KALETRA
-
lopinavir and
ritonavir capsule, liquid filled
Abbott Laboratories
----------
KALETRA®(lopinavir/ritonavir) capsules
DESCRIPTION
KALETRA (lopinavir/ritonavir) is a co-formulation of lopinavir and ritonavir. Lopinavir is an inhibitor of the HIV protease. As co-formulated in KALETRA, ritonavir inhibits the CYP3A-mediated metabolism of lopinavir, thereby providing increased plasma levels of lopinavir.
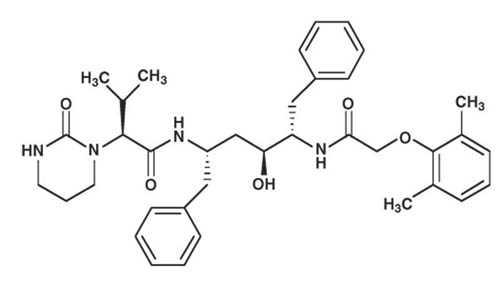
Lopinavir is chemically designated as [1S-[1R*,(R*), 3R*, 4R*]]-N-[4-[[(2,6-dimethylphenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-alpha-(1-methylethyl)-2-oxo-1(2H)-pyrimidineacetamide. Its molecular formula is C37H48N4O5, and its molecular weight is 628.80. Lopinavir has the following structural formula:
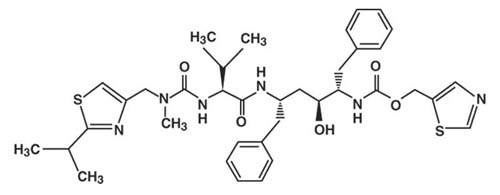
Ritonavir is chemically designated as 10-Hydroxy-2-methyl-5-(1-methylethyl)-1- [2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid, 5-thiazolylmethyl ester, [5S-(5R*,8R*,10R*,11R*)]. Its molecular formula is C37H48N6O5S2, and its molecular weight is 720.95. Ritonavir has the following structural formula:
Lopinavir is a white to light tan powder. It is freely soluble in methanol and ethanol, soluble in isopropanol and practically insoluble in water.
KALETRA capsules are available for oral administration in a strength of 133.3 mg lopinavir and 33.3 mg ritonavir with the following inactive ingredients: FD&C Yellow No. 6, gelatin, glycerin, oleic acid, polyoxyl 35 castor oil, propylene glycol, sorbitol special, titanium dioxide, and water.
KALETRA oral solution is available for oral administration as 80 mg lopinavir and 20 mg ritonavir per milliliter with the following inactive ingredients: Acesulfame potassium, alcohol, artificial cotton candy flavor, citric acid, glycerin, high fructose corn syrup, Magnasweet-110 flavor, menthol, natural & artificial vanilla flavor, peppermint oil, polyoxyl 40 hydrogenated castor oil, povidone, propylene glycol, saccharin sodium, sodium chloride, sodium citrate, and water.
KALETRA oral solution contains 42.4% alcohol (v/v).
CLINICAL PHARMACOLOGY
Microbiology
Mechanism of Action
Lopinavir, an inhibitor of the HIV protease, prevents cleavage of the Gag-Pol polyprotein, resulting in the production of immature, non-infectious viral particles.
Antiviral Activity
The antiviral activity of lopinavir against laboratory HIV strains and clinical HIV isolates was evaluated in acutely infected lymphoblastic cell lines and peripheral blood lymphocytes, respectively. In the absence of human serum, the mean 50% effective concentration (EC50) values of lopinavir against five different HIV-1 subtype B laboratory strains ranged from 10-27 nM (0.006-0.017 µg/mL, 1 µg/mL = 1.6 µM) and ranged from 4-11 nM (0.003-0.007 µg/mL) against several HIV-1 subtype B clinical isolates (n = 6). In the presence of 50% human serum, the mean EC50 values of lopinavir against these five HIV-1 laboratory strains ranged from 65-289 nM (0.04-0.18 µg/mL), representing a 7- to 11-fold attenuation. Combination antiviral drug activity studies with lopinavir in cell cultures demonstrated additive to antagonistic activity with nelfinavir and additive to synergistic activity with amprenavir, atazanavir, indinavir, saquinavir and tipranavir. The EC50 values of lopinavir against three different HIV-2 strains ranged from 12-180 nM (0.008-113 μg/mL).
Resistance
HIV-1 isolates with reduced susceptibility to lopinavir have been selected in cell culture. The presence of ritonavir does not appear to influence the selection of lopinavir-resistant viruses in cell culture.
The selection of resistance to KALETRA in antiretroviral treatment-naive patients has not yet been characterized. In a Phase III study of 653 antiretroviral treatment-naive patients (Study 863), plasma viral isolates from each patient on treatment with plasma HIV >400 copies/mL at Week 24, 32, 40 and/or 48 were analyzed. No evidence of resistance to KALETRA was observed in 37 evaluable KALETRA-treated patients (0%). The selection of resistance to KALETRA in antiretroviral treatment-naive pediatric patients (Study 940) appears to be consistent with that seen in adult patients (Study 863).
Resistance to KALETRA has been noted to emerge in patients treated with other protease inhibitors prior to KALETRA therapy. In Phase II studies of 227 antiretroviral treatment-naive and protease inhibitor experienced patients, isolates from 4 of 23 patients with quantifiable (>400 copies/mL) viral RNA following treatment with KALETRA for 12 to 100 weeks displayed significantly reduced susceptibility to lopinavir compared to the corresponding baseline viral isolates. Three of these patients had previously received treatment with a single protease inhibitor (indinavir, nelfinavir, or saquinavir) and one patient had received treatment with multiple protease inhibitors (indinavir, ritonavir, and saquinavir). All four of these patients had at least 4 mutations associated with protease inhibitor resistance immediately prior to KALETRA therapy. Following viral rebound, isolates from these patients all contained additional mutations, some of which are recognized to be associated with protease inhibitor resistance. However, there are insufficient data at this time to identify lopinavir-associated mutational patterns in isolates from patients on KALETRA therapy. The assessment of these mutational patterns is under study.
Cross-resistance – Preclinical Studies
Varying degrees of cross-resistance have been observed among HIV protease inhibitors. Little information is available on the cross-resistance of viruses that developed decreased susceptibility to lopinavir during KALETRA therapy.
The antiviral activity in cell culture of lopinavir against clinical isolates from patients previously treated with a single protease inhibitor was determined. Isolates that displayed >4-fold reduced susceptibility to nelfinavir (n = 13) and saquinavir (n = 4), displayed <4-fold reduced susceptibility to lopinavir. Isolates with >4-fold reduced susceptibility to indinavir (n = 16) and ritonavir (n = 3) displayed a mean of 5.7- and 8.3-fold reduced susceptibility to lopinavir, respectively. Isolates from patients previously treated with two or more protease inhibitors showed greater reductions in susceptibility to lopinavir, as described in the following paragraph.
Clinical Studies – Antiviral Activity of KALETRA in Patients with Previous Protease Inhibitor Therapies
The clinical relevance of reduced susceptibility in cell culture to lopinavir has been examined by assessing the virologic response to KALETRA therapy in treatment-experienced patients, with respect to baseline viral genotype in three studies and baseline viral phenotype in one study.
Virologic response to KALETRA has been shown to be affected by the presence of three or more of the following amino acid substitutions in protease at baseline: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, and I84V. Table 1 shows the 48-week virologic response (HIV RNA <400 copies/mL) according to the number of the above protease inhibitor resistance mutations at baseline in studies 888 and 765 (see INDICATIONS AND USAGE) and study 957 (see below).
| Number of protease inhibitor mutations at baseline1 | Study 888 (Single protease inhibitor-experienced2, NNRTI-naïve) n=130 | Study 765 (Single protease inhibitor-experienced 3, NNRTI-naïve) n=56 | Study 957 (Multiple protease inhibitor-experienced4, NNRTI-naïve) n=50 |
|
1 Substitutions considered in the analysis included L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T, and I84V. |
|||
| 0-2 | 76/103 (74%) | 34/45 (76%) | 19/20 (95%) |
| 3-5 | 13/26 (50%) | 8/11 (73%) | 18/26 (69%) |
| 6 or more | 0/1 (0%) | n/a | 1/4 (25%) |
Virologic response to KALETRA therapy with respect to phenotypic susceptibility to lopinavir at baseline was examined in Study 957. In this study 56 NNRTI-naïve patients with HIV RNA >1,000 copies/mL despite previous therapy with at least two protease inhibitors selected from indinavir, nelfinavir, ritonavir, and saquinavir were randomized to receive one of two doses of KALETRA in combination with efavirenz and nucleoside reverse transcriptase inhibitors (NRTIs). The EC50 values of lopinavir against the 56 baseline viral isolates ranged from 0.5- to 96-fold the wild-type EC50 value. Fifty-five percent (31/56) of these baseline isolates displayed > 4-fold reduced susceptibility to lopinavir. These 31 isolates had a median reduction in lopinavir susceptibility of 18-fold. Response to therapy by baseline lopinavir susceptibility is shown in Table 2.
| Lopinavir susceptibility2 at baseline | HIV RNA < 400 copies/mL (%) | HIV RNA < 50 copies/mL (%) |
|
1 Lopinavir susceptibility was determined by recombinant phenotypic technology performed by Virologic. 2 Fold change in susceptibility from wild type. |
||
| <10 fold | 25/27 (93%) | 22/27 (81%) |
| >10 and <40 fold | 11/15 (73%) | 9/15 (60%) |
| ≥40 fold | 2/8 (25%) | 2/8 (25%) |
Pharmacokinetics
The pharmacokinetic properties of lopinavir co-administered with ritonavir have been evaluated in healthy adult volunteers and in HIV-infected patients; no substantial differences were observed between the two groups. Lopinavir is essentially completely metabolized by CYP3A. Ritonavir inhibits the metabolism of lopinavir, thereby increasing the plasma levels of lopinavir. Across studies, administration of KALETRA 400/100 mg twice-daily yields mean steady-state lopinavir plasma concentrations 15- to 20-fold higher than those of ritonavir in HIV-infected patients. The plasma levels of ritonavir are less than 7% of those obtained after the ritonavir dose of 600 mg twice-daily. The in vitro antiviral EC50 of lopinavir is approximately 10-fold lower than that of ritonavir. Therefore, the antiviral activity of KALETRA is due to lopinavir.
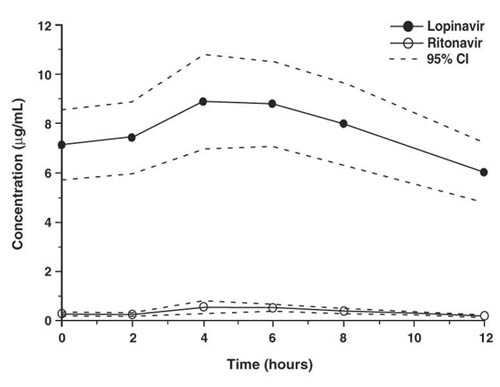
Figure 1 displays the mean steady-state plasma concentrations of lopinavir and ritonavir after KALETRA 400/100 mg twice-daily with food for 3 weeks from a pharmacokinetic study in HIV-infected adult subjects (n = 19).
Figure 1. Mean Steady-state Plasma Concentrations with 95% Confidence Intervals (CI) for HIV-Infected Adult Subjects (N = 19)
Absorption
In a pharmacokinetic study in HIV-positive subjects (n = 19), multiple dosing with 400/100 mg KALETRA twice-daily with food for 3 weeks produced a mean ± SD lopinavir peak plasma concentration (Cmax) of 9.8 ± 3.7 µg/mL, occurring approximately 4 hours after administration. The mean steady-state trough concentration prior to the morning dose was 7.1 ± 2.9 µg/mL and minimum concentration within a dosing interval was 5.5 ± 2.7 µg/mL. Lopinavir AUC over a 12 hour dosing interval averaged 92.6 ± 36.7 µg• h/mL. The absolute bioavailability of lopinavir co-formulated with ritonavir in humans has not been established. Under nonfasting conditions (500 kcal, 25% from fat), lopinavir concentrations were similar following administration of KALETRA co-formulated capsules and liquid. When administered under fasting conditions, both the mean AUC and Cmax of lopinavir were 22% lower for the KALETRA liquid relative to the capsule formulation.
Effects of Food on Oral Absorption
Administration of a single 400/100 mg dose of KALETRA capsules with a moderate fat meal (500-682 kcal, 23 to 25% calories from fat) was associated with a mean increase of 48 and 23% in lopinavir AUC and Cmax, respectively, relative to fasting. For KALETRA oral solution, the corresponding increases in lopinavir AUC and Cmax were 80 and 54%, respectively. Relative to fasting, administration of KALETRA with a high fat meal (872 kcal, 56% from fat) increased lopinavir AUC and Cmax by 97 and 43%, respectively, for capsules, and 130 and 56%, respectively, for oral solution. To enhance bioavailability and minimize pharmacokinetic variability KALETRA should be taken with food.
