ZENAPAX
-
daclizumab injection, solution, concentrate
Genentech, Inc.
----------
ZENAPAX®(daclizumab)
STERILE CONCENTRATE FOR INJECTION
WARNING
Only physicians experienced in immunosuppressive therapy and management of organ transplant patients should prescribe ZENAPAX® (daclizumab). The physician responsible for ZENAPAX administration should have complete information requisite for the follow-up of the patient. ZENAPAX should only be administered by healthcare personnel trained in the administration of the drug who have available adequate laboratory and supportive medical resources.
DESCRIPTION
ZENAPAX® (daclizumab) is an immunosuppressive, humanized IgG1 monoclonal antibody produced by recombinant DNA technology that binds specifically to the alpha subunit (p55 alpha, CD25, or Tac subunit) of the human high-affinity interleukin-2 (IL-2) receptor that is expressed on the surface of activated lymphocytes.
Daclizumab is a composite of human (90%) and murine (10%) antibody sequences. The human sequences were derived from the constant domains of human IgG1 and the variable framework regions of the Eu myeloma antibody. The murine sequences were derived from the complementarity-determining regions of a murine anti-Tac antibody. The molecular weight predicted from the DNA sequence is 144 kilodaltons.
ZENAPAX 25 mg/5 mL is supplied as a clear, sterile, colorless concentrate for further dilution and intravenous administration. Each milliliter of ZENAPAX contains 5 mg of daclizumab and 3.6 mg sodium phosphate monobasic monohydrate, 11 mg sodium phosphate dibasic heptahydrate, 4.6 mg sodium chloride, 0.2 mg polysorbate 80, and may contain hydrochloric acid or sodium hydroxide to adjust the pH to 6.9. No preservatives are added.
CLINICAL PHARMACOLOGY
General
Mechanism of Action
Daclizumab functions as an IL-2 receptor antagonist that binds with high-affinity to the Tac subunit of the high-affinity IL-2 receptor complex and inhibits IL-2 binding. Daclizumab binding is highly specific for Tac, which is expressed on activated but not resting lymphocytes. Administration of ZENAPAX inhibits IL-2-mediated activation of lymphocytes, a critical pathway in the cellular immune response involved in allograft rejection.
While in the circulation, ZENAPAX impairs the response of the immune system to antigenic challenges. Whether the ability to respond to repeated or ongoing challenges with those antigens returns to normal after ZENAPAX is cleared is unknown (see PRECAUTIONS).
Pharmacokinetics
Adults
In clinical trials involving renal allograft patients treated with a 1 mg/kg IV dose of ZENAPAX every 14 days for a total of five doses, peak serum concentration (mean ± SD) rose between the first dose (21 ± 14 µg/mL) and fifth dose (32 ± 22 µg/mL). The mean trough serum concentration before the fifth dose was 7.6 ± 4.0 µg/mL. Population pharmacokinetic analysis of the data using a two-compartment open model gave the following values for a reference patient (45-year-old male Caucasian patient with a body weight of 80 kg and no proteinuria): systemic clearance = 15 mL/hour, volume of central compartment = 2.5 liter, volume of peripheral compartment = 3.4 liter. The estimated terminal elimination half-life for the reference patient was 20 days (480 hours), which is similar to the terminal elimination half-life for human IgG (18 to 23 days). Bayesian estimates of terminal elimination half-life ranged from 11 to 38 days for the 123 patients included in the population analysis. The influence of body weight on systemic clearance supports the dosing of ZENAPAX on a milligram per kilogram (mg/kg) basis. For patients studied, this dosing maintained drug exposure within 30% of the reference exposure. Covariate analyses showed that no dosage adjustments based on age, race, gender or degree of proteinuria, are required for renal allograft patients. The estimated interpatient variability (percent coefficient of variation) in systemic clearance and central volume of distribution were 15% and 27%, respectively.
