LUPRON DEPOT
-
leuprolide acetate injection, powder, lyophilized, for suspension
Abbott Laboratories
----------
LUPRON DEPOT® 7.5 mg(leuprolide acetate for depot suspension)
Rx only
DESCRIPTION
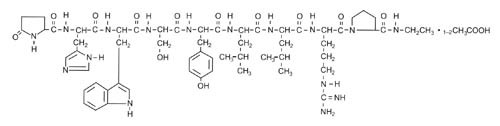
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin-releasing hormone (GnRH or LH-RH). The analog possesses greater potency than the natural hormone. The chemical name is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
LUPRON DEPOT is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, becomes a suspension intended as a monthly intramuscular injection.
The front chamber of LUPRON DEPOT 7.5 mg prefilled dual-chamber syringe contains leuprolide acetate (7.5 mg), purified gelatin (1.3 mg), DL-lactic and glycolic acids copolymer (66.2 mg), and D-mannitol (13.2 mg). The second chamber of diluent contains carboxymethylcellulose sodium (5 mg), D-mannitol (50 mg), polysorbate 80 (1 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
During the manufacture of LUPRON DEPOT 7.5 mg, acetic acid is lost, leaving the peptide.
CLINICAL PHARMACOLOGY
Leuprolide acetate, an LH-RH agonist, acts as a potent inhibitor of gonadotropin secretion when given continuously and in therapeutic doses. Animal and human studies indicate that following an initial stimulation, chronic administration of leuprolide acetate results in suppression of ovarian and testicular steroidogenesis. This effect is reversible upon discontinuation of drug therapy. Administration of leuprolide acetate has resulted in inhibition of the growth of certain hormone dependent tumors (prostatic tumors in Noble and Dunning male rats and DMBA-induced mammary tumors in female rats) as well as atrophy of the reproductive organs.
In humans, administration of leuprolide acetate results in an initial increase in circulating levels of luteinizing hormone (LH) and follicle stimulating hormone (FSH), leading to a transient increase in levels of the gonadal steroids (testosterone and dihydrotestosterone in males, and estrone and estradiol in premenopausal females). However, continuous administration of leuprolide acetate results in decreased levels of LH and FSH. In males, testosterone is reduced to castrate levels. In premenopausal females, estrogens are reduced to postmenopausal levels. These decreases occur within two to four weeks after initiation of treatment. Castrate levels of testosterone in prostatic cancer patients have been demonstrated for up to 10 years.
Leuprolide acetate is not active when given orally.
Pharmacokinetics
Absorption
Following a single injection of LUPRON DEPOT 7.5 mg to patients, mean plasma leuprolide concentration was almost 20 ng/mL at 4 hours and 0.36 ng/mL at 4 weeks. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study. Nondetectable leuprolide plasma concentrations have been observed during chronic LUPRON DEPOT 7.5 mg administration, but testosterone levels appear to be maintained at castrate levels.
Distribution
The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Metabolism
In healthy male volunteers, a 1 mg bolus of leuprolide administered intravenously revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
In rats and dogs, administration of 14C-labeled leuprolide was shown to be metabolized to smaller inactive peptides, a pentapeptide (Metabolite I), tripeptides (Metabolites II and III) and a dipeptide (Metabolite IV). These fragments may be further catabolized.
The major metabolite (M-I) plasma concentrations measured in 5 prostate cancer patients reached maximum concentration 2 to 6 hours after dosing and were approximately 6% of the peak parent drug concentration. One week after dosing, mean plasma M-I concentrations were approximately 20% of mean leuprolide concentrations.
Excretion
Following administration of LUPRON DEPOT 3.75 mg to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations
The pharmacokinetics of the drug in hepatically and renally impaired patients have not been determined.
Drug Interactions
No pharmacokinetic-based drug-drug interaction studies have been conducted with LUPRON DEPOT. However, because leuprolide acetate is a peptide that is primarily degraded by peptidase and the drug is only about 46% bound to plasma proteins, drug interactions would not be expected to occur.
CLINICAL STUDIES
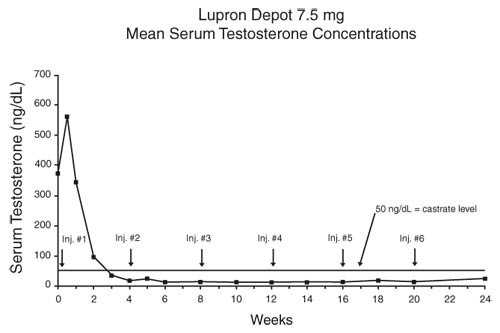
In an open-label, non-comparative, multicenter clinical study of LUPRON DEPOT 7.5 mg, 56 patients with stage D2 prostatic adenocarcinoma and no prior systemic treatment were enrolled. The objectives were to determine if a 7.5 mg depot formulation of leuprolide injected once every 4 weeks would reduce and maintain serum testosterone to castrate range (≤50 ng/dL), to evaluate objective clinical response, and to assess the safety of the formulation. During the initial 24 weeks, serum testosterone was measured weekly, biweekly, or every four weeks and objective tumor response assessments were performed at Weeks 12 and 24. Once the patient completed the initial 24-week treatment phase, treatment continued at the investigator's discretion. Data from the initial 24-week treatment phase are summarized in this section.
