LUPRON DEPOT - PED
-
leuprolide acetate injection, powder, lyophilized, for suspension
Abbott Laboratories
----------
LUPRON DEPOT-PED®(leuprolide acetate for depot suspension)
7.5 mg, 11.25 mg and 15 mg
Rx only
DESCRIPTION
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin-releasing hormone (GnRH or LH-RH). The analog possesses greater potency than the natural hormone. The chemical name is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
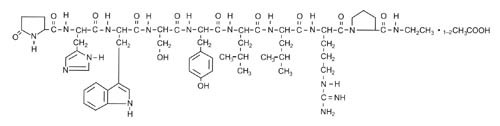
LUPRON DEPOT-PED is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as a single intramuscular injection.
The front chamber of LUPRON DEPOT-PED 7.5 mg, 11.25 mg, and 15 mg prefilled dual-chamber syringe contains leuprolide acetate (7.5/11.25/15 mg), purified gelatin (1.3/1.95/2.6 mg), DL-lactic and glycolic acids copolymer (66.2/99.3/132.4 mg), and D-mannitol (13.2/19.8/26.4 mg). The second chamber of diluent contains carboxymethylcellulose sodium (5 mg), D-mannitol (50 mg), polysorbate 80 (1 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
During the manufacture of LUPRON DEPOT-PED, acetic acid is lost, leaving the peptide.
CLINICAL PHARMACOLOGY
Leuprolide acetate, a GnRH agonist, acts as a potent inhibitor of gonadotropin secretion when given continuously and in therapeutic doses. Human studies indicate that following an initial stimulation of gonadotropins, chronic stimulation with leuprolide acetate results in suppression or "downregulation" of these hormones and consequent suppression of ovarian and testicular steroidogenesis. These effects are reversible on discontinuation of drug therapy.
Leuprolide acetate is not active when given orally.
Pharmacokinetics
Absorption
Following a single LUPRON DEPOT 7.5 mg injection to adult patients, mean peak leuprolide plasma concentration was almost 20 ng/mL at 4 hours and then declined to 0.36 ng/mL at 4 weeks. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study. Nondetectable leuprolide plasma concentrations have been observed during chronic LUPRON DEPOT 7.5 mg administration, but testosterone levels appear to be maintained at castrate levels.
Distribution
The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Metabolism
In healthy male volunteers, a 1 mg bolus of leuprolide administered intravenously revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
In rats and dogs, administration of 14C-labeled leuprolide was shown to be metabolized to smaller inactive peptides, a pentapeptide (Metabolite I), tripeptides (Metabolites II and III) and a dipeptide (Metabolite IV). These fragments may be further catabolized.
The major metabolite (M-I) plasma concentrations measured in 5 prostate cancer patients reached maximum concentration 2 to 6 hours after dosing and were approximately 6% of the peak parent drug concentration. One week after dosing, mean plasma M-I concentrations were approximately 20% of mean leuprolide concentrations.
Excretion
Following administration of LUPRON DEPOT 3.75 mg to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations
The pharmacokinetics of the drug in hepatically and renally impaired patients have not been determined.
CLINICAL STUDIES
In children with central precocious puberty (CPP), stimulated and basal gonadotropins are reduced to prepubertal levels. Testosterone and estradiol are reduced to prepubertal levels in males and females respectively. Reduction of gonadotropins will allow for normal physical and psychological growth and development. Natural maturation occurs when gonadotropins return to pubertal levels following discontinuation of leuprolide acetate.
The following physiologic effects have been noted with the chronic administration of leuprolide acetate in this patient population.
- Skeletal Growth. A measurable increase in body length can be noted since the epiphyseal plates will not close prematurely.
- Organ Growth. Reproductive organs will return to a prepubertal state.
- Menses. Menses, if present, will cease.
In a study of 22 children with central precocious puberty, doses of LUPRON DEPOT were given every 4 weeks and plasma levels were determined according to weight categories as summarized below:
| Patient Weight Range (kg) | Group Weight Average (kg) | Dose (mg) | Trough Plasma Leuprolide Level Mean ±SD (ng/mL)* |
|---|---|---|---|
|
|||
| 20.2 - 27.0 | 22.7 | 7.5 | 0.77±0.033 |
| 28.4 - 36.8 | 32.5 | 11.25 | 1.25±1.06 |
| 39.3 - 57.5 | 44.2 | 15.0 | 1.59±0.65 |
INDICATIONS AND USAGE
LUPRON DEPOT-PED is indicated in the treatment of children with central precocious puberty. Children should be selected using the following criteria:
- Clinical diagnosis of CPP (idiopathic or neurogenic) with onset of secondary sexual characteristics earlier than 8 years in females and 9 years in males.