Distribution
At steady state, lopinavir is approximately 98-99% bound to plasma proteins. Lopinavir binds to both alpha-1-acid glycoprotein (AAG) and albumin; however, it has a higher affinity for AAG. At steady state, lopinavir protein binding remains constant over the range of observed concentrations after 400/100 mg KALETRA twice-daily, and is similar between healthy volunteers and HIV-positive patients.
Metabolism
In vitro experiments with human hepatic microsomes indicate that lopinavir primarily undergoes oxidative metabolism. Lopinavir is extensively metabolized by the hepatic cytochrome P450 system, almost exclusively by the CYP3A isozyme. Ritonavir is a potent CYP3A inhibitor which inhibits the metabolism of lopinavir, and therefore increases plasma levels of lopinavir. A 14C-lopinavir study in humans showed that 89% of the plasma radioactivity after a single 400/100 mg KALETRA dose was due to parent drug. At least 13 lopinavir oxidative metabolites have been identified in man. Ritonavir has been shown to induce metabolic enzymes, resulting in the induction of its own metabolism. Pre-dose lopinavir concentrations decline with time during multiple dosing, stabilizing after approximately 10 to 16 days.
Elimination
Following a 400/100 mg 14C-lopinavir/ritonavir dose, approximately 10.4 ± 2.3% and 82.6 ± 2.5% of an administered dose of 14C-lopinavir can be accounted for in urine and feces, respectively, after 8 days. Unchanged lopinavir accounted for approximately 2.2 and 19.8% of the administered dose in urine and feces, respectively. After multiple dosing, less than 3% of the lopinavir dose is excreted unchanged in the urine. The apparent oral clearance (CL/F) of lopinavir is 5.98 ± 5.75 L/hr (mean ± SD, n = 19).
Once Daily Dosing
The pharmacokinetics of once daily KALETRA have been evaluated in HIV-infected subjects naïve to antiretroviral treatment. KALETRA 800/200 mg was administered in combination with emtricitabine 200 mg and tenofovir DF 300 mg as part of a once daily regimen. Multiple dosing of 800/200 mg KALETRA once-daily for 4 weeks with food (n = 24) produced a mean ± SD lopinavir peak plasma concentration (Cmax) of 11.8 ± 3.7 µg/mL, occurring approximately 6 hours after administration. The mean steady-state lopinavir trough concentration prior to the morning dose was 3.2 ± 2.1 µg/mL and minimum concentration within a dosing interval was 1.7 ± 1.6 µg/mL. Lopinavir AUC over a 24 hour dosing interval averaged 154.1 ± 61.4 µg• h/mL.
Special Populations
Gender, Race and Age
Lopinavir pharmacokinetics have not been studied in elderly patients. No gender related pharmacokinetic differences have been observed in adult patients. No clinically important pharmacokinetic differences due to race have been identified.
Pediatric Patients
The pharmacokinetics of KALETRA 300/75 mg/m2 twice-daily and 230/57.5 mg/m2 twice-daily have been studied in a total of 53 pediatric patients, ranging in age from 6 months to 12 years. The 230/57.5 mg/m2 twice-daily regimen without nevirapine and the 300/75 mg/m2 twice-daily regimen with nevirapine provided lopinavir plasma concentrations similar to those obtained in adult patients receiving the 400/100 mg twice-daily regimen (without nevirapine). KALETRA once-daily has not been evaluated in pediatric patients.
The mean steady-state lopinavir AUC, Cmax, and Cmin were 72.6 ± 31.1 µg•h/mL, 8.2 ± 2.9 and 3.4 ± 2.1 µg/mL, respectively after KALETRA 230/57.5 mg/m2 twice-daily without nevirapine (n = 12), and were 85.8 ± 36.9 µg• h/mL, 10.0 ± 3.3 and 3.6 ± 3.5 µg/mL, respectively, after 300/75 mg/m2 twice-daily with nevirapine (n = 12). The nevirapine regimen was 7 mg/kg twice-daily (6 months to 8 years) or 4 mg/kg twice-daily (> 8 years).
Renal Insufficiency
Lopinavir pharmacokinetics have not been studied in patients with renal insufficiency; however, since the renal clearance of lopinavir is negligible, a decrease in total body clearance is not expected in patients with renal insufficiency.
Hepatic Impairment
Lopinavir is principally metabolized and eliminated by the liver. Multiple dosing of KALETRA 400/100 mg twice-daily to HIV and HCV co-infected patients with mild to moderate hepatic impairment (n = 12) resulted in a 30% increase in lopinavir AUC and 20% increase in Cmax compared to HIV-infected subjects with normal hepatic function (n = 12). Additionally, the plasma protein binding of lopinavir was statistically significantly lower in both mild and moderate hepatic impairment compared to controls (99.09 vs. 99.31%, respectively). Caution should be exercised when administering KALETRA to subjects with hepatic impairment. KALETRA has not been studied in patients with severe hepatic impairment (see PRECAUTIONS ).
Drug-drug Interactions
See also CONTRAINDICATIONS , WARNINGS and PRECAUTIONS – Drug Interactions.
KALETRA is an inhibitor of the P450 isoform CYP3A in vitro. Co-administration of KALETRA and drugs primarily metabolized by CYP3A may result in increased plasma concentrations of the other drug, which could increase or prolong its therapeutic and adverse effects (see CONTRAINDICATIONS).
KALETRA does not inhibit CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 or CYP1A2 at clinically relevant concentrations.
KALETRA has been shown in vivo to induce its own metabolism and to increase the biotransformation of some drugs metabolized by cytochrome P450 enzymes and by glucuronidation.
KALETRA is metabolized by CYP3A. Drugs that induce CYP3A activity would be expected to increase the clearance of lopinavir, resulting in lowered plasma concentrations of lopinavir. Although not noted with concurrent ketoconazole, co-administration of KALETRA and other drugs that inhibit CYP3A may increase lopinavir plasma concentrations.
Drug interaction studies were performed with KALETRA and other drugs likely to be co-administered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of co-administration of KALETRA on the AUC, Cmax and Cmin are summarized in Table 3 (effect of other drugs on lopinavir) and Table 4 (effect of KALETRA on other drugs). The effects of other drugs on ritonavir are not shown since they generally correlate with those observed with lopinavir (if lopinavir concentrations are decreased, ritonavir concentrations are decreased) unless otherwise indicated in the table footnotes. For information regarding clinical recommendations, see Table 11 in PRECAUTIONS .
| Co-administered Drug | Dose of Co-administered Drug (mg) | Dose of KALETRA (mg) | n | Ratio (in combination with Co-administered drug-/alone) of Lopinavir Pharmacokinetic Parameters (90% CI); No Effect = 1.00 | |||
| Cmax | AUC | Cmin | |||||
|
All interaction studies conducted in healthy, HIV-negative subjects unless otherwise indicated. 1 The pharmacokinetics of ritonavir are unaffected by concurrent efavirenz. 2 Data extracted from the fosamprenavir package insert. 3 Study conducted in HIV-positive adult subjects. 4 Study conducted in HIV-positive pediatric subjects ranging in age from 6 months to 12 years. 5 Titrated to 800/200 BID as 533/133 BID x 1 d, 667/167 BID x 1 d, then 800/200 BID x 7 d, compared to 400/100 BID x 10 days alone. 6 Titrated to 400/400 BID as 400/200 BID x 1 d, 400/300 BID x 1 d, then 400/400 BID x 7 d, compared to 400/100 BID x 10 days alone. 7 Data extracted from the tenofovir package insert. 8 Intensive PK analysis. 9 Drug levels obtained at 8-16 hrs post-dose. * Parallel group design; n for KALETRA + co-administered drug, n for KALETRA alone. † NC = No change. |
|||||||
| Amprenavir | 750 BID, 10 d | 400/100 BID, 21 d | 12 | 0.72 (0.65, 0.79) | 0.62 (0.56, 0.70) | 0.43 (0.34, 0.56) |
|
| Atorvastatin | 20 QD, 4 d | 400/100 BID, 14 d | 12 | 0.90 (0.78, 1.06) | 0.90 (0.79, 1.02) | 0.92 (0.78, 1.10) |
|
| Efavirenz1 | 600 QHS, 9 d | 400/100 BID, 9 d | 11, 7* | 0.97 (0.78, 1.22) | 0.81 (0.64, 1.03) | 0.61 (0.38, 0.97) |
|
| Fosamprenavir2 | 700 BID plus ritonavir 100 BID, 14 d | 400/100 BID, 14 d | 18 | 1.30 (0.85, 1.47) | 1.37 (0.80, 1.55) | 1.52 (0.72, 1.82) |
|
| Ketoconazole | 200 single dose | 400/100 BID, 16 d | 12 | 0.89 (0.80, 0.99) | 0.87 (0.75, 1.00) | 0.75 (0.55, 1.00) |
|
| Nelfinavir | 1000 BID, 10 d | 400/100 BID, 21 d | 13 | 0.79 (0.70, 0.89) | 0.73 (0.63, 0.85) | 0.62 (0.49, 0.78) |
|
| Nevirapine | 200 BID, steady-state (> 1 yr)3 | 400/100 BID, steady-state | 22, 19* | 0.81 (0.62, 1.05) | 0.73 (0.53, 0.98) | 0.49 (0.28, 0.74) |
|
| 7 mg/kg or 4 mg/kg QD, 2 wk; BID 1 wk4 | (> 1 yr) 300/75 mg/m2 BID, 3 wk | 12, 15* | 0.86 (0.64, 1.16) | 0.78 (0.56, 1.09) | 0.45 (0.25, 0.81) |
||
| Omeprazole | 40 QD, 5 d | 400/100 tablet BID, 10 d | 12 | 1.08 (0.99, 1.17) | 1.07 (0.99, 1.15) | 1.03 (0.90, 1.18) |
|
| 40 QD, 5 d | 800/200 tablet QD, 10 d | 12 | 0.94 (0.88, 1.00) | 0.92 (0.86, 0.99) | 0.71 (0.57, 0.89) |
||
| Pravastatin | 20 QD, 4 d | 400/100 BID, 14 d | 12 | 0.98 (0.89, 1.08) | 0.95 (0.85, 1.05) | 0.88 (0.77, 1.02) |
|
| Rifabutin | 150 QD, 10 d | 400/100 BID, 20 d | 14 | 1.08 (0.97, 1.19) | 1.17 (1.04, 1.31) | 1.20 (0.96, 1.65) |
|
| Ranitidine | 150 single dose | 400/100 tablet BID, 10 d | 12 | 0.99 (0.95, 1.03) | 0.97 (0.93, 1.01) | 0.90 (0.85, 0.95) |
|
| 150 single dose | 800/200 tablet QD, 10 d | 10 | 0.97 (0.95, 1.00) | 0.95 (0.91, 0.99) | 0.82 (0.74, 0.91) |
||
| Rifampin | 600 QD, 10 d | 400/100 BID, 20 d | 22 | 0.45 (0.40, 0.51) | 0.25 (0.21, 0.29) | 0.01 (0.01, 0.02) |
|
| 600 QD, 14 d | 800/200 BID, 9 d5 | 10 | 1.02 (0.85, 1.23) | 0.84 (0.64, 1.10) | 0.43 (0.19, 0.96) |
||
| 600 QD, 14 d | 400/400 BID, 9 d6 | 9 | 0.93 (0.81, 1.07) | 0.98 (0.81, 1.17) | 1.03 (0.68, 1.56) |
||
| Co-administration of KALETRA and rifampin is not recommended. (See PRECAUTIONS – Table 10 and Table 11) |
|||||||
| Ritonavir3 | 100 BID, 3-4 wk | 400/100 BID, 3-4 wk | 8, 21* | 1.28 (0.94, 1.76) | 1.46 (1.04, 2.06) | 2.16 (1.29, 3.62) |
|
| Tenofovir7 | 300 mg QD, 14 d | 400/100 BID, 14 d | 24 | NC† | NC† | NC† | |
| Tipranavir/ritonavir3 | 500/200 mg BID (28 doses) | 400/100 capsule BID (27 doses) | 21 69 | 0.53 (0.40, 0.69)8 | 0.45 (0.32, 0.63)8 | 0.30 (0.17, 0.51)8
0.48 (0.40, 0.58)9 |
|
| Co-administered Drug | Dose of Co-administered Drug (mg) | Dose of KALETRA (mg) | n | Ratio (in combination with KALETRA/alone) of Co-administered Drug Pharmacokinetic Parameters (90% CI); No Effect = 1.00 | ||