In the majority of patients, serum testosterone increased by 50% or more above baseline during the first week of treatment. Serum testosterone suppressed to the castrate range within 30 days of the initial depot injection in 94% (51/54) of patients for whom testosterone suppression was achieved (2 patients withdrew prior to onset of suppression) and within 66 days in all 54 patients. Mean serum testosterone suppressed to castrate level by Week 3. The median dosing interval between injections was 28 days. One escape from suppression (2 consecutive testosterone values greater than 50 ng/dL after achieving castrate level) was noted at Week 18, associated with a substantial dosing delay. In this patient, serum testosterone returned to the castrate range at the next monthly measurement. Serum testosterone was minimally above the castrate range on a single occasion for 4 other patients. No clinical significance was attributed to these rises in testosterone.
Secondary efficacy endpoints evaluated included objective tumor response, assessed by clinical evaluations of tumor burden (complete response, partial response, objectively stable, and progression), as well as changes in local disease status, assessed by digital rectal examination, and changes in prostatic acid phosphatase (PAP). These evaluations were performed at Weeks 12 and 24. The objective tumor response analysis showed a "no progression" (ie. complete or partial response, or stable disease) in 77% (40/52) of patients at Week 12, and in 84% (42/50) of patients at Week 24. Local disease improved or remained stable in all (42) patients evaluated at Week 12 and in 98% (41/42) of patients elevated at Week 24. PAP normalized or decreased at Week 12 and/or 24 in the majority of patients with elevated baseline PAP.
Periodic monitoring of serum testosterone and PSA levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. It should be noted that results of testosterone determinations are dependent on assay methodology. It is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
INDICATIONS AND USAGE
LUPRON DEPOT 7.5 mg is indicated in the palliative treatment of advanced prostatic cancer.
CONTRAINDICATIONS
- Hypersensitivity to GnRH, GnRH agonist analogs or any of the excipients in LUPRON DEPOT. Reports of anaphylactic reactions to GnRH agonist analogs have been reported in the medical literature.1,2
- All formulations of LUPRON DEPOT are contraindicated in women who are or may become pregnant while receiving the drug. LUPRON DEPOT may cause fetal harm when administered to a pregnant woman. Major fetal abnormalities were observed in rabbits but not in rats after administration of LUPRON DEPOT throughout gestation. There was increased fetal mortality and decreased fetal weights in rats and rabbits. The effects on fetal mortality are expected consequences of the alterations in hormonal levels brought about by this drug. Therefore, the possibility exists that spontaneous abortion may occur. If this drug is administered during pregnancy or if the patient becomes pregnant while taking any formulation of LUPRON DEPOT, the patient should be apprised of the potential hazard to the fetus.
WARNINGS
Initially, LUPRON DEPOT, like other LH-RH agonists, causes increases in serum levels of testosterone to approximately 50% above baseline during the first week of treatment. Transient worsening of symptoms, or the occurrence of additional signs and symptoms of prostate cancer, may occasionally develop during the first few weeks of LUPRON DEPOT treatment. A small number of patients may experience a temporary increase in bone pain, which can be managed symptomatically. As with other LH-RH agonists, isolated cases of ureteral obstruction and spinal cord compression have been observed, which may contribute to paralysis with or without fatal complications.
For patients at risk, initiation of therapy with daily LUPRON® (leuprolide acetate) Injection (see DOSAGE AND ADMINISTRATION section in the LUPRON Injection labeling) for the first two weeks to facilitate withdrawal of treatment may be considered. If spinal cord compression or renal impairment develops, standard treatment of these complications should be instituted.
PRECAUTIONS
Information for Patients
An information pamphlet for patients is included with the product.
General
Patients with metastatic vertebral lesions and/or with urinary tract obstruction should be closely observed during the first few weeks of therapy (see WARNINGS section).
Laboratory Tests
Response to LUPRON DEPOT 7.5 mg should be monitored by measuring serum levels of testosterone as well as prostate-specific antigen. In the majority of patients, testosterone levels increased above baseline during the first week, declining thereafter to baseline levels or below by the end of the second week. Castrate levels were reached within two to four weeks and once achieved were maintained for the duration of treatment in all 54 patients. Minimal and transient increases to above the castrate level occurred in eight patients (see CLINICAL STUDIES section).
