lotronex (alosetron hydrochloride) tablet
[GlaxoSmithKline]
WARNING
Infrequent but serious gastrointestinal adverse events have been reported with the use of LOTRONEX. These events, including ischemic colitis and serious complications of constipation, have resulted in hospitalization, and rarely, blood transfusion, surgery, and death.
- The Prescribing Program for LOTRONEX™ was implemented to help reduce risks of serious gastrointestinal adverse events. Only physicians who have enrolled in GlaxoSmithKline’s Prescribing Program for LOTRONEX, based on their understanding of the benefits and risks,should prescribe LOTRONEX (see PRECAUTIONS: Prescribing Program for LOTRONEX).
- LOTRONEX is indicated only for women with severe diarrhea-predominant IBS who have not responded adequately to conventional therapy (see INDICATIONS AND USAGE). Before receiving the initial prescription for LOTRONEX, the patient must read and sign the Patient-Physician Agreement for LOTRONEX (see PRECAUTIONS: Information for Patients).
- LOTRONEX should be discontinued immediately in patients who develop constipation or symptoms of ischemic colitis. Patients should immediately report constipation or symptoms of ischemic colitis to their physician. LOTRONEX should not be resumed in patients who develop ischemic colitis. Patients who have constipation should immediately contact their physician if the constipation does not resolve after LOTRONEX is discontinued. Patients with resolved constipation should resume LOTRONEX only on the advice of their treating physician.
DESCRIPTION
The active ingredient in LOTRONEX Tablets is alosetron hydrochloride (HCl), a potent and selective antagonist of the serotonin 5-HT3 receptor type. Chemically, alosetron is designated as 2,3,4,5-tetrahydro-5-methyl-2-[(5-methyl-1H-imidazol-4-yl)methyl]-1H-pyrido[4,3-b]indol-1-one, monohydrochloride. Alosetron is achiral and has the empirical formula: C17H18N4O•HCl, representing a molecular weight of 330.8. Alosetron is a white to beige solid that has a solubility of 61 mg/mL in water, 42 mg/mL in 0.1M hydrochloric acid, 0.3 mg/mL in pH 6 phosphate buffer, and <0.1 mg/mL in pH 8 phosphate buffer. The chemical structure of alosetron is:
LOTRONEX Tablets are supplied for oral administration as 0.5-mg (white) and 1-mg (blue) tablets. The 0.5-mg tablet contains 0.562 mg alosetron HCl equivalent to 0.5 mg alosetron and the 1-mg tablet contains 1.124 mg alosetron HCl equivalent to 1 mg of alosetron. Each tablet also contains the inactive ingredients: lactose (anhydrous), magnesium stearate, microcrystalline cellulose, and pregelatinized starch. The white film-coat for the 0.5-mg tablet contains hypromellose, titanium dioxide, and triacetin. The blue film-coat for the 1-mg tablet contains hypromellose, titanium dioxide, triacetin, and indigo carmine.
CLINICAL PHARMACOLOGY
Pharmacodynamics
Mechanism of Action
Alosetron is a potent and selective 5-HT3 receptor antagonist. 5-HT3 receptors are ligand-gated cation channels that are extensively distributed on enteric neurons in the human gastrointestinal tract, as well as other peripheral and central locations. Activation of these channels and the resulting neuronal depolarization affect the regulation of visceral pain, colonic transit and gastrointestinal secretions, processes that relate to the pathophysiology of irritable bowel syndrome (IBS). 5-HT3 receptor antagonists such as alosetron inhibit activation of non-selective cation channels which results in the modulation of the enteric nervous system.
The cause of IBS is unknown. IBS is characterized by visceral hypersensitivity and hyperactivity of the gastrointestinal tract, which lead to abnormal sensations of pain and motor activity. Following distention of the rectum, IBS patients exhibit pain and discomfort at lower volumes than healthy volunteers. Following such distention, alosetron reduced pain and exaggerated motor responses, possibly due to blockade of 5-HT3 receptors.
In healthy volunteers and IBS patients, alosetron (2 mg orally, twice daily for 8 days) increased colonic transit time without affecting orocecal transit time. In healthy volunteers, alosetron also increased basal jejunal water and sodium absorption after a single 4-mg dose. In IBS patients, multiple oral dosages of alosetron (4 mg twice daily for 6.5 days) significantly increased colonic compliance.
Single oral doses of alosetron administered to healthy men produced a dose-dependent reduction in the flare response seen after intradermal injection of serotonin. Urinary 6-β-hydroxycortisol excretion decreased by 52% in elderly subjects after 27.5 days of alosetron 2 mg orally twice daily. This decrease was not statistically significant. In another study utilizing alosetron 1 mg orally twice daily for 4 days, there was a significant decrease in urinary 6-β-hydroxycortisol excretion. However, there was no change in the ratio of 6-β-hydroxycortisol to cortisol, indicating a possible decrease in cortisol production. The clinical significance of these findings is unknown.
Pharmacokinetics
The pharmacokinetics of alosetron have been studied after single oral doses ranging from 0.05 to 16 mg in healthy men. The pharmacokinetics of alosetron have also been evaluated in healthy women and men and in patients with IBS after repeated oral dosages ranging from 1 mg twice daily to 8 mg twice daily.
Absorption
Alosetron is rapidly absorbed after oral administration with a mean absolute bioavailability of approximately 50% to 60% (approximate range 30% to >90%). After administration of radiolabeled alosetron, only 1% of the dose was recovered in the feces as unchanged drug. Following oral administration of a 1-mg alosetron dose to young men, a peak plasma concentration of approximately 5 ng/mL occurs at 1 hour. In young women, the mean peak plasma concentration is approximately 9 ng/mL, with a similar time to peak.
Food Effects
Alosetron absorption is decreased by approximately 25% by co-administration with food, with a mean delay in time to peak concentration of 15 minutes (see DOSAGE AND ADMINISTRATION: Usual Dosage in Adults).
Distribution
Alosetron demonstrates a volume of distribution of approximately 65 to 95 L. Plasma protein binding is 82% over a concentration range of 20 to 4,000 ng/mL.
Metabolism and Elimination
Plasma concentrations of alosetron increase proportionately with increasing single oral doses up to 8 mg and more than proportionately at a single oral dose of 16 mg. Twice-daily oral dosing of alosetron does not result in accumulation. The terminal elimination half-life of alosetron is approximately 1.5 hours (plasma clearance is approximately 600 mL/min). Population pharmacokinetic analysis in IBS patients confirmed that alosetron clearance is minimally influenced by doses up to 8 mg.
Renal elimination of unchanged alosetron accounts for only 6% of the dose. Renal clearance is approximately 94 mL/min.