| Cmax | AUC | Cmin | ||||
|
All interaction studies conducted in healthy, HIV-negative subjects unless otherwise indicated. 1 Ratio of parameters for amprenavir, indinavir, nelfinavir, and saquinavir are not normalized for dose. 2 Desipramine is a probe substrate for assessing effects on CYP2D6-mediated metabolism. 3 Data extracted from the fosamprenavir package insert. 4 Effect on the dose-normalized sum of rifabutin parent and 25-O-desacetyl rifabutin active metabolite. 5 Data extracted from the rosuvastatin package insert and results presented at the 2007 Conference on Retroviruses and Opportunistic Infection (Hoody, et al, abstract L-107, poster #564). 6 Data extracted from the tenofovir package insert. * Parallel group design; n for KALETRA + co-administered drug, n for co-administered drug alone. N/A = Not available. † NC = No change. |
||||||
| Amprenavir1 | 750 BID, 10 d combo vs. 1200 BID, 14 d alone | 400/100 BID, 21 d | 11 | 1.12 (0.91, 1.39) | 1.72 (1.41, 2.09) | 4.57 (3.51, 5.95) |
| Atorvastatin | 20 QD, 4 d | 400/100 BID, 14 d | 12 | 4.67 (3.35, 6.51) | 5.88 (4.69, 7.37) | 2.28 (1.91, 2.71) |
| Desipramine2 | 100 single dose | 400/100 BID, 10 d | 15 | 0.91 (0.84, 0.97) | 1.05 (0.96, 1.16) | N/A |
| Efavirenz | 600 QHS, 9 d | 400/100 BID, 9 d | 11, 12* | 0.91 (0.72, 1.15) | 0.84 (0.62, 1.15) | 0.84 (0.58, 1.20) |
| Ethinyl Estradiol | 35 µg QD, 21 d (Ortho Novum®) | 400/100 BID, 14 d | 12 | 0.59 (0.52, 0.66) | 0.58 (0.54, 0.62) | 0.42 (0.36, 0.49) |
| Fosamprenavir3 | 700 BID plus ritonavir 100 BID, 14 d | 400/100 BID, 14 d | 18 | 0.42 (0.30, 0.58) | 0.37 (0.28, 0.49) | 0.35 (0.27, 0.46) |
| Indinavir1 | 600 BID, 10 d combo nonfasting vs. 800 TID, 5 d alone fasting | 400/100 BID, 15 d | 13 | 0.71 (0.63, 0.81) | 0.91 (0.75, 1.10) | 3.47 (2.60, 4.64) |
| Ketoconazole | 200 single dose | 400/100 BID, 16 d | 12 | 1.13 (0.91, 1.40) | 3.04 (2.44, 3.79) | N/A |
| Methadone | 5 single dose | 400/100 BID, 10 d | 11 | 0.55 (0.48, 0.64) | 0.47 (0.42, 0.53) | N/A |
| Nelfinavir1 | 1000 BID, 10 d combo vs. 1250 BID, 14 d alone | 400/100 BID, 21 d | 13 | 0.93 (0.82, 1.05) | 1.07 (0.95, 1.19) | 1.86 (1.57, 2.22) |
| M8 metabolite | 2.36 (1.91, 2.91) | 3.46 (2.78, 4.31) | 7.49 (5.85, 9.58) |
|||
| Nevirapine | 200 QD, 14 d; BID, 6 d | 400/100 BID, 20 d | 5, 6* | 1.05 (0.72, 1.52) | 1.08 (0.72, 1.64) | 1.15 (0.71, 1.86) |
| Norethindrone | 1 QD, 21 d (Ortho Novum®) | 400/100 BID, 14 d | 12 | 0.84 (0.75, 0.94) | 0.83 (0.73, 0.94) | 0.68 (0.54, 0.85) |
| Pravastatin | 20 QD, 4 d | 400/100 BID, 14 d | 12 | 1.26 (0.87, 1.83) | 1.33 (0.91, 1.94) | N/A |
| Rifabutin | 150 QD, 10 d; combo vs. 300 QD, 10 d; alone | 400/100 BID, 10 d | 12 | 2.12 (1.89, 2.38) | 3.03 (2.79, 3.30) | 4.90 (3.18, 5.76) |
| 25-O-desacetyl rifabutin | 23.6 (13.7, 25.3) | 47.5 (29.3, 51.8) | 94.9 (74.0, 122) |
|||
| Rifabutin + 25-O-desacetyl rifabutin4 | 3.46 (3.07, 3.91) | 5.73 (5.08, 6.46) | 9.53 (7.56, 12.01) |
|||
| Rosuvastatin5 | 20 mg QD, 7 d | 400/100 tablet BID, 7 d | 15 | 4.66 (3.4, 6.4) | 2.08 (1.66, 2.6) | 1.04 (0.9, 1.2) |
| Saquinavir1 | 800 BID, 10 d combo vs. 1200 TID, 5 d alone, | 400/100 BID, 15 d | 14 | 6.34 (5.32, 7.55) | 9.62 (8.05, 11.49) | 16.74 (13.73, 20.42) |
| 1200 BID, 5 d combo vs. 1200 TID, 5 d alone | 400/100 BID, 20 d | 10 | 6.44 (5.59, 7.41) | 9.91 (8.28, 11.86) | 16.54 (10.91, 25.08) |
|
| Tenofovir6 | 300 mg QD, 14 d | 400/100 BID, 14 d | 24 | NC† | 1.32 (1.26, 1.38) | 1.51 (1.32, 1.66) |
INDICATIONS AND USAGE
KALETRA is indicated in combination with other antiretroviral agents for the treatment of HIV-infection.
Once-daily administration of KALETRA is not recommended in therapy-experienced patients.
When initiating treatment with KALETRA in therapy-naïve patients, it should be noted that the incidence of diarrhea was greater for KALETRA once-daily compared to KALETRA twice-daily in Study 418 (57% vs. 35% - events of all grades and probably or possibly related to drug; 16% vs. 5% - events of at least moderate severity and probably or possibly related to drug) (see CLINICAL PHARMACOLOGY, ADVERSE REACTIONS, and DOSAGE AND ADMINISTRATION ).
Description of Clinical Studies
Patients Without Prior Antiretroviral Therapy
Study 863: KALETRA twice-daily + stavudine + lamivudine compared to nelfinavir three-times-daily + stavudine + lamivudine
Study 863 is an ongoing, randomized, double-blind, multicenter trial comparing treatment with KALETRA (400/100 mg twice-daily) plus stavudine and lamivudine versus nelfinavir (750 mg three-times-daily) plus stavudine and lamivudine in 653 antiretroviral treatment-naïve patients. Patients had a mean age of 38 years (range: 19 to 84), 57% were Caucasian, and 80% were male. Mean baseline CD4 cell count was 259 cells/mm3 (range: 2 to 949 cells/mm3) and mean baseline plasma HIV-1 RNA was 4.9 log10 copies/mL (range: 2.6 to 6.8 log10 copies/mL).
Treatment response and outcomes of randomized treatment are presented in Table 5.
| Outcome | KALETRA+d4T+3TC (N = 326) | Nelfinavir+d4T+3TC (N = 327) |
|
1 Patients achieved and maintained confirmed HIV RNA < 400 copies/mL through Week 48. 2 Includes confirmed viral rebound and failure to achieve confirmed < 400 copies/mL through Week 48. 3 Includes lost to follow-up, patient's withdrawal, non-compliance, protocol violation and other reasons. Overall discontinuation through Week 48, including patients who discontinued subsequent to virologic failure, was 17% in the KALETRA arm and 24% in the nelfinavir arm. |
||
| Responder1 | 75% | 62% |
| Virologic failure2
Rebound Never suppressed through Week 48 | 9% 7% 2% | 25% 15% 9% |
| Death | 2% | 1% |
| Discontinued due to adverse event | 4% | 4% |
| Discontinued for other reasons3 | 10% | 8% |
Through 48 weeks of therapy, there was a statistically significantly higher proportion of patients in the KALETRA arm compared to the nelfinavir arm with HIV RNA < 400 copies/mL (75% vs. 62%, respectively) and HIV RNA < 50 copies/mL (67% vs. 52%, respectively). Treatment response by baseline HIV RNA level subgroups is presented in Table 6.
| Baseline Viral Load (HIV-1 RNA copies/mL) | KALETRA +d4T+3TC | Nelfinavir +d4T+3TC | ||||
| < 400 copies/mL 1 | < 50 copies/mL 2 | n | < 400 copies/mL 1 | < 50 copies/mL 2 | n | |
|
1 Patients achieved and maintained confirmed HIV RNA < 400 copies/mL through Week 48. 2 Patients achieved HIV RNA < 50 copies/mL at Week 48. |
||||||
| < 30,000 | 74% | 71% | 82 | 79% | 72% | 87 |
| ≥ 30,000 to < 100,000 | 81% | 73% | 79 | 67% | 54% | 79 |
| ≥ 100,000 to < 250,000 | 75% | 64% | 83 | 60% | 47% | 72 |
| ≥ 250,000 | 72% | 60% | 82 | 44% | 33% | 89 |
Through 48 weeks of therapy, the mean increase from baseline in CD4 cell count was 207 cells/mm3 for the KALETRA arm and 195 cells/mm3 for the nelfinavir arm.
Study 418: KALETRA once-daily + tenofovir DF + emtricitabine compared to KALETRA twice-daily + tenofovir DF + emtricitabine
Study 418 is an ongoing, randomized, open-label, multicenter trial comparing treatment with KALETRA 800/200 mg once-daily plus tenofovir DF and emtricitabine versus KALETRA 400/100 mg twice-daily plus tenofovir DF and emtricitabine in 190 antiretroviral treatment-naïve patients. Patients had a mean age of 39 years (range: 19 to 75), 54% were Caucasian, and 78% were male. Mean baseline CD4 cell count was 260 cells/mm3 (range: 3 to 1006 cells/mm3) and mean baseline plasma HIV-1 RNA was 4.8 log10 copies/mL (range: 2.6 to 6.4 log10 copies/mL).
Treatment response and outcomes of randomized treatment are presented in Table 7.
| Outcome | KALETRA QD
+ TDF + FTC (n = 115) | KALETRA BID
+ TDF + FTC (n = 75) |
|
1 Patients achieved and maintained confirmed HIV RNA < 50 copies/mL through Week 48. 2 Includes confirmed viral rebound and failure to achieve confirmed < 50 copies/mL through Week 48. 3 Includes lost to follow-up, patient’s withdrawal, non-compliance, protocol violation and other reasons. |
||
| Responder1 | 71% | 65% |
| Virologic failure2
Rebound Never suppressed through Week 48 | 10% 6% 3% | 9% 5% 4% |
| Death | 0% | 1% |
| Discontinued due to an adverse event | 12% | 7% |
| Discontinued for other reasons3 | 7% | 17% |
Through 48 weeks of therapy, 71% in the KALETRA once-daily arm and 65% in the KALETRA twice-daily arm achieved and maintained HIV RNA < 50 copies/mL (95% confidence interval for the difference, -7.6% to 19.5%). Mean CD4 cell count increases at Week 48 were 185 cells/mm3 for the KALETRA once-daily arm and 196 cells/mm3 for the KALETRA twice-daily arm.
Patients with Prior Antiretroviral Therapy
Study 888: KALETRA twice-daily + nevirapine + NRTIs compared to investigator-selected protease inhibitor(s) + nevirapine + NRTIs
Study 888 is a randomized, open-label, multicenter trial comparing treatment with KALETRA (400/100 mg twice-daily) plus nevirapine and nucleoside reverse transcriptase inhibitors versus investigator-selected protease inhibitor(s) plus nevirapine and nucleoside reverse transcriptase inhibitors in 288 single protease inhibitor-experienced, non-nucleoside reverse transcriptase inhibitor (NNRTI)-naïve patients. Patients had a mean age of 40 years (range: 18 to 74), 68% were Caucasian, and 86% were male. Mean baseline CD4 cell count was 322 cells/mm3 (range: 10 to 1059 cells/mm3) and mean baseline plasma HIV-1 RNA was 4.1 log10 copies/mL (range: 2.6 to 6.0 log10 copies/mL).
Treatment response and outcomes of randomized treatment through Week 48 are presented in Table 8.
| Outcome | KALETRA + nevirapine + NRTIs (n = 148) | Investigator-Selected Protease Inhibitor(s) + nevirapine + NRTIs (n = 140) |
|
1 Patients achieved and maintained confirmed HIV RNA < 400 copies/mL through Week 48. 2 Includes confirmed viral rebound and failure to achieve confirmed < 400 copies/mL through Week 48. 3 Includes lost to follow-up, patient's withdrawal, non-compliance, protocol violation and other reasons. |
||
| Responder1 | 57% | 33% |
| Virologic Failure2 | 24% | 41% |
| Rebound Never suppressed through Week 48 | 11% 13% | 19% 23% |
| Death | 1% | 2% |
| Discontinued due to adverse events | 5% | 11% |
| Discontinued for other reasons3 | 14% | 13% |
Through 48 weeks of therapy, there was a statistically significantly higher proportion of patients in the KALETRA arm compared to the investigator-selected protease inhibitor(s) arm with HIV RNA < 400 copies/mL (57% vs. 33%, respectively).
