mannitol (Mannitol) injection
[B. Braun Medical Inc.]
For Therapeutic Use Only
DESCRIPTION
20% Mannitol Injection USP is a sterile, nonpyrogenic solution of Mannitol USP in a single dose container for intravenous administration. It contains no antimicrobial agents. Mannitol is a 6-carbon sugar alcohol prepared commercially by the reduction of dextrose. Although virtually inert metabolically in humans, it occurs naturally in fruits and vegetables. Mannitol is an obligatory osmotic diuretic.
Each 100 mL contains:
Mannitol USP 20 g; Water for Injection USP qs
pH: 5.3 (4.5–7.0); Calculated Osmolarity 1100 mOsmol/liter
Administration of substantially hypertonic solutions (≥ 600 mOsmol/L) may cause vein damage. Normal physiologic osmolarity range is approximately 280 to 310 mOsmol/L.
The formula of the active ingredient is:
| Ingredient | Molecular Formula | Molecular Weight |
|---|---|---|
| Mannitol USP | 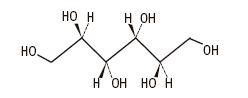 | 182.17 |
The EXCEL Container is Latex-free, PVC-free, and DEHP-free.
The plastic container is made from a multilayered film specifically developed for parenteral drugs. It contains no plasticizers. The solution contact layer is a rubberized copolymer of ethylene and propylene. Solutions in contact with the plastic container may leach out certain chemical components from the plastic in very small amounts; however, biological testing was supportive of the safety of the plastic container materials. The container-solution unit is a closed system and is not dependent upon entry of external air during administration. The container is overwrapped to provide protection from the physical environment and to provide an additional moisture barrier when necessary. Exposure to temperatures above 25°C/77°F during transport and storage will lead to minor losses in moisture content. Higher temperatures lead to greater losses. It is unlikely that these minor losses will lead to clinically significant changes within the expiration period.
The closure system has two ports; the one for the administration set has a tamper evident plastic protector and the other is a medication addition site. Refer to the Directions for Use of the container.
CLINICAL PHARMACOLOGY
Mannitol Injection USP is one of the nonelectrolyte, obligatory, osmotic diuretics. It is freely filterable at the renal glomerulus, only poorly reabsorbed by the renal tubule, not secreted by the tubule, and is pharmacologically inert.
Mannitol, when administered intravenously, exerts its osmotic effect as a solute of relatively small molecular size being largely confined to the extracellular space. Only relatively small amounts of the dose administered is metabolized. Mannitol is readily diffused through the glomerulus of the kidney over a wide range of normal and impaired kidney function. In this fashion, approximately 80% of a 100 gram dose of mannitol will appear in the urine in three hours with lesser amounts thereafter. Even at peak concentrations, mannitol will exhibit less than 10% of tubular reabsorption and is not secreted by tubular cells. Mannitol will hinder tubular reabsorption of water and enhance excretion of sodium and chloride by elevating the osmolarity of the glomerular filtrate.
This increase in extracellular osmolarity effected by the intravenous administration of mannitol will induce the movement of intracellular water to the extracellular and vascular spaces. This action underlies the role of mannitol in reducing intracranial pressure, intracranial edema, and elevated intraocular pressure.
INDICATIONS AND USAGE
20% Mannitol Injection USP is indicated for:
Promotion of diuresis, in the prevention and/or treatment of the oliguric phase of acute renal failure before irreversible renal failure becomes established.
Reduction of intracranial pressure and treatment of cerebral edema by reducing brain mass.
Reduction of elevated intraocular pressure when the pressure cannot be lowered by other means.
Promotion of urinary excretion of toxins.
CONTRAINDICATIONS
20% Mannitol Injection USP is contraindicated in patients with:
Well-established anuria due to severe renal disease.
Severe pulmonary congestion or frank pulmonary edema.
Active intracranial bleeding except during craniotomy.
Severe dehydration.
Progressive renal damage or dysfunction after institution of mannitol therapy, including increasing oliguria and azotemia.
Progressive heart failure or pulmonary congestion after institution of mannitol therapy.