Drug Interactions
(See Pharmacokinetics.)
Drug/Laboratory Test Interactions
Administration of LUPRON DEPOT in therapeutic doses results in suppression of the pituitary-gonadal system. Normal function is usually restored within three months after treatment is discontinued. Due to the suppression of the pituitary-gonadal system by LUPRON DEPOT, diagnostic tests of pituitary gonadotropic and gonadal functions conducted during treatment and for up to three months after discontinuation of LUPRON DEPOT may be affected.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at high daily doses (0.6 to 4 mg/kg). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males (highest incidence in the low dose group). In mice, no leuprolide acetate-induced tumors or pituitary abnormalities were observed at a dose as high as 60 mg/kg for two years. Patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Mutagenicity studies have been performed with leuprolide acetate using bacterial and mammalian systems. These studies provided no evidence of a mutagenic potential.
Clinical and pharmacologic studies in adults (≥ 18 years) with leuprolide acetate and similar analogs have shown reversibility of fertility suppression when the drug is discontinued after continuous administration for periods of up to 24 weeks.
Pregnancy Category X
See CONTRAINDICATIONS section.
Pediatric Use
See LUPRON DEPOT-PED® (leuprolide acetate for depot suspension) labeling for the safety and effectiveness of the monthly formulation in children with central precocious puberty.
Geriatric Use
In the clinical trials for LUPRON DEPOT, the majority (68%) of the subjects studied were at least 65 years of age. Therefore, the labeling reflects the pharmacokinetics, efficacy and safety of LUPRON DEPOT in this population.
Adverse Reactions
Clinical Trials
In the majority of patients testosterone levels increased above baseline during the first week, declining thereafter to baseline levels or below by the end of the second week of treatment.
Potential exacerbations of signs and symptoms during the first few weeks of treatment is a concern in patients with vertebral metastases and/or urinary obstruction or hematuria which, if aggravated, may lead to neurological problems such as temporary weakness and/or paresthesia of the lower limbs or worsening of urinary symptoms (see WARNINGS section).
In a clinical trial of LUPRON DEPOT 7.5 mg, the following adverse reactions were reported in 5% or more of the patients during the initial 24-week treatment period regardless of causality.
| LUPRON DEPOT 7.5 mg (N=56) | ||
|---|---|---|
| N | (%) | |
|
||
| Body as a Whole | ||
| General pain | 13 | (23.2) |
| Infection | 3 | (5.4) |
| Cardiovascular System | ||
| Hot flashes/sweats* | 32 | (57.1) |
| Digestive System | ||
| GI disorders | 8 | (14.3) |
| Metabolic and Nutritional Disorders | ||
| Edema | 8 | (14.3) |
| Nervous System | ||
| Libido decreased* | 3 | (5.4) |
| Respiratory System | ||
| Respiratory disorder | 6 | (10.7) |
| Urogenital System | ||
| Urinary disorder | 7 | (12.5) |
| Impotence* | 3 | (5.4) |
| Testicular atrophy* | 3 | (5.4) |
In this same study, the following adverse reactions were reported in less than 5% of the patients on LUPRON DEPOT 7.5 mg.
Body as a Whole - Asthenia, Cellulitis, Fever, Headache, Injection site reaction, Neoplasm; Cardiovascular System - Angina, Congestive heart failure; Digestive System - Anorexia, Dysphagia, Eructation, Peptic ulcer; Hemic and Lymphatic System - Ecchymosis; Musculoskeletal System - Myalgia; Nervous System - Agitation, Insomnia/sleep disorders, Neuromuscular disorders; Respiratory System - Emphysema, Hemoptysis, Lung edema, Sputum increased; Skin and Appendages - Hair disorder, Skin reaction; Urogenital System - Balanitis, Breast enlargement, Urinary tract infection.
Laboratory: Abnormalities of certain parameters were observed, but their relationship to drug treatment are difficult to assess in this population. The following were recorded in ≥5% of patients at final visit: Decreased albumin, decreased hemoglobin/hematocrit, decreased prostatic acid phosphatase, decreased total protein, decreased urine specific gravity, hyperglycemia, hyperuricemia, increased BUN, increased creatinine, increased liver function tests (AST, LDH), increased phosphorus, increased platelets, increased prostatic acid phosphatase, increased total cholesterol, increased urine specific gravity, leukopenia.
Postmarketing
During postmarketing surveillance, which includes other dosage forms and other patient populations, the following adverse events were reported.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely (incidence rate of about 0.002%) reported. Rash, urticaria, and photosensitivity reactions have also been reported.
