IPRIVASK
-
desirudin
CANYON PHARMACEUTICALS, INC
----------
IPRIVASK (DESIRUDIN FOR INJECTION 15MG)DESCRIPTIONIprivask [Desirudin for Injection] is a specific inhibitor of human thrombin. It has aprotein structure that is similar to that of hirudin, the naturally occurring anticoagulantpresent in the peripharyngeal glands in the medicinal leech, Hirudo medicinalis.Hirudin is a single polypeptide chain of 65 amino acids residues and containsthree disulfide bridges. Desirudin has a chemical formula of C287H440N80O110S6with a molecular weight of 6963.52. Desirudin, which is expressed in yeast(Saccharomyces cerevisiae, strain TR 1456) by recombinant DNA technology differs fromthe natural hirudin by lack of a sulfate group on Tyr-63. The biological activity ofdesirudin is determined through a chromogenic assay which measures the ability ofdesirudin to inhibit the hydrolysis of a chromogenic peptidic substrate by thrombin incomparison to a desirudin standard. One vial of desirudin contains 15.75 mgdesirudin corresponding to approximately 315,000 antithrombin units (ATU) or20,000 ATU per milligram of desirudin with reference to the WHO InternationalStandard (prepared 1991) for alphathrombin.
Iprivask 15 mg is supplied as a sterile, white, freeze dried powder for injection. Eachvial contains 15.75 mg desirudin and the following inactive ingredients: 1.31 mganhydrous magnesium chloride USP, sodium hydroxide for injection USP. Eachprefilled syringe of diluent for Iprivask contains 0.6 mL sterile Mannitol USP (3%)in Water for Injection and is preservative free. See DOSAGE ANDADMINISTRATION section for reconstitution instructions. Iprivask 15 mg isadministered by subcutaneous (SC) injection, preferably at an abdominal or thigh site.To prepare the reconstituted aqueous solution, 0.5 mL of the mannitol diluent is addedunder aseptic conditions to the vial containing the sterile powder. Shaking gently rapidlydisperses the drug. The reconstituted solution has a pH of 7.4.STRUCTURAL FORMULA

CLINICAL PHARMACOLOGYMechanism of Action: Desirudin is a selective inhibitor of free circulating andclot-bound thrombin. The anticoagulant properties of desirudin are demonstrated by itsability to prolong the clotting time of human plasma. One molecule of desirudin binds toone molecule of thrombin and thereby blocks the thrombogenic activity of thrombin.As a result, all thrombin-dependent coagulation assays are affected. Activatedpartial thromboplastin time (aPTT) is a measure of the anticoagulant activity ofdesirudin and increases in a dose-dependent fashion. The pharmacodynamic effect ofdesirudin on proteolytic activity of thrombin was assessed as an increase in aPTT. Amean peak aPTT prolongation of about 1.38 times baseline value (range 0.58 to 3.41)was observed following subcutaneous b.i.d. injections of 15 mg desirudin. Thrombintime (TT) frequently exceeds 200 seconds even at low plasma concentrations ofdesirudin, which renders this test unsuitable for routine monitoring of Iprivasktherapy. At therapeutic serum concentrations, desirudin has no effect on otherenzymes of the hemostatic system such as factors IXa, Xa, kallikrein, plasmin,tissue plasminogen activator, or activated protein C. In addition, it does not displayany effect on other serine proteases, such as the digestive enzymes trypsin, chymotrypsin,or on complement activation by the classical or alternative pathways.
Pharmacokinetic PropertiesPharmacokinetic parameters were calculated based on plasma concentration dataobtained by a nonspecific ELISA method that does not discriminate between nativedesirudin and its metabolites. It is not known if the metabolites are pharmacologicallyactive.
Absorption: The absorption of desirudin is complete when subcutaneouslyadministered at doses of 0.3 mg/kg or 0.5 mg/kg. Following subcutaneousadministration of single doses of 0.1 to 0.75 mg/kg, plasma concentrations of desirudinincreased to a maximum level (Cmax) between 1 and 3 hours. Both Cmax andarea-under-the-curve (AUC) values are dose proportional.