Pediatrics
Pharmacokinetic parameters were evaluated in 61 pediatric patients treated with a 1 mg/kg IV dose of ZENAPAX every 14 days for a total of five doses. Peak serum concentration (mean ± SD) rose between the first dose (16 ± 12 µg/mL) and fifth dose (21 ± 14 µg/mL). The mean trough serum concentration before the fifth dose was 5.0 ± 2.7 µg/mL. Population pharmacokinetic analysis of the data using a two-compartment open model gave the following values for a reference patient (Caucasian patient with a body weight of 29.7 kg): systemic clearance = 10 mL/hour, volume of central compartment = 2.0 liter, volume of peripheral compartment = 1.4 liter. The estimated terminal elimination half-life for the reference patient was 13 days (317 hours). For the patients studied, this dosing maintained drug exposure within 50% of the reference exposure. Covariate analyses suggested that disposition parameters were not influenced to a clinically relevant extent by race, gender or degree of proteinuria. The estimated interpatient variability (percent coefficient of variation) in systemic clearance and central volume of distribution were 30% and 40%, respectively.
Pharmacodynamics
In vitro and in vivo data suggest that serum levels of 5 to 10 µg/mL are necessary for saturation of the Tac subunit of the IL-2 receptors to block the responses of activated T lymphocytes. At the recommended dosage regimen, daclizumab saturates the Tac subunit of the IL-2 receptor for approximately 90 and 120 days posttransplant, respectively in pediatric and adult patients. The duration of clinically significant IL-2 receptor blockade after the recommended course of ZENAPAX is not known. No significant changes to circulating lymphocyte numbers or cell phenotypes were observed by flow cytometry. Cytokine release syndrome has not been observed after ZENAPAX administration.
CLINICAL STUDIES
The safety and efficacy of ZENAPAX for the prophylaxis of acute organ rejection in adult patients receiving their first cadaveric kidney transplant were assessed in two randomized, double-blind, placebo-controlled, multicenter trials. These trials compared a dose of 1.0 mg/kg of ZENAPAX with placebo when each was administered as part of standard immunosuppressive regimens containing either cyclosporine and corticosteroids (double-therapy trial, no US sites) or cyclosporine, corticosteroids, and azathioprine (triple-therapy trial, predominantly US sites) to prevent acute renal allograft rejection. ZENAPAX dosing was initiated within 24 hours pretransplant, with subsequent doses given every 14 days for a total of five doses.
The primary efficacy endpoint of both trials was the proportion of patients who developed a biopsy-proven acute rejection episode within the first 6 months following transplantation. As shown in Table 1, this incidence was significantly lower in the group treated with ZENAPAX in both the double-therapy and triple-therapy trials.
| Triple-therapy Regimen (cyclosporine, corticosteroids, and azathioprine) | Double-therapy Regimen (cyclosporine and corticosteroids) |
||||||
|---|---|---|---|---|---|---|---|
| Placebo | ZENAPAX | Placebo | ZENAPAX | ||||
| (N=134) | (N=126) | p-value | (N=134) | (N=141) | p-value | ||
| n.s. = not significant | |||||||
| Incidence of biopsy-proven acute rejection at 6 months | |||||||
| No. of patients | 47 (35%) | 28 (22%) | 0.03 | 63 (47%) | 39 (28%) | 0.001 | |
| Incidence of biopsy-proven acute rejection at 1 year | |||||||
| No. of patients | 51 (38%) | 35 (28%) | n.s. | 65 (49%) | 39 (28%) | <0.001 | |
| Graft survival at 3 years posttransplant | |||||||
| No. of patients with functioning graft | 111 (83%) | 106 (84%) | n.s. | 105 (78%) | 116 (82%) | n.s. | |
| Patient survival at 3 years posttransplant | |||||||
| No. of patients | 126 (94%) | 116 (92%) | n.s. | 118 (88%) | 135 (96%) | 0.02 | |
Treatment with ZENAPAX was associated with better patient survival up to 3 years posttransplant in the double-therapy study. No difference in patient survival was observed in the triple-therapy study between patients treated with ZENAPAX or placebo up to 3 years posttransplant. No difference was observed for graft survival between treatment groups in both studies at 3 years posttransplant.
