lupron depot (leuprolide acetate) injection, powder, lyophilized, for suspension
[TAP Pharmaceuticals Inc.]
Rx only
DESCRIPTION
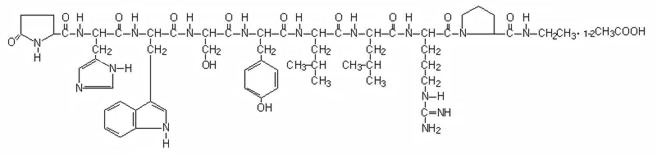
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin-releasing hormone (GnRH or LH-RH). The analog possesses greater potency than the natural hormone. The chemical name is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
LUPRON DEPOT is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as a monthly intramuscular injection.
The front chamber of LUPRON DEPOT 3.75 mg prefilled dual-chamber syringe contains leuprolide acetate (3.75 mg), purified gelatin (0.65 mg), DL-lactic and glycolic acids copolymer (33.1 mg), and D-mannitol (6.6 mg). The second chamber of diluent contains carboxymethylcellulose sodium (5 mg), D-mannitol (50 mg), polysorbate 80 (1 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
During the manufacture of LUPRON DEPOT 3.75 mg, acetic acid is lost, leaving the peptide.
CLINICAL PHARMACOLOGY
Leuprolide acetate is a long-acting GnRH analog. A single monthly injection of LUPRON DEPOT 3.75 mg results in an initial stimulation followed by a prolonged suppression of pituitary gonadotropins. Repeated dosing at monthly intervals results in decreased secretion of gonadal steroids; consequently, tissues and functions that depend on gonadal steroids for their maintenance become quiescent. This effect is reversible on discontinuation of drug therapy.
Leuprolide acetate is not active when given orally. Intramuscular injection of the depot formulation provides plasma concentrations of leuprolide over a period of one month.
Pharmacokinetics
Absorption
A single dose of LUPRON DEPOT 3.75 mg was administered by intramuscular injection to healthy female volunteers. The absorption of leuprolide was characterized by an initial increase in plasma concentration, with peak concentration ranging from 4.6 to 10.2 ng/mL at four hours postdosing. However, intact leuprolide and an inactive metabolite could not be distinguished by the assay used in the study. Following the initial rise, leuprolide concentrations started to plateau within two days after dosing and remained relatively stable for about four to five weeks with plasma concentrations of about 0.30 ng/mL.
Distribution
The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Metabolism
In healthy male volunteers, a 1 mg bolus of leuprolide administered intravenously revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
In rats and dogs, administration of 14C-labeled leuprolide was shown to be metabolized to smaller inactive peptides, a pentapeptide (Metabolite I), tripeptides (Metabolites II and III) and a dipeptide (Metabolite IV). These fragments may be further catabolized.
The major metabolite (M-I) plasma concentrations measured in 5 prostate cancer patients reached maximum concentration 2 to 6 hours after dosing and were approximately 6% of the peak parent drug concentration. One week after dosing, mean plasma M-I concentrations were approximately 20% of mean leuprolide concentrations.
Excretion
Following administration of LUPRON DEPOT 3.75 mg to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations
The pharmacokinetics of the drug in hepatically and renally impaired patients have not been determined.
Drug Interactions
No pharmacokinetic-based drug-drug interaction studies have been conducted with LUPRON DEPOT. However, because leuprolide acetate is a peptide that is primarily degraded by peptidase and not by cytochrome P-450 enzymes as noted in specific studies, and the drug is only about 46% bound to plasma proteins, drug interactions would not be expected to occur.
CLINICAL STUDIES
Endometriosis
In controlled clinical studies, LUPRON DEPOT 3.75 mg monthly for six months was shown to be comparable to danazol 800 mg/day in relieving the clinical sign/symptoms of endometriosis (pelvic pain, dysmenorrhea, dyspareunia, pelvic tenderness, and induration) and in reducing the size of endometrial implants as evidenced by laparoscopy. The clinical significance of a decrease in endometriotic lesions is not known at this time, and in addition laparoscopic staging of endometriosis does not necessarily correlate with the severity of symptoms.
LUPRON DEPOT 3.75 mg monthly induced amenorrhea in 74% and 98% of the patients after the first and second treatment months respectively. Most of the remaining patients reported episodes of only light bleeding or spotting. In the first, second and third post-treatment months, normal menstrual cycles resumed in 7%, 71% and 95% of patients, respectively, excluding those who became pregnant.
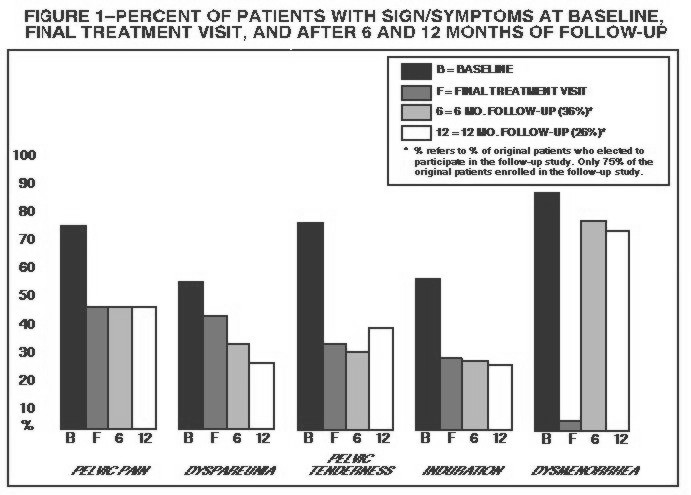
Figure 1 illustrates the percent of patients with symptoms at baseline, final treatment visit and sustained relief at 6 and 12 months following discontinuation of treatment for the various symptoms evaluated during two controlled clinical studies. This included all patients at end of treatment and those who elected to participate in the follow-up period. This might provide a slight bias in the results at follow-up as 75% of the original patients entered the follow-up study, and 36% were evaluated at 6 months and 26% at 12 months.