WARNINGS
In patients with severe impairment of renal function, a test dose should be utilized (see DOSAGE AND ADMINISTRATION). A second test dose may be tried if there is an inadequate response, but no more than two test doses should be attempted.
The obligatory diuretic response following rapid infusion of 20% Mannitol Injection USP may further aggravate preexisting hemoconcentration. Excessive loss of water and electrolytes may lead to serious imbalances. Serum sodium and potassium should be carefully monitored during mannitol administration.
If urine output continues to decline during mannitol infusion, the patient's clinical status should be closely reviewed and mannitol infusion suspended if necessary. Accumulation of mannitol may result in overexpansion of the extracellular fluid which may intensify existing or latent congestive heart failure.
Excessive loss of water and electrolytes may lead to serious imbalances. With rapid or prolonged administration of mannitol, loss of water in excess of electrolytes can cause hypernatremia. Electrolyte measurements including sodium and potassium are therefore of vital importance in monitoring the infusion of mannitol.
Osmotic nephrosis, a reversible vacuolization of the tubules of no known clinical significance, may proceed to severe irreversible nephrosis, requiring close monitoring during mannitol infusion.
PRECAUTIONS
General
Clinical evaluation and periodic laboratory determinations are necessary to monitor changes in fluid balance, electrolyte concentrations, and acid-base balance during parenteral therapy with a mannitol solution.
This solution should be used with care in patients with hypervolemia, renal insufficiency, urinary tract obstruction, or impending or frank cardiac decompensation.
The cardiovascular status of the patient should be carefully evaluated before rapidly administering mannitol since sudden expansion of the extracellular fluid may lead to fulminating congestive heart failure.
Shifting of sodium-free intracellular fluid into the extracellular compartment following mannitol infusion may lower serum sodium concentration and aggravate preexisting hyponatremia.
Mannitol administration may obscure and intensify inadequate hydration or hypovolemia by sustaining diuresis.
Electrolyte-free mannitol injection should not be given conjointly with blood. If it is essential that blood be given simultaneously, at least 20 mEq of sodium chloride should be added to each liter of mannitol solution to avoid pseudoagglutination. In no other instance should additions be made to 20% Mannitol Injection USP. The addition of sodium chloride to 20% mannitol solution may result in precipitation of mannitol. The final infusate should therefore be inspected for cloudiness or precipitation immediately after mixing, prior to administration, and periodically during administration.
Solutions of mannitol may crystallize when exposed to low temperatures. Concentrations greater than 15% have a greater tendency to crystallization. Inspect for crystals prior to administration. If crystals are observed, the container should be warmed by appropriate means to not greater than 60°C, shaken, then cooled to body temperature before administering. If all crystals cannot be completely redissolved, the container must be rejected. Administer intravenously using sterile, filter-type administration set.
Do not use plastic container in series connection.
If administration is controlled by a pumping device, care must be taken to discontinue pumping action before the container runs dry or air embolism may result.
This solution is intended for intravenous administration using sterile equipment. It is recommended that intravenous administration apparatus be replaced at least once every 24 hours.
Use only if solution is clear and container and seals are intact.
Laboratory Tests
Although blood levels of mannitol can be measured, there is little if any clinical virtue in doing so. The appropriate monitoring of blood levels of sodium and potassium; degree of hemoconcentration or hemodilution, if any; indices of renal, cardiac and pulmonary function are paramount in avoiding excessive fluid and electrolyte shifts. The routine features of physical examination and clinical chemistries suffice in achieving an adequate degree of appropriate patient monitoring.
Carcinogenesis, mutagenesis, impairment of fertility
Long term studies in animals to evaluate the carcinogenic and mutagenic potential or the effect on fertility of 20% Mannitol Injection USP have not been conducted.
Pregnancy
Teratogenic Effects
Pregnancy Category C. Animal reproduction studies have not been conducted with 20% Mannitol Injection USP. It is also not known whether 20% Mannitol Injection USP can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. 20% Mannitol Injection USP should be given to a pregnant woman only if clearly needed.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when 20% Mannitol Injection USP is administered to a nursing woman.