Through 48 weeks of therapy, the mean increase from baseline in CD4 cell count was 111 cells/mm3 for the KALETRA arm and 112 cells/mm3 for the investigator-selected protease inhibitor(s) arm.
Other Studies
Study 720: KALETRA twice-daily + stavudine + lamivudine
Study 765: KALETRA twice-daily + nevirapine + NRTIs
Study 720 (patients without prior antiretroviral therapy) and study 765 (patients with prior protease inhibitor therapy) are randomized, blinded, multi-center trials evaluating treatment with KALETRA at up to three dose levels (200/100 mg twice-daily [720 only], 400/100 mg twice-daily, and 400/200 mg twice-daily). In Study 720, all patients switched to 400/100 mg twice-daily between Weeks 48-72. Patients in study 720 had a mean age of 35 years, 70% were Caucasian, and 96% were male, while patients in study 765 had a mean age of 40 years, 73% were Caucasian, and 90% were male. Mean (range) baseline CD4 cell counts for patients in study 720 and study 765 were 338 (3-918) and 372 (72-807) cells/mm3, respectively. Mean (range) baseline plasma HIV-1 RNA levels for patients in study 720 and study 765 were 4.9 (3.3 to 6.3) and 4.0 (2.9 to 5.8) log10 copies/mL, respectively.
Through 360 weeks of treatment in study 720, the proportion of patients with HIV RNA < 400 (< 50) copies/mL was 61% (59%) [n = 100]. Among patients completing 360 weeks of treatment with CD4 cell count measurements [n=60], the mean (median) increase in CD4 cell count was 501 (457) cells/mm3. Thirty-nine patients (39%) discontinued the study, including 15 (15%) discontinuations due to adverse events and 1 (1%) death. Through 144 weeks of treatment in study 765, the proportion of patients with HIV RNA < 400 (< 50) copies/mL was 54% (50%) [n = 70], and the corresponding mean increase in CD4 cell count was 212 cells/mm3. Twenty-seven patients (39%) discontinued the study, including 9 (13%) discontinuations secondary to adverse events and 2 (3%) deaths.
CONTRAINDICATIONS
KALETRA is contraindicated in patients with known hypersensitivity to any of its ingredients, including ritonavir.
Co-administration of KALETRA is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events. These drugs are listed in Table 9.
| Drug Class | Drugs Within Class That Are Contraindicated With KALETRA** |
| Alpha 1- Adrenoreceptor antagonist | Alfuzosin |
| Antihistamines | Astemizole, Terfenadine |
| Ergot Derivatives | Dihydroergotamine, Ergonovine, Ergotamine, Methylergonovine |
| GI motility agent | Cisapride |
| Neuroleptic | Pimozide |
| PDE5 enzyme inhibitor | Sildenafil* (Revatio®) when used for the treatment of pulmonary arterial hypertension |
| Sedative/Hypnotics | Midazolam, Triazolam |
| * see WARNINGS – Drug Interactions and PRECAUTIONS, Table 11. Established and Other Potentially Significant Drug Interactions: Alteration in Dose or Regimen May Be Recommended Based on Drug Interaction Studies or Predicted Interaction for co-administration of sildenafil in patients with erectile dysfunction. ** For additional information for these contraindicated drugs, see also PRECAUTIONS – Table 10. Drugs That Should Not Be Co-administered With KALETRA. |
|
WARNINGS
ALERT: Find out about medicines that should NOT be taken with KALETRA. This statement is included on the product's bottle label.
Drug Interactions
KALETRA is an inhibitor of the P450 isoform CYP3A. Co-administration of KALETRA and drugs primarily metabolized by CYP3A may result in increased plasma concentrations of the other drug that could increase or prolong its therapeutic and adverse effects (see Pharmacokinetics – Drug-drug Interactions , CONTRAINDICATIONS –Table 9: Drugs That Are Contraindicated With KALETRA, PRECAUTIONS – Table 10: Drugs That Should Not Be Co-administered With KALETRA and Table 11: Established and Other Potentially Significant Drug Interactions).
Particular caution should be used when prescribing sildenafil, tadalafil, or vardenafil in patients receiving KALETRA. Co-administration of KALETRA with these drugs is expected to substantially increase their concentrations and may result in an increase in associated adverse events including hypotension, syncope, visual changes and prolonged erection (see PRECAUTIONS – Drug Interactions and the complete prescribing information for sildenafil, tadalafil, and vardenafil).
Concomitant use of KALETRA with lovastatin or simvastatin is not recommended. Caution should be exercised if HIV protease inhibitors, including KALETRA, are used concurrently with other HMG-CoA reductase inhibitors that are also metabolized by the CYP3A4 pathway (e.g., atorvastatin). The risk of myopathy, including rhabdomyolysis may be increased when HIV protease inhibitors, including KALETRA, are used in combination with these drugs.
Concomitant use of KALETRA and St. John's wort (hypericum perforatum), or products containing St. John's wort, is not recommended. Co-administration of protease inhibitors, including KALETRA, with St. John's wort is expected to substantially decrease protease inhibitor concentrations and may result in sub-optimal levels of lopinavir and lead to loss of virologic response and possible resistance to lopinavir or to the class of protease inhibitors.
A drug interaction study in healthy subjects has shown that ritonavir significantly increases plasma fluticasone propionate exposures, resulting in significantly decreased serum cortisol concentrations. Concomitant use of KALETRA and fluticasone propionate is expected to produce the same effects. Systemic corticosteroid effects including Cushing’s syndrome and adrenal suppression have been reported during postmarketing use in patients receiving ritonavir and inhaled or intranasally administered fluticasone propionate. Therefore, coadministration of fluticasone propionate and KALETRA is not recommended unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effects (see PRECAUTIONS – Drug Interactions ).
Pancreatitis
Pancreatitis has been observed in patients receiving KALETRA therapy, including those who developed marked triglyceride elevations. In some cases, fatalities have been observed. Although a causal relationship to KALETRA has not been established, marked triglyceride elevations is a risk factor for development of pancreatitis (see PRECAUTIONS – Lipid Elevations). Patients with advanced HIV disease may be at increased risk of elevated triglycerides and pancreatitis, and patients with a history of pancreatitis may be at increased risk for recurrence during KALETRA therapy.
Pancreatitis should be considered if clinical symptoms (nausea, vomiting, abdominal pain) or abnormalities in laboratory values (such as increased serum lipase or amylase values) suggestive of pancreatitis should occur. Patients who exhibit these signs or symptoms should be evaluated and KALETRA and/or other antiretroviral therapy should be suspended as clinically appropriate.
Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
PRECAUTIONS
Hepatic Impairment and Toxicity
KALETRA is principally metabolized by the liver; therefore, caution should be exercised when administering this drug to patients with hepatic impairment, because lopinavir concentrations may be increased (see CLINICAL PHARMACOLOGY – Hepatic Impairment).Patients with underlying hepatitis B or C or marked elevations in transaminases prior to treatment may be at increased risk for developing further transaminase elevations or hepatic decompensation. There have been postmarketing reports of hepatic dysfunction, including some fatalities. These have generally occurred in patients with advanced HIV disease taking multiple concomitant medications in the setting of underlying chronic hepatitis or cirrhosis. A causal relationship with KALETRA therapy has not been established. Increased AST/ALT monitoring should be considered in these patients, especially during the first several months of KALETRA treatment.
Resistance/Cross-resistance
Various degrees of cross-resistance among protease inhibitors have been observed. The effect of KALETRA therapy on the efficacy of subsequently administered protease inhibitors is under investigation (see Microbiology).
Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis, in patients with hemophilia type A and B treated with protease inhibitors. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced. A causal relationship between protease inhibitor therapy and these events has not been established.
Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and" cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Lipid Elevations
Treatment with KALETRA has resulted in large increases in the concentration of total cholesterol and triglycerides (see ADVERSE REACTIONS – Table 16). Triglyceride and cholesterol testing should be performed prior to initiating KALETRA therapy and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate. See PRECAUTIONS – Table 11: Established and Other Potentially Significant Drug Interactions for additional information on potential drug interactions with KALETRA and HMG-CoA reductase inhibitors.
Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including KALETRA. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis carinii pneumonia, or tuberculosis) which may necessitate further evaluation and treatment.
Information for Patients
A statement to patients and health care providers is included on the product's bottle label: "ALERT: Find out about medicines that should NOT be taken with KALETRA." A Patient Package Insert (PPI) for KALETRA is available for patient information.
Patients should be told that sustained decreases in plasma HIV RNA have been associated with a reduced risk of progression to AIDS and death. Patients should remain under the care of a physician while using KALETRA. Patients should be advised to take KALETRA and other concomitant antiretroviral therapy every day as prescribed. KALETRA must always be used in combination with other antiretroviral drugs. Patients should not alter the dose or discontinue therapy without consulting with their doctor. If a dose of KALETRA is missed patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped the patient should not double the next dose.
Patients should be informed that KALETRA is not a cure for HIV infection and that they may continue to develop opportunistic infections and other complications associated with HIV disease. The long-term effects of KALETRA are unknown at this time. Patients should be told that there are currently no data demonstrating that therapy with KALETRA can reduce the risk of transmitting HIV to others through sexual contact.
KALETRA may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John's wort.
Patients taking didanosine should take didanosine one hour before or two hours after KALETRA.
Patients receiving sildenafil, tadalafil, or vardenafil should be advised that they may be at an increased risk of associated adverse events including hypotension, visual changes, and sustained erection, and should promptly report any symptoms to their doctor. Concomitant use of sildenafil with KALETRA is contraindicated in patients with pulmonary arterial hypertension (PAH).
Patients receiving estrogen-based hormonal contraceptives should be instructed that additional or alternate contraceptive measures should be used during therapy with KALETRA.
KALETRA should be taken with food to enhance absorption.
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long term health effects of these conditions are not known at this time.
If the patient is taking or before the patient begins using Serevent® (salmeterol) and KALETRA, they should talk to their doctor about problems these two medications may cause when taken together. The doctor may choose not to keep someone on Serevent® (salmeterol). If the patient is taking or before the patient begins taking Advair® (salmeterol in combination with fluticasone propionate) and KALETRA, they should talk to their doctor about problems these two medications may cause when taken together. The doctor may choose not to keep someone on Advair® (salmeterol in combination with fluticasone propionate).
Drug Interactions
KALETRA is an inhibitor of CYP3A (cytochrome P450 3A) both in vitro and in vivo. Co-administration of KALETRA and drugs primarily metabolized by CYP3A (e.g., dihydropyridine calcium channel blockers, HMG-CoA reductase inhibitors, immunosuppressants and PDE5 inhibitors) may result in increased plasma concentrations of the other drugs that could increase or prolong their therapeutic and adverse effects (see Table 11. Established and Other Potentially Significant Drug Interactions). Agents that are extensively metabolized by CYP3A and have high first pass metabolism appear to be the most susceptible to large increases in AUC (> 3-fold) when co-administered with KALETRA.
KALETRA does not inhibit CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 or CYP1A2 at clinically relevant concentrations.
KALETRA has been shown in vivo to induce its own metabolism and to increase the biotransformation of some drugs metabolized by cytochrome P450 enzymes and by glucuronidation.
KALETRA is metabolized by CYP3A. Co-administration of KALETRA and drugs that induce CYP3A may decrease lopinavir plasma concentrations and reduce its therapeutic effect (see Table 11. Established and Other Potentially Significant Drug Interactions). Although not noted with concurrent ketoconazole, co-administration of KALETRA and other drugs that inhibit CYP3A may increase lopinavir plasma concentrations.
