RAPLON
-
rapacuronium bromide injection
Organon Pharmaceuticals USA
----------
RAPLON™(rapacuronium bromide) for Injection
THIS DRUG SHOULD BE ADMINISTERED BY ADEQUATELY-TRAINED INDIVIDUALS FAMILIAR WITH ITS ACTIONS, CHARACTERISTICS AND HAZARDS.
DESCRIPTION
RAPLON™ (rapacuronium bromide) for Injection is a nondepolarizing neuromuscular blocking agent. Its chemical name is 1-[(2β,3α,5α,16β,17β)-3-(Acetyloxy)-17-(1-oxopropoxy)-2-(1-piperidinyl)androstan-16-yl]-1-(2-propenyl) piperidinium bromide. The chemical formula of the bromide salt is C37H61BrN2O4 with a molecular weight of 677.78.
The structural formula is:
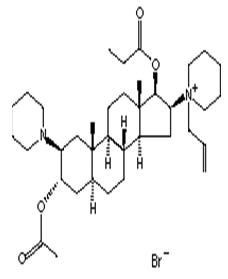
RAPLON™ is a synthetic steroid molecule with a mono-quaternary structure in the form of a bromide salt. This chemical structure has a basic steroid framework similar to other neuromuscular blocking agents like vecuronium, pancuronium, rocuronium, and pipecuronium. Rapacuronium bromide is distinguished as being a propenyl bromide ammonium salt with a 17-hydroxy propionate carboxyester that has the same basic steroid backbone as the rest of the family of steroid neuromuscular blockers.
RAPLON™ is supplied as a sterile, nonpyrogenic lyophilized cake in 5 mL and 10 mL vials. Each 5 mL vial contains 100 mg of rapacuronium bromide base, 35.8 mg of citric acid anhydrous, 7.5 mg of sodium phosphate dibasic anhydrous, 137.5 mg of mannitol, and sodium hydroxide and/or phosphoric acid to buffer and adjust the pH. When the 5 mL vial is reconstituted to a volume of 5 mL with sterile water for injection or bacteriostatic water for injection, an isotonic preparation for intravenous injection is obtained at a pH of 4.0 with a concentration of 20 mg of rapacuronium bromide base per mL. Each 10 mL vial contains 200 mg of rapacuronium bromide base, 71.5 mg of citric acid anhydrous, 15.0 mg of sodium phosphate dibasic anhydrous, 275.0 mg of mannitol, and sodium hydroxide and/or phosphoric acid to buffer and adjust the pH. When the 10 mL vial is reconstituted to a volume of 10 mL with sterile water for injection or bacteriostatic water for injection, an isotonic preparation for intravenous injection is obtained at a pH of 4.0 with a concentration of 20 mg of rapacuronium bromide base per mL.
CLINICAL PHARMACOLOGY
RAPLON™ (rapacuronium bromide) for Injection is a nondepolarizing neuromuscular blocking agent with a rapid onset of action (mean onset approximately 90 seconds; range 35–219 seconds) and a dose-dependent duration of action. The recommended dose of 1.5 mg/kg in adults has a short clinical duration of action (mean duration approximately 15 minutes; range 6–30 minutes) (see CLINICAL PHARMACOLOGY–Clinical Studies). Rapacuronium acts by competing for cholinergic receptors at the motor end plate. Profound neuromuscular blockade induced by RAPLON™ can be reversed by neostigmine (see CLINICAL PHARMACOLOGY–Early Reversal).
Pharmacodynamics
The neuromuscular block seen after the intravenous administration of 1.5 mg/kg RAPLON™ (rapacuronium bromide) for Injection is primarily due to rapacuronium. Plasma levels of the major active metabolite of rapacuronium (the 3-hydroxy metabolite) are relatively low compared to the parent at a dose of 1.5 mg/kg of RAPLON™. Pharmacokinetic and pharmacodynamic modeling studies were conducted after the separate administration of rapacuronium bromide and the 3-hydroxy metabolite. These studies evaluated the effect of plasma drug concentrations on the neuromuscular block achieved as measured by mechanomyography [MMG] of the adductor pollicis muscle to indirect supramaximal train-of-four stimulation of the ulnar nerve. These results are shown in Tables 1 and 2. Comparison of keo (rate constant for the equilibration between effect compartment and the outside effect compartment) and EC50 (concentration in the effect compartment at 50% drug effect) values of rapacuronium and the 3-hydroxy metabolite shows that the metabolite has slower onset and a higher potency than rapacuronium bromide. However, when comparing the ED90 (dose required to produce 90% suppression of the first [T1] mechanomyographic [MMG] response of the adductor pollicis muscle to indirect supramaximal train-of-four stimulation of the ulnar nerve) of rapacuronium bromide (1.03 mg/kg) and the 3-hydroxy metabolite (0.42 mg/kg) with the ED90 of rocuronium (0.3 mg/kg), vecuronium (0.05 mg/kg) and pancuronium (0.06 mg/kg), rapacuronium bromide and the 3-hydroxy metabolite may be viewed as low potency neuromuscular blocking drugs.
| Parameter | |
|---|---|
| keo, 1/minute | 0.44 (41) |
| γ | 2.97 (23) |
| EC50, mcg/mL | 4.44 (33) |
| ED90, mg/kg | 1.03 (33) |
| Parameter | |
|---|---|
| keo, 1/minute | 0.10 (40) |
| γ | 4.83 (44) |
| EC50, mcg/mL | 2.06 (55) |
| ED90, mg/kg | 0.46 (33) |
The ED50 for rapacuronium bromide (dose required to produce 50% suppression of the first [T1] mechanomyographic [MMG] response of the adductor pollicis muscle to indirect supramaximal train-of-four stimulation of the ulnar nerve) during opioid/nitrous oxide/oxygen anesthesia is approximately 0.3 mg/kg in adult (18 to 64 years) and geriatric (≥65 years) patients. The ED50 for rapacuronium bromide for pediatric patients (1 to 12 years) is 0.4 mg/kg and for infants (1 month to <1 year) is 0.3 mg/kg (see PRECAUTIONS–Pediatric Use).
Tables 3 and 4 present the neuromuscular function parameters following an initial dose of RAPLON™ in adult patients (18 to 64 years) and geriatric patients (≥65 years).
| Dosage | Time to Maximum Block* (sec) | Maximum Block† (%) | Clinical Duration (min)‡ | 25–75% T1 Recovery Index (min) | Time to 70% T4/T1 Recovery§ (min) |
|---|---|---|---|---|---|
| RAPLON™ | 88 (47) | 99 (2) | 15 (5) | 9 (5) | 34 (15) |
| 1.5 mg/kg | (n=32) | (n=49) | (n=57) | (n=38) | (n=47) |
In US clinical trials, in adult patients (18 to 64 years), the mean (SD) time to maximum block [time from injection to maximum block (peak effect)] following an initial 2.5 mg/kg dose of RAPLON™ was 72 (24) seconds (n=19). The mean (SD) clinical duration (time from injection to return to 25% of control T1) in adult patients was 24 (8) minutes (n=45) with a mean (SD) 25–75% T1 recovery index of 13 (7) minutes (n=23) and a mean (SD) time to 70% T4/T1 recovery (n=34) of 58 (15) minutes (time from injection to recovery of 70% T4/T1).
| Dosage | Time to Maximum Block* (sec) | Maximum Block† (%) | Clinical Duration‡ (min) | 25–75% T1 Recovery Index (min) | Time to 70% T4/T1 Recovery§ (min) |
|---|---|---|---|---|---|
| RAPLON™ | 89 (6) | 98 (5) | 17 (5) | 11 (6) | 38 (5) |
| 1.5 mg/kg | (n=6) | (n=17) | (n=16) | (n=4) | (n=12) |
In US clinical trials, in geriatric patients (≥65 years), the mean (SD) time to maximum block [time from injection to maximum block (peak effect)] following an initial 2.5 mg/kg dose of RAPLON™ was 51 (21) seconds (n=4). The mean (SD) clinical duration (time from injection to return to 25% of control T1) was 43 (37) minutes (n=13) with a mean (SD) 25–75% T1 recovery index of 17 (16) minutes (n=3) and a mean (SD) time to 70% T4/T1 recovery (n=9) of 76 (20) minutes (time from injection to recovery of 70% T4/T1).