The incidence of delayed graft function was not different between patients treated with placebo or ZENAPAX in either study. No difference in graft function was observed 1 year and 3 years posttransplant in either study between patients treated with placebo or ZENAPAX.
In a randomized, double-blind study to assess tolerability, pharmacokinetics, and drug interactions in renal allograft recipients, ZENAPAX (50 patients) or placebo (25 patients) was added to an immunosuppressive regimen of cyclosporine, mycophenolate mofetil, and corticosteroids. In this study, the addition of ZENAPAX did not result in an increased incidence of adverse events or a change in the types of adverse events reported. The incidence of the combined endpoint of biopsy-proven or clinically presumptive acute rejection was 20% (5 of 25 patients) in the placebo group and 12% (6 of 50 patients) in the ZENAPAX group. Although numerically lower, the difference in acute rejection was not significant. However, in a randomized, double-blind, placebo-controlled trial of ZENAPAX in cardiac transplant recipients (n=434) receiving concomitant cyclosporine, mycophenolate mofetil, and corticosteroids, mortality was increased in patients randomized to receive ZENAPAX compared with those randomized to receive placebo (see WARNINGS and ADVERSE REACTIONS).
INDICATION AND USAGE
ZENAPAX is indicated for the prophylaxis of acute organ rejection in patients receiving renal transplants. It is used as part of an immunosuppressive regimen that includes cyclosporine and corticosteroids.
The efficacy of ZENAPAX for the prophylaxis of acute rejection in recipients of other solid organ allografts has not been demonstrated.
CONTRAINDICATION
ZENAPAX is contraindicated in patients with known hypersensitivity to daclizumab or to any components of this product.
WARNINGS (see Boxed WARNING)
The use of ZENAPAX as part of an immunosuppressive regimen including cyclosporine, mycophenolate mofetil, and corticosteroids may be associated with an increase in mortality. In a randomized, double-blind, placebo-controlled trial of ZENAPAX for the prevention of allograft rejection in 434 cardiac transplant recipients receiving concomitant cyclosporine, mycophenolate mofetil, and corticosteroids, mortality at 6 and 12 months was increased in those patients receiving ZENAPAX compared to those receiving placebo (7% vs 5%, respectively at 6 months; 10% vs 6% respectively at 12 months). Some, but not all, of the increase in mortality appeared related to a higher incidence of severe infections. Concomitant use of anti-lymphocyte antibody therapy may also be a factor in some of the fatal infections.
ZENAPAX should be administered under qualified medical supervision. Patients should be informed of the potential benefits of therapy and the risks associated with administration of immunosuppressive therapy.
While the incidence of lymphoproliferative disorders and opportunistic infections in the limited clinical trial experience was no higher in patients treated with ZENAPAX compared with placebo-treated patients, patients on immunosuppressive therapy are at increased risk for developing lymphoproliferative disorders and opportunistic infections and should be monitored accordingly.
Hypersensitivity
Severe, acute (onset within 24 hours) hypersensitivity reactions including anaphylaxis have been observed both on initial exposure to ZENAPAX and following re-exposure. These reactions may include hypotension, bronchospasm, wheezing, laryngeal edema, pulmonary edema, cyanosis, hypoxia, respiratory arrest, cardiac arrhythmia, cardiac arrest, peripheral edema, loss of consciousness, fever, rash, urticaria, diaphoresis, pruritus, and/or injection site reactions. If a severe hypersensitivity reaction occurs, therapy with ZENAPAX should be permanently discontinued. Medications for the treatment of severe hypersensitivity reactions including anaphylaxis should be available for immediate use. Patients previously administered ZENAPAX should only be re-exposed to a subsequent course of therapy with caution. The potential risks of such re-administration, specifically those associated with immunosuppression, are not known.
PRECAUTIONS
General
It is not known whether ZENAPAX use will have a long-term effect on the ability of the immune system to respond to antigens first encountered during ZENAPAX-induced immunosuppression.
Re-administration of ZENAPAX after an initial course of therapy has not been studied in humans. The potential risks of such re-administration, specifically those associated with immunosuppression and/or the occurrence of anaphylaxis/anaphylactoid reactions, are not known.