Alosetron is extensively metabolized in humans. The biological activity of the metabolites is unknown. A mass balance study was performed utilizing an orally administered dose of unlabeled and 14C-labeled alosetron. On a molar basis, alosetron metabolites reached additive peak plasma concentrations 9-fold greater than alosetron, and the additive metabolite AUCs were 13-fold greater than the alosetron AUC. Plasma radioactivity declined with a half-life 2-fold longer than that of alosetron, indicating the presence of circulating metabolites. Approximately 73% of the radiolabeled dose was recovered in urine with another 24% of the dose recovered in feces. Only 7% of the dose was recovered as unchanged drug. At least 13 metabolites have been detected in urine. The predominant product in urine was a 6-hydroxy metabolite (15% of the dose). This metabolite was secondarily metabolized to a glucuronide that was also present in urine (14% of the dose). Smaller amounts of the 6-hydroxy metabolite and the 6-O-glucuronide also appear to be present in feces. A bis-oxidized dicarbonyl accounted for 14% of the dose, and its monocarbonyl precursor accounted for another 4% in urine and 6% in feces. No other urinary metabolite accounted for more than 4% of the dose. Glucuronide or sulfate conjugates of unchanged alosetron were not detected in urine.
In studies of Japanese men, an N-desmethyl metabolite was found circulating in plasma in all subjects and accounted for up to 30% of the dose in 1 subject when alosetron was administered with food. The clinical significance of this finding is unknown.
Alosetron is metabolized by human microsomal cytochrome P450 (CYP), shown in vitro to involve enzymes 2C9 (30%), 3A4 (18%), and 1A2 (10%). Non-CYP-mediated Phase I metabolic conversion also contributes to an extent of about 11%. However, in vivo data suggest that CYP1A2 plays a more prominent role in alosetron metabolism, based on correlation of alosetron clearance with in vivo CYP1A2 activity measured by probe substrate, increased clearance induced by smoking, and inhibition of clearance by fluvoxamine (see CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions).
Population Subgroups
Age
In some studies in healthy men or women, plasma concentrations were elevated by approximately 40% in individuals 65 years and older compared to young adults (see WARNINGS). However, this effect was not consistently observed in men.
Gender
Plasma concentrations are 30% to 50% lower and less variable in men compared to women given the same oral dose. Population pharmacokinetic analysis in IBS patients confirmed that alosetron concentrations were influenced by gender (27% lower in men).
Reduced Hepatic Function
A single 1-mg oral dose of alosetron was administered to 1 female and 5 male patients with moderate hepatic impairment (Child-Pugh score of 7 to 9) and to 1 female and 2 male patients with severe hepatic impairment (Child-Pugh score of >9). In comparison with historical data from healthy subjects, patients with severe hepatic impairment displayed higher systemic exposure to alosetron. The female with severe hepatic impairment displayed approximately 14-fold higher exposure, while the female with moderate hepatic impairment displayed approximately 1.6-fold higher exposure, than healthy females. Due to the small number of subjects and high intersubject variability in the pharmacokinetic findings, no definitive quantitative conclusions can be made. However, due to the greater exposure to alosetron in the female with severe hepatic impairment, alosetron should not be used in females with severe hepatic impairment (see CONTRAINDICATIONS, PRECAUTIONS: Hepatic Insufficiency, and DOSAGE AND ADMINISTRATION: Patients With Hepatic Impairment).
Reduced Renal Function
Renal impairment (creatinine clearance 4 to 56 mL/min) has no effect on the renal elimination of alosetron due to the minor contribution of this pathway to elimination. The effect of renal impairment on metabolite kinetics and the effect of end-stage renal disease have not been assessed (see DOSAGE AND ADMINISTRATION: Patients With Renal Impairment).
Drug Interactions
See CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions.
CLINICAL TRIALS
LOTRONEX 1 mg twice daily was studied in two 12-week U.S. multicenter, randomized, double-blind, placebo-controlled trials of identical design (Studies 1 and 2) in non-constipated women with IBS meeting the Rome Criteria1 for at least 6 months. Women with severe pain or a history of severe constipation were excluded. A 2-week run-in period established baseline IBS symptoms.
Of the 633 women on LOTRONEX and 640 on placebo, about two thirds had diarrhea-predominant IBS. Compared with placebo, 10% to 19% more women with diarrhea-predominant IBS who received LOTRONEX had adequate relief of IBS abdominal pain and discomfort during each month of the study.
Clinical studies have not been performed to adequately confirm the benefits of LOTRONEX in men or patients under the age of 18.
Starting Dosage
Data from a dose-ranging study of women (n = 85) who received 0.5 mg BID of alosetron, indicated that the incidence of constipation (14%) was lower than that experienced by women receiving 1 mg BID (29%). Therefore, to lower the risk of constipation, LOTRONEX should be started at a dosage of 0.5 mg twice a day. The efficacy of the 0.5-mg twice-daily dosage in treating severe diarrhea-predominant IBS has not been adequately evaluated in clinical trials.
Women With Severe Diarrhea-Predominant IBS
LOTRONEX is indicated only for women with severe diarrhea-predominant IBS (see INDICATIONS AND USAGE). The efficacy of LOTRONEX in this subset of the women studied in clinical trials is supported by prospective and retrospective analyses.
Prospective Analyses
In two 12-week, randomized, double-blind, placebo-controlled clinical trials of women with diarrhea-predominant IBS and bowel urgency on at least 50% of days at entry (Studies 3 and 4), a total of 778 women received LOTRONEX and 515 received placebo. Women receiving LOTRONEX had significant increases over placebo (13% to 16%) in the median percentage of days with urgency control.
The lower gastrointestinal functions of stool consistency, stool frequency, and sense of incomplete evacuation were also evaluated by patients’ daily reports. Stool consistency was evaluated on a scale of 1 to 5 (1 = very hard, 2 = hard, 3 = formed, 4 = loose, and 5 = watery). At baseline, average stool consistency was approximately 4 (loose) for both treatment groups. During the 12 weeks of treatment, the average stool consistency decreased to approximately 3.0 (formed) for patients who received LOTRONEX and 3.5 for the patients who received placebo in the two studies.
At baseline, average stool frequency was approximately 3.2 per day for both treatment groups. During the 12 weeks of treatment, the average daily stool frequency decreased to approximately 2.1 and 2.2 for patients receiving LOTRONEX and 2.7 and 2.8 for patients receiving placebo in the 2 studies.
There was no consistent effect upon the sense of incomplete evacuation during the 12 weeks of treatment for patients receiving LOTRONEX as compared to patients receiving placebo in either study.
Retrospective Analyses
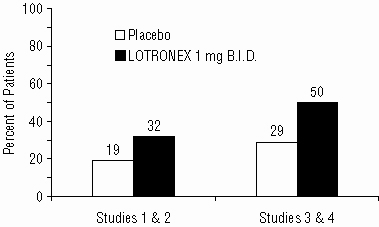
In analyses of patients from Studies 1 and 2 who had diarrhea-predominant IBS and indicated their baseline run-in IBS symptoms were severe at the start of the trial, LOTRONEX provided greater adequate relief of IBS pain and discomfort than placebo. In further analyses of Studies 1 and 2, 57% of patients had urgency at baseline on 5 or more days per week. In this subset, 32% of patients on LOTRONEX had urgency no more than 1 day in the last week of the trial, compared to 19% of patients on placebo.