Mean Desirudin Concentrations and Changes in APTT After A Single 15 mg Subcutaneous Dose in 12 Healthy Subjects
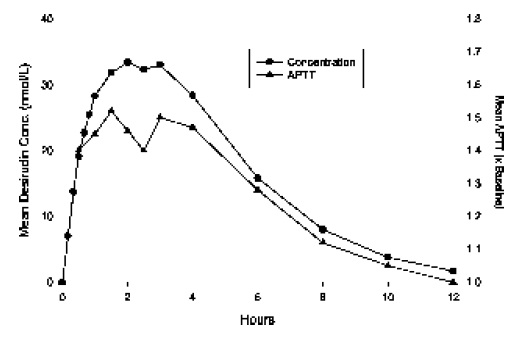
Distribution: The pharmacokinetic properties of desirudin following intravenous (IV) administration are well described by a two- or three- compartment disposition model. Desirudin is distributed in the extracellular space with a volume of distribution at steady state of 0.25 L/kg, independent of the dose. Desirudin binds specifically and directly to thrombin, forming an extremely tight, non-covalent complex with an inhibitionconstant of approximately 2.6 x 10-13 M. Thus, free or protein bound desirudinimmediately binds circulating thrombin. The pharmacological effect of desirudin is notmodified when co-administered with highly protein bound drugs (greater than 99%).Metabolism: Human and animal data suggest that desirudin is primarily eliminatedand metabolized by the kidney. The total urinary excretion of unchanged desirudinamounts to 40 to 50% of the administered dose. Metabolites lacking one or twoC-terminal amino acids constitute a minor proportion of the material recovered fromurine (less than 7%). There is no evidence for the presence of other metabolites. This indicatesthat desirudin is metabolized by stepwise degradation from the C-terminus probably catalyzed by carboxypeptidase(s) such as carboxypeptidase A, originating from thepancreas. Total clearance of desirudin is approximately 1.5 to 2.7 mL/min/kg followingeither subcutaneous or IV administration and is independent of dose. This clearancevalue is close to the glomerular filtration rate.Elimination: The elimination of desirudin from plasma is rapid after IVadministration, with approximately 90% of the dose disappearing from the plasmawithin 2 hours of the injection. Plasma concentrations of desirudin then decline with amean terminal elimination half-life of 2 to 3 hours. After subcutaneous administration,the mean terminal elimination half-life is also approximately 2 hours.Special Populations:Renal Insufficiency: In a pharmacokinetic study of renally impaired subjects,subjects with mild [creatinine clearance (CC) between 61 and 90 mL/min/1.73 m2body surface area], moderate (CC between 31 and 60 mL/min/1.73 m2 body surfacearea), and severe (CC below 31 mL/min/1.73 m2 body surface area) renalinsufficiency, were administered a single IV dose of 0.5, 0.25, or 0.125 mg/kg desirudin,respectively. This resulted in mean dose-normalized AUCeffect (AUC0-60th foraPTT prolongation) increases of approximately 3-, and 9-fold for the moderate andsevere renally impaired subjects, respectively, compared with healthy individuals. Insubjects with mild renal impairment, there was no increase in AUCeffect compared withhealthy individuals. In subjects with severe renal insufficiency, terminal eliminationhalf-lives were prolonged up to 12 hours compared with 2 to 4 hours in normalvolunteers or subjects with mild to moderate renal insufficiency (see WARNINGS).
Dose adjustments are recommended in certain circumstances in relation to the degree ofimpairment or degree of aPTT abnormality (see WARNINGS: Renal Insufficiency,PRECAUTIONS: Laboratory Tests, and DOSAGE AND ADMINISTRATION:Monitoring and Adjusting Therapy; Use in Renal Insufficiency).Hepatic Insufficiency: No pharmacokinetic studies have been conducted to investigatethe effects of Iprivask in hepatic insufficiency (see PRECAUTIONS, HepaticInsufficiency/Liver Injury and DOSAGE and ADMINISTRATION).