- Clinical diagnosis should be confirmed prior to initiation of therapy:
- Confirmation of diagnosis by a pubertal response to a GnRH stimulation test. The sensitivity and methodology of this assay must be understood.
- Bone age advanced one year beyond the chronological age.
- Baseline evaluation should also include:
- Height and weight measurements.
- Sex steroid levels.
- Adrenal steroid level to exclude congenital adrenal hyperplasia.
- Beta human chorionic gonadotropin level to rule out a chorionic gonadotropin-secreting tumor.
- Pelvic/adrenal/testicular ultrasound to rule out a steroid secreting tumor.
- Computerized tomography of the head to rule out intracranial tumor.
CONTRAINDICATIONS
- Hypersensitivity to GnRH, GnRH agonist analogs or any of the excipients in LUPRON DEPOT. Reports of anaphylactic reactions to GnRH agonist analogs have been reported in the medical literature.1,2
- LUPRON DEPOT-PED is contraindicated in women who are or may become pregnant while receiving the drug. When administered on day 6 of pregnancy at test dosages of 0.00024, 0.0024, and 0.024 mg/kg (1/1200 to 1/12 of the human pediatric dose) to rabbits, LUPRON DEPOT produced a dose-related increase in major fetal abnormalities. Similar studies in rats failed to demonstrate an increase in fetal malformations. There was increased fetal mortality and decreased fetal weights with the two higher doses of LUPRON DEPOT in rabbits and with the highest dose in rats. The effects on fetal mortality are logical consequences of the alterations in hormonal levels brought about by this drug. Therefore, the possibility exists that spontaneous abortion may occur if the drug is administered during pregnancy.
WARNINGS
During the early phase of therapy, gonadotropins and sex steroids rise above baseline because of the natural stimulatory effect of the drug. Therefore, an increase in clinical signs and symptoms may be observed. (See CLINICAL PHARMACOLOGY section.)
Noncompliance with drug regimen or inadequate dosing may result in inadequate control of the pubertal process. The consequences of poor control include the return of pubertal signs such as menses, breast development, and testicular growth. The long-term consequences of inadequate control of gonadal steroid secretion are unknown, but may include a further compromise of adult stature.
PRECAUTIONS
Laboratory Tests
Response to LUPRON DEPOT-PED should be monitored 1-2 months after the start of therapy with a GnRH stimulation test and sex steroid levels. Measurement of bone age for advancement should be done every 6-12 months.
Sex steroids may increase or rise above prepubertal levels if the dose is inadequate. (See WARNINGS section.) Once a therapeutic dose has been established, gonadotropin and sex steroid levels will decline to prepubertal levels.
Drug Interactions
No pharmacokinetic-based drug-drug interaction studies have been conducted. However, because leuprolide acetate is a peptide that is primarily degraded by peptidase and not by cytochrome P-450 enzymes as noted in specific studies, and the drug is only about 46% bound to plasma proteins, drug interactions would not be expected to occur.
Drug/Laboratory Test Interactions
Administration of LUPRON DEPOT 3.75 mg in women results in suppression of the pituitary-gonadal system. Normal function is usually restored within three months after treatment is discontinued. Therefore, diagnostic tests of pituitary gonadotropic and gonadal functions conducted during treatment and for up to three months after discontinuation of LUPRON DEPOT may be misleading.
Information for Parents
Prior to starting therapy with LUPRON DEPOT-PED, the parent or guardian must be aware of the importance of continuous therapy. Adherence to 4 week drug administration schedules must be accepted if therapy is to be successful.
- During the first 2 months of therapy, a female may experience menses or spotting. If bleeding continues beyond the second month, notify the physician.
- Any irritation at the injection site should be reported to the physician immediately.
- Report any unusual signs or symptoms to the physician.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at high daily doses (0.6 to 4 mg/kg). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males (highest incidence in the low dose group). In mice, no leuprolide acetate-induced tumors or pituitary abnormalities were observed at a dose as high as 60 mg/kg for two years. Adult patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Although no clinical studies have been completed in children to assess the full reversibility of fertility suppression, animal studies (prepubertal and adult rats and monkeys) with leuprolide acetate and other GnRH analogs have shown functional recovery. However, following a study with leuprolide acetate, immature male rats demonstrated tubular degeneration in the testes even after a recovery period. In spite of the failure to recover histologically, the treated males proved to be as fertile as the controls. Also, no histologic changes were observed in the female rats following the same protocol. In both sexes, the offspring of the treated animals appeared normal. The effect of the treatment of the parents on the reproductive performance of the F1 generation was not tested. The clinical significance of these findings is unknown.