Hormonal replacement therapy
Two clinical studies with a treatment duration of 12 months indicate that concurrent hormonal therapy (norethindrone acetate 5 mg daily) is effective in significantly reducing the loss of bone mineral density associated with LUPRON, without compromising the efficacy of LUPRON in relieving symptoms of endometriosis. (All patients in these studies received calcium supplementation with 1000 mg elemental calcium). One controlled, randomized and double-blind study included 51 women treated with LUPRON DEPOT alone and 55 women treated with LUPRON plus norethindrone acetate 5 mg daily. The second study was an open label study in which 136 women were treated with LUPRON plus norethindrone acetate 5 mg daily. This study confirmed the reduction in loss of bone mineral density that was observed in the controlled study. Suppression of menses was maintained throughout treatment in 84% and 73% of patients receiving LD/N in the controlled study and open label study, respectively. The median time for menses resumption after treatment with LD/N was 8 weeks.
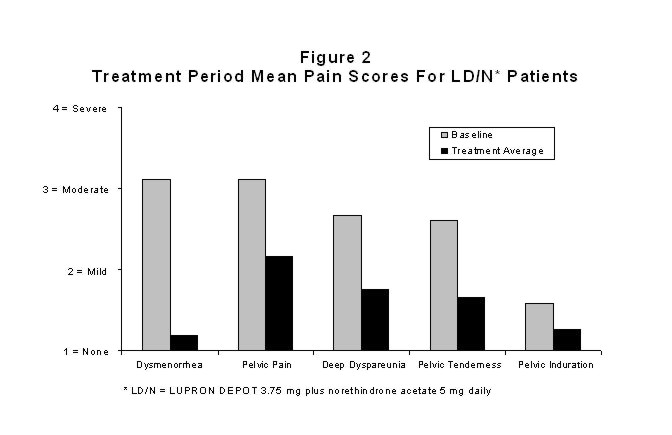
Figure 2 illustrates the mean pain scores for the LD/N group from the controlled study.
Uterine Leiomyomata (Fibroids)
In controlled clinical trials, administration of LUPRON DEPOT 3.75 mg for a period of three or six months was shown to decrease uterine and fibroid volume, thus allowing for relief of clinical symptoms (abdominal bloating, pelvic pain, and pressure). Excessive vaginal bleeding (menorrhagia and menometrorrhagia) decreased, resulting in improvement in hematologic parameters.
In three clinical trials, enrollment was not based on hematologic status. Mean uterine volume decreased by 41% and myoma volume decreased by 37% at final visit as evidenced by ultrasound or MRI. These patients also experienced a decrease in symptoms including excessive vaginal bleeding and pelvic discomfort. Benefit occurred by three months of therapy, but additional gain was observed with an additional three months of LUPRON DEPOT 3.75 mg. Ninety-five percent of these patients became amenorrheic with 61%, 25%, and 4% experiencing amenorrhea during the first, second, and third treatment months respectively.
Post-treatment follow-up was carried out for a small percentage of LUPRON DEPOT 3.75 mg patients among the 77% who demonstrated a ≥ 25% decrease in uterine volume while on therapy. Menses usually returned within two months of cessation of therapy. Mean time to return to pretreatment uterine size was 8.3 months. Regrowth did not appear to be related to pretreatment uterine volume.
In another controlled clinical study, enrollment was based on hematocrit ≤ 30% and/or hemoglobin ≤ 10.2 g/dL. Administration of LUPRON DEPOT 3.75 mg, concomitantly with iron, produced an increase of ≥ 6% hematocrit and ≥ 2 g/dL hemoglobin in 77% of patients at three months of therapy. The mean change in hematocrit was 10.1% and the mean change in hemoglobin was 4.2 g/dL. Clinical response was judged to be a hematocrit of ≥ 36% and hemoglobin of ≥ 12 g/dL, thus allowing for autologous blood donation prior to surgery. At three months, 75% of patients met this criterion.
At three months, 80% of patients experienced relief from either menorrhagia or menometrorrhagia. As with the previous studies, episodes of spotting and menstrual-like bleeding were noted in some patients.
In this same study, a decrease of ≥ 25% was seen in uterine and myoma volumes in 60% and 54% of patients respectively. LUPRON DEPOT 3.75 mg was found to relieve symptoms of bloating, pelvic pain, and pressure.
There is no evidence that pregnancy rates are enhanced or adversely affected by the use of LUPRON DEPOT 3.75 mg.
INDICATIONS AND USAGE
Endometriosis
LUPRON DEPOT 3.75 mg is indicated for management of endometriosis, including pain relief and reduction of endometriotic lesions. LUPRON DEPOT monthly with norethindrone acetate 5 mg daily is also indicated for initial management of endometriosis and for management of recurrence of symptoms. (Refer also to norethindrone acetate prescribing information for WARNINGS, PRECAUTIONS, CONTRAINDICATIONS and ADVERSE REACTIONS associated with norethindrone acetate). Duration of initial treatment or retreatment should be limited to 6 months.