Patient-reported subjective outcomes related to IBS were assessed by questionnaires obtained at baseline and week 12. Patients in the more severe subset who received LOTRONEX reported less difficulty sleeping, less tiredness, fewer eating problems, and less interference with social activities and work/main activities due to IBS symptoms or problems compared to those who received placebo. Change in the impact of IBS symptoms and problems on emotional and mental distress, and on physical and sexual activity in women who received LOTRONEX were not statistically different from those reported by women who received placebo.
In Studies 3 and 4, 66% of patients had urgency at baseline on 5 or more days per week. In this subset, 50% of patients on LOTRONEX had urgency no more than 1 day in the last week of the trial, compared to 29% of patients on placebo. Moreover, in the same subset, 12% on LOTRONEX had urgency no more than 2 days per week in any of the 12 weeks on treatment compared to 1% of placebo patients.
Figure 1. Percent of Patients With Urgency On >5 Days/Week At Baseline Who Improved to No More Than 1 Day in the Final Week
Long-Term Use
In a 48-week multinational, double-blind, placebo-controlled study, LOTRONEX 1 mg twice daily was evaluated in 714 women with non-constipated IBS. A retrospective analysis of the subset of women with severe diarrhea-predominant IBS (urgency on at least 10 days during the 2-week baseline period) was performed. Of the 417 patients with severe d-IBS enrolled, 62% completed the trial.
LOTRONEX (n = 198) provided a greater average rate of adequate relief of IBS pain and discomfort (52% vs. 41%) and a greater average rate of satisfactory control of bowel urgency (60% vs. 48%) compared with placebo (n = 219). Significant improvement of these symptoms occurred for most of the 48-week treatment period with no evidence of tachyphylaxis.
INDICATIONS AND USAGE
LOTRONEX is indicated only for women with severe diarrhea-predominant irritable bowel syndrome (IBS) who have:
- chronic IBS symptoms (generally lasting 6 months or longer),
- had anatomic or biochemical abnormalities of the gastrointestinal tract excluded, and
- not responded adequately to conventional therapy.
Diarrhea-predominant IBS is severe if it includes diarrhea and one or more of the following:
- frequent and severe abdominal pain/discomfort
- frequent bowel urgency or fecal incontinence
- disability or restriction of daily activities due to IBS
Because of infrequent but serious gastrointestinal adverse events associated with LOTRONEX, the indication is restricted to those patients for whom the benefit-to-risk balance is most favorable.
Clinical studies have not been performed to adequately confirm the benefits of LOTRONEX in men.
CONTRAINDICATIONS
LOTRONEX should not be initiated in patients with constipation (see WARNINGS).
LOTRONEX is contraindicated in patients with a history of the following:
- chronic or severe constipation or sequelae from constipation
- intestinal obstruction, stricture, toxic megacolon, gastrointestinal perforation, and/or adhesions
- ischemic colitis, impaired intestinal circulation, thrombophlebitis, or hypercoagulable state
- Crohn’s disease or ulcerative colitis
- diverticulitis
-
severe hepatic impairment
- hypersensitivity to any component of the product
LOTRONEX should not be used by patients who are unable to understand or comply with the Patient-Physician Agreement for LOTRONEX.
Concomitant administration of alosetron with fluvoxamine is contraindicated. Fluvoxamine, a known strong inhibitor of CYP1A2, has been shown to increase mean alosetron plasma concentrations (AUC) approximately 6–fold and prolong the half-life by approximately 3–fold (see PRECAUTIONS: Drug Interactions).
WARNINGS (See BOXED WARNING and DOSAGE AND ADMINISTRATION.)
Some patients have experienced serious complications of constipation or ischemic colitis without warning.
Constipation
Serious complications of constipation including obstruction, ileus, impaction, toxic megacolon, and secondary bowel ischemia have been reported with use of LOTRONEX during clinical trials. In addition, rare cases of perforation and death have been reported from postmarketing clinical practice. In some cases, complications of constipation required intestinal surgery, including colectomy. In IBS clinical trials, approximately 10% of patients on LOTRONEX withdrew prematurely because of constipation. The incidence of serious complications of constipation was approximately 0.1% (1 per 1,000 patients) in women receiving either LOTRONEX or placebo. Patients who are elderly, debilitated, or taking additional medications that decrease gastrointestinal motility may be at greater risk for complications of constipation.
LOTRONEX should be discontinued immediately in patients who develop constipation (see BOXED WARNING).
Ischemic Colitis
Ischemic colitis has been reported in patients receiving LOTRONEX in clinical trials as well as during marketed use of the drug. In IBS clinical trials, the cumulative incidence of ischemic colitis in women receiving LOTRONEX was 0.2% (2 per 1,000 patients, 95% confidence interval 1 to 3) through 3 months and was 0.3% (3 per 1,000 patients, 95% confidence interval 1 to 4) through 6 months. Ischemic colitis was not reported in women receiving placebo. The patient experience in controlled clinical trials is insufficient to estimate the incidence of ischemic colitis in patients taking LOTRONEX for longer than 6 months.
LOTRONEX should be discontinued immediately in patients with signs of ischemic colitis such as rectal bleeding, bloody diarrhea, or new or worsening abdominal pain. Because ischemic colitis can be life-threatening, patients with signs or symptoms of ischemic colitis should be evaluated promptly and have appropriate diagnostic testing performed. Treatment with LOTRONEX should not be resumed in patients who develop ischemic colitis.
PRECAUTIONS
Prescribing Program for LOTRONEX
To prescribe LOTRONEX, the physician must be enrolled in the Prescribing Program for LOTRONEX. To enroll, physicians must understand the benefits and risks of treatment with LOTRONEX for severe diarrhea-predominant IBS, including the information in the Prescribing Information, Medication Guide, and Patient-Physician Agreement for LOTRONEX. Physicians need to be able to:
- Diagnose and manage IBS, ischemic colitis, constipation and complications of constipation, or refer patients to specialists as needed.
- Educate patients on the benefits and risks of treatment with LOTRONEX, provide them with the Medication Guide, instruct them to read it, and encourage them to ask questions when first considering LOTRONEX. Patients may be educated by the enrolled physician or a healthcare provider under a physician’s direction.
- Prior to the initial prescription of LOTRONEX, obtain the patient’s signature on the Patient-Physician Agreement form, sign it, place the original signed form in the patient’s medical record, and give a copy to the patient.
- Affix program stickers to all prescriptions for LOTRONEX (i.e., the original and all subsequent prescriptions). Stickers will be provided as part of the GlaxoSmithKline Prescribing Program for LOTRONEX. No telephone, facsimile, or computerized prescriptions are permitted with this program. Refills are permitted to be written on prescriptions.