Age/Gender: The mean plasma clearance of desirudin in patientsgreater than or equal to 65 years ofage (n=12; 110 mL/min) is approximately 28% lower than in patients less than 65 years of age(n=8; 153 mL/min). Population pharmacokinetics conducted in 301 patients undergoingelective total hip replacement indicate that age or gender do not affect the systemicclearance of desirudin when renal creatinine clearance is considered. This drug issubstantially excreted by the kidney, and the risk of adverse events due to it may begreater in patients with impaired renal function. Because elderly patients are morelikely to have decreased renal function, care should be taken in dose selection, and itmay be useful to monitor renal function. Dosage adjustment in the case of moderate andsevere renal impairment is necessary. (See CLINICAL PHARMACOLOGY, SpecialPopulations, Renal Insufficiency, DOSAGE and ADMINISTRATION, Use in RenalInsufficiency).
CLINICAL TRIALSIprivask was evaluated in two controlled, randomized, multicenter, clinicalefficacy trials and a controlled, double-blind, dose-finding study. In the efficacystudies, Iprivask was compared to subcutaneously administered unfractionated heparinor enoxaparin sodium for the reduction of the risk of venous thromboembolic events(VTE) in patients undergoing total hip replacement surgery. In all studies Iprivask wasinitiated prior to surgery and continued for 8 to 12 days postoperatively (median duration10 days). Patients who received Iprivask had a lower incidence of VTE. The efficacystudies are described below.
In the first study, Iprivask 15 mg subcutaneously administered every 12 hours wascompared to unfractionated heparin 5000 IU subcutaneously administered every 8hours. A total of 445 patients were randomized in the study, 436 patients were treated,and 85 of the treated patients were excluded from efficacy analysis, mainly because ofno phlebography or inadequate reading of phlebography. Patients ranged in age from 34to 89 years (mean age 68.4 years) with 41.8% men and 58.2% women. All enrolledpatients were Caucasian. Iprivask significantly reduced the number of total VTEcompared to unfractionated heparin: Evaluable population: Iprivask, 13/174 (7.5%) vs.heparin, 41/177 (23.2%); p value less than0.001; Intent-to-Treat population: Iprivask13/225 (5.8%) vs. heparin 42/220 (19.1%);p value less than 0.0001.]. Significantly fewerpatients in the group treated with Iprivask experienced proximal DVT than thosepatients treated with heparin: Evaluable population: Iprivask 6/174 (3.4%) vs. heparin29/177 (16.4%); p value less than 0.001: Intent-to-Treat population: Iprivask 6/225 (2.7%)vs. heparin 30/220 (13.6%); p value less than 0.0001.
In a second study, Iprivask 15 mg subcutaneously administered every 12 hours wascompared to enoxaparin sodium 40 mg subcutaneously administered every 24 hours. Atotal of 2079 patients were randomized in the study, 2049 patients were treated, and508 of the treated patients were excluded from efficacy analysis mainly because of nophlebography or inadequate reading of phlebography. Patients ranged in age from 18 to90 years (mean age 68.5 years) with 41.7% men and 58.5% women. All enrolledpatients were Caucasian. In the both the evaluable patient population and theintent-to-treat population, patients who received Iprivask had a lower incidence of majorVTE, total VTE, and proximal DVT than did patients who received enoxaparin (seetable below).
| Efficacy of Iprivask in Hip Replacement Surgery Patients | Efficacy of Iprivask in Hip Replacement Surgery Patients | Efficacy of Iprivask in Hip Replacement Surgery Patients | Efficacy of Iprivask in Hip Replacement Surgery Patients |
| Dosing Regimen | Dosing Regimen | Dosing Regimen | Dosing Regimen |
| Iprivask a15 mg q12h SC | Enoxaparin40 mg qd | ||
| Evaluable HipReplacementSurgery Patients | n equal to 773 | n equal to 768 | |
| n (%) | n (%) | p Value | |
| Treatment failuresMajor VTEb,cTotal VTEeProximal DVT | 39 (4. 9) d | 61 (7.9) | p less than 0.02 |
| Treatment failures Major VTEb,c Total VTEe Proximal DVT | 145 (18.8) | 197 (25.7) | p less than 0.001 |
| Treatment failures Major VTEb,c Total VTEe Proximal DVT | 36 (4.5) | 59 (7.7) | p equal to 0.012 |
| Intent-to-Treat HipReplacementSurgery Patients | n equal to 1043 | n equal to 1036 | |
| Treatment FailuresMajor VTEbTotal VTEeProximal DVT | n (%) | n (%) | p Value |
| Treatment FailuresMajor VTEbTotal VTEeProximal DVT | 39 (3.7)f | 61 (5.9) | p equal to 0.024 |
| Treatment FailuresMajor VTEbTotal VTEeProximal DVT | 145 (13.9) | 199 (19.2) | p equal to 0.001 |
| Treatment FailuresMajor VTEbTotal VTEeProximal DVT | 36 (3.5) | 59 (5.7) | p equal to 0.012 |
INDICATIONS AND USAGEIprivask is indicated for the prophylaxis of deep vein thrombosis, which may leadto pulmonary embolism, in patients undergoing elective hip replacement surgery.