Uterine Leiomyomata (Fibroids)
LUPRON DEPOT 3.75 mg concomitantly with iron therapy is indicated for the preoperative hematologic improvement of patients with anemia caused by uterine leiomyomata. The clinician may wish to consider a one-month trial period on iron alone inasmuch as some of the patients will respond to iron alone. (See Table 1.) LUPRON may be added if the response to iron alone is considered inadequate. Recommended duration of therapy with LUPRON DEPOT 3.75 mg is up to three months.
Experience with LUPRON DEPOT in females has been limited to women 18 years of age and older.
| Treatment Group | Week 4 | Week 8 | Week 12 |
|---|---|---|---|
| LUPRON DEPOT 3.75 mg with Iron | 41* | 71† | 79* |
| Iron Alone | 17 | 40 | 56 |
CONTRAINDICATIONS
- Hypersensitivity to GnRH, GnRH agonist analogs or any of the excipients in LUPRON DEPOT.
- Undiagnosed abnormal vaginal bleeding.
- LUPRON DEPOT is contraindicated in women who are or may become pregnant while receiving the drug. LUPRON DEPOT may cause fetal harm when administered to a pregnant woman. Major fetal abnormalities were observed in rabbits but not in rats after administration of LUPRON DEPOT throughout gestation. There was increased fetal mortality and decreased fetal weights in rats and rabbits. (See Pregnancy section.) The effects on fetal mortality are expected consequences of the alterations in hormonal levels brought about by the drug. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
- Use in women who are breast-feeding. (See Nursing Mothers section.)
- Norethindrone acetate is contraindicated in women with the following conditions:
- –
- Thrombophlebitis, thromboembolic disorders, cerebral apoplexy, or a past history of these conditions
- –
- Markedly impaired liver function or liver disease
- –
- Known or suspected carcinoma of the breast
WARNINGS
Safe use of leuprolide acetate or norethindrone acetate in pregnancy has not been established clinically. Before starting treatment with LUPRON DEPOT, pregnancy must be excluded.
When used monthly at the recommended dose, LUPRON DEPOT usually inhibits ovulation and stops menstruation. Contraception is not insured, however, by taking LUPRON DEPOT. Therefore, patients should use non-hormonal methods of contraception. Patients should be advised to see their physician if they believe they may be pregnant. If a patient becomes pregnant during treatment, the drug must be discontinued and the patient must be apprised of the potential risk to the fetus.
During the early phase of therapy, sex steroids temporarily rise above baseline because of the physiologic effect of the drug. Therefore, an increase in clinical signs and symptoms may be observed during the initial days of therapy, but these will dissipate with continued therapy.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely reported post-marketing.
The following applies to co-treatment with LUPRON and norethindrone acetate:
Norethindrone acetate treatment should be discontinued if there is a sudden partial or complete loss of vision or if there is sudden onset of proptosis, diplopia, or migraine. If examination reveals papilledema or retinal vascular lesions, medication should be withdrawn.
Because of the occasional occurrence of thrombophlebitis and pulmonary embolism in patients taking progestogens, the physician should be alert to the earliest manifestations of the disease in women taking norethindrone acetate.
Assessment and management of risk factors for cardiovascular disease is recommended prior to initiation of add-back therapy with norethindrone acetate. Norethindrone acetate should be used with caution in women with risk factors, including lipid abnormalities or cigarette smoking.
PRECAUTIONS
Information for Patients
An information pamphlet for patients is included with the product. Patients should be aware of the following information:
- Since menstruation usually stops with effective doses of LUPRON DEPOT, the patient should notify her physician if regular menstruation persists. Patients missing successive doses of LUPRON DEPOT may experience breakthrough bleeding.
- Patients should not use LUPRON DEPOT if they are pregnant, breast feeding, have undiagnosed abnormal vaginal bleeding, or are allergic to any of the ingredients in LUPRON DEPOT.
- Safe use of the drug in pregnancy has not been established clinically. Therefore, a non-hormonal method of contraception should be used during treatment. Patients should be advised that if they miss successive doses of LUPRON DEPOT, breakthrough bleeding or ovulation may occur with the potential for conception. If a patient becomes pregnant during treatment, she should discontinue treatment and consult her physician.
- Adverse events occurring in clinical studies with LUPRON DEPOT that are associated with hypoestrogenism include: hot flashes, headaches, emotional lability, decreased libido, acne, myalgia, reduction in breast size, and vaginal dryness. Estrogen levels returned to normal after treatment was discontinued.
- Patients should be counseled on the possibility of the development or worsening of depression and the occurrence of memory disorders.
- The induced hypoestrogenic state also results in a loss in bone density over the course of treatment, some of which may not be reversible. For a period up to six months, this bone loss should not be clinically significant. Clinical studies show that concurrent hormonal therapy with norethindrone acetate 5 mg daily is effective in reducing loss of bone mineral density that occurs with LUPRON. (All patients received calcium supplementation with 1000 mg elemental calcium.) (See Changes in Bone Density section).
- If the symptoms of endometriosis recur after a course of therapy, retreatment with a six-month course of LUPRON DEPOT and norethindrone acetate 5 mg daily may be considered. Retreatment beyond this one six month course cannot be recommended. It is recommended that bone density be assessed before retreatment begins to ensure that values are within normal limits. Retreatment with LUPRON DEPOT alone is not recommended.