Drugs that are contraindicated and not recommended for co-administration with KALETRA are included in Table 10. Drugs That Should Not Be Co-administered With KALETRA. These recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious events or loss of efficacy.
| Drug Class: Drug Name | Clinical Comment |
| Alpha 1- Adrenoreceptor antagonist: Alfuzosin | CONTRAINDICATED due to potentially increased alfuzosin concentrations that can result in hypotension. |
| Antihistamines: astemizole, terfenadine | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| Antimycobacterial: rifampin | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors or other co-administered antiretroviral agents. (See Table 10 for further details). |
| Ergot Derivatives: dihydroergotamine, ergonovine, ergotamine, methylergonovine | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues. |
| GI Motility Agent: cisapride | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| Herbal Products: St. John's wort (hypericum perforatum) | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors. |
| HMG-CoA Reductase Inhibitors: lovastatin, simvastatin | Potential for serious reactions such as risk of myopathy including rhabdomyolysis. |
| Neuroleptic: pimozide | CONTRAINDICATED due to the potential for serious and/or life-threatening reactions such as cardiac arrhythmias. |
| PDE5 enzyme inhibitor: Sildenafil* (Revatio®) | CONTRAINDICATED as a safe and effective dose has not been established when used with KALETRA. There is an increased potential for sildenafil-associated adverse events, including visual abnormalities, hypotension, prolonged erection, and syncope. |
| Sedative/Hypnotics: midazolam, triazolam | CONTRAINDICATED due to potential for serious and/or life-threatening reactions such as prolonged or increased sedation or respiratory depression. |
| * see WARNINGS – Drug Interactions and PRECAUTIONS – Table 11. Established and Other Potentially Significant Drug Interactions: Alteration in Dose or Regimen May Be Recommended Based on Drug Interaction Studies or Predicted Interaction for co-administration of sildenafil in patients with erectile dysfunction. | |
| Concomitant Drug Class:
Drug Name | Effect on Concentration of lopinavir or Concomitant Drug | Clinical Comment |
|
* See CLINICAL PHARMACOLOGY for Magnitude of Interaction – Table 3 and Table 4. |
||
| HIV-Antiviral Agents | ||
| Non-nucleoside Reverse Transcriptase Inhibitors: efavirenz*, nevirapine* | ↓ Lopinavir | A dose increase of KALETRA to 533/133 mg (4 capsules or 6.5 mL) twice daily taken with food is recommended when used in combination with efavirenz or nevirapine (see DOSAGE AND ADMINISTRATION ). KALETRA should not be administered once-daily in combination with efavirenz or nevirapine. NOTE: Efavirenz and nevirapine induce the activity of CYP3A and thus have the potential to decrease plasma concentrations of other protease inhibitors when used in combination with KALETRA. |
| Non-nucleoside Reverse Transcriptase Inhibitor: delavirdine | ↑ Lopinavir | Appropriate doses of the combination with respect to safety and efficacy have not been established. |
| Nucleoside Reverse Transcriptase Inhibitor: didanosine | It is recommended that didanosine be administered on an empty stomach; therefore, didanosine should be given one hour before or two hours after KALETRA (given with food). | |
| Nucleoside Reverse Transcriptase Inhibitor: tenofovir | ↑ Tenofovir | KALETRA increases tenofovir concentrations. The mechanism of this interaction is unknown. Patients receiving KALETRA and tenofovir should be monitored for tenofovir-associated adverse events. |
| HIV-Protease Inhibitor: amprenavir* | ↑ Amprenavir (amprenavir 750 mg BID + KALETRA produces ↑ AUC, similar Cmax, ↑ Cmin, relative to amprenavir 1200 mg BID ↓ Lopinavir | Increase KALETRA dose to 533/133 mg and decrease amprenavir dose to amprenavir 750 mg BID, when co-administered. (see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY – Table 3 and Table 4). KALETRA should not be administered once-daily in combination with amprenavir. Appropriate doses of the combination of fosamprenavir and KALETRA have not been established. |
| HIV-Protease Inhibitor: fosamprenavir | ↓ Amprenavir ↓ Lopinavir | An increased rate of adverse events has been observed with co-administration of these medications. Appropriate doses of the combinations with respect to safety and efficacy have not been established. |
| HIV-Protease Inhibitor: indinavir* | ↑ Indinavir (indinavir 600 mg BID + KALETRA produces similar AUC, ↓ Cmax, ↑ Cmin relative to indinavir 800 mg TID | Decrease indinavir dose to 600 mg BID, when co-administered with KALETRA 400/100 mg BID (see CLINICAL PHARMACOLOGY – Table 4). KALETRA once-daily has not been studied in combination with indinavir. |
| HIV-Protease Inhibitor: nelfinavir* | ↑ Nelfinavir (nelfinavir 1000 mg BID + KALETRA produces similar AUC, similar Cmax, ↑ Cmin relative to nelfinavir 1250 mg BID) ↑ M8 metabolite of nelfinavir ↓ Lopinavir | Increase KALETRA dose to 533/133 mg and decrease nelfinavir dose to 1000 mg BID, when co-administered (see DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY – Table 3 and Table 4). KALETRA should not be administered once-daily in combination with nelfinavir. |
| HIV-Protease Inhibitor: saquinavir* | ↑ Saquinavir (saquinavir 800 mg BID + KALETRA produces ↑ AUC, ↑ Cmax, ↑ Cmin relative to saquinavir 1200 mg TID) | Decrease saquinavir dose to 800 mg BID, when co-administered with KALETRA 400/100 mg BID (see CLINICAL PHARMACOLOGY – Table 4). KALETRA once-daily has not been studied in combination with saquinavir. |
| HIV-Protease Inhibitor: tipranavir | ↓ Lopinavir AUC and Cmin | KALETRA should not be administered with tipranavir (500 mg twice-daily) co-administered with ritonavir (200 mg twice-daily). |
| HIV-Protease Inhibitor: ritonavir* | ↑ Lopinavir | Appropriate doses of additional ritonavir in combination with KALETRA with respect to safety and efficacy have not been established. |
| Other Agents | ||
| Antiarrhythmics: amiodarone, bepridil, lidocaine (systemic), and quinidine | ↑ Antiarrhythmics | Caution is warranted and therapeutic concentration monitoring is recommended for antiarrhythmics when co-administered with KALETRA, if available. |
| Anticoagulant: warfarin | Concentrations of warfarin may be affected. It is recommended that INR (international normalized ratio) be monitored. | |
| Anticonvulsants: carbamazepine, phenobarbital, phenytoin | ↓ Lopinavir | Use with caution. KALETRA may be less effective due to decreased lopinavir plasma concentrations in patients taking these agents concomitantly. KALETRA should not be administered once-daily in combination with carbamazepine, phenobarbital, or phenytoin. |
| Antidepressant: trazodone | ↑ Trazodone | Concomitant use of trazodone and KALETRA may increase concentrations of trazodone. Adverse events of nausea, dizziness, hypotension and syncope have been observed following co-administration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as ritonavir, the combination should be used with caution and a lower dose of trazodone should be considered. |
| Anti-infective: clarithromycin | ↑ Clarithromycin | For patients with renal impairment, the following dosage adjustments should be considered:
No dose adjustment for patients with normal renal function is necessary. |
| Antifungals: ketoconazole*, itraconazole, voriconazole | ↑ Ketoconazole ↑ Itraconazole Voriconazole effect is unknown. | High doses of ketoconazole or itraconazole (> 200 mg/day) are not recommended. Co-administration of voriconazole with KALETRA has not been studied. However, administration of voriconazole with ritonavir 400 mg every 12 hours decreased voriconazole steady-state AUC by an average of 82%. The effect of lower ritonavir doses on voriconazole is not known at this time. Until data are available, voriconazole should not be administered to patients receiving KALETRA. |
| Anti-gout colchicine | ↑ Colchicine | Patients with renal or hepatic impairment should not be given colchicine with KALETRA. Treatment of gout flares - co-administration of colchicine in patients on KALETRA: 0.6 mg (1 tablet) x 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days. Prophylaxis of gout flares - co-administration of colchicine in patients on KALETRA: If the original colchicine regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day. If the original colchicine regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day. Treatment of familial Mediterranean fever (FMF) - co-administration of colchicine in patients on KALETRA: Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day). |
| Antimycobacterial: rifabutin* | ↑ Rifabutin and rifabutin metabolite | Dosage reduction of rifabutin by at least 75% of the usual dose of 300 mg/day is recommended (i.e., a maximum dose of 150 mg every other day or three times per week). Increased monitoring for adverse events is warranted in patients receiving the combination. Further dosage reduction of rifabutin may be necessary. |
| Antimycobacterial: Rifampin | ↓ Lopinavir | May lead to loss of virologic response and possible resistance to KALETRA or to the class of protease inhibitors or other co-administered antiretroviral agents. A study evaluated combination of rifampin 600 mg QD, with KALETRA 800/200 mg BID or KALETRA 400/100 mg + ritonavir 300 mg BID. Pharmacokinetic and safety results from this study do not allow for a dose recommendation. Nine subjects (28%) experienced a ≥ grade 2 increase in ALT/AST, of which seven (21%) prematurely discontinued study per protocol. Based on the study design, it is not possible to determine whether the frequency or magnitude of the ALT/AST elevations observed is higher than what would be seen with rifampin alone. (See CLINICAL PHARMACOLOGY for Magnitude of Interaction – Table 3). |
| Antiparasitic: atovaquone | ↓ Atovaquone | Clinical significance is unknown; however, increase in atovaquone doses may be needed. |
| Calcium Channel Blockers, Dihydropyridine: e.g., felodipine, nifedipine, nicardipine | ↑ Dihydropyridine calcium channel blockers | Caution is warranted and clinical monitoring of patients is recommended. |
| Corticosteroid: Dexamethasone | ↓ Lopinavir | Use with caution. KALETRA may be less effective due to decreased lopinavir plasma concentrations in patients taking these agents concomitantly. |
| Disulfiram/metronidazole | KALETRA oral solution contains alcohol, which can produce disulfiram-like reactions when co-administered with disulfiram or other drugs that produce this reaction (e.g., metronidazole). | |
| Endothelin receptor antagonists: bosentan | ↑ Bosentan | Co-administration of bosentan in patients on KALETRA:
In patients who have been receiving KALETRA for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability. Co-administration of KALETRA in patients on bosentan: Discontinue use of bosentan at least 36 hours prior to initiation of KALETRA. After at least 10 days following the initiation of KALETRA, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability. |
| PDE5 inhibitors: sildenafil, tadalafil, vardenafil | ↑ Sildenafil ↑ Tadalafil ↑ Vardenafil | Particular caution should be used when prescribing sildenafil, tadalafil, or vardenafil in patients receiving KALETRA. Co-administration of KALETRA with these drugs is expected to substantially increase their concentrations and may result in an increase in PDE5 inhibitor associated adverse reactions including hypotension, syncope, visual changes, and prolonged erection. Use of PDE5 inhibitors for pulmonary arterial hypertension (PAH): Sildenafil (Revatio®) is contraindicated when used for the treatment of pulmonary arterial hypertension (PAH) because a safe and effective dose has not been established when used with KALETRA [see Contraindications (4)]. The following dose adjustments are recommended for use of tadalafil (AdcircaTM) with KALETRA: Co-administration of ADCIRCA in patients on KALETRA: In patients receiving KALETRA for at least one week, start ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Co-administration of KALETRA in patients on ADCIRCA: Avoid use of ADCIRCA during the initiation of KALETRA. Stop ADCIRCA at least 24 hours prior to starting KALETRA. After at least one week following the initiation of KALETRA, resume ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability. Use of PDE5 inhibitors for the treatment of erectile dysfunction: It is recommended not to exceed the following doses:
|
| HMG-CoA Reductase Inhibitors: atorvastatin* rosuvastatin | ↑ atorvastatin ↑ rosuvastatin | Use lowest possible dose of atorvastatin or rosuvastatin with careful monitoring, or consider other HMG-CoA reductase inhibitors such as pravastatin or fluvastatin in combination with KALETRA. |
| Immunosuppressants: cyclosporine, tacrolimus, rapamycin | ↑ Immunosuppressants | Therapeutic concentration monitoring is recommended for immunosuppressant agents when co-administered with KALETRA. |
| Inhaled Steroid: fluticasone | ↑ Fluticasone | Concomitant use of fluticasone propionate and KALETRA may increase plasma concentrations of fluticasone propionate, resulting in significantly reduced serum cortisol concentrations. Co-administration of fluticasone propionate and KALETRA is not recommended unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effect (see WARNINGS ) |
| Long-acting beta-adrenoceptor agonist: salmeterol | ↑ salmeterol | Concurrent administration of salmeterol and KALETRA is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia. |
| Narcotic Analgesic: Methadone* | ↓ Methadone | Dosage of methadone may need to be increased when co-administered with KALETRA. |
| Oral Contraceptive: ethinyl estradiol* | ↓ Ethinyl estradiol | Because contraceptive steroid concentrations may be altered when KALETRA is co-administered with oral contraceptives or with the contraceptive patch, alternative methods of nonhormonal contraception are recommended. |
Other Drugs
Drug interaction studies reveal no clinically significant interaction between KALETRA and desipramine (CYP2D6 probe), pravastatin, stavudine, lamivudine, omeprazole or ranitidine.
Based on known metabolic profiles, clinically significant drug interactions are not expected between KALETRA and fluvastatin, dapsone, trimethoprim/sulfamethoxazole, azithromycin, erythromycin, or fluconazole.
Zidovudine and Abacavir: KALETRA induces glucuronidation; therefore, KALETRA has the potential to reduce zidovudine and abacavir plasma concentrations. The clinical significance of this potential interaction is unknown.
Carcinogenesis, Mutagenesis and Impairment of Fertility
Lopinavir/ritonavir combination was evaluated for carcinogenic potential by oral gavage administration to mice and rats for up to 104 weeks. Results showed an increase in the incidence of benign hepatocellular adenomas and an increase in the combined incidence of hepatocellular adenomas plus carcinoma in both males and females in mice and males in rats at doses that produced approximately 1.6-2.2 times (mice) and 0.5 times (rats) the human exposure (based on AUC0-24hr measurement) at the recommended dose of 400/100 mg KALETRA twice-daily. Administration of lopinavir/ritonavir did not cause a statistically significant increase in the incidence of any other benign or malignant neoplasm in mice or rats.