Table 5 presents the neuromuscular function parameters following an initial dose of RAPLON™ in pediatric patients under halothane anesthesia.
| Age Group | Dosage | Time to Maximum Block*
(sec) | Maximum Block†
(%) | Clinical Duration‡
(min) | 25–75% T1 Recovery Index (min) | Time to 70% T4/T1 Recovery§
(min) |
|---|---|---|---|---|---|---|
| Infants (1 mo to < 2 yrs) | RAPLON™ 1 mg/kg (n=14) | 88 (73) | 96 (12) | 9 (3) (n=13) | 7 (4) (n=9) | 20 (7) (n=12) |
| RAPLON™ 2 mg/kg (n=16) | 84 (66) | 99 (3) | 16 (7) | 13 (11) (n=8) | 34 (13) | |
| Children (2 to 12 yrs) | RAPLON™ 2 mg/kg (n=23) | 53 (16) | 100 (1) | 14 (7) | 6 (4) (n=19) | 26 (9) (n=21) |
| RAPLON™ 3 mg/kg (n=21) | 67 (44) | 100 (2) | 18 (3) (n=20) | 11 (6) (n=12) | 37 (9) (n=19) |
|
Cardiac Patients
Hemodynamic parameters were assessed in patients with coronary artery and valvular disease receiving 1.5 mg/kg of RAPLON™ in one US (n=14) and one European (n=18) placebo controlled trial. Overall, there were mild to moderate changes in hemodynamic parameters (e.g., mean arterial pressure, heart rate, mean pulmonary artery pressure, pulmonary capillary wedge pressure, central venous pressure, cardiac index, and systemic vascular resistance index) measured invasively, in cardiac patients (valvular disease or coronary artery disease) receiving 1.5 mg/kg RAPLON™.
Obese Patients
Obese patients with a body mass index (BMI) ≥30 kg/m2 were compared to normal weight subjects in a European study in which they received 1.5 mg/kg of RAPLON™ as part of a rapid sequence induction of anesthesia using either fentanyl/thiopental or alfentanil/propofol. Patients were dosed based on actual body weight. Acceptable (excellent or good) intubating conditions following 1.5 mg/kg of RAPLON™ were similar in obese (86% under fentanyl/thiopental, 92% under alfentanil/propofol) and normal weight subjects (87% under fentanyl/thiopental, 91% under alfentanil/propofol) at 60 seconds. The percent of excellent scores under fentanyl/thiopental or alfentanil/propofol were 48% and 65%, respectively in obese patients, and 44% and 52%, respectively, in normal weight patients.
Repeat Dosing in Adults
In three controlled clinical trials, after an initial intubating dose of RAPLON™ of 1.5 mg/kg, 3 additional doses of 0.5 to 0.55 mg/kg were administered at 25% recovery of T1 or at the reappearance of T3 (n=76). The duration of action of maintenance doses of 0.5 to 0.55 mg/kg ranged from 3 to 35 minutes. A statistically significant increase in the duration of action of RAPLON™ was noted with subsequent maintenance doses (see Table 6).
| Study 1 RAPLON™ 0.55 mg/kg | Study 2 RAPLON™ 0.5 mg/kg | Study 3 RAPLON™ 0.5 mg/kg |
|
|---|---|---|---|
| Dose No. 1 | (n=15) | (n=28) | (n=33) |
| Mean (SD) | 7 (3) | 12 (3) | 13 (3) |
| Median | 6 | 12 | 13 |
| Range | 3–12 | 6–19 | 7–20 |
| Dose No. 2 | (n=15) | (n=28) | (n=33) |
| Mean (SD) | 8 (2) | 14 (4) | 15 (5) |
| Median | 8 | 14 | 14 |
| Range | 5–12 | 6–22 | 8–29 |
| Dose No. 3 | (n=14) | (n=25) | Not measured |
| Mean (SD) | 8 (2) | 16 (6) | |
| Median | 8 | 15 | |
| Range | 5–13 | 6–35 |
Early Reversal
Administration of neostigmine (50 or 70 mcg/kg at 2 or 5 minutes) at profound neuromuscular block (≥90%) following administration of either 1.5 or 2.5 mg/kg of RAPLON™ in adults reduced the recovery time by approximately 50%. After early reversal with neostigmine, a decrease in neuromuscular function did not occur over the clinical trial period.
Table 7 presents the recovery parameters following reversal of profound block from a US study of adult patients. Anesthesia consisted of premedication with midazolam, induction with fentanyl and propofol, and maintenance with N2O supplemented with fentanyl and propofol.
| RAPLON™ Dose | Neostigmine Dose | Time of Neostigmine Administration | Clinical Duration (min) | 25–75% T1 Recovery Index (min) | Time to 70% T4/T1 Recovery (min) | Time to 80% T4/T1 Recovery (min) |
|---|---|---|---|---|---|---|
| 1.5 mg/kg | None | N/A (n=11) | 17 (5)* | 12 (5)* | 38 (10) * | 43 (12) * |
| 50 mcg/kg | 2 min (n=7) | 8 (1) | 5 (1) | 17 (4) | 20 (5) | |
| 5 min (n=12) | 9 (1) | 5 (3) | 17 (3) | 19 (4) | ||
| 70 mcg/kg | 2 min (n=10) | 8 (1) | 7 (4) | 15 (3) | 21 (7) | |
| 5 min (n=9) | 9 (1) | 6 (2) | 19 (8) | 24 (8) | ||
| 2.5 mg/kg | None | N/A (n=10) | 24 (5) * | 15 (6) * | 56 (13) * | 60 (11) * |
| 50 mcg/kg | 2 min (n=12) | 12 (2) | 9 (4) | 26 (7) | 31 (8) | |
| 5 min (n=8) | 12 (3) | 8 (3) | 32 (13) | 38 (18) | ||
| 70 mcg/kg | 2 min (n=9) | 12 (2) | 12 (5)† | 35 (8) | 41 (10) | |
| 5 min (n=9) | 12 (2) | 8 (3) | 28 (9) | 36 (12) | ||
Hemodynamics
After the administration of RAPLON™, dose-related increases in heart rate were observed, peaking within the first few minutes after RAPLON™ administration. These changes in heart rate were generally mild to moderate and were stable or near baseline levels within 5 to 10 minutes of RAPLON™ administration. After the administration of RAPLON™, dose-related decreases in mean arterial pressure (MAP) were observed. Decreases in MAP occurred after all doses of RAPLON™. These changes were observed to peak within 5 minutes after the administration of RAPLON™, returning toward baseline by 10 minutes.
Increases in heart rate and decreases in mean blood pressure were also observed in the pediatric population. In neonates, infants, and children treated with RAPLON™, the observed changes of increased heart rate and decreased mean blood pressure were generally small in magnitude (see CLINICAL PHARMACOLOGY–Clinical Studies).
Dose- and duration-related adverse ECG changes were observed in nonclinical studies in dogs. These changes included prolongation of the QT interval after dosing 2 times per week over 4 weeks, at a total dosage of 18 mg/kg/day given in 3 divided doses, and prolongation of QT interval, sinus arrhythmia, lengthened PR intervals, P wave widening, and AV dissociation following a bolus dose of 27 mg/kg given at 30 minutes after an uneventful first dose of 13.5 mg/kg. In the cat, right bundle branch block pattern and prolonged PR intervals were observed following a bolus dose of 26 mg/kg given at 30 minutes after an uneventful first dose of 13 mg/kg. Therefore, potential adverse ECG effects in humans should be considered when RAPLON™ is given in a high bolus dose or following prolonged infusion.
Electrocardiogram parameters (QT, QTc, and RR intervals) were assessed in patients during a 15 minute observation period after receiving 1.5 mg/kg RAPLON™ (n=18) and placebo (n=16) in a European study. Mean QT interval decreases up to 0.015 second from baseline were observed in the RAPLON™ group while small increases of up to 0.082 second were observed in the placebo group. Mean changes in the QTc interval during the 15-minute period ranged from a decrease of 0.025 second to an increase of 0.052 second from baseline in the RAPLON™ group compared to increases of up to 0.04 second in the placebo group. Mean changes in the RR interval ranged from 0.101 to 0.024 second in the RAPLON™ group and up to 0.113 second in the placebo group. The clinical significance of these changes is unknown.