Drug Interactions
The following medications have been administered with ZENAPAX in clinical trials in renal allograft patients with no incremental increase in adverse reactions: cyclosporine, mycophenolate mofetil, ganciclovir, acyclovir, azathioprine, and corticosteroids. Very limited experience exists in these patients with the use of ZENAPAX concomitantly with tacrolimus, muromonab-CD3, antithymocyte globulin, and anti-lymphocyte globulin.
In renal allograft recipients (n=50) treated with ZENAPAX and mycophenolate mofetil, no pharmacokinetic interaction between ZENAPAX and mycophenolic acid, the active metabolite of mycophenolate mofetil, was observed.
However, in a large clinical study in cardiac transplant recipients (n=434), the use of ZENAPAX as part of an immunosuppression regimen including cyclosporine, mycophenolate mofetil, and corticosteroids was associated with an increase in mortality, particularly in patients receiving concomitant anti-lymphocyte antibody therapy and in patients who developed severe infections (see WARNINGS and ADVERSE REACTIONS: Incidence of Infectious Episodes).
Carcinogenesis, Mutagenesis and Impairment of Fertility
Long-term studies to evaluate the carcinogenic potential of ZENAPAX have not been performed. ZENAPAX was not genotoxic in the Ames or the V79 chromosomal aberration assays, with or without metabolic activation. The effect of ZENAPAX on fertility is not known, because animal reproduction studies have not been conducted with ZENAPAX (see WARNINGS and ADVERSE REACTIONS).
Pregnancy
Pregnancy Category C: A preclinical developmental toxicity study with ZENAPAX has shown an increased risk of early prenatal loss in cynomolgus monkeys compared to placebo. However, the clinical experience of ZENAPAX exposed pregnancies is still limited. In general, IgG molecules are known to cross the placental barrier. ZENAPAX should not be used in pregnant women unless the potential benefit justifies the potential risk to the fetus. Women of childbearing potential should use effective contraception before beginning ZENAPAX therapy, during therapy, and for 4 months after completion of ZENAPAX therapy.
Nursing Mothers
It is not known whether ZENAPAX is excreted in human milk. However, in preclinical developmental toxicity studies with ZENAPAX, four out of seven lactating cynomolgus monkeys given a 5-10 fold multiple (10mg/kg) of the normal human dose were found to secrete very low levels of ZENAPAX (0.17 – 0.28% of maternal serum levels) in breast milk. Because many drugs are excreted in human milk, including human antibodies, and because of the potential for adverse reactions, a decision should be made to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
The safety and effectiveness of ZENAPAX have been established in pediatric patients from 11 months to 17 years of age. Use of ZENAPAX in this age group is supported by evidence from adequate and well-controlled studies of ZENAPAX in adults with additional pediatric pharmacokinetic data (see CLINICAL PHARMACOLOGY). Data from the pediatric pharmacokinetic study were also analyzed for efficacy, immunogenicity and safety. In an open-label study, 60 pediatric renal transplant recipients [median age of 10 years] received standard immunosuppressive agents in addition to a regimen of ZENAPAX administered at a dose of 1.0 mg/kg at intervals of 14 days for a total of 5 doses, starting immediately before transplantation. In this study, the combined incidence of biopsy-proven and clinically presumptive acute rejection at 1 year posttransplant was 17% (10/60). Patient and graft survival at 1 year posttransplant were 100% and 96.7%, respectively. The incidence of anti-daclizumab antibodies (34%) observed in the first 3 months posttransplant was higher than the incidence previously observed in adult patients (14%) (see ADVERSE REACTIONS: Immunogenicity).
The safety profile of ZENAPAX in pediatric transplant patients was shown to be comparable with that in adult transplant patients with the exception of the following adverse events, which occurred more frequently in pediatric patients (>15% difference in incidence): diarrhea, post-operative pain, fever, vomiting, aggravated hypertension, pruritus, and infections of the upper respiratory tract and urinary tract.