- In patients with major risk factors for decreased bone mineral content such as chronic alcohol and/or tobacco use, strong family history of osteoporosis, or chronic use of drugs that can reduce bone mass such as anticonvulsants or corticosteroids, LUPRON DEPOT therapy may pose an additional risk. In these patients, the risks and benefits must be weighed carefully before therapy with LUPRON DEPOT alone is instituted, and concomitant treatment with norethindrone acetate 5 mg daily should be considered. Retreatment with gonadotropin-releasing hormone analogs, including LUPRON is not advisable in patients with major risk factors for loss of bone mineral content.
- Because norethindrone acetate may cause some degree of fluid retention, conditions which might be influenced by this factor, such as epilepsy, migraine, asthma, cardiac or renal dysfunctions require careful observation during norethindrone acetate add-back therapy.
- Patients who have a history of depression should be carefully observed during treatment with norethindrone acetate and norethindrone acetate should be discontinued if severe depression occurs.
Laboratory Tests
See ADVERSE REACTIONS section.
Drug Interactions
See CLINICAL PHARMACOLOGY, Pharmacokinetics.
Drug/Laboratory Test Interactions
Administration of LUPRON DEPOT in therapeutic doses results in suppression of the pituitary-gonadal system. Normal function is usually restored within three months after treatment is discontinued. Therefore, diagnostic tests of pituitary gonadotropic and gonadal functions conducted during treatment and for up to three months after discontinuation of LUPRON DEPOT may be misleading.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at high daily doses (0.6 to 4 mg/kg). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males (highest incidence in the low dose group). In mice, no leuprolide acetate-induced tumors or pituitary abnormalities were observed at a dose as high as 60 mg/kg for two years. Patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Mutagenicity studies have been performed with leuprolide acetate using bacterial and mammalian systems. These studies provided no evidence of a mutagenic potential.
Clinical and pharmacologic studies in adults (>18 years) with leuprolide acetate and similar analogs have shown reversibility of fertility suppression when the drug is discontinued after continuous administration for periods of up to 24 weeks. Although no clinical studies have been completed in children to assess the full reversibility of fertility suppression, animal studies (prepubertal and adult rats and monkeys) with leuprolide acetate and other GnRH analogs have shown functional recovery.
Pregnancy
Teratogenic Effects
Pregnancy Category X (see CONTRAINDICATIONS section).
When administered on day 6 of pregnancy at test dosages of 0.00024, 0.0024, and 0.024 mg/kg (1/300 to 1/3 of the human dose) to rabbits, LUPRON DEPOT produced a dose-related increase in major fetal abnormalities. Similar studies in rats failed to demonstrate an increase in fetal malformations. There was increased fetal mortality and decreased fetal weights with the two higher doses of LUPRON DEPOT in rabbits and with the highest dose (0.024 mg/kg) in rats.
Nursing Mothers
It is not known whether LUPRON DEPOT is excreted in human milk. Because many drugs are excreted in human milk, and because the effects of LUPRON DEPOT on lactation and/or the breast-fed child have not been determined, LUPRON DEPOT should not be used by nursing mothers.
Pediatric Use
Experience with LUPRON DEPOT 3.75 mg for treatment of endometriosis has been limited to women 18 years of age and older. See LUPRON DEPOT-PED® (leuprolide acetate for depot suspension) labeling for the safety and effectiveness in children with central precocious puberty.
Geriatric Use
This product has not been studied in women over 65 years of age and is not indicated in this population.
ADVERSE REACTIONS
Clinical Trials
Estradiol levels may increase during the first weeks following the initial injection of LUPRON, but then decline to menopausal levels. This transient increase in estradiol can be associated with a temporary worsening of signs and symptoms (see WARNINGS section).
As would be expected with a drug that lowers serum estradiol levels, the most frequently reported adverse reactions were those related to hypoestrogenism.
The monthly formulation of LUPRON DEPOT 3.75 mg was utilized in controlled clinical trials that studied the drug in 166 endometriosis and 166 uterine fibroids patients. Adverse events reported in ≥5% of patients in either of these populations and thought to be potentially related to drug are noted in the following table.