Carcinogenicity studies in mice and rats have been carried out on ritonavir. In male mice, there was a dose dependent increase in the incidence of both adenomas and combined adenomas and carcinomas in the liver. Based on AUC measurements, the exposure at the high dose was approximately 4-fold for males that of the exposure in humans with the recommended therapeutic dose (400/100 mg KALETRA twice-daily). There were no carcinogenic effects seen in females at the dosages tested. The exposure at the high dose was approximately 9-fold for the females that of the exposure in humans. There were no carcinogenic effects in rats. In this study, the exposure at the high dose was approximately 0.7-fold that of the exposure in humans with the 400/100 mg KALETRA twice-daily regimen. Based on the exposures achieved in the animal studies, the significance of the observed effects is not known. However, neither lopinavir nor ritonavir was found to be mutagenic or clastogenic in a battery of in vitro and in vivo assays including the Ames bacterial reverse mutation assay using S. typhimurium and E. coli, the mouse lymphoma assay, the mouse micronucleus test and chromosomal aberration assays in human lymphocytes.
Lopinavir in combination with ritonavir at a 2:1 ratio produced no effects on fertility in male and female rats at levels of 10/5, 30/15 or 100/50 mg/kg/day. Based on AUC measurements, the exposures in rats at the high doses were approximately 0.7-fold for lopinavir and 1.8-fold for ritonavir of the exposures in humans at the recommended therapeutic dose (400/100 mg twice-daily).
Pregnancy
Pregnancy Category C
No treatment-related malformations were observed when lopinavir in combination with ritonavir was administered to pregnant rats or rabbits. Embryonic and fetal developmental toxicities (early resorption, decreased fetal viability, decreased fetal body weight, increased incidence of skeletal variations and skeletal ossification delays) occurred in rats at a maternally toxic dosage. Based on AUC measurements, the drug exposures in rats at the toxic doses were approximately 0.7-fold for lopinavir and 1.8-fold for ritonavir for males and females that of the exposures in humans at the recommended therapeutic dose (400/100 mg twice-daily). In a peri- and postnatal study in rats, a developmental toxicity (a decrease in survival in pups between birth and postnatal Day 21) occurred.
No embryonic and fetal developmental toxicities were observed in rabbits at a maternally toxic dosage. Based on AUC measurements, the drug exposures in rabbits at the toxic doses were approximately 0.6-fold for lopinavir and 1.0-fold for ritonavir that of the exposures in humans at the recommended therapeutic dose (400/100 mg twice-daily). There are, however, no adequate and well-controlled studies in pregnant women. KALETRA should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Antiretroviral Pregnancy Registry
To monitor maternal-fetal outcomes of pregnant women exposed to KALETRA, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
Nursing Mothers
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV. Studies in rats have demonstrated that lopinavir is secreted in milk. It is not known whether lopinavir is secreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breast-feed if they are receiving KALETRA.
Geriatric Use
Clinical studies of KALETRA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, appropriate caution should be exercised in the administration and monitoring of KALETRA in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pediatric Use
The safety and pharmacokinetic profiles of KALETRA in pediatric patients below the age of 6 months have not been established. In HIV-infected patients age 6 months to 12 years, the adverse event profile seen during a clinical trial was similar to that for adult patients. The evaluation of the antiviral activity of KALETRA in pediatric patients in clinical trials is ongoing.
Study 940 is an ongoing open-label, multicenter trial evaluating the pharmacokinetic profile, tolerability, safety and efficacy of KALETRA oral solution containing lopinavir 80 mg/mL and ritonavir 20 mg/mL in 100 antiretroviral naive (44%) and experienced (56%) pediatric patients. All patients were non-nucleoside reverse transcriptase inhibitor naive. Patients were randomized to either 230 mg lopinavir/57.5 mg ritonavir per m2 or 300 mg lopinavir/75 mg ritonavir per m2. Naive patients also received lamivudine and stavudine. Experienced patients received nevirapine plus up to two nucleoside reverse transcriptase inhibitors.
Safety, efficacy and pharmacokinetic profiles of the two dose regimens were assessed after three weeks of therapy in each patient. After analysis of these data, all patients were continued on the 300 mg lopinavir/75 mg ritonavir per m2 dose. Patients had a mean age of 5 years (range 6 months to 12 years) with 14% less than 2 years. Mean baseline CD4 cell count was 838 cells/mm3 and mean baseline plasma HIV-1 RNA was 4.7 log10 copies/mL.
Through 48 weeks of therapy, the proportion of patients who achieved and sustained an HIV RNA < 400 copies/mL was 80% for antiretroviral naive patients and 71% for antiretroviral experienced patients. The mean increase from baseline in CD4 cell count was 404 cells/mm3 for antiretroviral naive and 284 cells/mm3 for antiretroviral experienced patients treated through 48 weeks. At 48 weeks, two patients (2%) had prematurely discontinued the study. One antiretroviral naive patient prematurely discontinued secondary to an adverse event attributed to KALETRA, while one antiretroviral experienced patient prematurely discontinued secondary to an HIV-related event.
Dose selection for patients 6 months to 12 years of age was based on the following results. The 230/57.5 mg/m2 twice-daily regimen without nevirapine and the 300/75 mg/m2 twice-daily regimen with nevirapine provided lopinavir plasma concentrations similar to those obtained in adult patients receiving the 400/100 mg twice-daily regimen (without nevirapine). KALETRA once-daily has not been evaluated in pediatric patients.
ADVERSE REACTIONS
Adults
Treatment-Emergent Adverse Events
KALETRA has been studied in 891 patients as combination therapy in Phase I/II and Phase III clinical trials. The most common adverse event associated with KALETRA therapy was diarrhea, which was generally of mild to moderate severity. Rates of discontinuation of randomized therapy due to adverse events were 5.8% in KALETRA-treated and 4.9% in nelfinavir-treated patients in Study 863. The incidence of diarrhea was greater for KALETRA once-daily compared to KALETRA twice-daily in Study 418 (see Table 12 and INDICATIONS AND USAGE).
Treatment-Emergent clinical adverse events of moderate or severe intensity in ≥ 2% of patients treated with combination therapy for up to 48 weeks (Phase III) and for up to 360 weeks (Phase I/II) are presented in Table 12. For other information regarding observed or potentially serious adverse events, please see WARNINGS and PRECAUTIONS.
| Study 863 (48 Weeks) | Study 418 (48 Weeks) | Study 720 (360 Weeks) |
|||
| KALETRA 400/100 mg BID + d4T + 3TC
(N=326) | Nelfinavir 750 mg TID + d4T + 3TC
(N=327) | KALETRA 800/200 mg QD + TDF + FTC
(N=115) | KALETRA 400/100 mg BID + TDF + FTC
(N=75) | KALETRA BID2 + d4T + 3TC
(N=100) |
|
|
1 Includes adverse events of possible, probable, or unknown relationship to study drug. 2 Includes adverse event data from dose group I (200/100 mg BID [N=16] and 400/100 mg BID [N=16]) and dose group II (400/100 mg BID [N=35] and 400/200 mg BID [N=33]). Within dosing groups, moderate to severe nausea of probable/possible relationship to KALETRA occurred at a higher rate in the 400/200 mg dose arm compared to the 400/100 mg dose arm in group II. |
|||||
| Body as a Whole | |||||
| Abdominal Pain | 4% | 3% | 3% | 3% | 11% |
| Asthenia | 4% | 3% | 0% | 0% | 9% |
| Headache | 2% | 2% | 3% | 3% | 6% |
| Cardiovascular System | |||||
| Vein distended | 0% | 0% | 0% | 0% | 3% |
| Digestive System | |||||
| Anorexia | 1% | <1% | <1% | 1% | 2% |
| Diarrhea | 16% | 17% | 16% | 5% | 28% |
| Dyspepsia | 2% | <1% | 0% | 1% | 6% |
| Flatulence | 2% | 1% | 2% | 1% | 4% |
| Nausea | 7% | 5% | 9% | 8% | 16% |
| Vomiting | 2% | 2% | 3% | 4% | 6% |
| Metabolic and Nutritional | |||||
| Weight Loss | 1% | <1% | 0% | 0% | 2% |
| Musculoskeletal | |||||
| Myalgia | 1% | 1% | 0% | 0% | 2% |
| Nervous System | |||||
| Depression | 1% | 2% | 1% | 0% | 0% |
| Insomnia | 2% | 1% | 0% | 0% | 3% |
| Libido decreased | <1% | <1% | 0% | 1% | 2% |
| Paresthesia | 1% | 1% | 0% | 0% | 2% |
| Respiratory | |||||
| Bronchitis | 0% | 0% | 0% | 0% | 2% |
| Skin and Appendages | |||||
| Rash | 1% | 2% | 1% | 0% | 5% |
| Urogenital | |||||
| Hypogonadism male | 0% | 0% | 0% | 0% | 2% |
| Study 888 (48 Weeks) | Study 9572 and Study 7653 (84-144 Weeks) | ||
| KALETRA 400/100 mg BID + NVP + NRTIs
(N=148) | Investigator-selected protease inhibitor(s) + NVP + NRTIs
(N=140) | KALETRA BID + NNRTI + NRTIs
(N=127) |
|
|
1 Includes adverse events of possible, probable, or unknown relationship to study drug. 2 Includes adverse event data from patients receiving 400/100 mg BID (n=29) or 533/133 mg BID (n=28) for 84 weeks. Patients receiving KALETRA in combination with NRTIs and efavirenz. 3 Includes adverse event data from patients receiving 400/100 mg BID (n=36) or 400/200 mg BID (n=34) for 144 weeks. Patients received KALETRA in combination with NRTIs and nevirapine. |
|||
| Body as a Whole | |||
| Abdominal Pain | 2% | 2% | 4% |
| Asthenia | 3% | 6% | 9% |
| Chills | 2% | 0% | 0% |
| Fever | 2% | 1% | 2% |
| Headache | 2% | 3% | 2% |
| Cardiovascular | |||
| Hypertension | 0% | 0% | 2% |
| Digestive System | |||
| Anorexia | 1% | 3% | 0% |
| Diarrhea | 7% | 9% | 23% |
| Dyspepsia | 1% | 1% | 2% |
| Dysphagia | 2% | 1% | 0% |
| Flatulence | 1% | 2% | 2% |
| Nausea | 7% | 16% | 5% |
| Vomiting | 4% | 12% | 2% |
| Metabolic and Nutritional | |||
| Weight loss | 0% | 1% | 3% |
| Musculoskeletal | |||
| Myalgia | 1% | 1% | 2% |
| Nervous System | |||
| Depression | 1% | 2% | 2% |
| Insomnia | 0% | 2% | 2% |
| Paresthesia | 1% | 0% | 2% |
| Skin and Appendages | |||
| Rash | 2% | 1% | 2% |
Treatment-emergent adverse events occurring in less than 2% of adult patients receiving KALETRA in all phase II/III clinical trials and considered at least possibly related or of unknown relationship to treatment with KALETRA and of at least moderate intensity are listed below by body system.
Body as a Whole
Allergic reaction, back pain, chest pain, chest pain substernal, cyst, drug interaction, drug level increased, face edema, flu syndrome, hypertrophy, infection bacterial, malaise, neoplasm, and viral infection.
Cardiovascular System
Atrial fibrillation, cerebral infarct, deep thrombophlebitis, deep vein thrombosis, migraine, myocardial infarct, palpitation, postural hypotension, thrombophlebitis, varicose vein, and vasculitis.
Digestive System
Cholangitis, cholecystitis, constipation, dry mouth, enteritis, enterocolitis, eructation, esophagitis, fecal incontinence, gastritis, gastroenteritis, hemorrhagic colitis, hepatitis, hepatomegaly, increased appetite, jaundice, liver fatty deposit, liver tenderness, mouth ulceration, pancreatitis, periodontitis, sialadenitis, stomatitis, and ulcerative stomatitis.
Endocrine System
Cushing's syndrome, diabetes mellitus, and hypothyroidism.
Hemic and Lymphatic System
Anemia, leukopenia, and lymphadenopathy.
Metabolic and Nutritional Disorders
Avitaminosis, dehydration, edema, glucose tolerance decreased, lactic acidosis, obesity, peripheral edema, and weight gain.
Musculoskeletal System
Arthralgia, arthrosis, bone necrosis, joint disorder, and myasthenia.
Nervous System
Abnormal dreams, agitation, amnesia, anxiety, apathy, ataxia, confusion, convulsion, dizziness, dyskinesia, emotional lability, encephalopathy, extrapyramidal syndrome, facial paralysis, hypertonia, nervousness, neuropathy, peripheral neuritis, somnolence, thinking abnormal, tremor, and vertigo.
Respiratory System
Asthma, cough increased, dyspnea, lung edema, pharyngitis, rhinitis, and sinusitis.
Skin and Appendages
Acne, alopecia, dry skin, eczema, exfoliative dermatitis, furunculosis, maculopapular rash, nail disorder, pruritis, seborrhea, skin benign neoplasm, skin discoloration, skin striae, skin ulcer, and sweating.