Histamine Release
Plasma histamine release was assessed following administration of RAPLON™ (1.0, 2.0, and 3.0 mg/kg) in a US study (n=46). Increases in plasma histamine levels peaked at 1 minute following 2.0 and 3.0 mg/kg of RAPLON™. The elevation in histamine levels was dose-related; 1/16, 2/15, and 6/15 subjects in the 1.0 mg/kg, 2.0 mg/kg, and 3.0 mg/kg groups, respectively, demonstrated clinically significant elevations of histamine levels (clinical significance defined as ≥1 ng/mL or 100% increase from baseline). Two of six patients in the 3.0 mg/kg group with clinically significant elevations of histamine levels had ≥30% increase in heart rate and ≥30% decrease in blood pressure after the administration of RAPLON™.
Events possibly related to histamine release (e.g., erythema, bronchospasm) occurred in 29 (5.1%) of 564 adult patients in US studies and in 43 (5.8%) of 736 adult patients in European studies.
Intraocular Pressure
In a clinical study, intraocular pressure following a single bolus dose of 1.5 mg/kg of RAPLON™ (n=8) decreased by a maximum of 15% at 3 minutes.
Pharmacokinetics
Data from the in vivo pharmacokinetic studies were used to develop population estimates of the parameters for the subpopulations represented (e.g., geriatric, pediatric, renal insufficiency, and hepatic insufficiency). These population-based estimates and a measure of the estimated variability are contained in the following sections.
Following intravenous administration of RAPLON™ (rapacuronium bromide) for Injection, plasma concentration data were best described by a three-compartment model. The pharmacokinetic model was parameterized in clearances and volumes. Estimates of these parameters were used subsequently to calculate volume of distribution at steady state and half-lives. Table 8 presents the results of a population pharmacokinetic analysis from 206 adult patients (18 to 83 years), including patients with end-stage renal disease (n=7) and cirrhosis (n=8). The variability for these parameters is not available from this analysis. See Tables 11 and 12 for variability estimates for these parameters.
| PK Parameter | Estimate | CV† |
|---|---|---|
| Plasma Clearance (mL/kg/min) | 6.56 | 2.5% |
| Volume of Distribution at Steady State (mL/kg) | 292 | ND‡ |
Distribution
The mean volume of distribution of RAPLON™ at steady state was 292 mL/kg in adult patients. The mean rapid distribution half-life was 4.56 minutes and the mean slow distribution half-life was 27.8 minutes.
Metabolism
Rapacuronium bromide undergoes hydrolysis of the acetyloxy-ester bond at the 3-position to form the 3-hydroxy metabolite, the major and active metabolite of rapacuronium. Relative to its parent, the 3-hydroxy metabolite has greater potency and a slower onset of action. This hydrolysis is non-specific and can occur at physiological temperature and pH. This hydrolysis may also be catalyzed by esterases of unknown identity and at unknown sites. The cytochrome P450 enzyme system does not appear to be involved in the hydrolysis of rapacuronium bromide. A mass balance study suggests that there may be seven additional minor metabolites of unknown identity in addition to the 3-hydroxy metabolite.
Elimination
A mass balance study using 1.5 mg/kg of [14C] rapacuronium bromide demonstrated that urine and feces are the main routes of elimination of [14C] rapacuronium bromide (Table 9). The mean combined excretion in urine and feces at the end of the continuous 13.5-day collection period was approximately 56% (range: 50–64%), with approximately 28% excreted in urine samples and 28% in feces. Measurable concentrations of radiocarbon were also detected in urine samples collected once a week over four weeks after the end of the continuous 13.5-day collection period.
The estimated radioactivity excreted in expired CO2 over 24 hours was approximately 0.6% of the administered dose. The apparent elimination half-life of radioactivity was estimated to be approximately 22 days, suggesting that complete excretion can take several weeks.
| Source | Percentage Recovery Mean (SD) | Duration of Sampling |
|---|---|---|
| Urine | 28.4 (4.3) | 13.5 days |
| Stool | 27.7 (4.2) | 13.5 days |
| Exhaled Gas† | 0.6 (0.07) | 24 hours |
| Total‡ | 56 (5) (range 50–64%) | 13.5 days |
Rapacuronium bromide, in addition to undergoing hydrolysis to its 3-hydroxy metabolite, is also excreted unchanged in urine and feces. The 3-hydroxy metabolite is excreted unchanged in urine and feces without further biotransformation. Approximately 8% of the administered rapacuronium bromide dose was recovered from urine up to 48 hours after dosing as unchanged rapacuronium bromide and approximately 5% as the 3-hydroxy metabolite (Table 10).
| Compound Excreted in Urine Mean (SD) | Time After RAPLON™ 1.5 mg/kg Bolus | |
|---|---|---|
| 0–24 hours | 0–48 hours | |
| Rapacuronium Bromide (% excreted) | 7.96 (2) | 8.12 (2) |
| 3-hydroxy Metabolite (% excreted) | 3.43 (1) | 4.96 (1.2) |
The mean plasma clearance of rapacuronium bromide in adult patients was 6.56 mL/kg/min and the mean plasma elimination half-life (t1/2β) was 141 minutes. However, this half-life may not represent the terminal elimination of rapacuronium bromide from the body as characterized in the mass balance study.
Protein Binding
Plasma protein binding of rapacuronium was studied in vitro for human plasma by equilibrium dialysis. The protein binding was variable and ranged between 50% and 88%, which was at least partly due to hydrolysis of rapacuronium bromide to its 3-hydroxy metabolite. The specific plasma protein to which rapacuronium binds is unknown. Plasma protein binding of the 3-hydroxy metabolite was not determined.
Special Populations
Geriatrics
In the pooled population pharmacokinetic analysis based on 206 adult patients ages 18 to 83 years, the analysis of covariates showed that total plasma clearance of rapacuronium bromide decreases with increasing age. However, as these changes were not clinically significant, no dosage adjustment is recommended for geriatric patients.
Pediatrics
Pharmacokinetic parameters in pediatric patients (n=49) ranging in age from 1 month to 12 years (median 3 years) were estimated using population pharmacokinetic (PK) analyses. The plasma concentration data were best described by a three-compartment model in which all PK parameters were proportional to body weight. The mean plasma clearance was 10.6 mL/kg/min. The mean volume of distribution at steady state was 495 mL/kg and the mean elimination half-life was 262 minutes (see PRECAUTIONS–Pediatric Use).
Gender
In general, studies in normal adult subjects did not reveal any differences in the pharmacokinetics of RAPLON™ (rapacuronium bromide) for Injection due to gender.
Race
Race was not examined as a covariate in the pooled population pharmacokinetic analysis of RAPLON™.
Renal Insufficiency
Table 11 summarizes the results of conventional PK analyses from a US study of normal volunteers and patients with end-stage renal disease (ESRD) receiving a single bolus dose of 1.5 mg/kg of RAPLON™. Patients with renal insufficiency had a mean 30% reduction in clearance compared with normal adult patients. The volume of distribution was more variable in patients with renal insufficiency compared to normal volunteers.
Comparison of the concentration of the 3-hydroxy metabolite relative to that of rapacuronium bromide up to eight hours after rapacuronium bromide administration and the plasma concentration versus time profiles between the normal volunteer group and patients with ESRD showed that the pharmacokinetics of the 3-hydroxy metabolite were altered in patients with ESRD. In normal volunteers, the ratio of the 3-hydroxy metabolite to rapacuronium increased steadily through the 6-hour period but decreased by the 8-hour time point (from 0.03 at 3 minutes to 4.5 at 8 hours). In patients with ESRD, this ratio showed an increasing trend even at the 8-hour time point (from 0.02 at 3 minutes to 7.5 at 8 hours). The decrease in the plasma concentration of the 3-hydroxy metabolite in patients with ESRD was only 35% from peak levels (265 to 171 ng/mL) compared to an 87% decrease in the normal volunteer group (381 to 49 ng/mL). No clear elimination phase was observed at the end of 8 hours. Despite the persistence of the 3-hydroxy metabolite, neuromuscular function recovered completely with a mean time course to 70% T4/T1 only slightly longer in patients with ESRD, compared to that in healthy volunteers with normal renal function after a single 1.5 mg/kg bolus. However, it is likely that recovery from supplemental doses of RAPLON™ will be prolonged in patients with renal failure.