| Endometriosis (2 Studies) | Uterine Fibroids (4 Studies) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LUPRON DEPOT 3.75 mg N=166 | Danazol N=136 | Placebo N=31 | LUPRON DEPOT 3.75 mg N=166 | Placebo N=163 |
||||||
| N | (%) | N | (%) | N | (%) | N | (%) | N | (%) | |
| In these same studies, symptoms reported in <5% of patients included: Body as a Whole - Body odor, Flu syndrome, Injection site reactions; Cardiovascular System - Palpitations, Syncope, Tachycardia; Digestive System - Appetite changes, Dry mouth, Thirst; Endocrine System - Androgen-like effects; Hemic and Lymphatic System - Ecchymosis, Lymphadenopathy; Nervous System– Anxiety*, Insomnia/Sleep disorders*, Delusions, Memory disorder, Personality disorder; Respiratory System - Rhinitis; Skin and Appendages - Alopecia, Hair disorder, Nail disorder; Special Senses - Conjunctivitis, Ophthalmologic disorders*, Taste perversion; Urogenital System - Dysuria*, Lactation, Menstrual disorders. | ||||||||||
|
||||||||||
| Body as a Whole | ||||||||||
| Asthenia | 5 | (3) | 9 | (7) | 0 | (0) | 14 | (8.4) | 8 | (4.9) |
| General pain | 31 | (19) | 22 | (16) | 1 | (3) | 14 | (8.4) | 10 | (6.1) |
| Headache* | 53 | (32) | 30 | (22) | 2 | (6) | 43 | (25.9) | 29 | (17.8) |
| Cardiovascular System | ||||||||||
| Hot flashes/sweats* | 139 | (84) | 77 | (57) | 9 | (29) | 121 | (72.9) | 29 | (17.8) |
| Gastrointestinal System | ||||||||||
| Nausea/vomiting | 21 | (13) | 17 | (13) | 1 | (3) | 8 | (4.8) | 6 | (3.7) |
| GI disturbances* | 11 | (7) | 8 | (6) | 1 | (3) | 5 | (3.0) | 2 | (1.2) |
| Metabolic and Nutritional Disorders | ||||||||||
| Edema | 12 | (7) | 17 | (13) | 1 | (3) | 9 | (5.4) | 2 | (1.2) |
| Weight gain/loss | 22 | (13) | 36 | (26) | 0 | (0) | 5 | (3.0) | 2 | (1.2) |
| Endocrine System | ||||||||||
| Acne | 17 | (10) | 27 | (20) | 0 | (0) | 0 | (0) | 0 | (0) |
| Hirsutism | 2 | (1) | 9 | (7) | 1 | (3) | 1 | (0.6) | 0 | (0) |
| Musculoskeletal System | ||||||||||
| Joint disorder* | 14 | (8) | 11 | (8) | 0 | (0) | 13 | (7.8) | 5 | (3.1) |
| Myalgia* | 1 | (1) | 7 | (5) | 0 | (0) | 1 | (0.6) | 0 | (0) |
| Nervous System | ||||||||||
| Decreased libido* | 19 | (11) | 6 | (4) | 0 | (0) | 3 | (1.8) | 0 | (0) |
| Depression/emotional lability* | 36 | (22) | 27 | (20) | 1 | (3) | 18 | (10.8) | 7 | (4.3) |
| Dizziness | 19 | (11) | 4 | (3) | 0 | (0) | 3 | (1.8) | 6 | (3.7) |
| Nervousness* | 8 | (5) | 11 | (8) | 0 | (0) | 8 | (4.8) | 1 | (0.6) |
| Neuromuscular disorders* | 11 | (7) | 17 | (13) | 0 | (0) | 3 | (1.8) | 0 | (0) |
| Paresthesias | 12 | (7) | 11 | (8) | 0 | (0) | 2 | (1.2) | 1 | (0.6) |
| Skin and Appendages | ||||||||||
| Skin reactions | 17 | (10) | 20 | (15) | 1 | (3) | 5 | (3.0) | 2 | (1.2) |
| Urogenital System | ||||||||||
| Breast changes/tenderness/pain* | 10 | (6) | 12 | (9) | 0 | (0) | 3 | (1.8) | 7 | (4.3) |
| Vaginitis* | 46 | (28) | 23 | (17) | 0 | (0) | 19 | (11.4) | 3 | (1.8) |
In one controlled clinical trial utilizing the monthly formulation of LUPRON DEPOT, patients diagnosed with uterine fibroids received a higher dose (7.5 mg) of LUPRON DEPOT. Events seen with this dose that were thought to be potentially related to drug and were not seen at the lower dose included glossitis, hypesthesia, lactation, pyelonephritis, and urinary disorders. Generally, a higher incidence of hypoestrogenic effects was observed at the higher dose.
Table 3 lists the potentially drug-related adverse events observed in at least 5% of patients in any treatment group during the first 6 months of treatment in the add-back clinical studies.
In the controlled clinical trial, 50 of 51 (98%) patients in the LD group and 48 of 55 (87%) patients in the LD/N group reported experiencing hot flashes on one or more occasions during treatment. During Month 6 of treatment, 32 of 37 (86%) patients in the LD group and 22 of 38 (58%) patients in the LD/N group reported having experienced hot flashes. The mean number of days on which hot flashes were reported during this month of treatment was 19 and 7 in the LD and LD/N treatment groups, respectively. The mean maximum number of hot flashes in a day during this month of treatment was 5.8 and 1.9 in the LD and LD/N treatment groups, respectively.