- Report all serious adverse events with LOTRONEX to GlaxoSmithKline at 1-888-825-5249 or to the Food and Drug Administration’s MedWatch Program at 1-800-FDA-1088.
To enroll in the Prescribing Program for LOTRONEX call 1-888-825-5249 or visit www.lotronex.com to complete the Physician Enrollment Form.
Information for Patients
Patients should be fully counseled on and understand the risks and benefits of LOTRONEX before an initial prescription is written. The patient may be educated by the enrolled physician or a healthcare provider under a physician’s direction.
PHYSICIANS MUST:
- Counsel patients for whom LOTRONEX is appropriate about the benefits and risks of LOTRONEX and discuss the impact of IBS symptoms on the patient’s life.
- Give the patient a copy of the Medication Guide, which outlines the benefits and risks of LOTRONEX, and instruct the patient to read it carefully. Answer all questions the patient may have about LOTRONEX. The complete text of the Medication Guide is printed at the end of this document.
- Review the Patient-Physician Agreement for LOTRONEX with the patient, answer all questions, and give a copy of the signed agreement to the patient.
- Provide each patient with appropriate instructions for taking LOTRONEX.
Copies of the Patient-Physician Agreement for LOTRONEX and additional copies of the Medication Guide are available by contacting GlaxoSmithKline at 1-888-825-5249 or visiting www.lotronex.com.
PATIENTS WHO ARE PRESCRIBED LOTRONEX SHOULD BE INSTRUCTED TO:
- Read the Medication Guide before starting LOTRONEX and each time they refill their prescription.
- Not start taking LOTRONEX if they are constipated.
- Immediately discontinue LOTRONEX and contact their physician if they become constipated, or have symptoms of ischemic colitis such as new or worsening abdominal pain, bloody diarrhea, or blood in the stool. Contact their physician again if their constipation does not resolve after discontinuation of LOTRONEX. Resume LOTRONEX only if their constipation has resolved and after discussion with and the agreement of their treating physician.
- Stop taking LOTRONEX and contact their physician if LOTRONEX does not adequately control IBS symptoms after 4 weeks of taking 1 mg twice a day.
Drug Interactions
Because alosetron is metabolized by a variety of hepatic CYP drug-metabolizing enzymes, inducers or inhibitors of these enzymes may change the clearance of alosetron.
Fluvoxamine is a known strong inhibitor of CYP1A2 and also inhibits CYP3A4, CYP2C9, and CYP2C19. In a pharmacokinetic study, 40 healthy female subjects received fluvoxamine in escalating doses from 50 to 200 mg per day for 16 days, with coadministration of alosetron 1 mg on the last day. Fluvoxamine increased mean alosetron plasma concentrations (AUC) approximately 6–fold and prolonged the half-life by approximately 3–fold. Concomitant administration of alosetron and fluvoxamine is contraindicated (see CONTRAINDICATIONS).
Concomitant administration of alosetron and moderate CYP1A2 inhibitors, including quinolone antibiotics and cimetidine, has not been evaluated, but should be avoided unless clinically necessary because of similar potential drug interactions.
Ketoconazole is a known strong inhibitor of CYP3A4. In a pharmacokinetic study, 38 healthy female subjects received ketoconazole 200 mg twice daily for 7 days, with coadministration of alosetron 1 mg on the last day. Ketoconazole increased mean alosetron plasma concentrations (AUC) by 29%. Caution should be used when alosetron and ketoconazole are administered concomitantly. Coadministration of alosetron and strong CYP3A4 inhibitors, such as clarithromycin, telithromycin, protease inhibitors, voriconazole, and itraconazole has not been evaluated but should be undertaken with caution because of similar potential drug interactions. The effect of induction or inhibition of other pathways on exposure to alosetron and its metabolites is not known.
In vitro human liver microsome studies and an in vivo metabolic probe study demonstrated that alosetron did not inhibit CYP enzymes 2D6, 3A4, 2C9, or 2C19. In vitro, at total drug concentrations 27-fold higher than peak plasma concentrations observed with the 1-mg dose, alosetron inhibited CYP enzymes 1A2 (60%) and 2E1 (50%). In an in vivo metabolic probe study, alosetron did not inhibit CYP2E1 but did produce 30% inhibition of both CYP1A2 and N-acetyltransferase. Although not studied with alosetron, inhibition of N-acetyltransferase may have clinically relevant consequences for drugs such as isoniazid, procainamide, and hydralazine. The effect on CYP1A2 was explored further in a clinical interaction study with theophylline and no effect on metabolism was observed. Another study showed that alosetron had no clinically significant effect on plasma concentrations of the oral contraceptive agents ethinyl estradiol and levonorgestrel (CYP3A4 substrates). A clinical interaction study was also conducted with alosetron and the CYP3A4 substrate cisapride. No significant effects on cisapride metabolism or QT interval were noted. The effects of alosetron on monoamine oxidases and on intestinal first pass secondary to high intraluminal concentrations have not been examined. Based on the above data from in vitro and in vivo studies, it is unlikely that alosetron will inhibit the hepatic metabolic clearance of drugs metabolized by the major CYP enzyme 3A4, as well as the CYP enzymes 2D6, 2C9, 2C19, 2E1, or 1A2.
Alosetron does not appear to induce the major cytochrome P450 (CYP) drug metabolizing enzyme 3A. Alosetron also does not appear to induce CYP enzymes 2E1 or 2C19. It is not known whether alosetron might induce other enzymes.
Hepatic Insufficiency
Due to the extensive hepatic metabolism of alosetron, increased exposure to alosetron and/or its metabolites is likely to occur in patients with hepatic insufficiency. Alosetron should not be used in patients with severe hepatic impairment and should be used with caution in patients with mild or moderate hepatic impairment (see CLINICAL PHARMACOLOGY: Population Subgroups: Reduced Hepatic Function).
Carcinogenesis, Mutagenesis, Impairment of Fertility
In 2-year oral studies, alosetron was not carcinogenic in mice at doses up to 30 mg/kg/day or in rats at doses up to 40 mg/kg/day. These doses are, respectively, about 60 to 160 times the recommended human dose of alosetron of 2 mg/day (1 mg twice daily) based on body surface area. Alosetron was not genotoxic in the Ames tests, the mouse lymphoma cell (L5178Y/TK±) forward gene mutation test, the human lymphocyte chromosome aberration test, the ex vivo rat hepatocyte unscheduled DNA synthesis (UDS) test, or the in vivo rat micronucleus test for mutagenicity. Alosetron at oral doses up to 40 mg/kg/day (about 160 times the recommended daily human dose based on body surface area) was found to have no effect on fertility and reproductive performance of male or female rats.