| PK Parameter | Normal Volunteers* | Patients with ESRD* |
|---|---|---|
| Elimination Half-Life† (t1/2β, min) | 240 (97) | 198 (141)‡ |
| Volume of Distribution at Steady State (mL/kg) | 431.7 (78) | 440.3 (347) |
| Plasma Clearance (mL/kg/min) | 9.4 (2.2) | 6.1 (1.7)‡ |
Hepatic Insufficiency
The pharmacokinetic (PK) parameters for patients with mild to moderate hepatic insufficiency and patients with normal liver function are presented in Table 12. These estimates are based on conventional PK analyses. Plasma clearance and volume of distribution at steady-state are greater in patients with cirrhosis compared to patients with normal liver function. Pharmacokinetics of rapacuronium bromide in patients with severe hepatic impairment have not been evaluated.
| PK Parameter | Normal Liver Function* | Hepatic Insufficiency (cirrhotic)* |
|---|---|---|
|
||
| Elimination Half-Life† (t1/2β, min) | 84 (4) | 88 (6) |
| Volume of Distribution at Steady State (mL/kg) | 252 (77) | 465 (82)‡ |
| Plasma Clearance (mL/kg/min) | 6.6 (1.7) | 9.0 (1.4)‡ |
Drug-Drug Interactions
There were no specific pharmacokinetic studies conducted to examine the drug-drug interactions of RAPLON™ (see PRECAUTIONS).
Clinical Studies
In US studies, 929 patients received RAPLON™ (rapacuronium bromide) for Injection including 219 pediatric, 146 geriatric, and 20 obstetric patients. In European studies, 964 patients received RAPLON™ including 165 pediatric and 63 geriatric patients. The majority of patients, 91%, were ASA (American Society of Anesthesiologists) Class I or II, approximately 8% were ASA Class III, and approximately 1% were ASA Class IV.
Neuromuscular function parameters following administration of RAPLON™, succinylcholine, and mivacurium were compared in one clinical study. In direct comparison with succinylcholine and mivacurium, RAPLON™ at the recommended 1.5 mg/kg dose had a mean (SD) onset of action of 98 (46) seconds compared with 67 (27) seconds for succinylcholine and 127 (50) seconds for mivacurium. RAPLON™ had a mean (SD) clinical duration of 15 (6) minutes compared with 9 (3) minutes for succinylcholine and 21 (5) minutes for mivacurium. Table 13 presents these neuromuscular function parameters in patients 18 years of age and older.
| Drug/ Dosage | Time to Maximum Block*
(sec) | Maximum Block†
(%) | Clinical Duration‡
(min) | 25–75% T1 Recovery Index (min) | Time to 70% T4/T1 Recovery§
(min) |
|---|---|---|---|---|---|
|
|||||
| RAPLON™ 1.5 mg/kg | |||||
| n | 28 | 28 | 25 | 22 | 16 |
| Mean (SD) | 98 (46) | 99 (2) | 15 (6) | 8 (5) | 38 (21) |
| Range | 35–219 | 94–100 | 7–30 | 2–21 | 21–101 |
| Succinylcholine 1 mg/kg | |||||
| n | 30 | 30 | 29 | 29 | N/A |
| Mean (SD) | 67 (27) | 99 (2) | 9 (3) | 2 (1) | |
| Range | 31–138 | 90–100 | 5–15 | 1–4 | |
| Mivacurium 0.25 mg/kg¶ | |||||
| n | 25 | 25 | 21 | 24 | 18 |
| Mean (SD) | 127 (50) | 100 (0.4) | 21 (5) | 9 (4) | 32 (7) |
| Range | 64–261 | 99–100 | 14–29 | 4–20 | 22–45 |
Intubating conditions following administration of RAPLON™ and succinylcholine were compared in three randomized, multicenter trials conducted in the US, France, and Germany. A blinded rater assessed intubating conditions on the Viby-Mogensen scale (see Table 14) 50 seconds after administration of the neuromuscular blocking agent. Several different anesthetic techniques were used. In the US study, fentanyl was given about 5 minutes before intubation followed by propofol one to two minutes before intubation. The French study was similar except that thiopental was used instead of propofol. The German study tested a rapid sequence, with initiation of administration of the neuromuscular blocking agent within a few seconds after the end of the injection of the hypnotic, either fentanyl and thiopental or alfentanil and propofol were used at random. Tables 15 and 16 show the intubation scores in adults (18 to 64 years) and in geriatric patients (≥65 years).
| CLINICALLY ACCEPTABLE | |||
|---|---|---|---|
| Excellent* | Good* | Poor* | |
| Vocal Cord Position | Abducted | Intermediate | Closed |
| Vocal Cord Movement | None | Moving | Closing |
| Easiness of Laryngoscopy† | Easy | Fair | Difficult |
| Airway Reaction | None | Diaphragm | Sustained >10sec |
| Movement of Limbs | None | Slight | Vigorous |
Intubating dosages of 1.5 and 2.5 mg/kg of RAPLON™ were evaluated in 784 patients. A population of patients undergoing Cesarean section was also studied (see below).
| Study | US | France | Germany | |||
|---|---|---|---|---|---|---|
| Drug/Dosage | RAPLON™ 1.5 mg/kg n=124 | Succinylcholine 1.0 mg/kg n=112 | RAPLON™ 1.5 mg/kg n=128 | Succinylcholine 1.0 mg/kg n=128 | RAPLON™ 1.5 mg/kg n=160 | Succinylcholine 1.0 mg/kg n=166 |
| Excellent | 43% | 67% | 30% | 48% | 51% | 73% |
| Good | 44% | 29% | 55% | 41% | 39% | 24% |
| Poor | 13% | 4% | 9% | 9% | 11% | 3% |
| Impossible | 0% | 2% | 5% | 2% | 0% | 0% |
| Study | US | France | ||
|---|---|---|---|---|
| Drug/Dosage | RAPLON™ 1.5 mg/kg n=26 | Succinylcholine 1.0 mg/kg n=28 | RAPLON™ 1.5 mg/kg n=25 | Succinylcholine 1.0 mg/kg n=26 |
| Excellent | 50% | 79% | 32% | 62% |
| Good | 46% | 21% | 48% | 35% |
| Poor | 4% | 0% | 4% | 0% |
| Impossible | 0% | 0% | 16% | 4% |
Intubating conditions were also studied in pediatric patients (≥1 month to ≤12 years) in one non-comparative European study. Patients were premedicated with midazolam and induced with thiopental. Results are presented in Table 17.
| Infants (1 mo to 1 yr) 2.0 mg/kg n=9 | Children (1 to 12 yrs) 2.0 mg/kg n=17 |
|
|---|---|---|
| Excellent | 100% | 59% |
| Good | 0% | 41% |
| Poor | 0% | 0% |
| Impossible | 0% | 0% |
Cesarean Section
In a controlled clinical trial, patients undergoing rapid sequence induction of anesthesia for Cesarean section received thiopental 4–6 mg/kg followed by 2.5 mg/kg of RAPLON™ or 1.5 mg/kg of succinylcholine. Laryngoscopy was initiated 50 seconds after the muscle relaxant was administered and intubation completed by 60 seconds in all patients. Acceptable (excellent or good) intubating conditions were achieved in 14/15 (93%) patients receiving RAPLON™ and in 17/19 (89%) patients receiving succinylcholine. Excellent scores were recorded in 10/15 (67%) RAPLON™ patients and 13/19 (68%) succinylcholine patients.
No neonates born of mothers who received RAPLON™ during Cesarean section had APGAR scores below 6 at 5 minutes post-delivery or NAC (Neurological and Adaptive Capacity) scores <30 at 24 hours post-delivery.
The venous umbilical/maternal concentrations of RAPLON™ (median 8.4%, range of 4.4 to 16.1%) and its 3-hydroxy metabolite (median 10.2%, range of 4.6 to 19.9%) demonstrated that there is some placental transfer of the drug from the maternal blood to the fetal blood at delivery.
Individualization of Dosage
DOSES OF RAPLON™ (rapacuronium bromide) FOR INJECTION SHOULD BE INDIVIDUALIZED AND A PERIPHERAL NERVE STIMULATOR SHOULD BE USED TO MEASURE NEUROMUSCULAR FUNCTION DURING RAPLON™ ADMINISTRATION IN ORDER TO MONITOR DRUG EFFECT, DETERMINE THE NEED FOR ADDITIONAL DOSES, AND CONFIRM RECOVERY FROM NEUROMUSCULAR BLOCK.