Special Senses
Abnormal vision, eye disorder, otitis media, taste loss, taste perversion, and tinnitus.
Urogenital System
Abnormal ejaculation, amenorrhea, breast enlargement, gynecomastia, impotence, kidney calculus, nephritis, and urine abnormality.
Post-marketing Experience
The following adverse reactions have been reported during post-marketing use of KALETRA. Because these reactions are reported voluntarily from a population of unknown size, it is not possible to reliably estimate their frequency or establish a causal relationship to KALETRA exposure.
Body as a Whole
Redistribution/accumulation of body fat has been reported (see PRECAUTIONS – Fat Redistribution ).
Cardiovascular
Bradyarrhythmias.
Skin and Appendages
Stevens Johnson Syndrome and erythema multiforme.
Laboratory Abnormalities
The percentages of adult patients treated with combination therapy with Grade 3-4 laboratory abnormalities are presented in Table 14 and Table 15.
| Study 863 (48 Weeks) | Study 418 (48 Weeks) | Study 720 (360 Weeks) | ||||
| Variable | Limit1 | KALETRA 400/100 mg BID + d4T +3TC (N=326) | Nelfinavir 750 mg TID + d4T + 3TC (N=327) | KALETRA 800/200 mg QD + TDF + FTC (N=115) | KALETRA 400/100 mg BID + TDF + FTC (N=75) | KALETRA BID + d4T + 3TC (N=100) |
|
1 ULN = upper limit of the normal range; N/A = Not Applicable. |
||||||
| Chemistry | High | |||||
| Glucose | >250 mg/dL | 2% | 2% | 3% | 1% | 4% |
| Uric Acid | >12 mg/dL | 2% | 2% | 0% | 3% | 5% |
| SGOT/ AST | >180 U/L | 2% | 4% | 5% | 3% | 10% |
| SGPT/ ALT | >215 U/L | 4% | 4% | 4% | 3% | 11% |
| GGT | >300 U/L | N/A | N/A | N/A | N/A | 10% |
| Total Cholesterol | >300 mg/dL | 9% | 5% | 3% | 3% | 27% |
| Triglycerides | >750 mg/dL | 9% | 1% | 5% | 4% | 29% |
| Amylase | >2 x ULN | 3% | 2% | 7% | 5% | 4% |
| Hematology | Low | |||||
| Neutrophils | 0.75 x 109/L | 1% | 3% | 5% | 1% | 5% |
| Study 888 (48 Weeks) | Study 9572 and Study 7653 (84-144 Weeks) | |||
| Variable | Limit1 | KALETRA 400/100 mg BID + NVP + NRTIs (N=148) | Investigator-selected protease inhibitor(s) + NVP + NRTIs (N=140) | KALETRA BID + NNRTI + NRTIs (N=127) |
|
1 ULN = upper limit of the normal range; N/A = Not Applicable. 2 Includes clinical laboratory data from patients receiving 400/100 mg BID (n=29) or 533/133 mg BID (n=28) for 84 weeks. Patients received KALETRA in combination with NRTIs and efavirenz. 3 Includes clinical laboratory data from patients receiving 400/100 mg BID (n=36) or 400/200 mg BID (n=34) for 144 weeks. Patients received KALETRA in combination with NRTIs and nevirapine. |
||||
| Chemistry | High | |||
| Glucose | >250 mg/dL | 1% | 2% | 5% |
| Total Bilirubin | >3.48 mg/dL | 1% | 3% | 1% |
| SGOT/AST | >180 U/L | 5% | 11% | 8% |
| SGPT/ALT | >215 U/L | 6% | 13% | 10% |
| GGT | >300 U/L | N/A | N/A | 29% |
| Total Cholesterol | >300 mg/dL | 20% | 21% | 39% |
| Triglycerides | >750 mg/dL | 25% | 21% | 36% |
| Amylase | >2 x ULN | 4% | 8% | 8% |
| Chemistry | Low | |||
| Inorganic Phosphorus | <1.5 mg/dL | 1% | 0% | 2% |
| Hematology | Low | |||
| Neutrophils | 0.75 x 109/L | 1% | 2% | 4% |
Pediatrics
Treatment-Emergent Adverse Events
KALETRA has been studied in 100 pediatric patients 6 months to 12 years of age. The adverse event profile seen during a clinical trial was similar to that for adult patients.
Taste aversion, vomiting, and diarrhea were the most commonly reported drug related adverse events of any severity in pediatric patients treated with combination therapy including KALETRA for up to 48 weeks in Study 940. A total of 8 children experienced moderate or severe adverse events at least possibly related to KALETRA. Rash (reported in 3%) was the only drug-related clinical adverse event of moderate to severe intensity observed in ≥ 2% of children enrolled.
Laboratory Abnormalities
The percentages of pediatric patients treated with combination therapy including KALETRA with Grade 3-4 laboratory abnormalities are presented in Table 16.
| Variable | Limit1 | KALETRA BID+ RTIs (N = 100) |
|
1 ULN = upper limit of the normal range. 2 Subjects with Grade 3-4 amylase confirmed by elevations in pancreatic amylase. |
||
| Chemistry | High | |
| Sodium | > 149 mEq/L | 3% |
| Total Bilirubin | ≥ 3.0 x ULN | 3% |
| SGOT/AST | > 180 U/L | 8% |
| SGPT/ALT | > 215 U/L | 7% |
| Total Cholesterol | > 300 mg/dL | 3% |
| Amylase | > 2.5 x ULN | 7%2 |
| Chemistry | Low | |
| Sodium | < 130 mEq/L | 3% |
| Hematology | Low | |
| Platelet Count | < 50 x 109/L | 4% |
| Neutrophils | < 0.40 x 109/L | 2% |
OVERDOSAGE
KALETRA oral solution contains 42.4% alcohol (v/v). Accidental ingestion of the product by a young child could result in significant alcohol-related toxicity and could approach the potential lethal dose of alcohol.
Human experience of acute overdosage with KALETRA is limited. Treatment of overdose with KALETRA should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with KALETRA. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage. Administration of activated charcoal may also be used to aid in removal of unabsorbed drug. Since KALETRA is highly protein bound, dialysis is unlikely to be beneficial in significant removal of the drug.
DOSAGE AND ADMINISTRATION
KALETRA capsules and oral solution must be taken with food.
The recommended oral dose of KALETRA is as follows: (Please also refer to INDICATIONS AND USAGE and ADVERSE REACTIONS)
Adults
Therapy-Naïve Patients
- KALETRA 400/100 mg (3 capsules or 5.0 mL) twice-daily taken with food.
- KALETRA 800/200 mg (6 capsules or 10 mL) once-daily taken with food.
Therapy-experienced Patients
- KALETRA 400/100 mg (3 capsules or 5.0 mL) twice-daily taken with food.
Once-daily administration of KALETRA is not recommended in therapy-experienced patients.
Concomitant therapy: Efavirenz, nevirapine, amprenavir or nelfinavir
A dose increase of KALETRA to 533/133 mg (4 capsules or 6.5 mL) twice-daily taken with food is recommended when used in combination with efavirenz, nevirapine, amprenavir or nelfinavir (see CLINICAL PHARMACOLOGY– Drug-drug Interactions and/or PRECAUTIONS – Table 11).
KALETRA should not be administered as a once-daily regimen in combination with efavirenz, nevirapine, amprenavir or nelfinavir.
Pediatric Patients
In children 6 months to 12 years of age, the recommended dosage of KALETRA oral solution is 12/3 mg/kg for those 7 to < 15 kg and 10/2.5 mg/kg for those 15 to 40 kg (approximately equivalent to 230/57.5 mg/m2) twice-daily taken with food, up to a maximum dose of 400/100 mg in children > 40 kg (5.0 mL or 3 capsules) twice-daily. KALETRA once-daily has not been evaluated in pediatric patients. It is preferred that the prescriber calculate the appropriate milligram dose for each individual child ≤ 12 years old and determine the corresponding volume of solution or number of capsules. However, as an alternative, the following table contains dosing guidelines for KALETRA oral solution based on body weight. When possible, dose should be administered using a calibrated dosing syringe.
| Weight (kg) | Dose (mg/kg)* | Volume of oral solution BID (80 mg lopinavir/20 mg ritonavir per mL) |
|
* Dosing based on the lopinavir component of lopinavir/ritonavir solution (80 mg/20 mg per mL). |
||
| Without nevirapine, efavirenz or amprenavir | ||
| 7 to < 15 kg | 12 mg/kg BID | |
| 7 to 10 kg | 1.25 mL | |
| > 10 to < 15 kg | 1.75 mL | |
| 15 to 40 kg | 10 mg/kg BID | |
| 15 to 20 kg | 2.25 mL | |
| > 20 to 25 kg | 2.75 mL | |
| > 25 to 30 kg | 3.5 mL | |
| > 30 to 35 kg | 4.0 mL | |
| > 35 to 40 kg | 4.75 mL | |
| > 40 kg | Adult dose | 5 mL (or 3 capsules) |
Note: Use adult dosage recommendation for children > 12 years of age.
Concomitant Therapy: Efavirenz, nevirapine or amprenavir
A dose increase of KALETRA oral solution to 13/3.25 mg/kg for those 7 to < 15 kg and 11/2.75 mg/kg for those 15 to 45 kg (approximately equivalent to 300/75 mg/m2) twice-daily taken with food, up to a maximum dose of 533/133 mg in children > 45 kg twice-daily is recommended when used in combination with efavirenz, nevirapine or amprenavir in children 6 months to 12 years of age. The following table contains dosing guidelines for KALETRA oral solution based on body weight, when used in combination with efavirenz, nevirapine or amprenavir in children (see CLINICAL PHARMACOLOGY– Drug-drug Interactions and/or PRECAUTIONS – Table 11 ).
| Weight (kg) | Dose (mg/kg)* | Volume of oral solution BID (80 mg lopinavir/20 mg ritonavir per mL) |
|
* Dosing based on the lopinavir component of lopinavir/ritonavir solution (80 mg/20 mg per mL). |
||
| With nevirapine, efavirenz or amprenavir | ||
| 7 to < 15 kg | 13 mg/kg BID | |
| 7 to 10 kg | 1.5 mL | |
| > 10 to < 15 kg | 2.0 mL | |
| 15 to 45 kg | 11 mg/kg BID | |
| 15 to 20 kg | 2.5 mL | |
| > 20 to 25 kg | 3.25 mL | |
| > 25 to 30 kg | 4.0 mL | |
| > 30 to 35 kg | 4.5 mL | |
| > 35 to 40 kg | 5.0 mL (or 3 capsules) | |
| > 40 to 45 kg | 5.75 mL | |
| > 45 kg | Adult dose | 6.5 mL (or 4 capsules) |
Note: Use adult dosage recommendation for children > 12 years of age.
HOW SUPPLIED
KALETRA (lopinavir/ritonavir) capsules are orange soft gelatin capsules imprinted with the corporate Abbott “A” logo and the Abbo-Code PK. KALETRA is available as 133.3 mg lopinavir/33.3 mg ritonavir capsules in the following package sizes:
Bottles of 180 capsules each….…………… (NDC 0074-3959-77)
Recommended storage: Store KALETRA soft gelatin capsules at 36°F - 46°F (2°C - 8°C) until dispensed. Avoid exposure to excessive heat. For patient use, refrigerated KALETRA capsules remain stable until the expiration date printed on the label. If stored at room temperature up to 77°F (25°C), capsules should be used within 2 months.
KALETRA (lopinavir/ritonavir) oral solution is a light yellow to orange colored liquid supplied in amber-colored multiple-dose bottles containing 400 mg lopinavir/100 mg ritonavir per 5 mL (80 mg lopinavir/20 mg ritonavir per mL) packaged with a marked dosing cup in the following size:
160 mL bottle……………………………….(NDC 0074-3956-46)
Recommended storage: Store KALETRA oral solution at 36°F - 46°F (2°C - 8°C) until dispensed. Avoid exposure to excessive heat. For patient use, refrigerated KALETRA oral solution remains stable until the expiration date printed on the label. If stored at room temperature up to 77°F (25°C), oral solution should be used within 2 months.
Abbott Laboratories
North Chicago, IL 60064, U.S.A.
Rev. 04/2010
--------------------------------------------(Perforation)-----------------------------------------
KALETRA®
(lopinavir/ritonavir) capsules
(lopinavir/ritonavir) oral solution
ALERT: Find out about medicines that should NOT be taken with KALETRA. Please also read the section "MEDICINES YOU SHOULD NOT TAKE WITH KALETRA."
Patient Information
KALETRA (kuh-LEE-tra)
Generic Name: lopinavir/ritonavir (lop-IN-uh-veer/rit-ON-uh-veer)
Read this leaflet carefully before you start taking KALETRA. Also, read it each time you get your KALETRA prescription refilled, in case something has changed. This information does not take the place of talking with your doctor when you start this medicine and at check ups. Ask your doctor if you have any questions about KALETRA.