Localized reactions including induration and abscess have been reported at the site of injection.
Symptoms consistent with fibromyalgia (eg, joint and muscle pain, headaches, sleep disorders, gastrointestinal distress, and shortness of breath) have been reported individually and collectively.
Cardiovascular System - Hypotension, Myocardial infarction, Pulmonary embolism; Hemic and Lymphatic System - Decreased WBC; Central/Peripheral Nervous System - Convulsion, Peripheral neuropathy, Spinal fracture/paralysis; Endocrine System – Diabetes; Musculoskeletal System - Tenosynovitis-like symptoms; Urogenital System - Prostate pain.
Changes in Bone Density: Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with an LH-RH agonist analog. In a clinical trial, 25 men with prostate cancer, 12 of whom had been treated previously with leuprolide acetate for at least six months, underwent bone density studies as a result of pain. The leuprolide-treated group had lower bone density scores than the nontreated control group. It can be anticipated that long periods of medical castration in men will have effects on bone density.
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
See other LUPRON DEPOT and LUPRON Injection package inserts for other events reported in women and pediatric populations.
OVERDOSAGE
In clinical trials using daily subcutaneous leuprolide acetate in patients with prostate cancer, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
DOSAGE AND ADMINISTRATION
LUPRON DEPOT Must Be Administered Under The Supervision Of A Physician.
The recommended dose of LUPRON DEPOT is 7.5 mg, incorporated in a depot formulation. The lyophilized microspheres are to be reconstituted and administered monthly as a single intramuscular injection. For optimal performance of the prefilled dual chamber syringe (PDS), read and follow the following instructions:
- The LUPRON DEPOT powder should be visually inspected and the syringe should NOT BE USED if clumping or caking is evident. A thin layer of powder on the wall of the syringe is considered normal. The diluent should appear clear.
- To prepare for injection, screw the white plunger into the end stopper until the stopper begins to turn.
- Hold the syringe UPRIGHT. Release the diluent by SLOWLY PUSHING (6 to 8 seconds) the plunger until the first stopper is at the blue line in the middle of the barrel.
- Keep the syringe UPRIGHT. Gently mix the microspheres (powder) thoroughly to form a uniform suspension. The suspension will appear milky. If the powder adheres to the stopper or caking/clumping is present, tap the syringe with your finger to disperse. DO NOT USE if any of the powder has not gone into suspension.
- Hold the syringe UPRIGHT. With the opposite hand pull the needle cap upward without twisting.
- Keep the syringe UPRIGHT. Advance the plunger to expel the air from the syringe.
- Inject the entire contents of the syringe intramuscularly at the time of reconstitution. The suspension settles very quickly following reconstitution; therefore, LUPRON DEPOT should be mixed and used immediately.
NOTE: Aspirated blood would be visible just below the luer lock connection if a blood vessel is accidentally penetrated. If present, blood can be seen through the transparent LuproLoc™ safety device.
AFTER INJECTION
- Withdraw the needle. Immediately activate the LuproLoc™ safety device by pushing the arrow forward with the thumb or finger until the device is fully extended and a CLICK is heard or felt.
Since the product does not contain a preservative, the suspension should be discarded if not used immediately.
As with other drugs administered by injection, the injection site should be varied periodically.
HOW SUPPLIED
Each LUPRON DEPOT 7.5 mg kit (NDC 0074-3642-03) contains:
- one prefilled dual-chamber syringe
- one plunger
- two alcohol swabs
- instructions for how to mix and administer
- an information pamphlet for patients
- a complete prescribing information enclosure
The prefilled dual-chamber syringe contains sterile lyophilized microspheres of leuprolide acetate incorporated in a biodegradable lactic acid/glycolic acid copolymer. When mixed with 1 mL of accompanying diluent, LUPRON DEPOT 7.5 mg is administered as a single monthly intramuscular injection.
Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [See USP Controlled Room Temperature]
REFERENCES
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006; 63; 1172-1193.
- Polovich, M., White, J.M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. Ed.) Pittsburgh, PA: Oncology Nursing Society.
Manufactured for
Abbott Laboratories
North Chicago, IL 60064
By Takeda Pharmaceutical Company Limited
Osaka, JAPAN 540-8645
™—Trademark
®—Registered Trademark
(No. 3642)
Rev. 07/2010
©2008, Abbott Laboratories
NDC 0074-3642-03
For Adult Use 7.5mg
Lupron Depot®
Single Dose Administration Kit with prefilled dual-chamber syringe.
Leuprolide Acetate for depot suspension 7.5mg Rx only

| LUPRON DEPOT
leuprolide acetate for depot suspension injection, powder, lyophilized, for suspension |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019732 | 08/23/2010 | |
| Labeler - Abbott Laboratories (001307602) |