CONTRAINDICATIONS
Iprivask is contraindicated in patients with known hypersensitivity to natural orrecombinant hirudins, and in patients with active bleeding and/or irreversiblecoagulation disorders.
WARNINGS
Renal Insufficiency: Iprivask must be used with caution in patients with renalimpairment, particularly in those with moderate and severe renalimpairment (creatinine clearance ≤60 mL/min/1.73 m2 body surface area)(see CLINICAL PHARMACOLOGY, Special Populations, Renal Insufficiency).
Dose reductions by factors of three and nine are recommended for pat ients wi thmoderate and severe renal impairment respect ively (see DOSAGE ANDADMINISTRATION). In addition, daily aPTT and serum creatinine monitoringare recommended for patients with moderate or severe renal impairment (seePRECAUTIONS, Laboratory Tests).
Hemorrhagic Events: Iprivask is not intended for intramuscular injection aslocal hematoma formation may result.
Iprivask, like other anticoagulants, should be used with caution in patients withincreased risks of hemorrhage such as those with recent major surgery, organ biopsy orpuncture of a non-compressible vessel within the last month; a history ofhemorrhagic stroke, intracranial or intraocular bleeding including diabetic(hemorrhagic) retinopathy; recent ischemic stroke, severe uncontrolledhypertension, bacterial endocarditis, a known hemostatic disorder (congenital oracquired, e.g. hemophilia, liver disease) or a history of gastrointestinal orpulmonary bleeding within the past 3 months.
Bleeding can occur at any site during therapy with Iprivask. An unexplained fall inhematocrit or blood pressure should lead to a search for a bleeding site.Spinal/Epidural Anesthesia: As with other anticoagulants, there is a risk of neuraxialhematoma formation with the concurrent use of desirudin and spinal/epidural anesthesia,which has the potential to result in long term or permanent paralysis. The risk may begreater with the use of post-operative indwelling catheters or the concomitant use ofadditional drugs affecting hemostasis such as NSAIDs (Non-SteroidalAnti-Inflammatory Drugs), platelet inhibitors or other anticoagulants (see BoxedWARNING and PRECAUTIONS, Drug Interactions). The risk may also be increasedby traumatic or repeated neuraxial puncture.
To reduce the potential risk of bleeding associated with the concurrent use of desirudinand epidural or spinal anesthesia/analgesia, the pharmacokinetic profile of thedrug should be considered (see CLINICAL PHARMACOLOGY, Pharmacokinetic properties) when scheduling or using epidural or spinal anesthesia inproximity to desirudin administration. The physician should consider placement of thecatheter prior to initiating desirudin and removal of the catheter when the anticoagulanteffect of desirudin is low (see DOSAGE and ADMINISTRATION).
Should the physician decide to administer anticoagulation in the context ofepidural/spinal anesthesia, extreme vigilance and frequent monitoring must be exercisedto detect any signs and symptoms of neurological impairment such as midline back pain,sensory and motor deficits (numbness or weakness in lower limbs), bowel and/or bladderdysfunction. Patients should be instructed to inform their physician immediately if theyexperience any of the above signs or symptoms. If signs or symptoms of spinalhematoma are suspected, urgent diagnosis and treatment including spinal corddecompression should be initiated.
The physician should consider the potential benefit versus risk before neuraxialintervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis(see also WARNINGS, Hemorrhage, and PRECAUTIONS, Drug Interactions).Iprivask cannot be used interchangeably with other hirudins as they differ inmanufacturing process and specific biological activity (ATUs). Each of these medicineshas its own instructions for use.