Based on the known actions of rapacuronium bromide and other neuromuscular blocking agents, the following factors should be considered when administering RAPLON™.
Renal or Hepatic Impairment
A slight delay in the onset of neuromuscular block and prolongation of duration of block were observed in patients with ESRD when compared to normal volunteers. A greater variability of onset and duration was also observed in patients with renal insufficiency. The mean time to 25–75% T1 recovery was greater in patients with renal and hepatic impairment. Although dosage adjustments are not recommended in patients with renal or hepatic impairment, RAPLON™ should be used with caution in these patient populations.
The results of studies in patients with renal failure and hepatic dysfunction do not suggest any additional safety concerns in these patients when RAPLON™ is administered as a single dose.
Reduced Plasma Cholinesterase Activity
RAPLON™ metabolism does not depend on plasma cholinesterase, therefore, no differences in clinical effect are expected in patients with reduced or normal plasma cholinesterase activity.
Drugs or Conditions Causing Potentiation of, or Resistance to, Neuromuscular Block
Use of inhalation anesthetics (enflurane, isoflurane, halothane, desflurane, sevoflurane) has been shown to enhance the activity of other neuromuscular blocking agents (see PRECAUTIONS–Drug Interactions–Inhalational Anesthetics).
Magnesium salts, lithium, local anesthetics, procainamide, quinidine, and certain antibiotics have been shown to increase the duration of neuromuscular block and decrease infusion requirements of other neuromuscular blocking agents. In patients in whom potentiation of neuromuscular block may be anticipated, a decrease from the recommended initial dose of RAPLON™ should be considered (see PRECAUTIONS–Drug Interactions–Other).
Severe acid-base and/or electrolyte abnormalities may potentiate or cause resistance to the neuromuscular blocking action of RAPLON™.
Resistance to non-depolarizing agents, consistent with up-regulation of skeletal muscle acetylcholine receptors, is associated with burns, disuse atrophy, denervation, and direct muscle trauma. Receptor up-regulation may also contribute to the resistance to nondepolarizing muscle relaxants that sometimes develops in patients with cerebral palsy and in patients with chronic exposure to anticonvulsant or nondepolarizing agents (see PRECAUTIONS–Drug Interactions).
Other nondepolarizing neuromuscular blocking agents have been found to exhibit profound effects in cachectic or debilitated patients, patients with neuromuscular diseases, and patients with carcinomatosis. In these or other patients in whom potentiation of neuromuscular block or difficulty with reversal may be anticipated, a decrease from the recommended initial dose of RAPLON™ should be considered.
Obesity
In obese patients, the initial dose of RAPLON™ should be based upon the patient's actual body weight (see CLINICAL PHARMACOLOGY–Obese Patients).
The neuromuscular blocking effect of 1.5 mg/kg RAPLON™ was determined in a group of obese patients (body mass index ≥30) and a group of non-obese patients (body mass index 20–28). When dosed by actual body weight, the obese group had approximately the same percentage of patients with acceptable (excellent or good) intubating conditions as the non-obese group.
Although RAPLON™ has not been formally studied in morbidly obese patients (body mass index ≥40), clinicians should consider dosing this patient population based on ideal body weight. As with other neuromuscular blocking drugs, RAPLON™ may exhibit prolonged duration and delayed spontaneous recovery when the morbidly obese are dosed based on actual body weight.
Burns
Patients with burns are known to develop resistance to nondepolarizing neuromuscular blocking agents, probably due to up-regulation of post-synaptic skeletal muscle cholinergic receptors.
INDICATIONS AND USAGE
RAPLON™ (rapacuronium bromide) for Injection is indicated as an adjunct to general anesthesia to facilitate tracheal intubation, and to provide skeletal muscle relaxation during surgical procedures.
CONTRAINDICATIONS
RAPLON™ (rapacuronium bromide) for Injection is contraindicated in patients known to have hypersensitivity to rapacuronium bromide.
WARNINGS
RAPLON™ (rapacuronium bromide) FOR INJECTION SHOULD BE ADMINISTERED IN CAREFULLY ADJUSTED DOSAGE BY OR UNDER THE SUPERVISION OF EXPERIENCED CLINICIANS WHO ARE FAMILIAR WITH THE DRUG'S ACTIONS AND THE POSSIBLE COMPLICATIONS OF ITS USE. THE DRUG SHOULD NOT BE ADMINISTERED UNLESS PERSONNEL AND FACILITIES FOR RESUSCITATION AND LIFE SUPPORT (TRACHEAL INTUBATION, ARTIFICIAL VENTILATION, OXYGEN THERAPY), AND AN ANTAGONIST OF RAPLON™ ARE IMMEDIATELY AVAILABLE. IT IS RECOMMENDED THAT ADEQUATE NEUROMUSCULAR MONITORING EQUIPMENT, SUCH AS A PERIPHERAL NERVE STIMULATOR, BE USED TO MEASURE NEUROMUSCULAR FUNCTION DURING THE ADMINISTRATION OF RAPLON™ IN ORDER TO MONITOR DRUG EFFECT, DETERMINE THE NEED FOR ADDITIONAL DOSES, AND CONFIRM RECOVERY FROM NEUROMUSCULAR BLOCK.
RAPLON™ HAS NO KNOWN EFFECT ON CONSCIOUSNESS, PAIN THRESHOLD, OR CEREBRATION. TO AVOID DISTRESS TO THE PATIENT, NEUROMUSCULAR BLOCK SHOULD NOT BE INDUCED BEFORE UNCONSCIOUSNESS. THEREFORE, ADMINISTRATION OF RAPLON™ MUST BE ACCOMPANIED BY ADEQUATE ANESTHESIA OR SEDATING AGENTS.
Anaphylaxis
Severe anaphylactic reactions to neuromuscular blocking agents, including RAPLON™, have been reported. These reactions have, in some cases, been life-threatening and fatal. Due to the potential severity of these reactions, the necessary precautions, such as the immediate availability of appropriate emergency treatment, should be taken. Precautions should also be taken in those individuals who have had previous anaphylactic reactions to other neuromuscular blocking agents, since cross-reactivity between neuromuscular blocking agents, both depolarizing and non-depolarizing, has been reported.
Long-Term Use
RAPLON™ (rapacuronium bromide) FOR INJECTION SHOULD NOT BE ADMINISTERED BY INFUSION, PARTICULARLY IN THE INTENSIVE CARE UNIT (ICU) OR DURING LONG SURGICAL PROCEDURES. In an infusion study (n=90) with RAPLON™, one patient displayed a deterioration of neuromuscular function after attaining evidence of adequate spontaneous recovery. Six minutes after spontaneously recovering to a T4/T1 of 70%, the T4/T1 decreased to 64%. Thirteen minutes later T4/T1 recovered to 80%. This patient did not receive neostigmine and had no respiratory problems that required reintubation or mask assisted breathing.
Radioisotope studies have demonstrated that only 56% of the original RAPLON™ dose had been excreted two weeks following a single IV bolus. Excretion of 14C-labeled rapacuronium continued for at least six weeks. Accumulation of rapacuronium bromide following repeat dosing is likely to occur but has not been studied.
ECG abnormalities have been observed in animals following repeat doses and large single doses of rapacuronium bromide (see CLINICAL PHARMACOLOGY–Hemodynamics).
During pregnancy there is passage of low levels of rapacuronium across the placenta and slow elimination following a single maternal dose (see CLINICAL PHARMACOLOGY–Pharmacokinetics). The risk to the developing fetus from extended low-dose intrauterine exposure to a neuromuscular blocking agent is unknown. Because of these concerns and because animal reproduction studies are not always predictive of human response, this drug should not be used during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus. This warning does not extend to use of RAPLON™ during Cesarean section, but appropriate monitoring of the infant is recommended after delivery (see CLINICAL PHARMACOLOGY–Clinical Studies).
In patients with myasthenia gravis or myasthenic (Eaton-Lambert) syndrome, small doses of nondepolarizing neuromuscular blocking agents may have profound effects. In such patients, a peripheral nerve stimulator and use of a small test dose may be of value in monitoring the response to administration of muscle relaxants.