| Controlled Study | Open Label Study | |||||
|---|---|---|---|---|---|---|
| LD - Only*
N=51 | LD/N†
N=55 | LD/N†
N=136 |
||||
| Adverse Events | N | (%) | N | (%) | N | (%) |
| Any Adverse Event | 50 | (98) | 53 | (96) | 126 | (93) |
| Body as a Whole | ||||||
| Asthenia | 9 | (18) | 10 | (18) | 15 | (11) |
| Headache/Migraine | 33 | (65) | 28 | (51) | 63 | (46) |
| Injection Site Reaction | 1 | (2) | 5 | (9) | 4 | (3) |
| Pain | 12 | (24) | 16 | (29) | 29 | (21) |
| Cardiovascular System | ||||||
| Hot flashes/sweats | 50 | (98) | 48 | (87) | 78 | (57) |
| Digestive System | ||||||
| Altered Bowel Function | 7 | (14) | 8 | (15) | 14 | (10) |
| Changes in Appetite | 2 | (4) | 0 | (0) | 8 | (6) |
| GI Disturbance | 2 | (4) | 4 | (7) | 6 | (4) |
| Nausea/Vomiting | 13 | (25) | 16 | (29) | 17 | (13) |
| Metabolic and Nutritional Disorders | ||||||
| Edema | 0 | (0) | 5 | (9) | 9 | (7) |
| Weight Changes | 6 | (12) | 7 | (13) | 6 | (4) |
| Nervous System | ||||||
| Anxiety | 3 | (6) | 0 | (0) | 11 | (8) |
| Depression/Emotional Lability | 16 | (31) | 15 | (27) | 46 | (34) |
| Dizziness/Vertigo | 8 | (16) | 6 | (11) | 10 | (7) |
| Insomnia/Sleep Disorder | 16 | (31) | 7 | (13) | 20 | (15) |
| Libido Changes | 5 | (10) | 2 | (4) | 10 | (7) |
| Memory Disorder | 3 | (6) | 1 | (2) | 6 | (4) |
| Nervousness | 4 | (8) | 2 | (4) | 15 | (11) |
| Neuromuscular Disorder | 1 | (2) | 5 | (9) | 4 | (3) |
| Skin and Appendages | ||||||
| Alopecia | 0 | (0) | 5 | (9) | 4 | (3) |
| Androgen-Like Effects | 2 | (4) | 3 | (5) | 24 | (18) |
| Skin/Mucous Membrane Reaction | 2 | (4) | 5 | (9) | 15 | (11) |
| Urogenital System | ||||||
| Breast Changes/Pain/Tenderness | 3 | (6) | 7 | (13) | 11 | (8) |
| Menstrual Disorders | 1 | (2) | 0 | (0) | 7 | (5) |
| Vaginitis | 10 | (20) | 8 | (15) | 11 | (8) |
Changes in Bone Density
In controlled clinical studies, patients with endometriosis (six months of therapy) or uterine fibroids (three months of therapy) were treated with LUPRON DEPOT 3.75 mg. In endometriosis patients, vertebral bone density as measured by dual energy x-ray absorptiometry (DEXA) decreased by an average of 3.2% at six months compared with the pretreatment value. Clinical studies demonstrate that concurrent hormonal therapy (norethindrone acetate 5 mg daily) and calcium supplementation is effective in significantly reducing the loss of bone mineral density that occurs with LUPRON treatment, without compromising the efficacy of LUPRON in relieving symptoms of endometriosis.
LUPRON DEPOT 3.75 mg plus norethindrone acetate 5 mg daily was evaluated in two clinical trials. The results from this regimen were similar in both studies. LUPRON DEPOT 3.75 mg was used as a control group in one study. The bone mineral density data of the lumbar spine from these two studies are presented in Table 4.
| LUPRON DEPOT 3.75mg | LUPRON DEPOT 3.75 mg plus norethindrone acetate 5 mg daily | |||||
|---|---|---|---|---|---|---|
| Controlled Study | Controlled Study | Open Label Study | ||||
| N | Change | N | Change | N | Change | |
| Week 24* | 41 | -3.2% | 42 | -0.3% | 115 | -0.2% |
| Week 52† | 29 | -6.3% | 32 | -1.0% | 84 | -1.1% |
When LUPRON DEPOT 3.75 mg was administered for three months in uterine fibroid patients, vertebral trabecular bone mineral density as assessed by quantitative digital radiography (QDR) revealed a mean decrease of 2.7% compared with baseline. Six months after discontinuation of therapy, a trend toward recovery was observed. Use of LUPRON DEPOT for longer than three months (uterine fibroids) or six months (endometriosis) or in the presence of other known risk factors for decreased bone mineral content may cause additional bone loss and is not recommended.
Changes in Laboratory Values During Treatment
Plasma Enzymes
Endometriosis
During early clinical trials with LUPRON DEPOT 3.75 mg, regular laboratory monitoring revealed that AST levels were more than twice the upper limit of normal in only one patient. There was no clinical or other laboratory evidence of abnormal liver function.
In two other clinical trials, 6 of 191 patients receiving LUPRON DEPOT 3.75 mg plus norethindrone acetate 5 mg daily for up to 12 months developed an elevated (at least twice the upper limit of normal) SGPT or GGT. Five of the 6 increases were observed beyond 6 months of treatment. None were associated with elevated bilirubin concentration.
Uterine Leiomyomata (Fibroids)
In clinical trials with LUPRON DEPOT 3.75 mg, five (3%) patients had a post-treatment transaminase value that was at least twice the baseline value and above the upper limit of the normal range. None of the laboratory increases were associated with clinical symptoms.
Lipids
Endometriosis
In earlier clinical studies, 4% of the LUPRON DEPOT 3.75 mg patients and 1% of the danazol patients had total cholesterol values above the normal range at enrollment. These patients also had cholesterol values above the normal range at the end of treatment.
Of those patients whose pretreatment cholesterol values were in the normal range, 7% of the LUPRON DEPOT 3.75 mg patients and 9% of the danazol patients had post-treatment values above the normal range.
The mean (±SEM) pretreatment values for total cholesterol from all patients were 178.8 (2.9) mg/dL in the LUPRON DEPOT 3.75 mg groups and 175.3 (3.0) mg/dL in the danazol group. At the end of treatment, the mean values for total cholesterol from all patients were 193.3 mg/dL in the LUPRON DEPOT 3.75 mg group and 194.4 mg/dL in the danazol group. These increases from the pretreatment values were statistically significant (p<0.03) in both groups.