Pediatric Use
Safety and effectiveness in children below the age of 12 years have not been established.
Usage in Children
Dosage requirements for patients 12 years of age and under have not been established.
Geriatric Use
Clinical studies of 20% Mannitol Injection USP did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
ADVERSE REACTIONS
Reactions which may occur because of the solution or the technique of administration include febrile response, infection at the site of injection, venous thrombosis or phlebitis extending from the site of injection, extravasation and hypervolemia.
Extensive use of mannitol over the last several decades has produced recorded adverse events, in a variety of clinical settings, that are isolated or idiosyncratic in nature. None of these adverse reactions have occurred with any great frequency nor with any security in attributing them to mannitol.
The inability to clearly exclude the drug related nature of such events in these isolated reports prompts the necessity to list the reactions that have been observed in patients during or following mannitol infusion. In this fashion, patients have exhibited nausea, vomiting, rhinitis, local pain, skin necrosis and thrombophlebitis at the site of infection, chills, dizziness, urticaria, hypotension, hypertension, tachycardia, fever and angina-like chest pains.
Of far greater clinical significance is a variety of events that are related to inappropriate recognition and monitoring of fluid shifts. These are not intrinsic adverse reactions to the drug but the consequence of manipulating osmolarity by any agency in a therapeutically inappropriate manner. Failure to recognize severe impairment of renal function with the high likelihood of nondiuretic response can lead to aggravated dehydration of tissues and increased vascular fluid load. Induced diuresis in the presence of preexisting hemoconcentration and preexisting deficiency of water and electrolytes can lead to serious imbalances. Expansion of the extracellular space can aggravate cardiac decompensation or induce it in the presence of latent heart failure. Pulmonary congestion or edema can be seriously aggravated with the expansion of the extracellular and therefore intravascular fluid load. Hemodilution and dilution of the extracellular fluid space by osmotic shift of water can induce or aggravate preexisting hyponatremia.
If unrecognized, such fluid and/or electrolyte shift can produce the reported adverse reactions of pulmonary congestion, acidosis, electrolyte loss, dryness of mouth, thirst, edema, headache, blurred vision, convulsions and congestive cardiac failure.
These are not truly adverse reactions to the drug and can be appropriately prevented by evaluation of degree of renal failure with a test dose response to mannitol when indicated; evaluation of hypervolemia and hypovolemia; sodium and potassium levels; hemodilution or hemoconcentration; and evaluation of renal, cardiac and pulmonary function at the onset of therapy.
Too rapid infusion of hypertonic solutions may cause local pain and venous irritation. Rate of administration should be adjusted according to tolerance. Use of the largest peripheral vein and a small bore needle is recommended. (See DOSAGE AND ADMINISTRATION.)
The physician should also be alert to the possibility of adverse reactions to drug additives. Prescribing information for drug additives to be administered in this manner should be consulted.
If an adverse reaction does occur, discontinue the infusion, evaluate the patient, institute appropriate therapeutic countermeasures and save the remainder of the fluid for examination if deemed necessary.
OVERDOSAGE
In the event of a fluid or solute overload during parenteral therapy, reevaluate the patient's condition, and institute appropriate corrective treatment.
Larger doses than recommended may result in increased electrolyte excretion, particularly sodium, chloride, and potassium. Sodium depletion can result in orthostatic tachycardia and/or hypotension and decreased central venous pressure. Chloride metabolism closely follows that of sodium. Potassium deficit can impair neuromuscular function and cause intestinal dilatation and ileus.
Mannitol may cause pulmonary edema or water intoxication if urine flow is inadequate. See WARNINGS.
DOSAGE AND ADMINISTRATION
This solution is for intravenous use only.
The total dosage, concentration, and rate of administration should be governed by the nature and severity of the condition being treated, and the patient's fluid requirement and urinary output. The adult dosage ranges from 50 to 200 g in a 24-hour period, but in most cases an adequate response will be achieved at a usual dosage of approximately 100 g/24 hours. The rate of administration is usually adjusted to maintain a urine flow of at least 30 to 50 mL/hour. Lower mannitol concentrations and solutions containing sodium chloride are useful in preventing dehydration and electrolyte depletion. This outline of administration and dosage is only a general guide to therapy.