PRECAUTIONS
Antibodies/Re-exposure: Antibodies have been reported in patients treated withhirudins. Potential for cross-sensitivity to hirudin products cannot be excluded. Irritativeskin reactions were observed in 9/322 volunteers exposed to Iprivask by subcutaneousinjection or IV bolus or infusion in single or multiple administrations of the drug.
Allergic events were reported in less than 2% of patients who were administered desirudin inPhase III clinical trials. Allergic events were reported in 1% of patients receivingunfractionated heparin and 1% of patients receiving enoxaparin. Hirudin-specificIgE evaluations may not be indicative of sensitivity to Iprivask as this test was notalways positive in the presence of symptoms. Very rarely, anti-hirudin antibodies havebeen detected upon re-exposure to desirudin. (See ADVERSE REACTIONS,Non-hemorrhagic Events, Allergic Reactions). Fatal anaphylactoid reactions have beenreported during hirudin therapy.
Hepatic Insufficiency/Liver Injury: No information is available about the useof desirudin in patients with hepatic insufficiency/liver injury. Although Iprivask isnot significantly metabolized by the liver, hepatic impairment or serious liver injury(e.g., liver cirrhosis) may alter the anticoagulant effect of Iprivask due to coagulationdefects secondary to reduced generation of vitamin K-dependent coagulation factors.Iprivask should be used with caution in these patients.
Laboratory Tests: Activated partial thromboplastin time (aPTT) should bemonitored daily in patients with increased risk of bleeding and/or renal impairment. Serum creatinine should be monitored daily in patients with renalimpairment. Peak aPTT should not exceed two times control. Should peak aPTTexceed this level, dose reduction is advised based on the degree of aPTTabnormality (see DOSAGE and ADMINISTRATION, Initial Dosage, Use in RenalInsufficiency). If necessary, therapy with desirudin should be interrupted until aPTTfalls to less than two times control, at which time treatment with desirudin canbe resumed at a reduced dose. (See Drug Interactions for information on use ofIprivask in conjunction with other drugs affecting coagulation). Thrombin time (TT) isnot a suitable test for routine monitoring of Iprivask therapy (see CLINICALPHARMACOLOGY, Mechanism of Action). Dose adjustments based on serumcreatinine may be necessary (see DOSAGE AND ADMINISTRATION, Use in RenalInsufficiency).
Drug Interactions: Any agent which may enhance the risk of hemorrhage should bediscontinued prior to initiation of desirudin therapy. These agents include medicationssuch as Dextran 40, systemic glucocorticoids, thrombolytics, and anticoagulants. Ifco-administration cannot be avoided, close clinical and laboratory moni toringshould be conducted. During prophylaxis of venous thromboembolism,concomitant treatment with heparins (unfractionated and low-molecular weightheparins) or dextrans is not recommended. The effects of desirudin and unfractionatedheparins on prolongation of aPTT are additive.
As with other anticoagulants, desirudin should be used with caution in conjunction withdrugs which affect platelet function. These medications include systemic salicylates,NSAIDS including ketorolac, acetylsalicylic acid, ticlopidine, dipyridamole,sulfinpyrazone, clopidogrel, abciximab and other glycoprotein IIb/IIIa antagonists (seePRECAUTIONS, Laboratory Tests).
Use in patients switching from oral anticoagulants to Iprivask or from Iprivask to oralanticoagulants. The concomitant administration of warfarin did not significantly affectthe pharmacokinetic effects of desirudin. When warfarin and desirudin wereco-administered, greater inhibition of hemostasis measured by activated partialthromboplastin time (aPTT), prothrombin time (PT), and international normalized ratio(INR) was observed. If a patient is switched from oral anticoagulants to Iprivasktherapy or from Iprivask to oral anticoagulants, the anticoagulant activity shouldcontinue to be closely monitored with appropriate methods. That activity should betaken into account in the evaluation of the overall coagulation status of the patientduring the switch.