Reconstituted RAPLON™, which has an acid pH of 4.0, should not be mixed with alkaline solutions (e.g., barbiturate solutions) in the same syringe or administered simultaneously during intravenous infusion through the same needle.
PRECAUTIONS
Anaphylaxis
Since allergic cross-reactivity has been reported in this class, request information from your patients about previous anaphylactic reactions to other neuromuscular blocking agents. In addition, inform your patients that severe anaphylactic reactions to neuromuscular blocking agents, including RAPLON™, have been reported.
Repeat Dosing
It is strongly recommended that during administration of RAPLON™ (rapacuronium bromide) for Injection, neuromuscular transmission and recovery be monitored continuously using a nerve stimulator. Additional doses of RAPLON™ should not be given until there is a definite response (one twitch of the train-of-four) to nerve stimulation.
Repeat dosing in adults after intubating doses greater than 1.5 mg/kg, and repeat dosing in pediatric patients have not been studied, and are, therefore, not recommended.
The experience with a limited number of patients indicates that repeat bolus dosing of RAPLON™ may have a potential for prolonged block. Theoretically, increased histamine effects may result from slow elimination of the drug from the body; however, no studies to date have been conducted to substantiate this possibility (see CLINICAL PHARMACOLOGY–Pharmacokinetics).
In three European studies, adult patients were given an intubating dose of 1.5 mg/kg of RAPLON™ followed by three maintenance doses of RAPLON™ 0.5 mg/kg (n=61) or 0.55 mg/kg (n=19). In one study, median (range) clinical durations of the three doses of 0.55 mg/kg were 6 (3–12), 8 (5–12), and 8 (5–13) minutes. In the second study, the three maintenance doses of 0.5 mg/kg had median (range) clinical durations of 12 (6–19), 14 (6–22), and 15 (6–35) minutes. Neostigmine was administered to half the patients in this study after the third dose when T1 returned to 25%, and the median (range) time to recover to 70% T4/T1 was 6 (2–9) minutes (n=14). The remaining half of the patients had spontaneous recovery after the third dose. The median spontaneous recovery from 25% T1 to 70% T4/T1 was 57 minutes and ranged from 44 to 80 minutes (n=11). In the third study, the median (range) clinical durations of the first and second maintenance doses of 0.5 mg/kg were 13 (7–20) and 14 (8–29) minutes. Neostigmine (n=12) or edrophonium (n=13) were administered two minutes after the third dose of RAPLON™. Median (range) recovery time to 70% T4/T1 was 14 (7–24) minutes after neostigmine and 33 (19–49) minutes after edrophonium (see CLINICAL PHARMACOLOGY–Repeat Dosing in Adults).
In the 80 patients who received three maintenance doses following a bolus dose of 1.5 mg/kg of RAPLON™, adverse events reported in separate patients during or following the maintenance doses consisted of hypotension, tachycardia, respiratory depression, and bronchospasm.
Drug Interactions
Inhalation Anesthetics
Use of inhalation anesthetics (enflurane, isoflurane, halothane, desflurane, sevoflurane) have been shown to enhance the activity of other neuromuscular blocking agents and may enhance the activity of RAPLON™ (rapacuronium bromide) for Injection.
Intravenous Anesthetics
In clinical studies, the use of propofol for induction and maintenance of anesthesia did not alter the clinical duration or recovery characteristics of recommended doses of RAPLON™.
Anticonvulsants
As with other nondepolarizing neuromuscular blocking drugs, if RAPLON™ is administered to patients chronically receiving anticonvulsant agents such as carbamazepine or phenytoin, shorter durations of neuromuscular block may occur and infusion rates may be higher due to the development of resistance to nondepolarizing muscle relaxants. While the mechanism for development of this resistance is not known, receptor up-regulation may be a contributing factor.
Antibiotics
Certain antibiotics (e.g., aminoglycosides, vancomycin, tetracyclines, bacitracin, polymyxin, and colistin) may enhance the neuromuscular blocking action of nondepolarizing agents such as RAPLON™. If these antibiotics are used in conjunction with RAPLON™, prolongation of neuromuscular block should be considered a possibility.
Other
Magnesium salts, administered for the management of toxemia of pregnancy, may enhance neuromuscular blockade. Experience concerning injection of quinidine during recovery from use of other muscle relaxants suggests that recurrent paralysis may occur. This possibility must also be considered for RAPLON™.
Other drugs that may possibly enhance the neuromuscular blocking action of nondepolarizing muscle relaxants, such as RAPLON™, include lithium, local anesthetics, procainamide, and quinidine.
Acid-base and/or serum electrolyte abnormalities may potentiate or antagonize the action of neuromuscular blocking agents.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies in animals to evaluate carcinogenic potential or impairment of fertility with rapacuronium bromide have not been performed. Mutagenicity studies conducted with rapacuronium using the Ames test and the Mouse Lymphoma L5178Y cell assay were negative. An in vivo rat bone marrow micronucleus assay for clastogenic activity was also negative for rapacuronium. Two in vitro human lymphocyte chromosomal aberration assays for clastogenic potential were conducted with rapacuronium. Both assays were negative in the presence of metabolic activation, while in the absence of metabolic activation the first assay was inconclusive and the second assay was positive.
Pregnancy
Pregnancy Category C
Reproduction studies have been performed in pregnant nonventilated New Zealand White rabbits and nonventilated Sprague Dawley rats. Throughout gestation days 6–18, rabbits received 0.75, 1.5, or 3 mg/kg/day of rapacuronium bromide by continuous infusion. Rats, during gestation days 6–17, received intravenous doses of 0.75, 1.5, or 2.25 mg/kg/day of rapacuronium bromide in 3 divided doses at 30 minute intervals on each treatment day. No teratogenic effects were observed in rabbits or rats at the highest doses tested. The high doses of 3 and 2.25 mg/kg are approximately 0.3 and 0.1 times the maximum recommended human intravenous dose for adults on a mg/m2 basis, respectively. Post-implantation losses, as evidenced by increased resorption, were observed in rabbits at and above the lowest dose of 0.75 mg/kg, which is approximately 0.1 times the maximum recommended human intravenous dose for adults on a mg/m2 basis.
Fetotoxicity, as evidenced by increased fetal deaths and subsequent resorption, was observed in rats at the high dose of 2.25 mg/kg, which is approximately 0.1 times the maximum recommended human intravenous dose for adults on a mg/m2 basis. There are no adequate and well-controlled studies in pregnant women.
During pregnancy there is passage of low levels of rapacuronium across the placenta and slow elimination following a single maternal dose (see CLINICAL PHARMACOLOGY–Clinical Studies–Cesarean Section). The risk to the developing fetus from extended low-dose intrauterine exposure to a neuromuscular blocking agent is unknown. Because of these concerns and because animal reproduction studies are not always predictive of human response, this drug should not be used during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus.
Labor and Delivery
The use of RAPLON™ (rapacuronium bromide) for Injection in Cesarean section has been studied in a limited number of patients (see CLINICAL PHARMACOLOGY–Clinical Studies).
Nursing Mothers
It is not known whether this drug is excreted in human milk or what effects it may have after oral administration. Since many drugs are excreted in human milk, caution should be exercised when RAPLON™ (rapacuronium bromide) for Injection is administered to nursing mothers.
Pediatric Use
RAPLON™ (rapacuronium bromide) for Injection single bolus dose administration has been studied in 397 pediatric patients, the majority of whom were ASA Class I and II.
The use of RAPLON™ has not been studied in pediatric and adolescent patients aged 13 to 17 years.
There are insufficient data to recommend the use of RAPLON™ in infants <1 month of age until more is known about the safety of RAPLON™ in this population.
The intravenous administration of RAPLON™ has been studied in pediatric patients from 1 month up to 12 years of age (see CLINICAL PHARMACOLOGY–Clinical Studies and DOSAGE AND ADMINISTRATION). Initial doses of 2 mg/kg intravenously in pediatric patients (ages 1 month to 12 years) under halothane anesthesia produce acceptable intubating conditions within 60 seconds. Mean maximum block occurred within 90 seconds in most pediatric patients and had a mean clinical duration of 15 minutes. Intubating doses of 3.0 mg/kg in children (2 to 12 years) provided maximum block within 90 seconds and a mean clinical duration of 18 minutes. Sufficient numbers of pediatric patients 1 month of age and older have received RAPLON™ to establish the safety of single-dose administration in this age group.
