimated plasma exposures of humans receiving the maximum recommended daily dose of entacapone (MRDD = 1600 mg). Mice were treated once daily by oral gavage with doses of 20, 100 or 600 mg/kg of entacapone (0.05, 0.3, and 2 times the MRDD for humans on a mg/m2 basis). Because of a high incidence of premature mortality in mice receiving the highest dose of entacapone, the mouse study is not an adequate assessment of carcinogenicity. Although no treatment related tumors were observed in animals receiving the lower doses, the carcinogenic potential of entacapone has not been fully evaluated. The carcinogenic potential of entacapone administered in combination with levodopa/carbidopa has not been evaluated.
Mutagenesis
Entacapone was mutagenic and clastogenic in the in vitro mouse lymphoma/thymidine kinase assay in the presence and absence of metabolic activation, and was clastogenic in cultured human lymphocytes in the presence of metabolic activation. Entacapone, either alone or in combination with levodopa/carbidopa, was not clastogenic in the in vivo mouse micronucleus test or mutagenic in the bacterial reverse mutation assay (Ames test).
Impairment of Fertility
Entacapone did not impair fertility or general reproductive performance in rats treated with up to 700 mg/kg/day (plasma AUCs 28 times those in humans receiving the MRDD). Delayed mating, but no fertility impairment, was evident in female rats treated with 700 mg/kg/day of entacapone.
Pregnancy
Pregnancy Category C
In embryofetal development studies, entacapone was administered to pregnant animals throughout organogenesis at doses of up to 1000 mg/kg/day in rats and 300 mg/kg/day in rabbits. Increased incidences of fetal variations were evident in litters from rats treated with the highest dose, in the absence of overt signs of maternal toxicity. The maternal plasma drug exposure (AUC) associated with this dose was approximately 34 times the estimated plasma exposure in humans receiving the maximum recommended daily dose (MRDD) of 1600 mg. Increased frequencies of abortions and late/total resorptions and decreased fetal weights were observed in the litters of rabbits treated with maternotoxic doses of 100 mg/kg/day (plasma AUCs 0.4 times those in humans receiving the MRDD) or greater. There was no evidence of teratogenicity in these studies.
However, when entacapone was administered to female rats prior to mating and during early gestation, an increased incidence of fetal eye anomalies (macrophthalmia, microphthalmia, anophthalmia) was observed in the litters of dams treated with doses of 160 mg/kg/day (plasma AUCs 7 times those in humans receiving the MRDD) or greater, in the absence of maternotoxicity. Administration of up to 700 mg/kg/day (plasma AUCs 28 times those in humans receiving the MRDD) to female rats during the latter part of gestation and throughout lactation, produced no evidence of developmental impairment in the offspring.
Entacapone is always given concomitantly with levodopa/carbidopa, which is known to cause visceral and skeletal malformations in rabbits. The teratogenic potential of entacapone in combination with levodopa/carbidopa was not assessed in animals.
There is no experience from clinical studies regarding the use of Comtan in pregnant women. Therefore, Comtan should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Women
In animal studies, entacapone was excreted into maternal rat milk.
It is not known whether entacapone is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when entacapone is administered to a nursing woman.
Pediatric Use
There is no identified potential use of entacapone in pediatric patients.
ADVERSE REACTIONS
During the pre-marketing development of entacapone, 1450 patients with Parkinson's Disease were treated with entacapone. Included were patients with fluctuating symptoms, as well as those with stable responses to levodopa therapy. All patients received concomitant treatment with levodopa preparations, however, and were similar in other clinical aspects.
The most commonly observed adverse events (>5%) in the double-blind, placebo-controlled trials (N=1003) associated with the use of Comtan (entacapone) and not seen at an equivalent frequency among the placebo-treated patients were: dyskinesia/hyperkinesia, nausea, urine discoloration, diarrhea, and abdominal pain.
Approximately 14% of the 603 patients given entacapone in the double-blind, placebo-controlled trials discontinued treatment due to adverse events compared to 9% of the 400 patients who received placebo. The most frequent causes of discontinuation in decreasing order are: psychiatric reasons (2% vs. 1%), diarrhea (2% vs. 0%), dyskinesia/hyperkinesia (2% vs. 1%), nausea (2% vs. 1%), abdominal pain (1% vs. 0%), and aggravation of Parkinson's Disease symptoms (1% vs. 1%).