Animal Pharmacology and Toxicology: General Toxicity Desirudin produced bleeding, local inflammation, and granulation at injection sitesin rat and dog toxicity studies. In a 28-day study in Rhesus monkeys, there was also evidence of subcutaneous bleeding and local inflammation at the injection sites. In addition, desirudin was immunogenic in dogs and formed antibody complexes resulting in prolonged half-life and accumulation. Desirudin showed sensitization potential in guinea pig immediate and delayed hypersensitivity models. Carcinogenesis, Mutagenesis, Impairment of Fertility. No long-term studies in animals have been performed to evaluate the carcinogenic potential of desirudin.
Desirudin was not genotoxic in the Ames test, the Chinese hamster lung cell (V79/HGPRT) forward mutation test or the rat micronucleus test. It was, however, equivocal in its genotoxic effect in Chinese hamster ovarian cell (CCL 61) chromosome aberration tests. Desirudin at subcutaneous doses up to 10mg/kg/day (about 2.7 times the recommended human dose based on body surface area) was found to have no effect on fertility andreproductive performance of male and female rats.
Pregnancy: Teratogenic Effects: Pregnancy Category C. Teratology studies have been performed in rats at subcutaneous doses in a range of 1 to 15 mg/kg/day (about 0.3 to 4 times the recommended human dose based on body surface area) and in rabbits at IV doses in a range of 0.6 to 6mg/kg/day (about 0.3 to 3 times the recommended human dose based on body surface area) and have revealed desirudin to be teratogenic.
Observed teratogenic findings were: omphalocele, asymmetric and fused sternebrae, edema, shortened hind limbs, etc. in rats; and spina bifida, malrotated hind limb, hydrocephaly, gastroschisis, etc. in rabbits. There are no adequate and well controlled studies in pregnant women. Iprivask should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: It is not known whether desirudin is excreted in human milk.Because many drugs are excreted in human milk, caution should be exercised whendesirudin is administered to a nursing woman.
Pediatric Use: Safety and effectiveness in pediatric patients have not been established.Geriatric Use: In three clinical studies of Iprivask, the percentage of patients greater than 65 years of age treated with 15 mg of Iprivask subcutaneously every 12 hours was 58.5%, while 20.8% were 75 years of age or older. Elderly patients treated with Iprivask had a reduction in the incidence of VTE similar to that observed in the younger patients, and a slightly lower incidence of VTE compared to those patients treated with heparin or enoxaparin.Regarding safety, in the clinical studies the incidence of hemorrhage (major or otherwise) in patients 65 years of age or older was similar to that in patients less than 65 years of age. In addition, the elderly had a similar incidence of total, treatment-related, or serious adverse events compared to those patients less than 65 years of age. Serious adverse events occurred more frequently in patients 75 years of age or older as compared to those less than 65 years of age. In general, 15 mg desirudin every 12 hours can be used safely in the geriatric population as in the population of patients less than 65 years of age so long as renal function is adequate (see CLINICAL PHARMACOLOGY, Special Populations, Renal Insufficiency, DOSAGE and ADMINISTRATION, Use in Renal Insufficiency).
ADVERSE REACTIONSIn the Phase II and III clinical studies, desirudin was administered to 2159 patients undergoing elective hip replacement surgery to determine the safety and efficacy of Iprivask in preventing VTE in this population. Below is the safety profile of the Iprivask 15 mg (q12h) regimen from these 5 multicenter clinical trials. Hemorrhagic Events: The following rates of hemorrhagic events have been reported during clinical trials.