It is not known whether the immune response to vaccines, infection, and other antigenic stimuli administered or encountered during ZENAPAX therapy is impaired or whether such response will remain impaired after ZENAPAX therapy.
Also see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION.
Geriatric Use
Clinical studies of ZENAPAX did not include sufficient numbers of subjects age 65 and older to determine whether they respond differently from younger subjects. Caution must be used in giving immunosuppressive drugs to elderly patients.
ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug. Rates observed in clinical studies may not reflect those observed in clinical practice. Adverse reaction information obtained in clinical trials does, however, provide a basis for identifying adverse events that appear to be related to drug use and for approximating the rate of occurrence.
The safety of ZENAPAX was determined in four clinical studies of renal allograft rejection, three of which were randomized controlled clinical trials, in 629 patients receiving renal allografts of whom 336 received ZENAPAX and 293 received placebo. All patients received concomitant cyclosporine and corticosteroids. In these clinical trials, ZENAPAX did not appear to alter the pattern, frequency or severity of known major toxicities associated with the use of immunosuppressive drugs.
The use of ZENAPAX was associated with a higher incidence of mortality when compared to placebo in a large (n=434) randomized controlled study of patients receiving cardiac transplants (see WARNINGS and Incidence of Infectious Episodes).
Adverse events were reported by 95% of the patients in the placebo-treated group and 96% of the patients in the group treated with ZENAPAX. The proportion of patients prematurely withdrawn from the combined studies because of adverse events was 8.5% in the placebo-treated group and 8.6% in the group treated with ZENAPAX.
ZENAPAX did not increase the number of serious adverse events observed compared with placebo. The most frequently reported adverse events were gastrointestinal disorders, which were reported with equal frequency in ZENAPAX- (67%) and placebo-treated (68%) patient groups.
The incidence and types of adverse events were similar in both placebo-treated patients and patients treated with ZENAPAX. The following adverse events occurred in ≥5% of patients treated with ZENAPAX. These events included: Gastrointestinal System: constipation, nausea, diarrhea, vomiting, abdominal pain, pyrosis, dyspepsia, abdominal distention, epigastric pain not food-related; Metabolic and Nutritional: edema extremities, edema; Central and Peripheral Nervous System: tremor, headache, dizziness; Urinary System: oliguria, dysuria, renal tubular necrosis; Body as a Whole - General: posttraumatic pain, chest pain, fever, pain, fatigue; Autonomic Nervous System: hypertension, hypotension, aggravated hypertension; Respiratory System: dyspnea, pulmonary edema, coughing; Skin and Appendages: impaired wound healing without infection, acne; Psychiatric: insomnia; Musculoskeletal System: musculoskeletal pain, back pain; Heart Rate and Rhythm: tachycardia; Vascular Extracardiac: thrombosis; Platelet, Bleeding and Clotting Disorders: bleeding; Hemic and Lymphatic: lymphocele.
The following adverse events occurred in <5% and ≥2% of patients treated with ZENAPAX. These included: Gastrointestinal System: flatulence, gastritis, hemorrhoids; Metabolic and Nutritional: fluid overload, diabetes mellitus, dehydration; Urinary System: renal damage, hydronephrosis, urinary tract bleeding, urinary tract disorder, renal insufficiency; Body as a Whole - General: shivering, generalized weakness; Central and Peripheral Nervous System: urinary retention, leg cramps, prickly sensation; Respiratory System: atelectasis, congestion, pharyngitis, rhinitis, hypoxia, rales, abnormal breath sounds, pleural effusion; Skin and Appendages: pruritus, hirsutism, rash, night sweats, increased sweating; Psychiatric: depression, anxiety; Musculoskeletal System: arthralgia, myalgia; Vision: vision blurred; Application Site: application site reaction.
Incidence of Malignancies
One and 3 years posttransplant, the incidence of malignancies was 2.7% and 7.8%, respectively, in the placebo group compared with 1.5% and 6.4%, respectively, in the ZENAPAX group. Addition of ZENAPAX did not increase the number of posttransplant lymphomas up to 3 years posttransplant. Lymphomas occurred at a frequency of ≤1.5% in both placebo-treated and ZENAPAX-treated groups.