Adverse Event Incidence in Controlled Clinical Studies
Table 4 lists treatment emergent adverse events that occurred in at least 1% of patients treated with entacapone participating in the double-blind, placebo-controlled studies and that were numerically more common in the entacapone group, compared to placebo. In these studies, either entacapone or placebo was added to levodopa/carbidopa (or levodopa/benserazide).
| SYSTEM ORGAN CLASS | Comtan | Placebo |
|---|---|---|
| Preferred term | (n = 603) % of patients | (n = 400)) % of patients |
| SKIN AND APPENDAGES DISORDERS | ||
| Sweating increased | 2 | 1 |
| MUSCULOSKELETAL SYSTEM DISORDERS | ||
| Back pain | 2 | 1 |
| CENTRAL & PERIPHERAL NERVOUS SYSTEM DISORDERS | ||
| Dyskinesia | 25 | 15 |
| Hyperkinesia | 10 | 5 |
| Hypokinesia | 9 | 8 |
| Dizziness | 8 | 6 |
| SPECIAL SENSES, OTHER DISORDERS | ||
| Taste perversion | 1 | 0 |
| PSYCHIATRIC DISORDERS | ||
| Anxiety | 2 | 1 |
| Somnolence | 2 | 0 |
| Agitation | 1 | 0 |
| GASTROINTESTINAL SYSTEM DISORDERS | ||
| Nausea | 14 | 8 |
| Diarrhea | 10 | 4 |
| Abdominal pain | 8 | 4 |
| Constipation | 6 | 4 |
| Vomiting | 4 | 1 |
| Mouth dry | 3 | 0 |
| Dyspepsia | 2 | 1 |
| Flatulence | 2 | 0 |
| Gastritis | 1 | 0 |
| Gastrointestinal disorders nos | 1 | 0 |
| RESPIRATORY SYSTEM DISORDERS | ||
| Dyspnea | 3 | 1 |
| PLATELET, BLEEDING & CLOTTING DISORDERS | ||
| Purpura | 2 | 1 |
| URINARY SYSTEM DISORDERS | ||
| Urine discoloration | 10 | 0 |
| BODY AS A WHOLE - GENERAL DISORDERS | ||
| Back pain | 4 | 2 |
| Fatigue | 6 | 4 |
| Asthenia | 2 | 1 |
| RESISTANCE MECHANISM DISORDERS | ||
| Infection bacterial | 1 | 0 |
The prescriber should be aware that these figures cannot be used to predict the incidence of adverse events in the course of usual medical practice where patient characteristics and other factors differ from those that prevailed in the clinical studies. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures do, however, provide the prescriber with some basis for estimating the relative contribution of drug and nondrug factors to the adverse events observed in the population studied.
Myocardial infarction (MI) and Ischemic heart disease (IHD) events other than MI in clinical studies
The frequencies of myocardial infarction (MI) and other ischemic heart disease (IHD) events are derived from an analysis of 15 double-blind studies (13 studies in Parkinson's disease patients with end-of-dose wearing-off symptoms and 2 studies in early levodopa-naive Parkinson's disease patients). These 15 clinical studies included 2663 subjects in the entacapone arms and 2169 patients in the placebo arms. Twenty patients (0.75 %) taking entacapone reported MI as compared with 7 patients (0.32%) taking placebo. Other IHD was reported by 42 patients (1.58%) taking entacapone as compared with 28 patients (1.29%) on placebo. Risk factors for MI and IHD were present in the majority of patients affected.
Effects of gender and age on adverse reactions
No differences were noted in the rate of adverse events attributable to entacapone by age or gender.
DRUG ABUSE AND DEPENDENCE
Comtan (entacapone) is not a controlled substance. Animal studies to evaluate the drug abuse and potential dependence have not been conducted. Although clinical trials have not revealed any evidence of the potential for abuse, tolerance or physical dependence, systematic studies in humans designed to evaluate these effects have not been performed.
OVERDOSAGE
There have been no reported cases of either accidental or intentional overdose with entacapone tablets. However, COMT inhibition by entacapone treatment is dose-dependent. A massive overdose of Comtan (entacapone) may theoretically produce a 100% inhibition of the COMT enzyme in people, thereby preventing the metabolism of endogenous and exogenous catechols.
The highest single dose of entacapone administered to humans was 800 mg, resulting in a plasma concentration of 14.1 µg/mL. The highest daily dose given to humans was 2400 mg, administered in one study as 400 mg six times daily with levodopa/carbidopa for 14 days in 15 Parkinson's Disease patients, and in another study as 800 mg t.i.d. for 7 days in 8 healthy volunteers. At this daily dose, the peak plasma concentrations of entacapone averaged 2.0 µg/mL (at 45 min., compared to 1.0 and 1.2 µg/mL with 200 mg entacapone at 45 min.). Abdominal pain and loose stools were the most commonly observed adverse events during this study. Daily doses as high as 2000 mg Comtan have been administered as 200 mg 10 times daily with levodopa/carbidopa or levodopa/benserazide for at least 1 year in 10 patients, for at least 2 years in 8 patients and for at least 3 years in 7 patients. Overall, however, clinical experience with daily doses above 1600 mg is limited.