| Dosing Regimen | Dosing Regimen | Dosing Regimen | |
| Iprivask | Heparin | Enoxaparin | |
| 15 mg q12h SC N=1561 n (%) | 5000 IU q8h SCN=501 n (%) | 40 mg QD SCN=1036n (%) | |
| Patients with Any Hemorrhagea | 464 (30) | 111 (22) | 341 (33) |
| Patients with Serious Hemorrhageb | 41 (3) | 15 (3) | 21 (2) |
| Patients with Major Hemorrhagec | 13 (less than 1) | 0 (0) | 2 (less than 1) |
| Iprivask | Heparin | Enoxaparin | |
| Body System (Preferred Term) | 15 mg q12h SC N=1561 n (%) | 5000 IU q8h SC N=501n (%) | 40 mg QD SCN=1036n (%) |
| Injection Site Mass | 56 (4) | 32 (6) | 7 (less than 1) |
| Wound Secretion | 59 (4) | 23 (5) | 34 (3) |
| Anemia | 51 (3) | 11 (2) | 37 (4) |
| Deep Thrombophlebitis | 24 (2) | 41 (8) | 22 (2) |
| Nausea | 24 (2) | 5 (less than 1) | 10 (less than 1) |
OVERDOSAGE
In case of overdose, most likely reflected in hemorrhagic complications or suggested by excessively high aPTT values, Iprivask therapy should be discontinued. Emergency procedures should be instituted as appropriate (for example, determination of aPTT and other coagulation levels, hemoglobin, the use of blood transfusion or plasma expanders).No specific antidote for Iprivask is available; however, the anticoagulant effect of desirudin is partially reversible using thrombin-rich plasma concentrates while aPTT levels can be reduced by the IV administration of 0.3 Mg/kg DDAVP (desmopressin). The clinical effectiveness of DDAVP in treating bleeding due to desirudin overdose has not been studied. In an open, pilot, dose-ascending study to assess safety, the highest dose of desirudin (40 mg q12h) caused excessive hemorrhage.
DOSAGE AND ADMINISTRATION
All patients should be evaluated for bleeding disorder risk before prophylactic administration of Iprivask (see PRECAUTIONS, Drug Interactions).Initial Dosage: In patients undergoing hip replacement surgery, the recommended dose of Iprivask is 15 mg every 12 hours administered by subcutaneous injection with the initial dose given up to 5 to 15 minutes prior to surgery, but after induction of regional block anesthesia, if used (see WARNINGS, Spinal/Epidural Anesthesia). Up to 12 days administration (average duration 9 to 12 days) of Iprivask has been well tolerated in controlled clinical trials.
| Degree of RenalInsufficiency | CreatinineClearance[mL/min/1.73m2 bodysurface area] | aPTT Monitoring and Dosing Instructions |
| Moderate | greater than or equal to 31 to 60 | Initiate therapy at 5 mg every 12 hours by subcutaneous injection.Monitor aPTT and serum creatinine at least daily. If aPTTexceeds 2 times control:1. Interrupt therapy until the value returns to less than 2 timescontrol.2. Resume therapy at a reduced dose guided by the initial degreeof aPTT abnormality |
| Severea | less than 31 | Initiate therapy at 1.7 mg every 12 hours by subcutaneousinjection. Monitor aPTT and serum creatinine at least daily. IfaPTT exceeds 2 times control:1. Interrupt therapy until the value returns to less than 2 timescontrol.2. Consider further dose reductions guided by the initial degreeof aPTT abnormality |
Administration:
Directions on Preparation
Use Iprivask before the expiration date given on the carton and container.
1. Reconstitution should be carried out under sterile conditions.
2. Remove plastic flip-top cap from IprivaskVial. Remove back cover of Vial Adapter package. Attach Vial Adapter to Vial by using the outer package to handle Adapter. Push Adapter down onto Vial until spike pierces rubber stopper and snaps into place. Discard Vial Adapter package.
3. Remove Syringe cap by twisting and pulling gently. Attach Syringe to Adapter on Vial by twisting. Slowly push plunger down to completely transfer solution into Vial. Leaving Syringe connected to Vial, gently swirl. Round tablet in Vial will dissolve within 10 seconds.
4. With Syringe still connected to Vial, turn Vial upside down and withdraw entire contents of Vial back into Syringe. Remove Syringe from Vial and hold it with plunger-end down.
5. You must use enclosed Eclipse™ needle. Attach Needle to Syringe by twisting. Pull pink lever down and uncap needle. You are ready to inject Iprivask. After injection, flip up pink lever to cover needle until it snaps into place. Dispose of the used Syringe in a Sharps® container.
Iprivask should not be mixed with other injections, solvents, or infusions. Iprivask is administered by subcutaneous injection. It must not be administered by intramuscular injection. Subcutaneous Injection Technique: Select a syringe with a 26 or 27 gauge needle which is approximately ½ inch in length for administration of Iprivask. Withdraw the entire reconstituted solution (15.75 mg desirudin/0.5 mL) into thesyringe and inject the total volume subcutaneously.