Pregnancy
Teratogenic Effects
Pregnancy Category B. Reproduction studies have been performed in rats at doses up to 40 mg/kg/day (about 160 times the recommended human dose based on body surface area) and rabbits at oral doses up to 30 mg/kg/day (about 240 times the recommended daily human dose based on body surface area). These studies have revealed no evidence of impaired fertility or harm to the fetus due to alosetron. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, LOTRONEX should be used during pregnancy only if clearly needed.
Nursing Mothers
Alosetron and/or metabolites of alosetron are excreted in the breast milk of lactating rats. It is not known whether alosetron is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when LOTRONEX is administered to a nursing woman.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Postmarketing experience suggests that elderly patients may be at greater risk for complications of constipation (see WARNINGS).
ADVERSE REACTIONS
Table 1 summarizes adverse events from 22 repeat-dose studies in patients with IBS who were treated with 1 mg of LOTRONEX twice daily for 8 to 24 weeks. The adverse events in Table 1 were reported in 1% or more of patients who received LOTRONEX and occurred more frequently on LOTRONEX than on placebo. A statistically significant difference was observed for constipation in patients treated with LOTRONEX compared to placebo (p<0.0001).
|
Body System Adverse Event |
LOTRONEX 1 mg B.I.D. (n = 8,328) |
Placebo (n = 2,363) |
|
Gastrointestinal |
||
|
Constipation |
29% |
6% |
|
Abdominal discomfort and pain |
7% |
4% |
|
Nausea |
6% |
5% |
|
Gastrointestinal discomfort and pain |
5% |
3% |
|
Abdominal distention |
2% |
1% |
|
Regurgitation and reflux |
2% |
2% |
|
Hemorrhoids |
2% |
1% |
Gastrointestinal
Constipation is a frequent and dose-related side effect of treatment with LOTRONEX (see WARNINGS). In clinical studies constipation was reported in approximately 29% of IBS patients treated with LOTRONEX 1 mg twice daily (n = 9,316). This effect was statistically significant compared to placebo (p<0.0001). Eleven percent (11%) of patients treated with LOTRONEX 1 mg twice daily withdrew from the studies due to constipation. Although the number of IBS patients treated with LOTRONEX 0.5 mg twice daily is relatively small (n = 243), only 11% of those patients reported constipation and 4% withdrew from clinical studies due to constipation. Among the patients treated with LOTRONEX 1 mg twice daily who reported constipation, 75% reported a single episode and most reports of constipation (70%) occurred during the first month of treatment with the median time to first report of constipation onset of 8 days. Occurrences of constipation in clinical trials were generally mild to moderate in intensity, transient in nature, and resolved either spontaneously with continued treatment or with an interruption of treatment. However, serious complications of constipation have been reported in clinical studies and in postmarketing experience (see BOXED WARNING and WARNINGS). In Studies 1 and 2, 9% of patients treated with LOTRONEX reported constipation and 4 consecutive days with no bowel movement (see CLINICAL TRIALS). Following interruption of treatment, 78% of the affected patients resumed bowel movements within a 2-day period and were able to re-initiate treatment with LOTRONEX.
Hepatic
A similar incidence in elevation of ALT (>2–fold) was seen in patients receiving LOTRONEX or placebo (1.0% vs. 1.2%). A single case of hepatitis (elevated ALT, AST, alkaline phosphatase, and bilirubin) without jaundice was reported in a 12-week study. A causal association with LOTRONEX has not been established.
Long-Term Safety
Patient experience in controlled clinical trials is insufficient to estimate the incidence of ischemic colitis in patients taking LOTRONEX for longer than 6 months.
Other Events Observed During Clinical Evaluation of LOTRONEX
During its assessment in clinical trials, multiple and single doses of LOTRONEX were administered resulting in 11,874 subject-exposures in 86 completed clinical studies. The conditions, dosages, and duration of exposure to LOTRONEX varied between trials, and the studies included healthy male and female volunteers as well as male and female patients with IBS and other indications.
In the listing that follows, reported adverse events were classified using a standardized coding dictionary. Only those events that an investigator believed were possibly related to alosetron, occurred in at least 2 patients, and occurred at a greater frequency during treatment with LOTRONEX than during placebo administration are presented. Serious adverse events occurring in at least 1 patient for whom an investigator believed there was reasonable possibility that the event was related to alosetron treatment and occurring at a greater frequency in LOTRONEX than placebo-treated patients are also presented.
In the following listing, events are categorized by body system. Within each body system, events are presented in descending order of frequency. The following definitions are used: Infrequent adverse events are those occurring on one or more occasion in 1/100 to 1/1,000 patients; Rare adverse events are those occurring on one or more occasion in fewer than 1/1,000 patients.
Although the events reported occurred during treatment with LOTRONEX, they were not necessarily caused by it.
Blood and Lymphatic
Rare
Quantitative red cell or hemoglobin defects, hemorrhage, and lymphatic signs and symptoms.
Cardiovascular
Infrequent
Tachyarrhythmias.
Rare
Arrhythmias, increased blood pressure, and extrasystoles.
Drug Interaction, Overdose, and Trauma
Rare
Contusions and hematomas.
Ear, Nose, and Throat
Rare
Ear, nose, and throat infections; viral ear, nose, and throat infections; and laryngitis.
Endocrine and Metabolic
Rare
Disorders of calcium and phosphate metabolism, hyperglycemia, hypothalamus/pituitary hypofunction, hypoglycemia, and fluid disturbances.
Eye
Rare
Light sensitivity of eyes.
Gastrointestinal
Infrequent
Hyposalivation, dyspeptic symptoms, gastrointestinal spasms, ischemic colitis (see WARNINGS), and gastrointestinal lesions.
Rare
Abnormal tenderness, colitis, gastrointestinal signs and symptoms, proctitis, diverticulitis, positive fecal occult blood, hyperacidity, decreased gastrointestinal motility and ileus, gastrointestinal obstructions, oral symptoms, gastrointestinal intussusception, gastritis, gastroduodenitis, gastroenteritis, and ulcerative colitis.
Hepatobiliary Tract and Pancreas
Rare
Abnormal bilirubin levels and cholecystitis.
Lower Respiratory
Infrequent
Breathing disorders.
Rare
Viral respiratory infections.
Musculoskeletal
Rare
Muscle pain; muscle stiffness, tightness and rigidity; and bone and skeletal pain.
Neurological
Infrequent
Hypnagogic effects.
Rare
Memory effects, tremors, dreams, cognitive function disorders, disturbances of sense of taste, disorders of equilibrium, confusion, sedation, and hypoesthesia.
Non-Site Specific
Infrequent
Malaise and fatigue, cramps, pain, temperature regulation disturbances.
Rare
General signs and symptoms, non-specific conditions, burning sensations, hot and cold sensations, cold sensations, and fungal infections.
Psychiatry
Infrequent
Anxiety.
Rare
Depressive moods.
Reproduction
Rare
Sexual function disorders, female reproductive tract bleeding and hemorrhage, reproductive infections, and fungal reproductive infections.
Skin
Infrequent
Sweating and urticaria.