Hyperglycemia
No differences in abnormal hematologic or chemical laboratory test results were seen between groups treated with placebo or ZENAPAX with the exception of fasting blood glucose. Fasting blood glucose was measured in a small number of patients treated with placebo or ZENAPAX. A total of 16% (10 of 64 patients) of placebo-treated and 32% (28 of 88 patients) of patients treated with ZENAPAX had high fasting blood glucose values. Most of these high values occurred either on the first day posttransplant when patients received high doses of corticosteroids or in patients with diabetes.
Incidence of Infectious Episodes
The overall incidence of infectious episodes, including viral infections, fungal infections, bacteremia and septicemia, and pneumonia, was not higher in patients treated with ZENAPAX than in placebo-treated patients in trials of renal transplantation. In a large randomized study of ZENAPAX used for the prevention of allograft rejection in patients receiving cardiac allografts, more patients receiving ZENAPAX experienced severe or fatal infections after 12 months of therapy when compared to those receiving placebo (10% vs 7%, respectively). The risks of infection or death may be increased in patients receiving concomitant anti-lymphocyte antibody therapy (see WARNINGS).
The types of infections reported in trials of renal transplantation were similar in both the ZENAPAX-treated and the placebo-treated groups. Cytomegalovirus infection was reported in 16% of the patients in the placebo group and 13% of the patients in the ZENAPAX group. One exception was cellulitis and wound infections, which occurred in 4.1% of placebo-treated patients and 8.4% of patients treated with ZENAPAX. At 1 year posttransplant, 7 placebo patients and 1 patient treated with ZENAPAX had died of an infection. At 3 years posttransplant, 8 placebo patients and 4 patients treated with ZENAPAX had died of infection.
Immunogenicity
Low titers of anti-idiotype antibodies to daclizumab were detected in the adult patients treated with ZENAPAX with an overall incidence of 14%. The incidence of anti-daclizumab antibodies observed in the pediatric patients was 34%. No antibodies that affected efficacy, safety, serum daclizumab levels or any other clinically relevant parameter examined were detected. The data reflect the percentage of patients whose test results were considered positive for antibodies to daclizumab in an ELISA assay and are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in the assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of the incidence of antibodies to daclizumab with the incidence of antibodies to other products may be misleading.
Post-Marketing Experience
The following adverse reactions have been identified and reported during post-approval use of ZENAPAX (daclizumab). Because the reports of these reactions are voluntary and the population is of uncertain size, it is not always possible to reliably estimate the frequency of the reaction or establish a causal relationship to drug exposure.
Severe acute hypersensitivity reactions including anaphylaxis characterized by hypotension, bronchospasm, wheezing, laryngeal edema, pulmonary edema, cyanosis, hypoxia, respiratory arrest, cardiac arrhythmia, cardiac arrest, peripheral edema, loss of consciousness, fever, rash, urticaria, diaphoresis, pruritus, and/or injection site reactions, as well as cytokine release syndrome, have been reported during post-marketing experience with ZENAPAX. The relationship between these reactions and the development of antibodies to ZENAPAX is unknown.
OVERDOSAGE
There have not been any reports of overdoses with ZENAPAX. A maximum tolerated dose has not been determined in patients. A dose of 1.5 mg/kg has been administered to bone marrow transplant recipients without any associated adverse events.
DOSAGE AND ADMINISTRATION
ZENAPAX is used as part of an immunosuppressive regimen that includes cyclosporine and corticosteroids. The recommended dose for ZENAPAX in adult and pediatric patients is 1.0 mg/kg (see PRECAUTIONS: Pediatric Use). The calculated volume of ZENAPAX should be mixed with 50 mL of sterile 0.9% sodium chloride solution and administered via a peripheral or central vein over a 15-minute period.
Based on the clinical trials, the standard course of ZENAPAX therapy is five doses. The first dose should be given no more than 24 hours before transplantation. The four remaining doses should be given at intervals of 14 days.