Triglycerides were increased above the upper limit of normal in 12% of the patients who received LUPRON DEPOT 3.75 mg and in 6% of the patients who received danazol.
At the end of treatment, HDL cholesterol fractions decreased below the lower limit of the normal range in 2% of the LUPRON DEPOT 3.75 mg patients compared with 54% of those receiving danazol. LDL cholesterol fractions increased above the upper limit of the normal range in 6% of the patients receiving LUPRON DEPOT 3.75 mg compared with 23% of those receiving danazol. There was no increase in the LDL/HDL ratio in patients receiving LUPRON DEPOT 3.75 mg but there was approximately a two-fold increase in the LDL/HDL ratio in patients receiving danazol.
In two other clinical trials, LUPRON DEPOT 3.75 mg plus norethindrone acetate 5 mg daily was evaluated for 12 months of treatment. LUPRON DEPOT 3.75 mg was used as a control group in one study. Percent changes from baseline for serum lipids and percentages of patients with serum lipid values outside of the normal range in the two studies are summarized in the tables below.
| LUPRON | LUPRON plus norethindrone acetate 5 mg daily | |||||
|---|---|---|---|---|---|---|
| Controlled Study (n=39) | Controlled Study (n=41) | Open Label Study (n=117) | ||||
| Baseline Value* | Wk 24 % Change | Baseline Value* | Wk 24 % Change | Baseline Value* | Wk 24 % Change |
|
| Total Cholesterol | 170.5 | 9.2% | 179.3 | 0.2% | 181.2 | 2.8% |
| HDL Cholesterol | 52.4 | 7.4% | 51.8 | -18.8% | 51.0 | -14.6% |
| LDL Cholesterol | 96.6 | 10.9% | 101.5 | 14.1% | 109.1 | 13.1% |
| LDL/HDL Ratio | 2.0† | 5.0% | 2.1† | 43.4% | 2.3† | 39.4% |
| Triglycerides | 107.8 | 17.5% | 130.2 | 9.5% | 105.4 | 13.8% |
Changes from baseline tended to be greater at Week 52. After treatment, mean serum lipid levels from patients with follow up data returned to pretreatment values.
| LUPRON | LUPRON plus norethindrone acetate 5 mg daily | |||||
|---|---|---|---|---|---|---|
| Controlled Study (n=39) | Controlled Study (n=41) | Open Label Study (n=117) | ||||
| Wk 0 | Wk 24* | Wk 0 | Wk 24* | Wk 0 | Wk 24* | |
|
||||||
| Total Cholesterol (>240 mg/dL) | 15% | 23% | 15% | 20% | 6% | 7% |
| HDL Cholesterol (<40 mg/dL) | 15% | 10% | 15% | 44% | 15% | 41% |
| LDL Cholesterol (>160 mg/dL) | 0% | 8% | 5% | 7% | 9% | 11% |
| LDL/HDL Ratio (>4.0) | 0% | 3% | 2% | 15% | 7% | 21% |
| Triglycerides (>200 mg/dL) | 13% | 13% | 12% | 10% | 5% | 9% |
Low HDL-cholesterol (<40 mg/dL) and elevated LDL-cholesterol (>160 mg/dL) are recognized risk factors for cardiovascular disease. The long-term significance of the observed treatment-related changes in serum lipids in women with endometriosis is unknown. Therefore assessment of cardiovascular risk factors should be considered prior to initiation of concurrent treatment with LUPRON and norethindrone acetate.
Uterine Leiomyomata (Fibroids)
In patients receiving LUPRON DEPOT 3.75 mg, mean changes in cholesterol (+11 mg/dL to +29 mg/dL), LDL cholesterol (+8 mg/dL to +22 mg/dL), HDL cholesterol (0 to +6 mg/dL), and the LDL/HDL ratio (-0.1 to +0.5) were observed across studies. In the one study in which triglycerides were determined, the mean increase from baseline was 32 mg/dL.
Other Changes
Endometriosis
The following changes were seen in approximately 5% to 8% of patients. In the earlier comparative studies, LUPRON DEPOT 3.75 mg was associated with elevations of LDH and phosphorus, and decreases in WBC counts. Danazol therapy was associated with increases in hematocrit, platelet count, and LDH. In the hormonal add-back studies LUPRON DEPOT in combination with norethindrone acetate was associated with elevations of GGT and SGPT.
Uterine Leiomyomata (Fibroids)
Hematology: (see CLINICAL STUDIES section) In LUPRON DEPOT 3.75 mg treated patients, although there were statistically significant mean decreases in platelet counts from baseline to final visit, the last mean platelet counts were within the normal range. Decreases in total WBC count and neutrophils were observed, but were not clinically significant.
Chemistry: Slight to moderate mean increases were noted for glucose, uric acid, BUN, creatinine, total protein, albumin, bilirubin, alkaline phosphatase, LDH, calcium, and phosphorus. None of these increases were clinically significant.
Postmarketing
During postmarketing surveillance, the following adverse events were reported. Like other drugs in this class, mood swings, including depression, have been reported. There have been rare reports of suicidal ideation and attempt. Many, but not all, of these patients had a history of depression or other psychiatric illness. Patients should be counseled on the possibility of development or worsening of depression during treatment with LUPRON.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely reported. Rash, urticaria, and photosensitivity reactions have also been reported.