No long-term follow-up data are available in pediatric patients exposed to RAPLON™. Studies have demonstrated small quantities of residual drug remaining in tissues of animals administered a single bolus injection of rapacuronium one week after injection. This small residual was primarily observed in kidney, heart, lung, and pituitary. Elimination kinetics in pediatric patients have not been studied, although elimination in adult humans is known to be slower than in animal species tested. Measurable concentrations of radiolabeled rapacuronium in human urine samples following single-dose administration in adults were detected over a period of 6 weeks. The effect of sequestered drug in tissues theoretically may affect development; however, no studies to date have been conducted to substantiate this possibility.
Geriatric Use
RAPLON™ (rapacuronium bromide) for Injection has been studied in 209 patients ≥65 years of age. Advanced age or other conditions associated with slower circulation time, e.g., cardiovascular disease, may be associated with a delay in onset time. Nevertheless, the recommended dosage of 1.5 mg/kg should not be increased in these patients to reduce onset time, as higher doses produce a longer duration of action (see CLINICAL PHARMACOLOGY–Pharmacodynamics–Special Populations).
RAPLON™ is known to be substantially excreted by the kidney, and the risk of prolonged effect or other toxic reactions to this drug may be greater in patients with impaired renal function. While elderly patients are more likely to have altered renal function, no dosage adjustments are recommended in geriatric patients.
Hepatic Disease
Resistance to neuromuscular blocking agents in patients with hepatic insufficiency has been ascribed to an increase in volume of distribution. RAPLON™ (rapacuronium bromide) for Injection at a dose of 1.5 mg/kg has been studied in a limited number of patients with cirrhosis (n=6) under isoflurane anesthesia. Following 1.5 mg/kg of RAPLON™, the median (range) of clinical duration and recovery rate in patients with cirrhosis were 14 (8–18) minutes and 14 (9–18) minutes, respectively. These times were similar to the median times of 16 minutes clinical duration and recovery rate of 14 minutes in patients with normal hepatic function. The plasma clearance of rapacuronium was faster and the volume of distribution was greater in patients with cirrhosis compared to normal controls.
Renal Failure
RAPLON™ (rapacuronium bromide) for Injection has been studied at a dose of 1.5 mg/kg in one US study, in patients with end-stage renal disease (n=9) under isoflurane anesthesia. The median (range) onset time in patients with ESRD [83 (38–180) seconds] was slow compared to normal volunteers (median onset time 66 seconds). The median clinical duration of 12 minutes (range 6 to 39 minutes) in patients with ESRD was similar to the median time of 13 minutes in normal volunteers. The recovery time from 25–75% T1 ranged from 6 to 68 minutes in patients with ESRD.
Malignant Hyperthermia (MH)
RAPLON™ (rapacuronium bromide) for Injection has not been studied in MH-susceptible patients. No subjects exposed to RAPLON™ developed MH or any other syndrome suggestive of MH during premarketing clinical studies. In a study with MH-susceptible swine, the administration of RAPLON™ did not trigger malignant hyperthermia. Since RAPLON™ is always used with other agents, and the occurrence of malignant hyperthermia during anesthesia is possible even in the absence of known triggering agents, clinicians should be prepared to diagnose and treat malignant hyperthermia during the administration of any anesthetic.
Use in Patients With Elevated Intracranial Pressure
In a clinical trial enrolling patients with head injury in which intracranial pressure was monitored, the effects of RAPLON™ (rapacuronium bromide) for Injection and vecuronium were compared. One patient in the RAPLON™-treated group developed an increase in intracranial pressure from 17 to 34 mm Hg two minutes after receiving 1.5 mg/kg of RAPLON™. In the same study, a patient in the vecuronium-treated group developed a rise in intracranial pressure from 26 to 45 mm Hg six minutes after receiving vecuronium 0.1 mg/kg. The results of this study were not conclusive.
ADVERSE REACTIONS
Premarketing Clinical Trial Experience
The safety of RAPLON™ (rapacuronium bromide) for Injection was evaluated in 2036 subjects in prospective clinical trials. The majority of use in clinical trials was single bolus intravenous exposure.
Incidence of Adverse Events in Controlled Clinical Trials
The most common adverse event with an incidence of >5% seen with RAPLON™ in controlled clinical trials was hypotension (5.2%). Table 18 lists treatment-emergent signs and symptoms that occurred in at least 1% of patients receiving RAPLON™ in controlled clinical trials that were numerically more frequent than in the active control.
| Body System Adverse Clinical Experience | RAPLON™ n=1956 | Succinylcholine n=572 | Other Active Controls*
n=141 |
|---|---|---|---|
|
|||
| Cardiovascular | |||
| Hypotension | 5.2% | 6.5% | 4.3% |
| Tachycardia | 3.2% | 0.52% | 1.4% |
| Bradycardia | 1.5% | 1% | 2.1% |
| Respiratory | |||
| Bronchospasm | 3.2% | 2.1% | 0.71% |
Incidence of Other Adverse Events During Premarketing Evaluation of RAPLON™–in All Treated Patients
In the following tabulation, the frequencies represent the proportion of the 1956 patients exposed to at least one dose of RAPLON™ who experienced an event of the type cited on at least one occasion while receiving RAPLON™. All events reported are included except those already listed in the previous table. Although events reported occurred during treatment with RAPLON™, a causal relationship has not necessarily been established.
Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Body as a Whole: Infrequent: fever, rigors, back pain, hypothermia, chest pain, peripheral edema, pain; Rare: asthenia, fatigue, non-inflammatory swelling, therapeutic response decrease.
Cardiovascular: Infrequent: hypertension, extrasystoles, abnormal ECG, arrhythmia, cerebrovascular disorder, ventricular fibrillation, atrial fibrillation, ventricular tachycardia; Rare: atria arrhythmia, cardiac failure, right cardiac failure, cardio-respiratory arrest, cardiac arrest, thrombophlebitis, supraventricular extrasystoles, supraventricular tachycardia, myocardial infarction, left bundle branch block.
Digestive: Frequent: vomiting, nausea; Infrequent: ileus, saliva increased; Rare: abdominal pain, cholelithiasis, nonspecific gastrointestinal disorder, rectal hemorrhage, esophagospasm, oral hemorrhage, tooth disorder.
Hemic and Lymphatic: Infrequent: thrombosis, post-operative bleeding; Rare: epistaxis, coagulation factor decrease, purpura, anemia, hemoperitoneum.
Metabolic and Nutritional: Rare: acidosis.
Musculoskeletal: Infrequent: myalgia; Rare: muscle weakness, neonatal hypotonia.
Nervous: Infrequent: hypoesthesia, hemiparesis, hypertonia, prolonged neuromuscular block, prolonged anesthesia emergence; Rare: headache, cerebral hemorrhage, intracranial pressure increased, migraine, ptosis, tetany, breath holding, confusion, anxiety.
Respiratory: Infrequent: hypoxia, increased airway pressure, hypoventilation, laryngismus, coughing, apnea, respiratory depression, upper airway obstruction, neonatal respiratory distress syndrome, pneumothorax, pulmonary edema, respiratory insufficiency, stridor; Rare: pharyngitis, larynx edema, dyspnea, neonatal respiratory depression, hyperventilation, rhinitis, sputum increase.
Skin: Frequent: erythematous rash; Infrequent: injection site reaction, injection site pain, rash, urticaria, pruritus, sweating increased; Rare: paravenous injection.
Special Senses: Rare: corneal ulceration, meiosis, decreased hearing.
Urogenital: Infrequent: urinary retention, oliguria; Rare: abnormal renal function, urinary tract infection, pelvic inflammation, vaginal bleeding.
Post-Marketing Experience
There have been post-marketing reports of severe allergic reactions (anaphylactic and anaphylactoid reactions) associated with the use of neuromuscular blocking agents, including RAPLON™. Some of these reactions have been life-threatening and fatal. Because these reactions were reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency (see WARNINGS and PRECAUTIONS).
OVERDOSAGE
In premarketing clinical studies, one case of accidental overdose with RAPLON™ (rapacuronium bromide) for Injection was reported. A 22-year-old obstetric patient received 5 mg/kg of RAPLON™ during rapid sequence induction for Cesarean section. The patient did not meet extubating criteria until more than two hours after administration of RAPLON™. Complete recovery was reached 19 minutes after the sixth dose of 1.0 mg of neostigmine. There was no evidence of recurarization or respiratory distress in the recovery room. The premature newborn did not demonstrate evidence of neuromuscular weakness.