Rare
Hair loss and alopecia; acne and folliculitis; disorders of sweat and sebum; allergic skin reaction; eczema; skin infections; dermatitis and dermatosis; and nail disorders.
Urology
Infrequent
Urinary frequency.
Rare
Bladder inflammation; polyuria and diuresis; and urinary tract hemorrhage.
Postmarketing Experience
The following events have been identified during use of LOTRONEX in clinical practice. Because they were reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These events have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to LOTRONEX.
Gastrointestinal
Constipation, ileus, impaction, obstruction, perforation, ulceration, ischemic colitis, small bowel mesenteric ischemia (see WARNINGS).
Neurological
Headache.
Skin
Rash.
DRUG ABUSE AND DEPENDENCE
LOTRONEX has no known potential for abuse or dependence.
OVERDOSAGE
There is no specific antidote for overdose of LOTRONEX. Patients should be managed with appropriate supportive therapy. Individual oral doses as large as 16 mg have been administered in clinical studies without significant adverse events. This dose is 8 times higher than the recommended total daily dose. Inhibition of the metabolic elimination and reduced first pass of other drugs might occur with overdoses of alosetron (see PRECAUTIONS: Drug Interactions). Single oral doses of LOTRONEX at 15 mg/kg in female mice and 60 mg/kg in female rats (30 and 240 times, respectively, the recommended human dose based on body surface area) were lethal. Symptoms of acute toxicity were labored respiration, subdued behavior, ataxia, tremors, and convulsions.
DOSAGE AND ADMINISTRATION
For safety reasons, only physicians who enroll in the GlaxoSmithKline Prescribing Program for LOTRONEX should prescribe LOTRONEX (see PRECAUTIONS: Prescribing Program for LOTRONEX).
Usual Dosage in Adults
To lower the risk of constipation, LOTRONEX should be started at a dosage of 0.5 mg twice a day. Patients well controlled on 0.5 mg twice a day may be maintained on this regimen. If, after 4 weeks, the 0.5-mg twice-daily dosage is well tolerated but does not adequately control IBS symptoms, then the dosage can be increased to up to 1 mg twice a day, the dose used in controlled clinical trials (see CLINICAL TRIALS). LOTRONEX should be discontinued in patients who have not had adequate control of IBS symptoms after 4 weeks of treatment with 1 mg twice a day.
LOTRONEX can be taken with or without food (see CLINICAL PHARMACOLOGY: Pharmacokinetics: Food Effects).
LOTRONEX should be discontinued immediately in patients who develop constipation or signs of ischemic colitis. LOTRONEX should not be restarted in patients who develop ischemic colitis.
Clinical trial and postmarketing experience suggest that debilitated patients or patients taking additional medications that decrease gastrointestinal motility may be at greater risk of serious complications of constipation. Therefore, appropriate caution and follow-up should be exercised if LOTRONEX is prescribed for these patients (see also Geriatric Patients).
Pediatric Patients
Safety and effectiveness have not been established in pediatric patients.
Geriatric Patients
Postmarketing experience suggests that elderly patients may be at greater risk for complications of constipation; therefore, appropriate caution and follow-up should be exercised if LOTRONEX is prescribed for these patients (see WARNINGS).
Patients With Renal Impairment
There are insufficient data available on the biological activity of the metabolites of LOTRONEX. It is unknown if dosage adjustment is needed in patients with renal impairment (see CLINICAL PHARMACOLOGY: Population Subgroups: Reduced Renal Function).
Patients With Hepatic Impairment
LOTRONEX is extensively metabolized by the liver and increased exposure to LOTRONEX is likely to occur in patients with hepatic impairment. Increased drug exposure may increase the risk of serious adverse events. LOTRONEX should be used with caution in patients with mild or moderate hepatic impairment and is contraindicated in patients with severe hepatic impairment (see CLINICAL PHARMACOLOGY: Population Subgroups: Reduced Hepatic Function, CONTRAINDICATIONS, and PRECAUTIONS: Hepatic Insufficiency).
Information for Pharmacists
LOTRONEX may be dispensed only on presentation of a prescription for LOTRONEX with a sticker for the Prescribing Program for LOTRONEX attached. A Medication Guide for LOTRONEX must be given to the patient each time LOTRONEX is dispensed as required by law. No telephone, facsimile, or computerized prescriptions are permitted with this program. Refills are permitted to be written on prescriptions.
HOW SUPPLIED
LOTRONEX Tablets, 0.5 mg (0.562 mg alosetron HCl equivalent to 0.5 mg alosetron) are white, oval, film-coated tablets debossed with GX EX1 on one face.
Bottles of 30 (NDC 0173-0738-00) with child-resistant closures.
LOTRONEX Tablets, 1 mg (1.124 mg alosetron HCl equivalent to 1 mg alosetron), are blue, oval, film-coated tablets debossed with GX CT1 on one face.
Bottles of 30 (NDC 0173-0690-05) with child-resistant closures.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Protect from light and moisture.
REFERENCE
1. Thompson WG, Creed F, Drossman DA, et al. Functional bowel disease and functional abdominal pain. Gastroenterol Int. 1992;5:75-91.
MEDICATION GUIDE
LOTRONEX® (LOW-trah-nex) Tablets
(alosetron hydrochloride)
Before using LOTRONEX for the first time, you should:
- Understand that LOTRONEX has serious risks for some people.
- Read and follow the directions in this Medication Guide.
- Sign a Patient-Physician Agreement with your doctor.
Read this Medication Guide carefully before you sign the Patient-Physician Agreement. You must sign the Patient-Physician Agreement before you start LOTRONEX. Read the Medication Guide you get with each refill for LOTRONEX. There may be new information. This Medication Guide does not take the place of talking with your doctor.
1. What is the most important information I should know about LOTRONEX?
LOTRONEX is a medicine only for some women with severe chronic IBS whose:
- main problem is diarrhea and
- IBS symptoms have not been helped enough by other treatments.
A. Some patients have developed serious bowel side effects while taking LOTRONEX.
Serious bowel (intestine) side effects can happen suddenly, including the following two:
1. Serious complications of constipation: About 1 out of every 1,000 women who take LOTRONEX may get serious complications of constipation. These complications may lead to a hospital stay, and in rare cases, blood transfusions, surgery, and death. People who are older, who are weak from illness, or who take other constipating medicines may be more likely to have serious constipation problems with LOTRONEX.
To lower your chances of getting serious complications of constipation do the following:
- If you are constipated, do not start taking LOTRONEX.
- If you get constipated while taking LOTRONEX, stop taking it right away and call your doctor.
- If your constipation does not get better after stopping LOTRONEX, call your doctor again.
- If you stopped taking LOTRONEX, do not start taking LOTRONEX again unless your doctor tells you to do so.