Before taking your medicine, make sure you have received the correct medicine. Compare the name above with the name on your bottle and the appearance of your medicine with the description provided below. Contact your pharmacist immediately if you believe a dispensing error has occurred.
What is KALETRA and how does it work?
KALETRA is a combination of two medicines. They are lopinavir and ritonavir. KALETRA is a type of medicine called an HIV (human immunodeficiency virus) protease (PRO-tee-ase) inhibitor. KALETRA is always used in combination with other anti-HIV medicines to treat people with human immunodeficiency virus (HIV) infection. KALETRA is for adults and for children age 6 months and older.
HIV infection destroys CD4 (T) cells, which are important to the immune system. After a large number of T cells are destroyed, acquired immune deficiency syndrome (AIDS) develops.
KALETRA blocks HIV protease, a chemical which is needed for HIV to multiply. KALETRA reduces the amount of HIV in your blood and increases the number of T cells. Reducing the amount of HIV in the blood reduces the chance of death or infections that happen when your immune system is weak (opportunistic infections).
Does KALETRA cure HIV or AIDS?
KALETRA does not cure HIV infection or AIDS. The long-term effects of KALETRA are not known at this time. People taking KALETRA may still get opportunistic infections or other conditions that happen with HIV infection. Some of these conditions are pneumonia, herpes virus infections, and Mycobacterium avium complex (MAC) infections.
Does KALETRA reduce the risk of passing HIV to others?
KALETRA does not reduce the risk of passing HIV to others through sexual contact or blood contamination. Continue to practice safe sex and do not use or share dirty needles.
How should I take KALETRA?
- You should stay under a doctor's care when taking KALETRA. Do not change your treatment or stop treatment without first talking with your doctor.
- You must take KALETRA every day exactly as your doctor prescribed it. The dose of KALETRA may be different for you than for other patients. Follow the directions from your doctor, exactly as written on the label.
- Dosing in adults (including children 12 years of age and older):
The usual dose for adults is 3 capsules (400/100 mg) or 5.0 mL of the oral solution twice a day (morning and night), in combination with other anti-HIV medicines.
The doctor may prescribe KALETRA as 6 capsules or 10.0 mL of oral solution (800/200 mg) once-daily in combination with other anti-HIV medicines for some patients who have not taken anti-HIV medications in the past.
- Dosing in children from 6 months to 12 years of age:
Children from 6 months to 12 years of age can also take KALETRA. The child's doctor will decide the right dose based on the child's weight. - Take KALETRA with food to help it work better.
- Do not change your dose or stop taking KALETRA without first talking with your doctor.
- When your KALETRA supply starts to run low, get more from your doctor or pharmacy. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to KALETRA and become harder to treat.
- Be sure to set up a schedule and follow it carefully.
- Only take medicine that has been prescribed specifically for you. Do not give KALETRA to others or take medicine prescribed for someone else.
What should I do if I miss a dose of KALETRA?
It is important that you do not miss any doses. If you miss a dose of KALETRA, take it as soon as possible and then take your next scheduled dose at its regular time. If it is almost time for your next dose, do not take the missed dose. Wait and take the next dose at the regular time. Do not double the next dose.
What happens if I take too much KALETRA?
If you suspect that you took more than the prescribed dose of this medicine, contact your local poison control center or emergency room immediately.
As with all prescription medicines, KALETRA should be kept out of the reach of young children. KALETRA liquid contains a large amount of alcohol. If a toddler or young child accidentally drinks more than the recommended dose of KALETRA, it could make him/her sick from too much alcohol. Contact your local poison control center or emergency room immediately if this happens.
Who should not take KALETRA?
Together with your doctor, you need to decide whether KALETRA is right for you.
- Do not take KALETRA if you are taking certain medicines. These could cause serious side effects that could cause death. Before you take KALETRA, you must tell your doctor about all the medicines you are taking or are planning to take. These include other prescription and non-prescription medicines and herbal supplements.
For more information about medicines you should not take with KALETRA, please read the section titled "MEDICINES YOU SHOULD NOT TAKE WITH KALETRA."
- Do not take KALETRA if you have an allergy to KALETRA or any of its ingredients, including ritonavir or lopinavir.
Can I take KALETRA with other medications?*
KALETRA may interact with other medicines, including those you take without a prescription. You must tell your doctor about all the medicines you are taking or planning to take before you take KALETRA.
KALETRA can be taken with acid reducing agents (such as omeprazole and ranitidine) with no dose adjustment.
MEDICINES YOU SHOULD NOT TAKE WITH KALETRA:
- Do not take the following medicines with KALETRA because they can cause serious problems or death if taken with KALETRA.
- Dihydroergotamine, ergonovine, ergotamine and methylergonovine such as Cafergot®, Migranal® D.H.E. 45®, Ergotrate Maleate, Methergine, and others
- Halcion® (triazolam)
- Hismanal® (astemizole)
- Orap® (pimozide)
- Propulsid® (cisapride)
- Seldane® (terfenadine)
- Versed® (midazolam)
- Uroxatral® (alfuzosin)
- Revatio® (sildenafil) only when used for the treatment of pulmonary arterial hypertension
- Do not take KALETRA with rifampin, also known as Rimactane®, Rifadin®, Rifater®, or Rifamate®. Rifampin may lower the amount of KALETRA in your blood and make it less effective.
- Do not take KALETRA with St. John's wort (hypericum perforatum), an herbal product sold as a dietary supplement, or products containing St. John's wort. Talk with your doctor if you are taking or planning to take St. John's wort. Taking St. John's wort may decrease KALETRA levels and lead to increased viral load and possible resistance to KALETRA or cross-resistance to other anti-HIV medicines.
- Do not take KALETRA with the cholesterol-lowering medicines Mevacor® (lovastatin) or Zocor® (simvastatin) because of possible serious reactions. There is also an increased risk of drug interactions between KALETRA and Lipitor® (atorvastatin); talk to your doctor before you take any of these cholesterol-reducing medicines with KALETRA.
Medicines that require dosage adjustments:
It is possible that your doctor may need to increase or decrease the dose of other medicines when you are also taking KALETRA. Remember to tell your doctor all medicines you are taking or plan to take.
Before you take Viagra ® (sildenafil), Cialis® (tadalafil), or Levitra ® (vardenafil) with KALETRA, talk to your doctor about problems these two medicines can cause when taken together. You may get increased side effects of VIAGRA, CIALIS, or LEVITRA such as low blood pressure, vision changes, and penis erection lasting more than 4 hours. If an erection lasts longer than 4 hours, get medical help right away to avoid permanent damage to your penis. Your doctor can explain these symptoms to you.
- If you are taking oral contraceptives ("the pill") or the contraceptive patch to prevent pregnancy, you should use an additional or different type of contraception since KALETRA may reduce the effectiveness of oral or patch contraceptives.
- Efavirenz (Sustiva™), nevirapine (Viramune®), Agenerase (amprenavir) and Viracept (nelfinavir) may lower the amount of KALETRA in your blood. Your doctor may increase your dose of KALETRA if you are also taking efavirenz, nevirapine, amprenavir or nelfinavir. KALETRA should not be taken once-daily with these medicines.
- If you are taking Mycobutin® (rifabutin), your doctor will lower the dose of Mycobutin.
- If you are taking Colcrys® (colchicine) for gout, your doctor will tell you what dose to use
- If you are taking Tracleer® (bosentan) or Adcirca® (tadalafil) to treat pulmonary arterial hypertension, your doctor will tell you what dose to use
- If you are taking inhaled medicines such as salmeterol (Serevent®) or salmeterol in combination with fluticasone propionate (Advair®), your doctor may need to change to a different medicine
A change in therapy should be considered if you are taking KALETRA with:
- Phenobarbital
- Phenytoin (Dilantin® and others)
- Carbamazepine (Tegretol® and others)
These medicines may lower the amount of KALETRA in your blood and make it less effective. KALETRA should not be taken once-daily with these medicines.
- If you are taking or before you begin using inhaled Flonase® (fluticasone propionate) talk to your doctor about problems these two medicines may cause when taken together. Your doctor may choose not to keep you on inhaled Flonase®.
-
Other Special Considerations:
KALETRA oral solution contains alcohol. Talk with your doctor if you are taking or planning to take metronidazole or disulfiram. Severe nausea and vomiting can occur. -
If you are taking both didanosine (Videx®) and KALETRA:
Didanosine (Videx®) should be taken one hour before or two hours after KALETRA.
What are the possible side effects of KALETRA?
- This list of side effects is not complete. If you have questions about side effects, ask your doctor, nurse, or pharmacist. You should report any new or continuing symptoms to your doctor right away. Your doctor may be able to help you manage these side effects.
- The most commonly reported side effects of moderate severity that are thought to be drug related are: abdominal pain, abnormal stools (bowel movements), diarrhea, feeling weak/tired, headache, and nausea. Children taking KALETRA may sometimes get a skin rash.
- Blood tests in patients taking KALETRA may show possible liver problems. People with liver disease such as Hepatitis B and Hepatitis C who take KALETRA may have worsening liver disease. Liver problems including death have occurred in patients taking KALETRA. In studies, it is unclear if KALETRA caused these liver problems because some patients had other illnesses or were taking other medicines.
- Some patients taking KALETRA can develop serious problems with their pancreas (pancreatitis), which may cause death. You have a higher chance of having pancreatitis if you have had it before. Tell your doctor if you have nausea, vomiting, or abdominal pain. These may be signs of pancreatitis.
- Some patients have large increases in triglycerides and cholesterol. The long-term chance of getting complications such as heart attacks or stroke due to increases in triglycerides and cholesterol caused by protease inhibitors is not known at this time.
- Diabetes and high blood sugar (hyperglycemia) occur in patients taking protease inhibitors such as KALETRA. Some patients had diabetes before starting protease inhibitors, others did not. Some patients need changes in their diabetes medicine. Others needed new diabetes medicine.
- Changes in body fat have been seen in some patients taking antiretroviral therapy. These changes may include increased amount of fat in the upper back and neck ("buffalo hump"), breast, and around the trunk. Loss of fat from the legs, arms and face may also happen. The cause and long term health effects of these conditions are not known at this time.
- Some patients with hemophilia have increased bleeding with protease inhibitors.
- There have been other side effects in patients taking KALETRA. However, these side effects may have been due to other medicines that patients were taking or to the illness itself. Some of these side effects can be serious.
What should I tell my doctor before taking KALETRA?
- If you are pregnant or planning to become pregnant: The effects of KALETRA on pregnant women or their unborn babies are not known.
- If you are breast-feeding: Do not breast-feed if you are taking KALETRA. You should not breast-feed if you have HIV. If you are a woman who has or will have a baby, talk with your doctor about the best way to feed your baby. You should be aware that if your baby does not already have HIV, there is a chance that HIV can be transmitted through breast- feeding.
- If you have liver problems: If you have liver problems or are infected with Hepatitis B or Hepatitis C, you should tell your doctor before taking KALETRA.
- If you have diabetes: Some people taking protease inhibitors develop new or more serious diabetes or high blood sugar. Tell your doctor if you have diabetes or an increase in thirst or frequent urination.
- If you have hemophilia: Patients taking KALETRA may have increased bleeding.
How do I store KALETRA?
- Keep KALETRA and all other medicines out of the reach of children.
- Refrigerated KALETRA capsules and oral solution remain stable until the expiration date printed on the label. If stored at room temperature up to 77°F (25°C), KALETRA capsules and oral solution should be used within 2 months.
- Avoid exposure to excessive heat.
Do not keep medicine that is out of date or that you no longer need. Be sure that if you throw any medicine away, it is out of the reach of children.
General advice about prescription medicines:
Talk to your doctor or other health care provider if you have any questions about this medicine or your condition. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. If you have any concerns about this medicine, ask your doctor. Your doctor or pharmacist can give you information about this medicine that was written for health care professionals. Do not use this medicine for a condition for which it was not prescribed. Do not share this medicine with other people.
* The brands listed are trademarks of their respective owners and are not trademarks of Abbott Laboratories. The makers of these brands are not affiliated with and do not endorse Abbott Laboratories or its products.
Abbott Laboratories
North Chicago, IL 60064, U.S.A.
Rev. 04/2010
Package Label.Principal Display Panel
NDC 0074-3959-77
180 Capsules
KALETRA™ (lopinavir/ritonavir) capsules
Each soft gelatin capsule contains: Lopinavir.....133.3mg Ritonavir......33.3mg
ALERT Find out about medicines that should NOT be taken with KALETRA
Note to Pharmacist: Do not cover ALERT box with pharmacy label.
Enclosure is provided with tear-off patient information.
Rx only Abbott

| KALETRA
lopinavir and ritonavir capsule, liquid filled |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA021226 | 12/12/2005 | 12/21/2005 |
| Labeler - Abbott Laboratories (001307602) |
| Registrant - Abbott Laboratories (001307602) |