The range of lethal plasma concentrations of entacapone based on animal data was 80 - 130 µg/mL in mice. Respiratory difficulties, ataxia, hypoactivity, and convulsions were observed in mice after high oral (gavage) doses.
Management of Overdose
Management of Comtan overdose is symptomatic; there is no known antidote to Comtan. Hospitalization is advised, and general supportive care is indicated. There is no experience with hemodialysis or hemoperfusion, but these procedures are unlikely to be of benefit, because Comtan is highly bound to plasma proteins. An immediate gastric lavage and repeated doses of charcoal over time may hasten the elimination of Comtan by decreasing its absorption/reabsorption from the GI tract. The adequacy of the respiratory and circulatory systems should be carefully monitored and appropriate supportive measures employed. The possibility of drug interactions, especially with catechol-structured drugs, should be borne in mind.
DOSAGE AND ADMINISTRATION
The recommended dose of Comtan (entacapone) is one 200 mg tablet administered concomitantly with each levodopa/carbidopa dose to a maximum of 8 times daily (200 mg × 8 = 1600 mg per day). Clinical experience with daily doses above 1600 mg is limited.
Comtan should always be administered in association with levodopa/carbidopa. Entacapone has no antiparkinsonian effect of its own.
In clinical trials, the majority of patients required a decrease in daily levodopa dose if their daily dose of levodopa had been ≥ 800 mg or if patients had moderate or severe dyskinesias before beginning treatment.
To optimize an individual patient's response, reductions in daily levodopa dose or extending the interval between doses may be necessary. In clinical trials, the average reduction in daily levodopa dose was about 25% in those patients requiring a levodopa dose reduction. (More than 58% of patients with levodopa doses above 800 mg daily required such a reduction.)
Comtan can be combined with both the immediate and sustained-release formulations of levodopa/carbidopa.
Comtan may be taken with or without food (see CLINICAL PHARMACOLOGY).
Patients With Impaired Hepatic Function
Patients with hepatic impairment should be treated with caution. The AUC and Cmax of entacapone approximately doubled in patients with documented liver disease, compared to controls. However, these studies were conducted with single-dose entacapone without levodopa/dopa decarboxylase inhibitor coadministration, and therefore the effects of liver disease on the kinetics of chronically administered entacapone have not been evaluated (see CLINICAL PHARMACOLOGY, Pharmacokinetics of Entacapone).
Withdrawing Patients from Comtan
Rapid withdrawal or abrupt reduction in the Comtan dose could lead to emergence of signs and symptoms of Parkinson's Disease (see CLINICAL PHARMACOLOGY, Clinical Studies), and may lead to Hyperpyrexia and Confusion, a symptom complex resembling the neuroleptic malignant syndrome (see PRECAUTIONS, Other Events Reported With Dopaminergic Therapy). This syndrome should be considered in the differential diagnosis for any patient who develops a high fever or severe rigidity. If a decision is made to discontinue treatment with Comtan, patients should be monitored closely and other dopaminergic treatments should be adjusted as needed. Although tapering Comtan has not been systematically evaluated, it seems prudent to withdraw patients slowly if the decision to discontinue treatment is made.
HOW SUPPLIED
Comtan (entacapone) is supplied as 200-mg film-coated tablets for oral administration. The oval-shaped tablets are brownish-orange, unscored, and embossed "Comtan" on one side. Tablets are provided in HDPE containers as follows:
| Bottles of 100 | NDC 0078-0327-05 |
Store at 25°C (77°F) excursions permitted to 15°-30°C (59°-86°F). [See USP Controlled Room Temperature.]
Comtan (entacapone) tablets are manufactured by Orion Corporation, Orion Pharma (Espoo, Finland) and marketed by Novartis Pharmaceuticals Corporation (East Hanover, N.J. 07936, U.S.A.).
T2010-95
REV: December 2010
Printed in U.S.A.
©2000 Novartis
PRINCIPAL DISPLAY PANEL - 200 mg Bottle Label
NOVARTIS
NDC 0078-0327-05
Comtan®
(entacapone)
tablets
200 mg
100
Tablets
Rx only

| COMTAN
entacapone tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020796 | 10/19/1999 | |
| Labeler - Novartis Pharmaceuticals Corporation (002147023) |