Dosage requirements for patients 12 years of age and under have not been established. As with adults, dose is dependent on weight, clinical condition, and laboratory results. Follow recommendations of appropriate pediatric reference text.
Test Dose
A test dose of mannitol should be given prior to instituting therapy for patients with marked oliguria or those believed to have inadequate renal function. Such test doses may be approximately 0.2 g/kg body weight (about 75 mL of a 20% solution) infused in a period of 3 to 5 minutes to produce a urine flow of at least 30 to 50 mL/hour. If urine flow does not increase, a second test dose may be given. If response is inadequate, the patient should be reevaluated.
Prevention of Acute Renal Failure (Oliguria)
When used during cardiovascular and other types of surgery, 50 to 100 g of mannitol may be given.
Treatment of Oliguria
The usual dose for treatment of oliguria is 100 g administered as a 20% solution.
Reduction of Intraocular Pressure
A dose of 1.5 to 2.0 g/kg as a 20% solution (7.5 to 10 mL/kg) may be given over a period as short as 30 minutes in order to obtain a prompt and maximal effect. When used preoperatively the dose should be given one to one and one-half hours before surgery to achieve maximal reduction of intraocular pressure before operation.
Reduction of Intracranial Pressure
Usually a maximum reduction in intracranial pressure in adults can be achieved with a dose of 0.25 g/kg given not more frequently than every six to eight hours. An osmotic gradient between the blood and cerebrospinal fluid of approximately 10 mOsmol will yield a satisfactory reduction in intracranial pressure.
Adjunctive Therapy for Intoxications
As an agent to promote diuresis in intoxications, mannitol is indicated. The concentration will depend upon the fluid requirement and urinary output of the patient.
Measurement of glomerular filtration rate by creatinine clearance may be useful for determination of dosage.
It is recommended that 20% Mannitol Injection USP be administered through a blood filter set to ensure against infusion of mannitol crystals.
When a hypertonic solution is to be administered peripherally, it should be slowly infused through a small bore needle, placed well within the lumen of a large vein to minimize venous irritation. Carefully avoid infiltration.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Use of a final filter is recommended during administration of all parenteral solutions, where possible.
This solution is intended for intravenous administration using sterile equipment.
The use of supplemental additive medication is not recommended.
HOW SUPPLIED
20% Mannitol Injection USP is supplied sterile and nonpyrogenic in EXCEL® Containers. The 500 mL and 250 mL containers are packaged 24 per case.
| NDC | Cat. No. | Size |
|---|---|---|
| 20% Mannitol Injection USP | ||
| 0264-7578-10 | L5781 | 500 mL |
| 0264-7578-20 | L5782 | 250 mL |
Exposure of pharmaceutical products to heat should be minimized. Avoid excessive heat. Protect from freezing. It is recommended that the product be stored at room temperature (25°C).
Rx only
Rev: December 2005
U.S. Patent No. 4,803,102
EXCEL® is a registered trademark of B. Braun Medical Inc.
Made in USA
Directions for Use of EXCEL® Container
Do not admix with other drugs.
Caution: Do not use plastic container in series connection.
To Open
Tear overwrap and remove solution container. Check for minute leaks by squeezing solution container firmly. If leaks are found, discard solution as sterility may be impaired.
NOTE: Before use, perform the following checks:
- Inspect each container. Read the label. Ensure solution is the one ordered and is within the expiration date.
- Invert container and carefully inspect the solution in good light for cloudiness, haze, or particulate matter. Any container which is suspect should not be used.
- Use only if solution is clear and container and seals are intact.
Preparation for Administration
- Remove plastic protector from sterile set port at bottom of container.
- Attach administration set. Refer to complete directions accompanying set.
B. Braun Medical Inc.
Irvine, CA USA 92614-5895
© 2005 B. Braun Medical Inc.
Y36-002-578
| Mannitol (Mannitol) | |||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
Revised: 12/2006