Overdosage with neuromuscular blocking agents may result in neuromuscular block extending beyond the time needed for surgery and anesthesia. The primary treatment is maintenance of a patent airway and controlled ventilation until recovery of neuromuscular function is assured.
ANTAGONISM OF NEUROMUSCULAR BLOCKADE
THE USE OF A NERVE STIMULATOR TO DOCUMENT RECOVERY AND ANTAGONISM OF NEUROMUSCULAR BLOCKADE IS RECOMMENDED. Patients should be evaluated for adequate clinical evidence of antagonism, e.g., 5 second head lift, ventilation, and upper airway maintenance. Ventilation must be supported until recovery of normal respiration is assured. A 1.5 mg/kg or 2.5 mg/kg dose of RAPLON™ (rapacuronium bromide) for Injection may be reversed 2 minutes after administration with neostigmine 50 mcg/kg in order to reduce the duration by approximately 50%.
Antagonism may be delayed in the presence of debilitation, carcinomatosis, and concomitant use of certain broad-spectrum antibiotics, anesthetic agents, and other drugs that enhance neuromuscular blockade or separately cause respiratory depression. Under such circumstances, clinical management is the same as that for prolonged neuromuscular blockade.
DOSAGE AND ADMINISTRATION
RAPLON™ (rapacuronium bromide) FOR INJECTION IS INTENDED FOR INTRAVENOUS USE ONLY. THIS DRUG SHOULD BE ADMINISTERED BY OR UNDER THE SUPERVISION OF EXPERIENCED CLINICIANS FAMILIAR WITH THE USE OF NEUROMUSCULAR BLOCKING AGENTS. THE DOSAGE INFORMATION PROVIDED BELOW IS INTENDED AS A GUIDE ONLY (see CLINICAL PHARMACOLOGY). THE USE OF ADEQUATE NEUROMUSCULAR MONITORING EQUIPMENT, SUCH AS A PERIPHERAL NERVE STIMULATOR, WILL PERMIT THE MOST ADVANTAGEOUS USE OF RAPLON™, MINIMIZE THE POSSIBILITY OF OVERDOSAGE OR UNDERDOSAGE, AND ASSIST IN THE EVALUATION OF RECOVERY.
Dose for Tracheal Intubation
The recommended initial dose of RAPLON™ (rapacuronium bromide) for Injection in adult and geriatric patients is 1.5 mg/kg for short surgical procedures. In US and European studies, acceptable intubation scores were present in at least 85% of patients within 60 seconds after administration of 1.5 mg/kg of RAPLON™. Maximum block was achieved in most patients by 90 seconds. This dose had a mean clinical duration of approximately 15 minutes. In patients undergoing Cesarean section, the recommended RAPLON™ intubating dose, with thiopental induction, is 2.5 mg/kg.
Repeat Dosing in Adults (Bolus)
Following an intubating dose of 1.5 mg/kg, up to three maintenance doses of 0.50 mg/kg of RAPLON™ (rapacuronium bromide) for Injection, administered at 25% recovery of control T1 provided a mean clinical duration of 12 to 16 minutes under opioid/nitrous oxide/oxygen anesthesia. The duration of neuromuscular blockade was noted to increase with each additional dose. Repeat dosing should always be guided based on the clinical duration of the previous dose and should not be administered until recovery of neuromuscular function is evident (see PRECAUTIONS–Repeat Dosing).
Use in Pediatrics
Initial doses of RAPLON™ (rapacuronium bromide) for Injection of 2.0 mg/kg intravenously in pediatric patients (ages 1 month to 12 years) under halothane anesthesia produced acceptable intubating conditions within 60 seconds. Mean maximum block occurred within 90 seconds in most pediatric patients and had a mean clinical duration of approximately 15 minutes. When administration is being considered for patients 13 to 17 years of age, clinicians should consider the physical maturity, height, and weight of the patient in determining the dose of RAPLON™. The adult (1.5 mg/kg), pediatric (2.0 mg/kg), and Cesarean section (2.5 mg/kg) dosing recommendations may serve as a general guideline in determining an intubating dose in this age group.
Use in Geriatrics
The clinical duration of RAPLON™ (rapacuronium bromide) for Injection is not prolonged in geriatric patients at a dose of 1.5 mg/kg, and the median spontaneous recovery time is not different from that in other adults. No dosage adjustment is recommended in elderly patients.
Compatibility
RAPLON™ (rapacuronium bromide) for Injection is compatible in solution with:
| 0.9% NaCl solution | sterile water for injection |
| 5% dextrose in water | lactated Ringers |
| 5% dextrose in saline | bacteriostatic water for injection |
Use within 24 hours of mixing with the above solutions. Prepared solutions may be stored at room temperature.
Studies have shown that RAPLON™ is physically compatible when mixed with the following drugs:
| alfentanil | lidocaine |
| aminophylline (compatible if used within 4 hours) | methohexital |
| atropine sulfate | metoclopramide |
| ceftazidime (compatible if used within 4 hours) | midazolam |
| droperidol | morphine |
| epinephrine | potassium chloride |
| fentanyl | propranolol |
| gentamicin | ranitidine |
| glycopyrrolate | remifentanil |
| heparin sulfate | sufentanil |
| ketamine | verapamil |
| labetalol |
RAPLON™ is physically incompatible when mixed with the following drugs:
| cefuroxime | nitroglycerin |
| danaparoid sodium | thiopental |
| diazepam |
Parenteral drug products should be inspected visually for particulate matter and clarity prior to administration, whenever solution and container permit. Do not use solution if particulate matter is present.
Safety and Handling
There is no specific work-exposure limit for RAPLON™ (rapacuronium bromide) for Injection. In case of eye contact, flush with water for at least 10 minutes.
HOW SUPPLIED
RAPLON™ (rapacuronium bromide) for Injection is available in the following:
5 mL vials containing 100 mg of rapacuronium bromide base and when reconstituted to 5 mL with sterile water for injection or bacteriostatic water for injection provides 20 mg of rapacuronium bromide base per milliliter (20 mg/mL) at a pH of 4.0
| Box of 10 | NDC 0052-0490-15 |
10 mL vials containing 200 mg of rapacuronium bromide base and when reconstituted to 10 mL with sterile water for injection or bacteriostatic water for injection provides 20 mg of rapacuronium bromide base per milliliter (20 mg/mL) at a pH of 4.0
| Box of 10 | NDC 0052-0495-16 |
The packaging of this product contains no natural rubber (latex).
Storage
Store at 2–25°C (36–77°F).
After Reconstitution
When reconstituted with sterile water for injection or other compatible I.V. solutions, keep vial at room temperature or refrigerated 2–25°C (36–77°F) and use within 24 hours. Discard unused portion. Single use only.
When reconstituted with bacteriostatic water for injection, keep vial at room temperature or refrigerated 2–25°C (36–77°F) and use within 24 hours. Bacteriostatic water for injection CONTAINS BENZYL ALCOHOL, WHICH IS NOT INTENDED FOR USE IN NEWBORNS.
Rx only

Organon Inc.
West Orange, NJ 07052
XXXXXXX
Rev. 11/10
PRINCIPAL DISPLAY PANEL - 6 Vial Professional Sample Carton
6 x 5 mL vials
NDC 0052-0490-86
Professional Sample — Not For Sale
Raplon™
(rapacuronium bromide) for injection
100 mg*
For IV use only
*20 mg/mL rapacuronium bromide base
when reconstituted to 5 mL
6 vials lyophilized powder—sterile
5322160
490-86 8/99 06
Storage
Store at 2° to 25°C (36° to 77°F).
Dosage: Read enclosed prescribing
information
Rx only
Warning: Raplon™ may cause
respiratory depression. Facilities
for artificial respiration must be
immediately available.
Organon Inc.
West Orange
NJ 07052

| RAPLON
rapacuronium bromide injection |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020984 | 08/18/1999 | 03/30/2001 |
| RAPLON
rapacuronium bromide injection |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020984 | 08/18/1999 | 03/30/2001 |
| Labeler - Organon Pharmaceuticals USA (002152858) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| ORGANON INC. | 942193160 | MANUFACTURE | |