2. Ischemic colitis (reduced blood flow to the bowel): About 3 out of every 1,000 women who take LOTRONEX over a 6-month period may get a serious problem where blood flow to parts of the large bowel is reduced. This is called ischemic colitis. The chance of getting ischemic colitis when you take LOTRONEX for more than 6 months is not known. Ischemic colitis may lead to a hospital stay, and in rare cases, blood transfusions, surgery, and death.
To lower your chances of getting serious complications of ischemic colitis,stop taking LOTRONEX and call your doctor right away if you get:
- new or worse pain in your stomach area (abdomen) or
- blood in your bowel movements.
B. Is LOTRONEX right for you?
LOTRONEX may be right for you if all of these things are true about you:
- Your doctor has told you that your symptoms are due to IBS.
- Your IBS bowel problem is diarrhea.
- Your IBS has lasted for 6 months or longer.
- You tried other IBS treatments and they didn’t give you the relief you need.
- Your IBS is severe.
You can tell if your IBS is severe ifat least 1 of the following is true for you:
- You have lots of painful stomach cramps or bloating.
- You often can’t control the need to have a bowel movement, or you have “accidents” where your underwear gets dirty from diarrhea or bowel movements.
- You can't lead a normal home or work life because you need to be near a bathroom.
Enough testing has not been done to confirm LOTRONEX works in men or children under age 18.
C. There is a special prescribing program for LOTRONEX.
Only doctors who have signed up with the company that makes LOTRONEX should write prescriptions for LOTRONEX. As part of signing up, these doctors have said that they understand about IBS and the possible side effects of LOTRONEX. They have agreed to use a special sticker on all prescriptions for LOTRONEX, so the pharmacist will know that the doctors have signed up with the company.
You may be taught about LOTRONEX by your doctor or healthcare provider under a doctor’s direction. Your doctor will ask you to sign a Patient-Physician Agreement after you read this Medication Guide for the first time. Signing the Agreement means that you understand the benefits and risks of LOTRONEX and that you have read and understand this Medication Guide.
2. What is LOTRONEX?
LOTRONEX is a medicine only for some women with severe chronic IBS whose:
- main problem is diarrhea and
- IBS symptoms have not been helped enough by other treatments.
LOTRONEX does not cure IBS, and it may not help every person who takes it. For those who are helped, LOTRONEX reduces lower stomach area (abdominal) pain and discomfort, the sudden need to have a bowel movement (bowel urgency), and diarrhea from IBS. If you stop taking LOTRONEX, your IBS symptoms may return within 1 or 2 weeks.
3. Who should not take LOTRONEX?
LOTRONEX is not right for everyone. Do not take LOTRONEX if any of the following apply to you:
- Your main IBS problem is constipation or you are constipated most of the time.
- You have had a serious problem from constipation.
- You have had serious bowel blockages.
- You have had blood flow problems to your bowels, such as ischemic colitis.
- You have had blood clots.
- You have had Crohn’s disease, ulcerative colitis, diverticulitis, or severe liver disease.
- You do not understand this Medication Guide or the Patient-Physician Agreement, or you are not willing to follow them.
- You are allergic to LOTRONEX or any of its ingredients. (See the list of ingredients at the end of this Medication Guide.)
- You are taking fluvoxamine (LUVOX®)
If you are constipated now, do not start taking LOTRONEX.
4. What should I talk about with my doctor before taking LOTRONEX?
Talk with your doctor:
- about the possible benefits and risks of LOTRONEX.
- about how much of a problem IBS is in your life and what treatments you have tried.
- about any other illnesses you have and medicines you take or plan to take. These include prescription and non-prescription medicines, supplements, and herbal remedies. Certain illnesses and medicines can increase your chance of getting serious side effects while taking LOTRONEX. Other medicines may interact with how the body handles LOTRONEX.
- if you are pregnant, planning to get pregnant, or breastfeeding.
5. How should I take LOTRONEX?
- Take LOTRONEX exactly as your doctor prescribes it. You can take LOTRONEX with or without food.
- Begin with 0.5 mg two times a day for 4 weeks to see how LOTRONEX affects you. You and your doctor may decide that you should keep taking this dose if you are doing well.
- Check with your doctor 4 weeks after
starting LOTRONEX:
- If you try 0.5 mg two times a day for 4 weeks, it may not control your symptoms. If you do not get constipation or other side effects from LOTRONEX, your doctor may increase your dose up to 1 mg two times a day.
- If 1 mg two times a day does not work after 4 weeks, LOTRONEX is not likely to help you. You should stop taking it and call your doctor.
- If you miss a dose of LOTRONEX, just skip that dose. Do not take 2 doses the next time. Wait until the next time you are supposed to take it and then take your normal dose.
- Follow the important instructions in the section“What is the most important information I should know about LOTRONEX?” about when you must stop taking the drug and when you should call your doctor.
- If you see other doctors about your IBS or side effects from LOTRONEX, let the doctor who prescribed LOTRONEX know.
6. What are the possible side effects of LOTRONEX?
Constipation is the most common side effect among women with IBS who take LOTRONEX. Some patients have developed serious bowel side effects while taking LOTRONEX. Read the section “What is the most important information I should know about LOTRONEX?”at the beginning of this Medication Guide for information about the serious side effects you may get with LOTRONEX.
This Medication Guide does not tell you about all the possible side effects of LOTRONEX. Your doctor or pharmacist can give you a more complete list.
7. General information about the safe and effective use of LOTRONEX
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you have any questions or concerns about LOTRONEX, ask your doctor. Do not use LOTRONEX for a condition for which it was not prescribed. Do not share your medicine with other people. It may harm them.
Your doctor or pharmacist can give you more information about LOTRONEX that was written for healthcare professionals. You can also contact the company that makes LOTRONEX (toll free) at 1-888-825-5249 or at www.lotronex.com.
8. What are the ingredients of LOTRONEX?
Active Ingredient:alosetron hydrochloride
Inactive Ingredients: lactose (anhydrous), magnesium stearate, microcrystalline cellulose, and pregelatinized starch. The white film-coat for the 0.5-mg tablet contains hypromellose, titanium dioxide, and triacetin. The blue film-coat for the 1-mg tablet contains hypromellose, titanium dioxide, triacetin, and indigo carmine.
This Medication Guide has been approved by the US Food and Drug Administration.
March 2006 MG-035
PATIENT-PHYSICIAN AGREEMENT FOR LOTRONEX
LOTRONEX® (alosetron hydrochloride) is only for women with severe irritable bowel syndrome (IBS) whose main problem is diarrhea and who did not get the relief needed from other treatments. LOTRONEX has not been shown to help men with IBS or patients under age 18.
My doctor, or a healthcare provider under a doctor’s direction, answered my questions about treatment with LOTRONEX. I have read and I understand the Medication Guide for LOTRONEX, and
- I understand that some patients using LOTRONEX have had serious bowel conditions (ischemic colitis and complications of constipation). I understand that these serious conditions can happen suddenly, and that they may lead to a hospital stay, and in rare cases, blood transfusions, surgery, and death. I also understand that certain patients may be more likely to develop a serious bowel condition while taking LOTRONEX. These include older patients, those who have other health problems and those who take other medicines that may cause constipation.