Patients should be sitting or lying down and Iprivask injection administered by deep subcutaneous injection. Administration should be alternated between the left and right anterolateral and left and right posterolateral thigh or abdominal wall. The whole length of the needle should be introduced into a skin fold held between the thumb and forefinger; the skin fold should be held throughout the injection. To minimize bruising, do not rub the injection site after completion of the injection.
HOW SUPPLIED
Iprivask [Desirudin for Injection] is supplied as a single dose (15.75 mg) lyophilized powder with an accompanying sterile, non-pyrogenic diluent [0.6 mL of Mannitol USP (3%) in Water for Injection].
Each Iprivask Vial contains 15.75 mg desirudin and the following inactive ingredients:1.31 mg anhydrous magnesium chloride USP, sodium hydroxide for injection USP.
Each carton of Iprivask [Desirudin for Injection] contains 10 individual doses of Iprivask,each in a separate tray.
Each tray of Iprivask
[Desirudin for Injection] contains:-
One (1) x 15.75 mg Single Dose Vial-
One (1) x 0.6 mL Prefilled syringe of Diluent-
One (1) Eclipse™ needle-
One (1) Vial Adapter
Each prefilled syringe of diluent contains 0.6 mL Mannitol USP (3% w/v) in Water for Injection provided for reconstitution of the desirudin lyophilized powder.
Distributed by:Canyon Pharmaceuticals11350 McCormick Rd., Ste. 400Hunt Valley, MD 21031 USA Rev 0 1 / 1 0
Storage: Protect from light.Unopened vials or prefilled syringes: Store at 25 degree Celsius (77 degree Fahrenheit); excursions permitted to 15–30 degree C (59-86 degree Fahrenheit). [See USP Controlled Room Temperature.]Once Iprivask is reconstituted it may be used for up to 24 hours, when stored as indicated above. After 24 hours discard the solution.
Keep this and all medicines out of the reach of children.
NDC 83055-111-12 Rx only
Directions for use: see insert.
Iprivask
(desirudin for injection) 15 mg*
FOR SUBCUTANEOUS INJECTION
Each Iprivask Vial contains
15.75 mg* desirudin to deliver 15 mg
Manufactured for and Distributed
by: Canyon Pharmaceuticals
Hunt Valley, MD 780-05329
Lot
Exp.
Rx only FOR SUBCUTANEOUS INJECTION NDC 83055-111-12
Iprivask
(desirudin for injection) 15 mg
Each vial of Iprivask also contains the following inactive ingredients:1.25 mg anhydrous magnesium chloride USP,sodium hydroxide for injection USP.
Store at 25 degree Centigrade (77 degree F): excursions permitted to 15-30 degree Centigrade (59-86 degree F).[See USP Controlled Room Temperature]. Protect from light.
Contents:
- One (1) 15.75 mg Single-DoseVial of Iprivask to deliver 15mg.- One (1) 0.6 mL Prefilled Syringe of Diluent- One (1) BD Eclipse Needle- One (1) Vial Adapter
Bar Code
750-05513
Iprivask is a licensed trademark of Canyon Pharmaceuticals Groupand its affillates. Manufactured for and Distributed by:Canyon PharmaceuticalsHunt Valley, MD 21031 USA
Lot
Exp.
Directions for use:See package insert
Rx only NDC 83055-121-01
Iprivask
(desirudin for injection) 15mg
Contents:Ten (10) pre-packaged trays each containing:- 15.75 mg Single-Dose Vial of Iprivask to dellver 15mg- 0.6 mL Prefilled Syringe of Diluent- BD Eclipse Needle- Vial Adapter
Rx only NDC 83055-131-01
Mannitol Injection 3% for use as aDiluent 0.6mL
contents: Mannitol USP (3% w/v) in Water for injection USP.
Directions for use: See Insert
Mfg. for and Distr. By: Canyon Pharmaceuticals,
780-05904 Hunt Valley MD



| IPRIVASK
desirudin kit |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021271 | 08/30/2010 | |
| Labeler - CANYON PHARMACEUTICALS, INC (167549414) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| WASSERBURGER ARZNEIMITTELWERK, GMBH | 326482247 | manufacture | |