Pregnancy
Teratogenic Effects
Pregnancy Category X
(See CONTRAINDICATIONS section.)
Nursing Mothers
It is not known whether leuprolide acetate is excreted in human milk. LUPRON should not be used by nursing mothers.
Geriatric Use
See also the labeling for LUPRON DEPOT 7.5 mg which is indicated for the palliative treatment of advanced prostate cancer. For LUPRON DEPOT-PED 11.25 mg and LUPRON DEPOT-PED 15 mg, no clinical information has been established for persons aged 65 and over.
ADVERSE REACTIONS
Clinical Trials
Potential exacerbation of signs and symptoms during the first few weeks of treatment (See PRECAUTIONS section.) is a concern in patients with rapidly advancing central precocious puberty.
In two studies of children with central precocious puberty, in 2% or more of the patients receiving the drug, the following adverse reactions were reported to have a possible or probable relationship to drug as ascribed by the treating physician. Reactions which are not considered drug-related are excluded.
| Number of Patients | ||
|---|---|---|
| N = 395 | (%) | |
| Body as a Whole | ||
| General Pain | 7 | (2) |
| Integumentary System | ||
| Acne/Seborrhea | 7 | (2) |
| Injection Site Reactions Including Abscess | 21 | (5) |
| Rash Including Erythema Multiforme | 8 | (2) |
| Urogenital System | ||
| Vaginitis/Bleeding/Discharge | 7 | (2) |
In those same studies, the following adverse reactions were reported in less than 2% of the patients.
Body as a Whole - Body Odor, Fever, Headache, Infection; Cardiovascular System - Syncope, Vasodilation; Digestive System - Dysphagia, Gingivitis, Nausea/Vomiting; Endocrine System - Accelerated Sexual Maturity; Metabolic and Nutritional Disorders - Peripheral Edema, Weight Gain; Nervous System - Emotional Lability, Nervousness, Personality Disorder, Somnolence; Respiratory System - Epistaxis; Integumentary System - Alopecia, Skin Striae; Urogenital System - Cervix Disorder, Gynecomastia/Breast Disorders, Urinary Incontinence.
Postmarketing
During postmarketing surveillance, which includes other dosage forms, the following adverse events were reported.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely reported. Rash, urticaria, and photosensitivity reactions have also been reported.
Localized reactions including induration and abscess have been reported at the site of injection.
Cardiovascular System - Hypotension; Hemic and Lymphatic System - Decreased WBC; Central/Peripheral Nervous System - Peripheral neuropathy, Spinal fracture/paralysis; Musculoskeletal System - Tenosynovitis-like symptoms; Urogenital System - Prostate pain.
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
See other LUPRON DEPOT and LUPRON Injection package inserts for other events reported in different patient populations.
OVERDOSAGE
In rats, subcutaneous administration of 125 to 250 times the recommended human pediatric dose, expressed on a per body weight basis, resulted in dyspnea, decreased activity, and local irritation at the injection site. There is no evidence at present that there is a clinical counterpart of this phenomenon. In early clinical trials using leuprolide acetate in adult patients, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
DOSAGE AND ADMINISTRATION
LUPRON DEPOT-PED must be administered under the supervision of a physician.
The dose of LUPRON DEPOT-PED must be individualized for each child. The dose is based on a mg/kg ratio of drug to body weight. Younger children require higher doses on a mg/kg ratio.
For each dosage form, after 1-2 months of initiating therapy or changing doses, the child must be monitored with a GnRH stimulation test, sex steroids, and Tanner staging to confirm downregulation. Measurements of bone age for advancement should be monitored every 6-12 months. The dose should be titrated upward until no progression of the condition is noted either clinically and/or by laboratory parameters.
The first dose found to result in adequate downregulation can probably be maintained for the duration of therapy in most children. However, there are insufficient data to guide dosage adjustment as patients move into higher weight categories after beginning therapy at very young ages and low dosages. It is recommended that adequate downregulation be verified in such patients whose weight has increased significantly while on therapy.
Discontinuation of LUPRON DEPOT-PED should be considered before age 11 for females and age 12 for males.