- My doctor and I agree that my IBS is severe and that other treatments have not given me the relief that I need. I also agree that I meet all of the requirements described in the section of the Medication Guide “What is the most important information I should know about LOTRONEX?” I understand that these requirements help to make sure that LOTRONEX is used only by patients who are likely to have more benefit from treatment than risk.
- I don’t have any problems listed in the section of the Medication Guide “Who should not take LOTRONEX?” that prevents me from taking LOTRONEX.
- I will follow instructions in the Medication Guide about:
- telling my doctor, before taking LOTRONEX, about any illnesses I have, or other medicines I am taking or planning to take.
- taking LOTRONEX exactly as my doctor prescribes it.
- stopping LOTRONEX and calling my doctor right away if I get constipated, if I have new or worse pain in my abdomen, or if I see blood in my bowel movements.
- calling my doctor again if the constipation I called about before has not gotten better.
- not starting LOTRONEX again unless my doctor tells me to do so, if I stopped taking it because I got constipated.
- talking with my doctor 4 weeks after starting LOTRONEX to recheck my IBS symptoms.
- stopping LOTRONEX and calling my doctor if my IBS symptoms have not improved after 4 weeks of taking 1 mg 2 times a day.
I understand that LOTRONEX should be prescribed only by doctors who have signed up with the company that makes the drug. Doctors in the program must:
- fully discuss the drug’s benefits and risks with each patient.
- sign this agreement with each patient before giving the initial prescription. It is not necessary to sign an agreement more than once.
- use a special sticker on all LOTRONEX prescriptions so that pharmacists know the doctor has signed up.
If I see other doctors about my IBS or possible side effects from LOTRONEX, I will let the doctor who prescribed LOTRONEX know.
My signature below indicates I have read, understood, and agree with all the statements made above. I would like to begin treatment with LOTRONEX.
|
________________________________ |
|
|
Name of Physician (print) |
|
|
________________________________ |
________________________________ |
|
Signature |
Date |
SECTION FOR THE PHYSICIAN
I am enrolled in the Prescribing Program for LOTRONEX, and I will continue to follow the requirements of the Program.
I, or a healthcare provider under a physician's direction, have given the patient named above:
- a copy of the Medication Guide for LOTRONEX, and instructed the patient to read it carefully before signing this Agreement, and to take it home.
- counseling about the benefits and risks of LOTRONEX.
- appropriate instructions for taking LOTRONEX.
- answers to all of the patient’s questions about treatment with LOTRONEX.
- a prescription for LOTRONEX that has the program sticker affixed on it to alert pharmacists I am enrolled in the Prescribing Program for LOTRONEX.
The patient signed the Patient-Physician Agreement in my presence after I counseled the patient, asked if the patient had any questions about treatment with LOTRONEX, and answered all questions to the best of my ability.
|
________________________________ |
|
|
Name of Physician (print) |
|
|
________________________________ |
________________________________ |
|
Signature |
Date |
After the patient and the physician sign this Patient-Physician Agreement, give a copy to the patient and put the original signed form in the patient’s medical record.
PRESCRIBING PROGRAM FOR LOTRONEX™
PHYSICIAN ENROLLMENT FORM
The Prescribing Program for LOTRONEX was implemented to help reduce risks of serious gastrointestinal adverse events, some fatal, associated with this medicine. The program is intended to help physicians and their patients understand the benefits and risks of treatment with LOTRONEX in order to make fully informed decisions.
I wish to participate in the Prescribing Program for LOTRONEX (PPL) and acknowledge that I have read the complete Prescribing Information for LOTRONEX and understand and will follow the requirements of the PPL described below.
- For safety reasons, LOTRONEX is approved only for women with severe,
diarrhea-predominant irritable bowel syndrome (D-IBS) who have:
- Chronic IBS symptoms (generally lasting for 6 months or longer),
- had anatomic or biochemical abnormalities of the gastrointestinal tract excluded, and
- not responded adequately to conventional therapy.
Diarrhea-predominant IBS is severe if it includes diarrhea and one or more of the following:
- Frequent and severe abdominal pain/discomfort
- Frequent bowel urgency or fecal incontinence
- Disability or restriction of daily activities due to IBS
- Physicians who enroll in the PPL should be able to diagnose and manage IBS, ischemic colitis, constipation, and complications of constipation, or refer patients to a specialist as needed.
- Patients considering treatment with LOTRONEX must be educated on the benefits and risks of the drug, given a copy of the Medication Guide, instructed to read it, and encouraged to ask questions. The patient may be educated by the enrolled physician or a healthcare provider under a physician’s direction.
- After reviewing the Medication Guide prior to the initial prescription, the physician and the patient must both sign the Patient-Physician Agreement form. The original signed form must be placed in the patient’s medical record, and a copy given to the patient.
- Program stickers must be affixed to all prescriptions for LOTRONEX (i.e., the original and all subsequent prescriptions). Stickers will be provided as part of the GlaxoSmithKline Prescribing Program for LOTRONEX. Refills are permitted to be written on prescriptions.
- All prescriptions for LOTRONEX must be written and not transmitted by telephone, facsimile, or computer.
- Prescribers must report all serious adverse events with LOTRONEX to
GlaxoSmithKline at 1-888-825-5249 or to the Food and Drug Administration at
1-800-FDA-1088.
________________________________
Name of Physician (print)
________________________________
________________________________
Signature
Date
DEA Number
________________________________
Office Address:
________________________________
________________________________
________________________________
Office Phone Number:
________________________________
Office Fax Number:
________________________________
Upon enrollment, you will receive a prescribing kit for LOTRONEX with the complete Prescribing Information, Prescribing Program for LOTRONEX stickers, multiple copies of the Medication Guide and Patient-Physician Agreement for LOTRONEX, and instructions for ordering additional supplies of Program materials.
You only need to enroll once, and you are under no obligation to prescribe LOTRONEX.
If you have any questions, please call the Prescribing Program for LOTRONEX at 1-888-825-5249 or visit www.lotronex.com.
TO ENROLL, VISIT WWW.LOTRONEX.COM OR PHONE 1-888-825-5249 OR COMPLETE THIS FORM IN ITS ENTIRETY AND MAIL OR FAX TO THE FOLLOWING ADDRESS:
Prescribing Program for Lotronex
Customer Response Center
Five Moore Drive
PO Box 13398
Research Triangle Park, NC 27709-3398
Fax Number: 1-866-698-7582
GlaxoSmithKline
Research Triangle Park, NC 27709
©2006, GlaxoSmithKline. All rights reserved.
March 2006 RL-2263
| LOTRONEX (alosetron hydrochloride) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
| LOTRONEX (alosetron hydrochloride) | ||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
Revised: 07/2006GlaxoSmithKline