Localized reactions including induration and abscess have been reported at the site of injection. Symptoms consistent with fibromyalgia (eg: joint and muscle pain, headaches, sleep disorder, gastrointestinal distress, and shortness of breath) have been reported individually and collectively. Other events reported are:
Cardiovascular System– Hypotension, Pulmonary embolism; Hemic and Lymphatic System - Decreased WBC; Central/Peripheral Nervous System - Peripheral neuropathy, Spinal fracture/paralysis; Musculoskeletal System - Tenosynovitis-like symptoms; Urogenital System - Prostate pain.
Pituitary apoplexy
During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
See other LUPRON DEPOT and LUPRON Injection package inserts for other events reported in different patient populations.
OVERDOSAGE
In rats subcutaneous administration of 250 to 500 times the recommended human dose, expressed on a per body weight basis, resulted in dyspnea, decreased activity, and local irritation at the injection site. There is no evidence that there is a clinical counterpart of this phenomenon. In early clinical trials using daily subcutaneous leuprolide acetate in patients with prostate cancer, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
DOSAGE AND ADMINISTRATION
LUPRON DEPOT Must Be Administered Under The Supervision Of A Physician.
Endometriosis
The recommended duration of treatment with LUPRON DEPOT 3.75 mg alone or in combination with norethindrone acetate is six months. The choice of LUPRON DEPOT alone or LUPRON DEPOT plus norethindrone acetate therapy for initial management of the symptoms and signs of endometriosis should be made by the health care professional in consultation with the patient and should take into consideration the risks and benefits of the addition of norethindrone to LUPRON DEPOT alone.
If the symptoms of endometriosis recur after a course of therapy, retreatment with a six-month course of LUPRON DEPOT monthly and norethindrone acetate 5 mg daily may be considered. Retreatment beyond this one six-month course cannot be recommended. It is recommended that bone density be assessed before retreatment begins to ensure that values are within normal limits. LUPRON DEPOT alone is not recommended for retreatment. If norethindrone acetate is contraindicated for the individual patient, then retreatment is not recommended.
An assessment of cardiovascular risk and management of risk factors such as cigarette smoking is recommended before beginning treatment with LUPRON DEPOT and norethindrone acetate.
Uterine Leiomyomata (Fibroids)
Recommended duration of therapy with LUPRON DEPOT 3.75 mg is up to 3 months. The symptoms associated with uterine leiomyomata will recur following discontinuation of therapy. If additional treatment with LUPRON DEPOT 3.75 mg is contemplated, bone density should be assessed prior to initiation of therapy to ensure that values are within normal limits.
The recommended dose of LUPRON DEPOT is 3.75 mg, incorporated in a depot formulation. The lyophilized microspheres are to be reconstituted and administered monthly as a single intramuscular injection. For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the following instructions:
- The LUPRON DEPOT powder should be visually inspected and the syringe should NOT BE USED if clumping or caking is evident. A thin layer of powder on the wall of the syringe is considered normal. The diluent should appear clear.
- To prepare for injection, screw the white plunger into the end stopper until the stopper begins to turn.
- Hold the syringe UPRIGHT. Release the diluent by SLOWLY PUSHING (6 to 8 seconds) the plunger until the first stopper is at the blue line in the middle of the barrel.
- Keep the syringe UPRIGHT. Gently mix the microspheres (powder) thoroughly to form a uniform suspension. The suspension will appear milky. If the powder adheres to the stopper or caking/clumping is present, tap the syringe with your finger to disperse. DO NOT USE if any of the powder has not gone into suspension.
- Hold the syringe UPRIGHT. With the opposite hand pull the needle cap upward without twisting.
- Keep the syringe UPRIGHT. Advance the plunger to expel the air from the syringe.
- Inject the entire contents of the syringe intramuscularly at the time of reconstitution. The suspension settles very quickly following reconstitution; therefore, LUPRON DEPOT should be mixed and used immediately.
NOTE: Aspirated blood would be visible just below the luer lock connection if a blood vessel is accidentally penetrated. If present, blood can be seen through the transparent LuproLoc™ safety device.
AFTER INJECTION
- 8.
- Withdraw the needle. Immediately activate the LuproLoc™ safety device by pushing the arrow forward with the thumb or finger until the device is fully extended and a CLICK is heard or felt.
Since the product does not contain a preservative, the suspension should be discarded if not used immediately.
As with other drugs administered by injection, the injection site should be varied periodically.
HOW SUPPLIED
LUPRON DEPOT 3.75 mg is packaged as follows:
Kit with prefilled dual-chamber syringe NDC 0300-3641-01
Each syringe contains sterile lyophilized microspheres, which is leuprolide incorporated in a biodegradable copolymer of lactic and glycolic acids. When mixed with diluent, LUPRON DEPOT 3.75 mg is administered as a single monthly IM injection.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [See USP Controlled Room Temperature]
U.S. Patent Nos. 4,652,441; 4,677,191; 4,728,721; 4,849,228; 4,917,893; 5,330,767; 5,476,663; 5,575,987; 5,631,020; 5,631,021; 5,716,640; 5,823,997; 5,980,488; and 6,036,976.
Other patents pending.
Manufactured for
TAP Pharmaceuticals Inc.
Lake Forest, IL 60045, U.S.A.
by Takeda Pharmaceutical Company Limited
Osaka, JAPAN 540-8645
™ - Trademark
® - Registered Trademark
(No. 3641)
03-5447-R20; Rev: October, 2005
©1990-2005, TAP Pharmaceutical Products Inc.
| Lupron Depot (leuprolide acetate) | |||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||
Revised: 08/2006