The recommended starting dose is 0.3 mg/kg/4 weeks (minimum 7.5 mg) administered as a single intramuscular injection. The starting dose will be dictated by the child's weight.
| ≤ 25 kg | 7.5 mg |
| > 25-37.5 kg | 11.25 mg |
| > 37.5 kg | 15 mg |
If total downregulation is not achieved, the dose should be titrated upward in increments of 3.75 mg every 4 weeks. This dose will be considered the maintenance dose.
The lyophilized microspheres are to be reconstituted and administered as a single intramuscular injection. For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the following instructions:
- The lyophilized microspheres are to be reconstituted and administered as a single intramuscular injection.
- Since LUPRON DEPOT-PED does not contain a preservative, the suspension should be discarded if not used immediately.
- The LUPRON DEPOT powder should be visually inspected and the syringe should NOT BE USED if clumping or caking is evident. A thin layer of powder on the wall of the syringe is considered normal. The diluent should appear clear.
- To prepare for injection, screw the white plunger into the end stopper until the stopper begins to turn.
- Hold the syringe UPRIGHT. Release the diluent by SLOWLY PUSHING (6 to 8 seconds) the plunger until the first stopper is at the blue line in the middle of the barrel.
- Keep the syringe UPRIGHT. Gently mix the microspheres (powder) thoroughly to form a uniform suspension. The suspension will appear milky. If the powder adheres to the stopper or caking/clumping is present, tap the syringe with your finger to disperse. DO NOT USE if any of the powder has not gone into suspension.
- Hold the syringe UPRIGHT. With the opposite hand pull the needle cap upward without twisting.
- Keep the syringe UPRIGHT. Advance the plunger to expel the air from the syringe.
NOTE: Aspirated blood would be visible just below the luer lock connection if a blood vessel is accidentally penetrated. If present, blood can be seen through the transparent LuproLoc™ safety device. If blood is present remove the needle immediately. Do not inject the medication.
- Inject the entire contents of the syringe intramuscularly at the time of reconstitution. The suspension settles very quickly following reconstitution; therefore, LUPRON DEPOT should be mixed and used immediately.
AFTER INJECTION
- Withdraw the needle. Immediately activate the LuproLoc™ safety device by pushing the arrow forward with the thumb or finger until the device is fully extended and a CLICK is heard or felt.
Since the product does not contain a preservative, the suspension should be discarded if not used immediately.
As with other drugs administered by injection, the injection site should be varied periodically.
HOW SUPPLIED
LUPRON DEPOT-PED is packaged as follows:
| Kit with prefilled dual-chamber syringe | 7.5 mg | NDC 0074-2108-03 |
| Kit with prefilled dual-chamber syringe | 11.25 mg | NDC 0074-2282-03 |
| Kit with prefilled dual-chamber syringe | 15 mg | NDC 0074-2440-03 |
Each syringe contains sterile lyophilized microspheres of leuprolide acetate incorporated in a biodegradable lactic acid/glycolic acid copolymer. When mixed with 1 milliliter of accompanying diluent, LUPRON DEPOT-PED is administered as a single intramuscular injection.
Each kit contains:
- one prefilled dual-chamber syringe containing 1½ inch needle with LuproLocTM safety device
- one plunger
- two alcohol swabs
- instructions for how to mix and administer
- an information pamphlet for patients
- a complete prescribing information enclosure
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [See USP Controlled Room Temperature]
REFERENCES
- Taylor, JD. Anaphylactic reaction to LHRH analogue, leuprorelin. Med J Australia 1994 Oct; 161(3): 455.
- Letterie GS, et al. Recurrent anaphylaxis to a depot form of GnRH analogue. Obstet Gynecol 1991 Nov; 78: 943–946.
Manufactured for
Abbott Laboratories
North Chicago, IL 60064
by Takeda Pharmaceutical Company Limited
Osaka, Japan 540-8645
™ -Trademark
® -Registered Trademark
(Nos. 2108, 2282, 2440)
Revised: July, 2010
© 2008, Abbott Laboratories
NDC 0074-2282-03
For Intramuscular Use Only. Single Dose Administration Kit with prefi lled dual-chamber syringe.
LUPRON DEPOT_PED® leuprolide acetate for depot suspension 11.25 mg

| LUPRON DEPOT - PED
leuprolide acetate for depot suspension injection, powder, lyophilized, for suspension |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020263 | 08/23/2010 | |
| LUPRON DEPOT - PED
leuprolide acetate for depot suspension injection, powder, lyophilized, for suspension |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020263 | 08/23/2010 | |
| LUPRON DEPOT - PED
leuprolide acetate for depot suspension injection, powder, lyophilized, for suspension |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA020263 | 08/23/2010 | |
| Labeler - Abbott Laboratories (001307602) |