No dosage adjustment is necessary for patients with severe renal impairment. No dosage adjustments based on other identified covariates (age, gender, proteinuria, race) are required for renal allograft patients. No data are available for administration in patients with severe hepatic impairment.
Instructions for Administration
- ZENAPAX IS NOT FOR DIRECT INJECTION. The calculated volume should be diluted in 50 mL of sterile 0.9% sodium chloride solution before intravenous administration to patients. When mixing the solution, gently invert the bag in order to avoid foaming; DO NOT SHAKE.
- Parenteral drug products should be inspected visually for particulate matter and discoloration before administration. If particulate matter is present or the solution colored, do not use.
- Care must be taken to assure sterility of the prepared solution, since the drug product does not contain any antimicrobial preservative or bacteriostatic agents.
- ZENAPAX is a colorless solution provided as a single-use vial; any unused portion of the drug should be discarded.
- Once the infusion is prepared, it should be administered intravenously within 4 hours. If it must be held longer, it should be refrigerated between 2° to 8°C (36° to 46°F) for up to 24 hours. After 24 hours, the prepared solution should be discarded.
- No incompatibility between ZENAPAX and polyvinyl chloride or polyethylene bags or infusion sets has been observed. No data are available concerning the incompatibility of ZENAPAX with other drug substances. Other drug substances should not be added or infused simultaneously through the same intravenous line.
- ZENAPAX should only be administered by healthcare personnel trained in the administration of the drug who have available adequate laboratory and supportive medical resources.
HOW SUPPLIED
ZENAPAX is supplied in single-use glass vials. Each vial contains 25 mg of daclizumab in 5 mL of solution (NDC 0004-0501-09). Vials should be stored between the temperatures of 2° to 8°C (36° to 46°F); do not shake or freeze. Protect undiluted solution against direct light. Diluted medication is stable for 24 hours at 4°C or for 4 hours at room temperature.
Roche Pharmaceuticals
Hoffmann-La Roche Inc.
340 Kingsland Street
Nutley, NJ07110–1199
US Govt. Lic. No. 0136
3609650 USA
27899043
Revised: September 2005
Copyright © 1999-2005 by Hoffmann-La Roche Inc. All rights reserved.
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
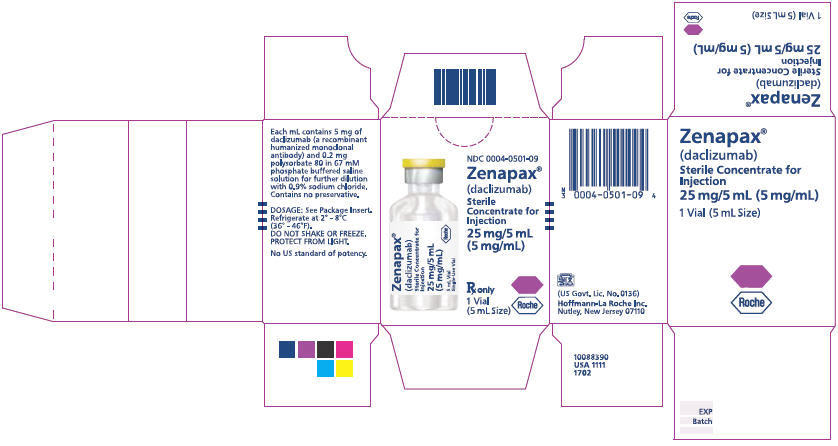
PRINCIPAL DISPLAY PANEL - 25 mg/5 mL Vial Label
NDC 0004-0501-09
Zenapax®
(daclizumab)
Sterile
Concentrate for
Injection
25 mg/5 mL
(5 mg/mL)
Rx only
1 Vial
(5 mL Size)
Roche
| ZENAPAX
daclizumab injection, solution, concentrate |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA103749 | 12/10/1997 | |
| Labeler - Genentech, Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| Hoffmann-La Roche Inc | 002191211 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| F. Hoffmann-La Roche Ltd | 482242971 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Operations |
| F. Hoffmann-La Roche Ltd | 485244961 | MANUFACTURE | |